DOI:
10.1039/D5RA01982F
(Paper)
RSC Adv., 2025,
15, 18358-18371
Square planar vs. pyramidal copper(II)-complexes with benzylal vs. naphthylal-based Schiff bases†‡
Received
20th March 2025
, Accepted 6th May 2025
First published on 5th June 2025
Abstract
Reaction of copper(II) acetate with (E)-2-(((2-benzoylphenyl)imino)methyl)phenol (HL1), (E)-2-(((2-benzoyl-5-chlorophenyl)imino)methyl)phenol (HL2) and (E)-1-(((2-benzoyl-5-chlorophenyl)imino)methyl)-naphthalen-2-ol (HL3) provided bis[(E)-2-(((2-benzoylphenyl)imino)methyl)phenolato-κ2N,O]copper(II) (1), bis[(E)-2-(((2-benzoyl-5-chlorophenyl)imino)methyl)phenolato-κ2N,O]copper(II) (2) and bis[(E)-1-(((2-benzoyl-5-chlorophenyl)imino)methyl)naphthalen-2-olato-κ2N,O]copper(II) (3). The molecular structure determinations revealed that the ligands existed as a usual (imine)N⋯H–O(phenol) (enolimine-form) in the solid state, which was further evidenced using 1H NMR studies in solution (CDCl3 and DMSO-d6). Unlike HL1 and HL2, two symmetry-independent molecules (A and B) were present in the unit cell of the HL3 crystal. The molecular structures showed that the two N,O-chelating ligands coordinate to the copper(II) ion through a square-planar (1), a distorted square-planar (2) and a square-pyramidal geometry (3). Each asymmetric unit of the crystal structure contained one-half of the molecule for 1, a single molecule for 2 and two symmetry-independent molecules for 3 (molecules A and B). Thermal investigations using DSC demonstrated an irreversible phase transition from a crystalline solid to an isotropic liquid (m.p.). Cyclic voltammogram results proved two quasi-reversible one-electron charge transfer process for 1 and 3 in DMF at 25 °C. Complexes 1 and 2 exhibited low and significant antibacterial activity, respectively, against E. coli and S. aureus, while 3 was completely inactive. Among the ligands, only HL2 exhibited medium activity against microorganisms. The electronic and molecular structures correlated well with the computational modeling performed using DFT/TD-DFT calculations.
Introduction
Copper is one of the most interesting and useful bio-metals, mainly because of its biological functions and potential synergistic activities with medicines.1–3 Moreover, it plays a vital role in cell physiology, acting as a catalyst in the redox chemistry of mitochondrial respiration and free radical scavenging.4 Alternatively, Schiff base ligands are considered some of the most versatile and useful bioactive compounds because of their capability to interact with a variety of metal ions.5,6 The presence of different substituents in Schiff bases not only affects their physicochemical properties but also strongly influences their coordination behavior and geometry of the resulting metal complexes.7–9 In this regard, benzophenone and its derivatives play crucial roles in organic chemistry, serving as intermediates in the synthesis of various pharmaceutical compounds, such as benzodiazepines10 and diazocines,11 and in photochemical reactions, such as photo-Fries and photo-Claisen rearrangements.12 These compounds are also significant for their biological and physicochemical characteristics, including electrochemical, spectroscopical, metal complexation and crystallographic behaviors.13–16 A key biological property of benzophenone/derivatives is their ability to absorb a wide range of UV radiation (200–350 nm). Owing to this characteristic, benzophenone derivatives, such as 2-amino-benzophenone, 2-hydroxy-4-methoxybenzophenone and 2-hydroxy-4-methoxy-4′-methoxy-benzophenone, are utilized as raw materials in the synthesis of sunscreen creams.17 Furthermore, 2-amino-benzophenone and its derivatives are important compounds in heterocyclic synthesis and medicines.18–21 The redox behaviors of these compounds in non-aqueous solvents are well-established.22–24
Benzophenone-derived Schiff bases have garnered significant attention in the synthesis of metal complexes owing to their widespread applications in both commercial and pharmaceutical sectors. Furthermore, they have been investigated as potent inhibitors of HIV, farnesyltransferase and reverse transcriptase.25–28 Investigations on structural properties of copper(II) and palladium(II) complexes with 2-amino-5-chlorobenzophenone and 2-(2-hydroxybenzylidene)amino-5-chlorobenzophenone-S-methyl-thiosemicarbazone have been reported.29 Indeed, some benzophenone-based Schiff bases, such as 5-chloro-2-(((2-hydroxy-phenyl)methylene)amino-phenyl)phenyl-methanone, [(Z)-(5-chloro-2((3,5-dichloro-2-hydroxybenzylidene)amino)phenyl)(phenyl)methanone and (Z)-(2((5-bromo-2-hydroxybenzylidene)amino-5-chlorophenyl)(phenyl)methanone)] and their copper(II) complexes, have been synthesized and structurally elucidated.30,31
The halogen atom (chlorine) was incorporated into the ligand framework to fine-tune the electronic and steric environment of the resulting complexes' geometry. Chlorine exerts an electron-withdrawing inductive effect, which can influence metal–ligand coordination and potentially alter the redox characteristics of the metal center. Furthermore, halogen substitution may induce subtle changes in the molecular geometry by affecting the spatial orientation of the ligands around the metal, thereby offering a means to modulate the structural and physicochemical properties of the complexes. The organic chlorine atom may also be involved in non-covalent C–H⋯Cl or Cl⋯Cl interactions in the crystal packing.
Building upon ongoing research in metal(II)-Schiff base complexes, herein, we report the syntheses and spectroscopic characterizations of Schiff base ligands (HL1, HL2 and HL3) and their copper complexes (1, 2 and 3), respectively. The molecular structures, redox potential, thermal and antibacterial activities of the Schiff bases and complexes were discussed. Furthermore, a thorough computational modeling using DFT/TD-DFT was applied to rationalise the experimental results.
Results and discussion
Treatment of the Schiff base ligands (HL1, HL2 and HL3) with copper(II) acetate provides complexes 1, 2 and 3, respectively (Scheme 1). We note that the nomenclature name for the three ligands are (E)-(2-((2-hydroxybenzylidene)amino)phenyl)(phenyl)-methanone (HL1), (E)-(5-chloro-2-((2-hydroxybenzylidene)amino)phenyl)(phenyl)-methanone (HL2) and (E)-(5-chloro-2-(((2-hydroxynaphthalen-1-yl)methylene)amino)phenyl)(phenyl)methanone (HL3). The vibrational spectra show two strong bands at the range of 1666–1659, 1622–1591 and 1579–1529 cm−1 for νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, νCN and νCC, respectively (Fig. S1‡). The very low molar conductance, Λm = 2.02 (1), 2.37 (2) and 1.98 S m2 mol−1 (3) suggests a non-electrolyte profile of the complexes in DMF at 25 °C. The EI-mass spectra show the parent ion peaks at m/z = 663 (1), 733 (2) and 828 (3) along with various ion peaks for the fragmented species of Schiff bases and complexes (Fig. S2‡).
O, νCN and νCC, respectively (Fig. S1‡). The very low molar conductance, Λm = 2.02 (1), 2.37 (2) and 1.98 S m2 mol−1 (3) suggests a non-electrolyte profile of the complexes in DMF at 25 °C. The EI-mass spectra show the parent ion peaks at m/z = 663 (1), 733 (2) and 828 (3) along with various ion peaks for the fragmented species of Schiff bases and complexes (Fig. S2‡).
 |
| | Scheme 1 Synthetic route for Schiff bases (HL1–HL3) and their complexes (1–3). | |
1H/13C NMR studies
1H NMR spectra (DMSO-d6) show a singlet at δ 8.90 (HL1), 8.92 (HL2) and 9.68 ppm (HL3) for the imine-proton (CHN) (Fig. S3a‡). The aromatic protons displayed several peaks in the range of δ 6.78–7.74 (HL1), 6.78–7.64 (HL2) and 6.98–8.50 ppm (HL3), which were fully assigned (Fig. S3a,‡ see data in Experimental section). The aromatic proton labeled as H3 shows a triplet at δ 7.34 ppm (JHH = 7.2 Hz) in HL1, a doublet of a triplet at δ 7.35 ppm (JHH =8.0, 7.6, 1.2 Hz) in HL2 and a multiplet at relatively downfield at δ 7.76–7.83 ppm in HL3. The phenolic-proton (O–H) shows a broad peak at the most downfield at δ 11.82 (HL1), 11.60 (HL2) and 14.05 ppm (HL3). In the protic solvent (CDCl3, Fig. S3b‡), the CHN peak shifted to a relatively downfield at δ 9.93 ppm (δ 8.92 ppm in DMSO-d6), while the O–H peak shifts to high field at δ 11.04 ppm (δ 11.60 ppm in DMSO-d6) in comparison with the aprotic solvent (DMSO-d6) for HL2. 1H NMR studies further confirmed the absence of any keto–enol tautomerism in solution as observed in the related Schiff base ligands.32–34
13C NMR spectra (Fig. S3c‡) feature resonances in the range of δ 116.95–137.24 (HL1), 116.98–136.76 (HL2) and 109.79–136.84 ppm (HL3), corresponding to the aromatic carbon atoms from benzoyl and benzylal or naphthylal moieties. The imine carbon (HCN) appears at δ 160.33 (HL1), 160.28 (HL2) and 159.96 ppm (HL3), consistent with a de-shielded sp2-hybridized carbon in a conjugated system. The phenolic carbon bearing the OH group resonates at δ 164.65 (HL1), 164.97 (HL2) and 165.38 ppm (HL3). Finally, the carbonyl carbon (CO) of the benzoyl fragment is observed at the most downfield at δ 197.17 (HL1), 195.69 (HL2) and 195.66 ppm (HL3), confirming the presence of ketone functionality. The 1H/13C NMR spectral results collectively affirm the formation of conjugated Schiff base ligands (Scheme 1).
Experimental and simulated electronic spectra
Electronic spectra (UV-vis.) show several strong bands or shoulders below ca. 400 nm (for HL1 and HL2) and ca. 500 nm (for HL3), which are attributed to intra-ligand n → π*/π → π* (LL) transitions (Fig. S6a‡). These bands shifted to higher wavelengths (red-shift) upon coordination with the copper(II) ion (Fig. 1, S6b‡). The spectra of the complexes feature a medium broad band at 400–500 nm (1 or 2) and 500–550 nm (3), which are assigned to the metal–ligand charge transfer (MLCT) transitions. The spectra further show a weak broadband at 550–1000 nm, due to combinations of various d–d electron transitions in metal, a classic band for the d9 copper(II) core electrons (Fig. 1 and S6b;‡ inset).33–38 To justify the experimental UV-vis. spectra, we calculated the spectra by DFT/TD-DFT with different arrangements of the functionals (e.g., B3LYP, cam-B3LYP, M06 and M06-2X) and the basis sets (e.g., 6-31G(d), SDD and SVP) with PCM in chloroform, respectively (Fig. 1 and S7–S9‡). The spectra thus obtained are almost identical in pattern with a small change in the positions of the bands, and match well with the experimental spectra (Fig. 1 and S7–S9‡). The relatively best-matching spectra are found for cam-B3LYP/SDD (Fig. 1, see Experimental section for details).
 |
| | Fig. 1 Experimental and simulated UV-vis. spectra for 1 (ca. 0.02 mM), 2 (ca. 0.05 mM) and 3 (ca. 0.04 mM) in chloroform at 25 °C. Simulated spectra were obtained using the cam-B3LYP/SDD method with PCM in chloroform (spectra at visible range are displayed in the inset). | |
X-ray crystal structures
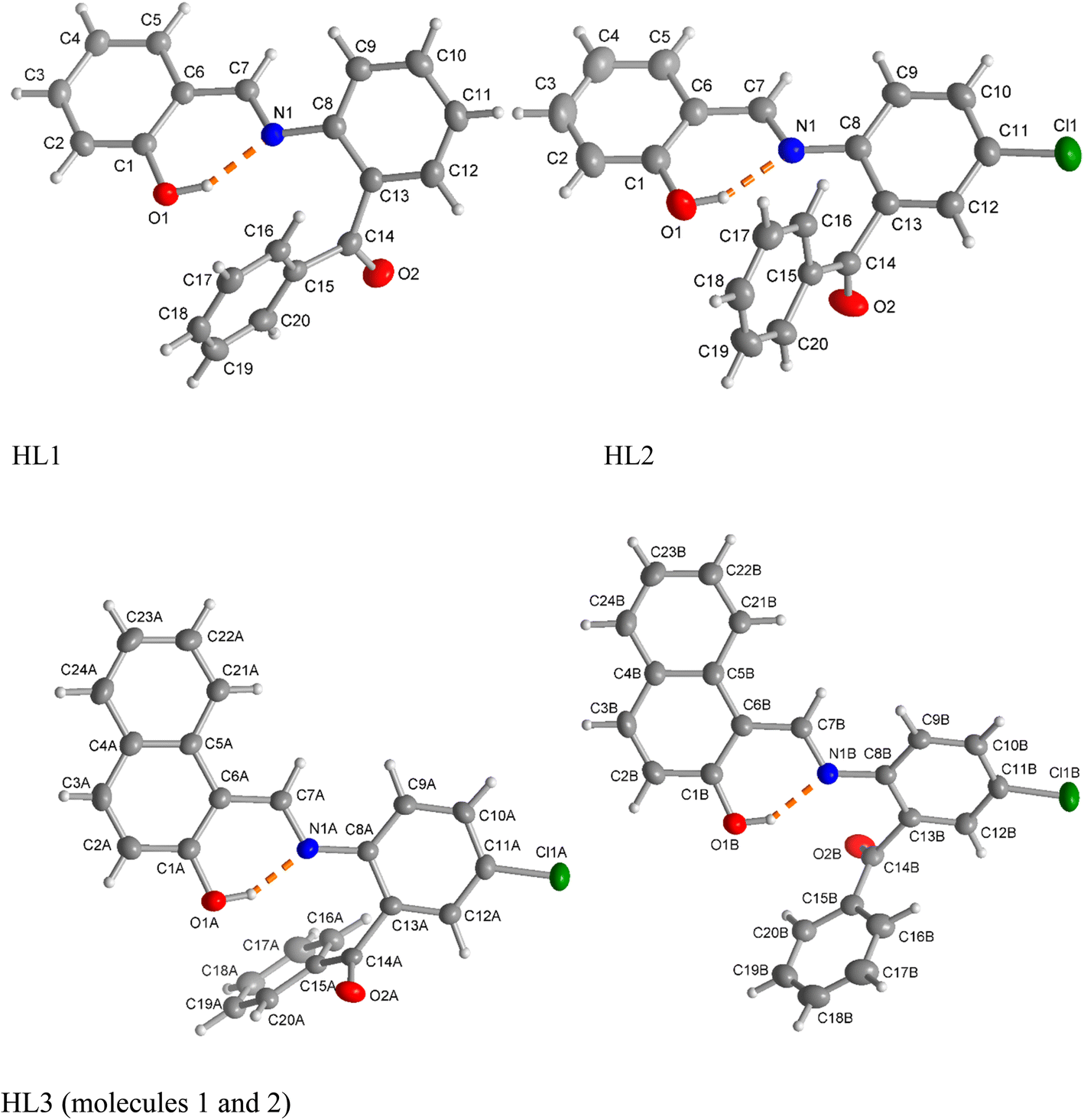
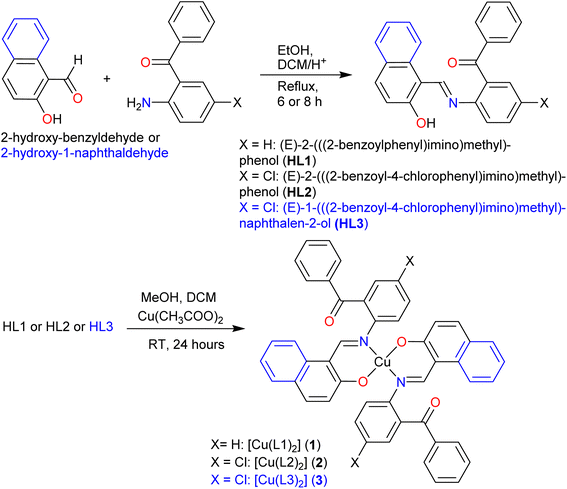
X-ray molecular structure (Fig. 2) determination shows the HL1 ligand to crystallize in the orthorhombic space group Pbcn, HL2 in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and HL3 in the orthorhombic space group P212121. The structures further reveal that all ligands exist as a usual (imine)N⋯H–O(phenol) (i.e., enol–amine form) via intramolecular hydrogen bonding in the solid state, in parallel to 1H NMR studies in solution. Unlike HL1 and HL2, there are two symmetry-independent molecules (A and B) in a unit cell of the crystal structure of HL3. The bond lengths and angles (Table 1) are comparable to those of the salicylaldimine or naphthalaldimine Schiff bases.32,33,38
and HL3 in the orthorhombic space group P212121. The structures further reveal that all ligands exist as a usual (imine)N⋯H–O(phenol) (i.e., enol–amine form) via intramolecular hydrogen bonding in the solid state, in parallel to 1H NMR studies in solution. Unlike HL1 and HL2, there are two symmetry-independent molecules (A and B) in a unit cell of the crystal structure of HL3. The bond lengths and angles (Table 1) are comparable to those of the salicylaldimine or naphthalaldimine Schiff bases.32,33,38
 |
| | Fig. 2 Molecular structures of the Schiff bases HL1, HL2 and HL3 (50% thermal ellipsoids, H atoms with arbitrary radii). The intramolecular O–H⋯N hydrogen bond is depicted as an orange dashed line. HL3 contains two symmetry-independent molecules (labelled as A and B) in the unit cell. For clarity, these two molecules are presented separately. | |
Table 1 Selected bond lengths (Å) and angles (°) in HL1, HL2 and HL3
| |
HL1 |
HL2 |
HL3 (A) |
HL3 (B) |
| O1–H1 |
0.925 (15) |
0.89 (3) |
0.90 (5) |
0.88 (5) |
| O1–C1 |
1.3516 (12) |
1.3533 (19) |
1.349 (3) |
1.336 (3) |
| O2–C14 |
1.2195 (12) |
1.2131 (18) |
1.221 (3) |
1.218 (3) |
| N1–C7 |
1.2871 (13) |
1.2855 (18) |
1.295 (3) |
1.291 (3) |
| N1–C8 |
1.4166 (12) |
1.4124 (18) |
1.408 (3) |
1.411 (3) |
| Cl1–C11 |
|
1.7371 (15) |
1.746 (2) |
1.741 (2) |
| C1–O1–H1 |
106.0 (9) |
107.5 (17) |
105 (3) |
108 (3) |
| C7–N1–C8 |
119.18 (8) |
122.11 (12) |
122.13 (19) |
121.31 (19) |
| O2–C14–C13 |
119.59 (9) |
118.50 (13) |
119.3 (2) |
120.1 (2) |
| O2–C14–C15 |
121.01 (9) |
122.12 (13) |
121.1 (2) |
122.2 (2) |
| C10–C11–Cl1 |
|
120.01 (11) |
118.87 (18) |
120.14 (18) |
| C12–C11–Cl1 |
|
119.00 (12) |
119.41 (18) |
117.75 (19) |
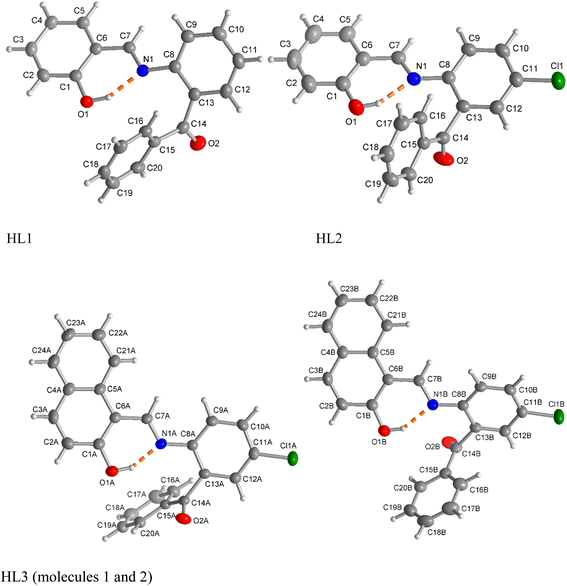
The complexes crystallize in the triclinic space group P for 1 and 3, and in the monoclinic space group P21/n for 2. An asymmetric unit of the crystal structure contains one half of the molecule for 1, a single molecule for 2 and two symmetry-independent molecules for 3 (Fig. 3). The molecular structures reveal that two N,O-chelating ligands coordinate to the copper(II) ion in a symmetry-imposed perfect square-planar geometry for 1 and a distorted square-planar geometry for 2 with a dihedral angle (θ/°) value of 24.2° between the two chelate rings (i.e., N1–Cu1–O1 and N2–Cu1–O3) (Fig. 3a and b). In 3, one of the benzophenone-oxygen is involved in axial coordination with the metal ion with a Jahn–Teller-distorted bond length of Cu1A⋯O2A = 2.726(2) Å in molecule A and Cu1B⋯O2B = 2.593(2) Å in molecule B, resulting in a square-pyramidal geometry (Fig. 3c). Molecules A and B in 3 mainly differ in this long Cu1–O2 bond and in the angle between the metal-chelate ring planes, which is 26.4° in molecule A and 20.6° in molecule B. An overlay of molecules A and B is shown in Fig. 4. The Cu–O/Cu–N and C–Cl bond lengths and N–Cu–O bond angles (Table 2) are in the range as expected from Cu(II)-salicylaldimine/naphthalaldimine analogues.34–42 The bond lengths and angles of the calculated structures (Fig. S4‡) were also comparable to the experimental data (Table 2). In addition to these differences, the two molecules A and B in 3 behave as enantiomeric mirror images. Because of the centrosymmetric space group P for 3, the mirror image molecules A and B in Fig. 3c are supplemented by inversion symmetric molecules A′ to A and B′ to B such that A′ and B (or A and B′) become almost superimposable (Fig. 4). The calculated structures for such close-to-enantiomeric molecules A and B in 3 are expected to be equi-energetic (Fig. S4‡).
 |
| | Fig. 3 Molecular structures of (a) 1, (b) 2 and (c) 3. Compound 3 contains two symmetry-independent molecules (labelled as A and B) in the unit cell. For clarity, these two molecules, which behave as left- and right-hand mirror images, are shown separately. Symmetry code for 1 (i) −x + 1, −y + 1, −z + 1. For fully atom-labeled structures, see Fig. S5 in the ESI.‡ | |
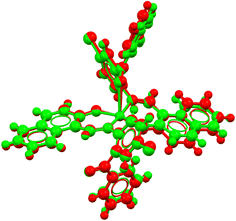 |
| | Fig. 4 Overlay of molecules A′ (red) and B (green) in 3. | |
Table 2 Selected bond lengths (Å) and angles (°) in 1, 2 and 3
| |
Complex 1 |
Complex 2 |
Complex 3 |
| X-ray structure |
Opt. structurea |
X-ray structure |
Opt. structureb |
Molecule A (Cu1A) |
Molecule B (Cu1B) |
| X-ray structure |
Opt. structurea |
X-ray structure |
| Optimized structures at B3LYP/SDD. B3LYP/6-31G(d). θ/° = dihedral angle between the chelate ring planes. |
| Cu1–O1 |
1.8776 (13) |
1.9128 |
1.8973 (18) |
1.9002 |
1.9068 (10) |
1.9456 |
1.9118 (10) |
| Cu1–O3 |
|
|
1.9072 (18) |
1.9137 |
1.9022 (11) |
1.9325 |
1.9036 (11) |
| Cu1–N1 |
2.0002 (15) |
2.0188 |
1.983 (2) |
1.9841 |
1.9562 (11) |
1.9995 |
1.9627 (11) |
| Cu1–N2 |
|
|
1.980 (2) |
1.9927 |
1.9561 (11) |
1.9878 |
1.9574 (11) |
| Cl1–C11 |
|
|
1.744 (3) |
1.7592 |
1.7442(14) |
1.8228 |
1.7455 (14) |
| Cl2–C31/35 |
|
|
1.749 (3) |
1.7563 |
1.7401 (15) |
1.8253 |
1.7383 (14) |
| O1–Cu1–O1i/O3 |
180.00 (6) |
179.98 |
161.75 (9) |
145.72 |
160.34 (5) |
153.52 |
164.94 (5) |
| O1–Cu1–N1 |
91.91 (6) |
91.04 |
91.98 (8) |
94.04 |
90.32 (5) |
89.41 |
89.59 (5) |
| O1–Cu1–N1i/N2 |
88.09 (6) |
88.96 |
89.38 (8) |
90.63 |
91.14 (4) |
93.19 |
91.38 (4) |
| O1i/O3–Cu1–N1 |
88.09 (6) |
88.96 |
91.32 (8) |
92.81 |
90.59 (5) |
90.61 |
89.16 (5) |
| O1i–Cu1–N1i/O3–Cu1–N2 |
91.91 (6) |
91.04 |
92.51 (8) |
92.03 |
92.24 (5) |
91.45 |
92.04 (5) |
| N1–Cu1–N1i/N2 |
180.0 |
179.99 |
163.53 (9) |
163.78 |
167.37 (5) |
169.76 |
171.65 (5) |
| θ/°c |
0 |
0 |
24.2 |
37.3 |
24.6 |
28.29 |
20.6 |
Hirshfeld surface analysis
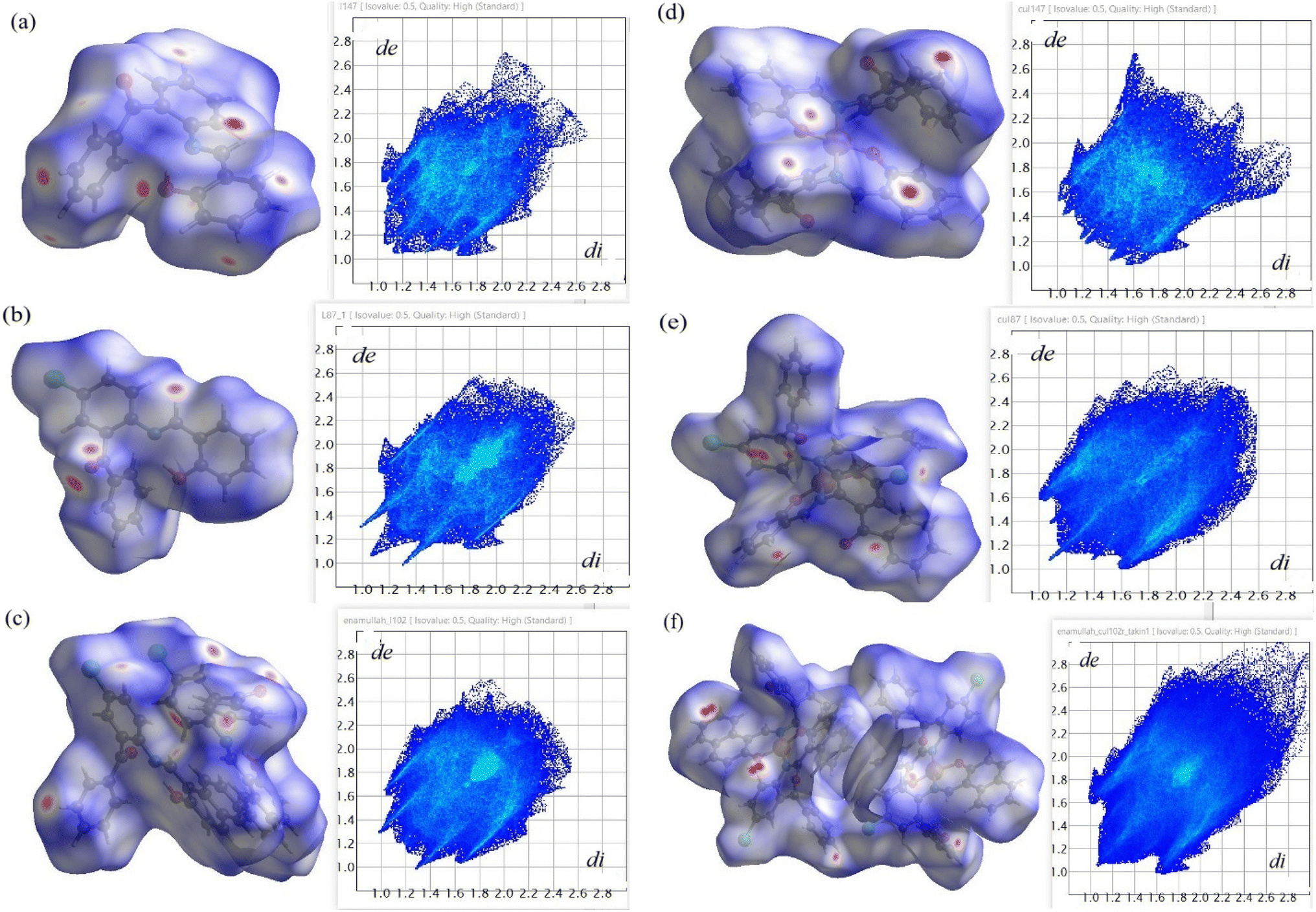
The analysis of the Hirshfeld surface provides a quantitative assessment of the intermolecular contacts contributing to crystal packing in the complexes. The 2D fingerprint plot of the Hirshfeld surface estimated by CrystalExplorer43,44 shows all possible closest contacts present in the compounds (Fig. 5, right: for details see Fig. 6 and S10‡). The highest contribution to crystal packing arises from H⋯H contacts (35.2–51.7% of the total surface), which features the dominance of van der Waals forces between the hydrogen atoms in organizing the crystal structure. The second-highest contribution comes from C⋯H contacts (17.7–33.0%), an indicator of C–H⋯π and C–H⋯C contacts. The O⋯H contacts (5.8–15.6%) originate from intermolecular C–H⋯O contacts. Finally, the Cl⋯H contacts (11.4–13.1%) indicate the influence of halogen bonding on the molecular assembly. All these interactions play an important role in stabilizing the crystal lattice and in enhancing the directional nature of crystal packing. The Hirshfeld surface mapped with the d-norm property displays the red- and blue-spots, indicating the nearest and distant contacts to the closest atom's interior and exterior to the surface, respectively (Fig. 5, left). The breakdown of all possible contributions to the Hirshfeld surface for different intermolecular contacts is illustrated in Fig. 6 (Fig. S10‡).
 |
| | Fig. 5 Hirshfeld surface mapped with the d-norm property (left: red-spots indicate the nearest contact and blue ones represent the farthest contacts) and 2D fingerprint plots (right) showing an overlay of all the possible close contacts in (a) HL1, (b) HL2, (c) HL3, (d) 1, (e) 2 and (f) 3. The abscissa (di) and ordinate (de) represent the distances from the surface to the closest atom's interior and exterior, respectively. | |
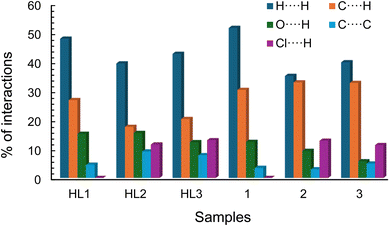 |
| | Fig. 6 Relative contributions of different intermolecular contacts (%) present in the compounds to the Hirshfeld surface area. | |
Phase transition and thermal stability
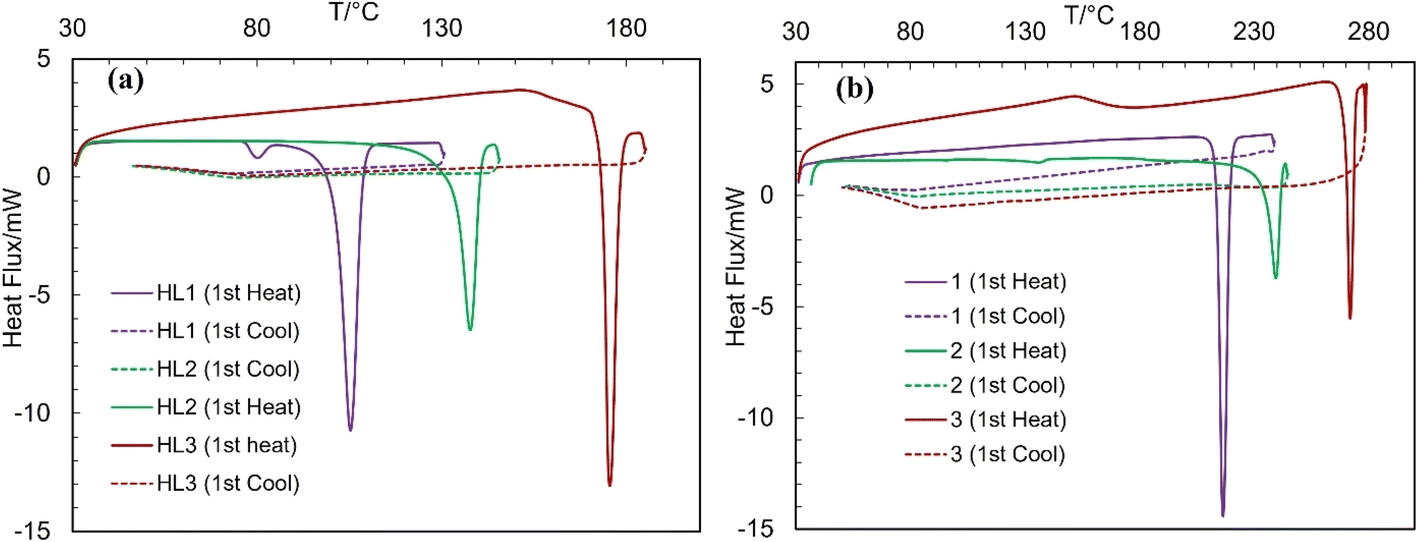
The differential scanning calorimetry (DSC) curves of the Schiff bases and complexes are presented in Fig. 7 (Table 3). The DSC curves feature an endothermic peak with substantial amount of enthalpy change (ΔH/kJ mol−1), corresponding to a simple phase transition from crystalline solid (Cr) to isotropic liquid (I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m.p.), and subsequently confirm the thermal stability of the compounds, as reported for the related Schiff bases and metal(II)-Schiff base complexes.34–40 The cooling curves show no corresponding peak in the reverse direction, indicating that the phase transition is irreversible. The repeated heating curves for the same sample (probe) during the second cycle also show no peaks. The phase transition temperatures for HL3 (ca. 176 °C) and 3 (ca. 272 °C) are considerably higher than those of HL2 (ca. 137 °C) and 2 (ca. 239 °C), whereas HL1 (ca. 105 °C) and 1 (ca. 217 °C) are the least, parallel to higher thermal stability as per their high molecular weight.
:m.p.), and subsequently confirm the thermal stability of the compounds, as reported for the related Schiff bases and metal(II)-Schiff base complexes.34–40 The cooling curves show no corresponding peak in the reverse direction, indicating that the phase transition is irreversible. The repeated heating curves for the same sample (probe) during the second cycle also show no peaks. The phase transition temperatures for HL3 (ca. 176 °C) and 3 (ca. 272 °C) are considerably higher than those of HL2 (ca. 137 °C) and 2 (ca. 239 °C), whereas HL1 (ca. 105 °C) and 1 (ca. 217 °C) are the least, parallel to higher thermal stability as per their high molecular weight.
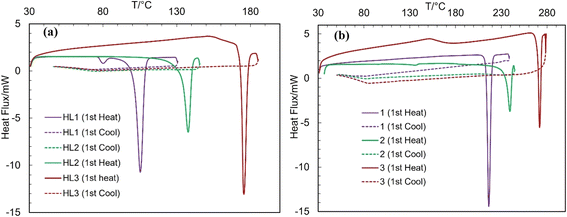 |
| | Fig. 7 DSC curves of (a) Schiff bases (HL1, HL2, and HL3) and (b) complexes (1, 2 and 3). | |
Table 3 Phase transition temperature (T/°C) and enthalpy change (ΔH/kJ mol−1) for the complexes
| Compounds |
Cycles |
T (°C)/ΔH (kJ mol−1) (heating curve) |
| HL1 |
1st |
80/−0.84 and 105/−22.43 (Cr ⇆ I) |
| HL2 |
1st |
138/−27.39 (Cr ⇆ I) |
| HL3 |
1st |
176/−32.73 (Cr ⇆ I) |
| 1 |
1st |
217/−48.61 (Cr ⇆ I) |
| 2 |
1st |
239/−41.29 (Cr ⇆ I) |
| 3 |
1st |
273/−34.18 (Cr ⇆ I) |
Cyclic voltammetry
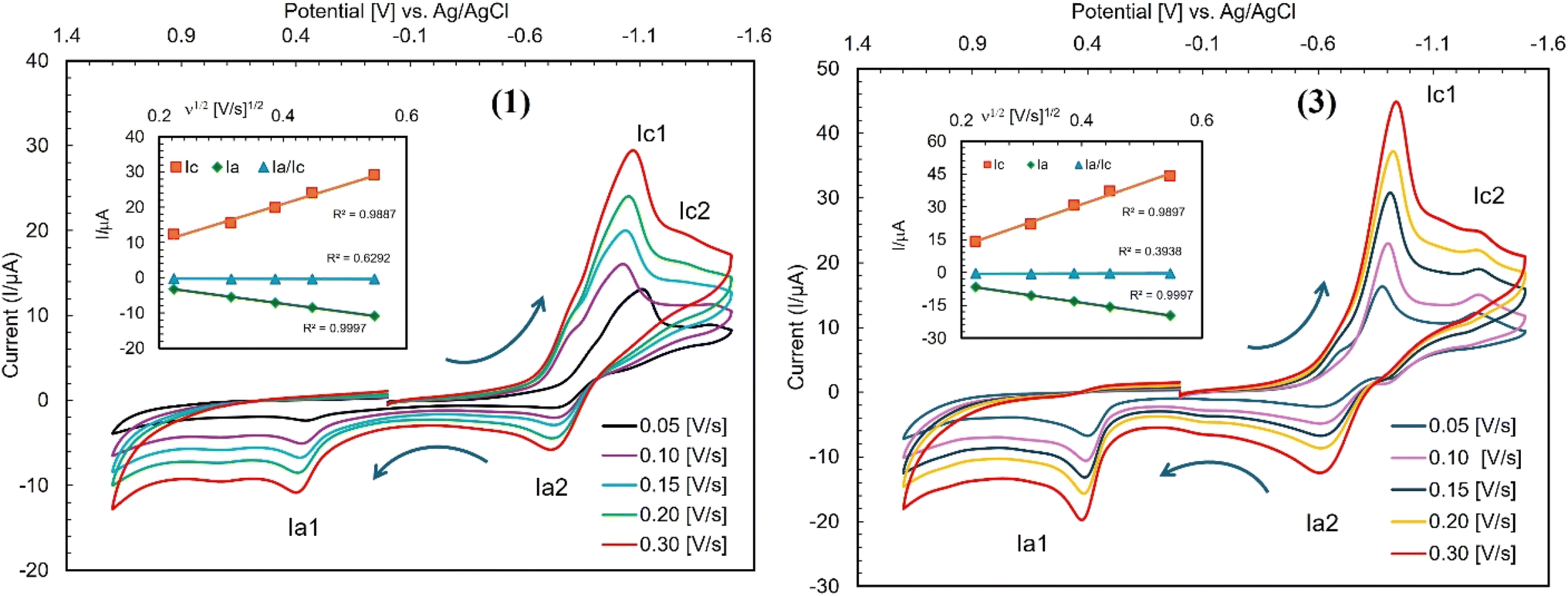
Cyclic voltammetry (CV) measurements of complexes 1 and 3 were conducted in DMF at 25 °C over a range of −1.5 to +1.2 V (versus Ag/AgCl), with scan rates varying from 0.05 to 0.3 V s−1 (Fig. 8 and Table S1‡). For the reduction scan, complex 1 showed peaks at Ec1 = −1.05 V (Ic1 = +15.48 μA) and Ec2 = −1.3 V (Ic2 = +11.19 μA), while complex 3 displayed peaks at Ec1 = −0.92 V (Ic1 = +21.94 μA) and Ec2 = −1.33 V (Ic2 = +14.25 μA) at a scan rate of 0.10 V s−1. On the other hand, for the oxidation scan, complex 1 showed peaks at Ea1 = +0.41 V (Ia1 = −4.85 μA) and Ea2 = −0.73 V (Ia2 = −2.05 μA), while complex 3 displayed peaks at Ea1 = +0.42 V (Ia1 = −10.53 μA) and Ea2 = −0.63 V (Ia2 = −3.82 μA). This redox process is attributed to two stepwise one-electron transfer processes corresponding to the Cu2+/Cu+ and Cu+/Cu0 couples and vice versa and diagnoses a quasi-reversible redox process, as reported for the analogous Cu(II)-Schiff base complexes.34,37,40,45,46 The voltammograms (Fig. 8) were recorded in the conventional direction (i.e., from cathodic to anodic). Analysis of CV data at varying scan rates exposes that the cathodic or anodic peak shifts to more negative or positive potentials with an increase in peak strength. The plots of Ic, Ia or Ia/Ic vs. ν1/2 exhibit an increase in Ic or a decrease in Ia values, whereas no change in Ia/Ic values (Fig. 8, inset). The straight correlation between the plots reflects a diffusion-controlled redox process for the complexes in solution.
 |
| | Fig. 8 Cyclic voltammograms of complexes 1 and 3 (ca. 0.5 mM) recorded in DMF at 25 °C containing 0.1 M TBAP at scan rates from 0.05 to 0.30 V s−1. The arrows indicate the scan direction starting from −0.1 V, and the inset shows the plot of Ic, Ia or Ia/Ic vs. ν1/2. | |
Magnetic measurements
The solid-state magnetic susceptibility (χm = 1.48 × 10−3 (1), 1.18 × 10−3 (2), 1.35 × 10−3 (3)) and moment (μeff. = 1.89 (1), 1.67 (2), 1.79 μB (3)) (diamagnetic contributions from ligand are ignored) indicate the paramagnetic nature of the copper(II) complexes.33,40,47–50 The theoretical spin-only magnetic moment for one unpaired electron (S = 1) for Cu(II) (d9) is 1.73 μB, and the remaining differences (i.e., +0.16 μB for 1 and −0.06/+0.06 μB for 2/3) with the measured values (μeff.) may arise from orbital angular magnetic moment and/or diamagnetism of the ligand.33,40,51–54
Anti-bacterial activity
The antibacterial activities of HL1, HL2, HL3, 1, 2, 3, chloramphenicol (standard: +ve control) and DMSO (blank: −ve control) are shown in Fig. 9.30,33,34 Complex 2 exhibits significant antibacterial activity against E. coli and S. aureus compared with standard chloramphenicol, whereas complex 1 exhibits low activity. On the other hand, complex 3 is fully inactive against microorganisms, likely due to the steric and electronic effects imparted by the bulky naphthyl group in the HL3 ligand, which may impede effective interaction with bacterial membranes or intracellular targets. In contrast to the square-planar geometry observed in 1 and 2, complex 3 adopts a square-pyramidal geometry, which alters the accessibility and redox potential of the copper center, thereby reducing its ability to generate reactive oxygen species (ROS), which are essential for antibacterial action. Furthermore, a reduced lipophilicity and altered electronic distribution in 3 may impair membrane permeability and target binding, leading to its lack of bioactivity. Although the ligand HL2 exhibits medium activity, HL1 and HL3 are fully inactive. The increased antibacterial activity of the complex compared with that of the free Schiff base was explained by the chelation of the ligand to the metal(II) ions.55–57
 |
| | Fig. 9 Antibacterial activities of the Schiff bases and Cu(II) complexes against E. coli and S. aureus: (a and b) HL1 (L147), CuL147 (1); (c and d) HL2 (L87), HL3 (L102), CuL87 (2), CuL102 (3) and (e) comparison plot of inhibition zone with respect to chloramphenicol (+ve control). | |
Conclusions
The molecular structure determination for the Schiff bases (HL1–HL3) shows a usual (imine)N⋯H–O(phenol) (enolimine) based structure in the solid state and solution (evidenced by 1H NMR studies). Each unit cell in the HL1 or HL2 crystals contains a single molecule, whereas two symmetry-independent molecules (A and B) are present in HL3. The structural analysis reveals that the two ligand molecules coordinate to the copper(II) ion through the N,O-chelation mode, resulting in a variable geometry from square-planar (1) to distorted square-planar (2) and to square-pyramidal (3) as a function of benzylal- vs. naphthylal-based Schiff base ligands. An asymmetric unit of the crystal structure holds half of the molecule for 1, a single molecule for 2 and two symmetry-independent molecules for 3 (A and B). CV results indicate two quasi-reversible one-electron charge transfer processes for 1 and 3 in DMF. Thermal analysis corresponds to a phase transition from a crystalline solid to an isotropic liquid (m.p.). The complexes were further authenticated by paramagnetism and non-electrolyte profile. Complex 2 exhibited substantial antibacterial activity against E. coli and S. aureus, whereas low or no activity was observed for 1 or 3. Among the ligands, only HL2 exhibited medium activity against microorganisms. Computational modeling corresponds well to experimental results for electronic and molecular structures.
Experimental
Materials and methods
FT-IR spectra were recorded using a Prestige-21 spectrophotometer (Shimadzu) as KBr discs at room temperature. UV-vis. spectra were collected using a UV-1800 spectrophotometer (Shimadzu) in chloroform at 25 °C. Differential scanning calorimeter (DSC) data were recorded on a DSC 60 (Shimadzu) at 30–300 °C with a heating rate of 10 K min−1. 1H/13C NMR spectra were run on a Bruker Avance (DPX 400 spectrometer) at 20 °C using CDCl3 and DMSO-d6 as solvents. EI-MS was collected on a Thermo-Finnigan TSQ-700 mass spectrometer. The isotopic scattering shapes of the 63/65Cu-containing mass ions are visible in the spectra. Elemental analyses were performed using a Vario EL instrument from Elementaranalysensysteme. The Magnetic Susceptibility Balance MSB Mk1 (Sherwood Scientific Ltd.) was used to measure the solid-state magnetic data at 25 °C. A Mettler Toledo Fivego (Model F3) conductivity meter was used to determine the molar conductance in DMF at 25 °C. A CV experiment was run on an Epsilon™ Instruments (BASi) using the supporting electrolyte of tetra-N-butyl-ammonium-hexafluorophosphate (TBAP) in acetonitrile at 25 °C. A three-electrode system consisting of a platinum disc electrode (working), a platinum wire electrode (auxiliary) and an Ag/AgCl electrode (reference) was used for collecting CV data. N2 gas was passed into the solution mixture for ca. 10 min before collecting the data.
Synthesis of the Schiff bases (HL1–HL3)
2-Hydroxy-1-benzaldehyde (salicylaldehyde: 1.2212 g, 10.0 mmol) or 2-hydroxy-1-naphthaldehyde (1.7800 g, 10.0 mmol) were dissolved in 10 mL of ethanol. Added 2–3 drops of concentrated H2SO4 into this solution and stirred the solution for 10–15 minutes at room temperature. Equimolar amounts of 2-amino-benzophenone (1.973 g, 10.0 mmol) or 2-amino-5-chloro-benzophenone (2.3168 g, 10.0 mmol) dissolved in 5 mL of ethanol were gently added to the mixture solution. The mixture solution was refluxed for 6–8 hours, color turned into a deep yellow. TLC was performed to track the reaction progress. After completing the reaction, the solvent was reduced to ca. 50% in a vacuum rotatory evaporator until precipitation occurred. This solution was left in air for one day to ensure complete precipitation. The precipitate was collected via filtration and washed with cold ethanol three times (2 mL in each). The products were dried in air for several days to obtain yellow and lemon microcrystals of HL1, HL2 and HL3, respectively. Single crystals suitable for X-ray diffraction were grown via slow evaporation of concentrated methanol solution for HL1, slow diffusion of ethanol into concentrated dichloromethane (or n-hexane into concentrated chloroform) solution for HL2, and slow diffusion of methanol into concentrated chloroform solution for HL3 after 2–3 days at room temperature.
(E)-2-(((2-Benzoylphenyl)imino)methyl)phenol or (E)-(2-((2-hydroxybenzylidene)amino)phenyl)(phenyl)methanone (HL1). Yield: 2.532 g (79.27% based on 2-hydroxy-benzaldehyde). IR (KBr, cm−1): ν = 3453 m (br, O–H), 3049, 3032, 2929 w (Ar–H), 1659 vs. (CO), 1614 vs. (CN) and 1566 vs. (CC) (see Fig. S1‡). UV-vis. (0.09 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 344 (7168) and 257 (16935). 1H NMR (400 MHz, DMSO-d6): δ/ppm = 6.79 (d, JHH = 8.0 Hz, 1H, H2), 6.91 (t, JHH = 7.6 Hz, 1H, H4), 7.34 (t, JHH = 7.2 Hz, 1H, H3),7.46–7.55 (m, 5H, H9,11,17,18,19), 7.59 (d, JHH = 8.0 Hz,1H, H5), 7.65–7.68 (m, 2H, H10,12), 7.73 (d, JHH = 7.6 Hz, 2H, H16,20), 8.90 (s, 1H, CNH) and 11.82 (s, H, OH) (Fig. S3a‡). 13C NMR (100 MHz, DMSO-d6) δ/ppm: 116.95 (C2), 119.30 (C6), 119.59 (C4), 119.60 (C9), 127.27 (C11), 128.69 (C17), 128.71 (C19), 128.97 (C16), 129.32 (C20), 129.93 (C13), 132.09 (C12), 133.17 (C5), 134.09 (C3), 134.16 (C18), 134.97 (C10), 137.24 (C15), 146.73 (C8), 160.33 (C7, CN), 164.65 (C1, OH) and 197.17 (C14, CO) (Fig. S3c‡). Anal. calcd for C20H15NO2: C, 79.72; H, 5.02; N, 4.65%. Found C, 79.44; H, 4.83; N, 4.82%.
(E)-2-(((2-Benzoyl-5-chlorophenyl)imino)methyl)phenol or (E)-(5-chloro-2-((2-hydroxybenzylidene)amino)phenyl)(phenyl)methanone (HL2). Yield: 2.924 g (82.65% based on 2-hydroxy-benzaldehyde). IR (KBr, cm−1): ν = 3421 m (br, O–H), 3059, 3037, 2989 w (Ar–H), 1664 vs. (CO), 1610, 1593 vs. (CN) and 1579 vs. (CC) (see Fig. S1‡). UV-vis. (0.05 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 348 (12240) and 261 (23000). 1H NMR (400 MHz, CDCl3): δ/ppm = 6.98–7.07 (m, 2H, H2,4), 7.33 (dd, JHH = 8.4, 1.6 Hz, 1H, H3), 7.46 (d, JHH = 2.0 Hz, 1H, H9), 7.48–7.52 (m, 3H, H17,18,19), 7.56–7.61 (m, 3H, H5,10,12), 7.67 (d, JHH = 7.6 Hz, 2H, H16,20), 9.93 (s, 1H, CHN) and 11.04 (s, H, OH) (Fig. S3b‡). 1HNMR (400 MHz, DMSO-d6): δ/ppm = 6.79 (d, JHH = 8.4 Hz, 1H, H2), 6.91 (t, JHH = 7.6 Hz, 1H, H4), 7.35 (dt, JHH = 8.0, 7.6, 1.2 Hz, 1H, H3), 7.54 (t, JHH = 8.0 Hz, 3H, H17,18,19), 7.61–7.69 (m, 3H, H5,9,10), 7.73–7.76 (m, 3H, H12,16,20), 8.92 (s, 1H, CHN) and 11.60 (s, H, OH) (Fig. S3a‡). 13C NMR (100 MHz, DMSO-d6) δ/ppm: 116.98 (C2), 119.55 (C6), 119.69 (C4), 121.18 (C9), 128.13 (C11), 128.88 (C17), 128.97 (C19), 129.42 (C16), 129.99 (C20), 131.64C13), 131.76 (C12), 133.12 (C5), 134.33 (C3), 134.48 (C18), 136.70 (C10), 136.76 (C15), 145.51 (C8), 160.28 (C7, CN), 164.97 (C1, OH) and 195.69 (C14, CO) (Fig. S3c‡). Anal. calcd for C20H14ClNO2: C, 71.54; H, 4.20; N, 4.17%. Found C, 71.43; H, 4.09; N, 4.04%.
(E)-1-(((2-Benzoyl-5-chlorophenyl)imino)methyl)naphthalen-2-ol or (E)-(5-chloro-2-(((2-hydroxynaphthalen-1-yl)methylene)amino)phenyl)(phenyl)methanone (HL3). Yield: 3.156 g (81.62% based on 2-hydroxy-1-napthaldehyde). IR (KBr, cm−1): ν = 3431 m (br, O–H), 3057, 2924 w (Ar–H), 1660 vs. (CO), 1622, 1602 vs. (CN) and 1577, 1550 vs. (CC) (see Fig. S1‡). UV-vis. (0.05 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 386 (5166), 322 (4466) and 256 (11900). 1HNMR (400 MHz, DMSO-d6): δ/ppm = 6.97 (d, JHH = 9.2 Hz, 1H, H2), 7.38 (t, JHH = 7.6 Hz, 1H, H23), 7.52–7.57 (m, 3H, H9,17,22), 7.61 (d, JHH = 2.4 Hz, 1H, H10), 7.69 (t, JHH = 7.6 Hz, 1H, H19), 7.76–7.83 (m, 4H, H3,12,18,21), 7.92–7.98 (m, 2H, H16,20), 8.49 (d, JHH = 8.4 Hz, 1H, H24), 9.68 (s, 1H, CHN) and 14.05 (s, H, OH) (Fig. S3a‡). 13C NMR (100 MHz, DMSO-d6) δ/ppm: 109.79 (C6), 120.42 (C21), 121.31 (C2), 121.86 (C23), 124.28 (C22), 124.29 (C9), 127.66 (C24), 128.35 (C17), 128.60 (C19), 129.42 (C4), 129.45 (C16), 130.15 (C20), 130.17 (C12), 131.16 (C3), 131.87 (C18), 133.06 (C11), 134.53 (C13), 135.77 (C5), 136.68 (C10), 136.84 (C15), 144.25 (C8), 159.96 (C7, CN), 165.38 (C1, OH) and 195.66 (C14, CO) (Fig. S3c‡). Anal. calcd for C24H16ClNO2: C, 74.71; H, 4.18; N, 3.63%. Found C, 74.57; H, 4.09; N, 3.78%.
Syntheses of the complexes (1–3)
Two equivalents of HL1 (602.5 mg, 2.0 mmol) or HL2 (670.0 mg, 2.0 mmol) or HL3 (772.0 mg, 2.0 mmol) were dissolved in 10 mL of methanol and 2 mL of dichloromethane (DCM), and the solution was stirred for ca. 10 min. One equivalent amount of copper(II) acetate (200.0 mg, 1.0 mmol) dissolved in 10 mL of methanol was added to this mixture solution. The color changed immediately to light green (for HL1) or light brown (for HL2 or HL3). The reaction mixture was stirred for ca. 24 hours at room temperature in the presence of N2 gas. The final color of the solution turned to green (for HL1) and deep brown (for HL2 or HL3). The solution was then fully dried in a vacuum rotary evaporator at ca. 40 °C. The products were washed four times with ethanol, followed by n-hexane (2 mL in each) to obtain green or deep brown microcrystals of 1, 2 and 3. The microcrystals were dried in air for 3–4 days at room temperature and then preserved under N2 gas. Single crystals suitable for X-ray diffraction were grown via slow diffusion of MeOH into concentrated DCM solution for 1 and slow evaporation of concentrated methanol solution of 2 or 3 after 2–3 days at room temperature.
Bis[(E)-2-(((2-benzoylphenyl)imino)methyl)phenolato-κ2N,O]copper(II), [(Cu(L1)2] (1). Yield: 0.538 g (67%). IR (KBr, cm−1): ν = 3048 and 2997 w (νC–H), 1661vs. (νCO), 1607, 1591v s (νCN) and 1530 vs. (νCC) (see Fig. S1‡). UV-vis. (0.04 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 552 (201) and 257 (33533). EI-MS: m/z (%) = 663 (30) [Cu(L1)2]+, 363 (100) [Cu(L1)2−L1]+, 301 (15) [HL1]+, 256 (10) [C18H11NO–H]+, 152 (22) [C11H7N–H]+ and 105 (18) [C7H5O]+ {[Cu(L1)2] = C40H28CuN2O4, HL1 = C20H15NO2} (see Fig. S2‡). Conductance (Λm) = 2.02 S m2 mol−1 in DMF at 25 °C. Solid state magnetic moment (μeff.) = 1.89 μB at 25 °C. Anal. calcd for C40H28CuN2O4: C, 72.33; H, 4.25; N, 4.22%. Found C, 72.18; H, 4.12; N, 4.12%.
Bis[(E)-2-(((2-benzoyl-4-chlorophenyl)imino)methyl)phenolato-κ2N,O]copper(II), [(Cu(L2)2] (2). Yield: 0.698 g (76%). IR (KBr, cm−1): ν = 3055 and 3020 w (νC–H), 1664 vs. (νCO), 1614 vs. (νCN) and 1531 vs. (νCC) (see Fig. S1‡). UV-vis. (0.05 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 572 (75) and 399 (11153). EI-MS: m/z (%) = 733 (5) [Cu(L2)2]+, 397 (100) [Cu(L2)2−HL2 + H]+, 335 (15) [HL2]+, 182 (22) [C7H5CuNO]+ and 105 (21) [C7H5O]+ {[Cu(L2)2] = C40 H26Cl2CuN2O4, HL2 = C20H14ClNO2} (see Fig. S2‡). Conductance (Λm) = 2.37 S m2 mol−1 in DMF at 25 °C. Solid-state magnetic moment (μeff.) = 1.67 μB at 25 °C. Anal. calcd for C40H26Cl2CuN2O4: C, 65.54; H, 3.57; N, 3.82%. Found C, 65.34; H, 3.38; N, 3.79%.
Bis[(E)-1-(((2-benzoyl-4-chlorophenyl)imino)methyl)naphthalen-2-olato-κ2N,O]copper(II), [(Cu(L3)2] (3). Yield: 0.644 g (78.12%). IR (KBr, cm−1): ν = 3049, 3028 w (νC–H), 1666 vs. (νCO), 1614, 1601 vs. (νCN) and 1571, 1529 vs. (νCC) (see Fig. S1‡). UV-vis. (0.016 mM, CHCl3): λmax/nm (εmax/L mol−1 cm−1) = 640 (80), 390 (1688) and 333 (3888). EI-MS: m/z (%) = 828 (15) [Cu(L3)2]+, 442 (100) [Cu(L3)2–L3+H]+, 384 (90) [L3–H]+, 277 (15) [C17H10ClNO–2H]+, 226 (10) [C11H7NNiO]+, 152 (45) [C11H8N–2H]+, 105 (48) [C7H5O]+ and 77 (25) [C6H5]+ {[Cu(L3)2] = C48H30Cl2CuN2O4, HL3 = C24H16ClNO2} (see Fig. S2‡). Conductance (Λm) = 1.98 S m2 mol−1 in DMF at 25 °C. Solid-state magnetic moment (μeff.) = 1.79 μB at 25 °C. Anal. calcd for C48H30Cl2CuN2O4: C, 69.19; H, 3.63; N, 3.36%. Found C, 70.00; H, 3.58; N, 3.23%.
X-ray crystallography
Data collection. A Rigaku XtaLAB Synergy (Rigaku, Tokyo, Japan), Dualflex, HyPix diffractometer with a micro-focus X-ray tube with Cu-Kα radiation (λ = 1.54182 Å) was used. A polarized-light Leica M80 microscope (Leica, Wetzlar, Germany) was used to select suitable single crystals, which were covered with oil on a cryo-loop. The temperatures for data collection were 150 K for HL1 and 1, HL2 at 220 K, HL3 at 251 K, 2 at 100 K and 3 at 200 K. Structure Analysis and Refinement: CRYSALISPRO was used for cell refinement, data reduction and absorption correction.58 The crystal structures were solved using OLEX2-1.5 (ref. 59) with SHELXT and refined with SHELXL.60,61 All non-hydrogen atoms were refined using anisotropic displacement parameters. All hydrogen atoms on C were positioned geometrically. Hydrogen atoms on O were found and refined with Uiso(H) = 1.5Ueq(O) for HL1, HL2 and HL3. The crystal data and structure refinement results are listed in Table 4. The intermolecular interactions were calculated, and the graphics were designed using DIAMOND.62 The CCDC numbers are 2425911–2425916 and data can be obtained free of charge from http://www.ccdc.cam.ac.uk/data_request/cif.
Table 4 Crystal data and structure refinement for compounds HL1, HL2, HL3, 1, 2 and 3
| Complexes |
HL1 |
HL2 |
HL3 |
1 |
2 |
3 |
| R1 = [Σ(‖Fo| − |Fc‖)/Σ|Fo|] and wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. Goodness-of-fit = [Σ[w(Fo2 – Fc2)2]/(n − p)]1/2. |
| Empirical formula |
C20H15NO2 |
C20H14ClNO2 |
C24H16ClNO2 |
C40H28CuN2O4 |
C40H27ClCuN2O4 |
C48 H30Cl2CuN2O4 |
| M (g mol−1) |
301.33 |
335.77 |
385.83 |
664.18 |
733.07 |
833.18 |
| Crystal size (mm) |
0.78 × 0.60 × 0.20 |
0.11 × 0.10 × 0.05 |
0.23 × 0.08 × 0.04 |
0.83 × 0.31 × 0.15 |
0.13 × 0.07 × 0.04 |
0.20 × 0.16 × 0.06 |
| Temperature (K) |
150 |
220 |
251 |
150 |
100 |
200 |
| θ range (°) |
2.57–77.98 |
4.25–76.09 |
4.008–76.843 |
4.63–77.69 |
3.10–78.4 |
2.73–76.81 |
| h; k; l range |
+21, −20; ±10; +25, −26 |
+9, −7; ±13; ±13 |
±9, +20, −12; +35, –36 |
±10; ±12; ±13 |
+20, −23; ±10; +22, −23 |
±19; ±21; +21, −22 |
| Crystal system |
Orthorhombic |
Triclinic |
Orthorhombic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
Pbcn |
P |
P212121 |
P |
P121/n1 |
P |
| a (Å) |
17.2049(1) |
7.3460(2) |
7.5166(10) |
8.4841(2) |
19.4435(3) |
15.8451(7) |
| b (Å) |
8.2984(1) |
10.7550(3) |
16.8538(2) |
10.2724(3) |
8.5568(2) |
17.0095(8) |
| c (Å) |
21.2414(1) |
10.8277(3) |
28.8531(4) |
10.9070(2) |
19.5887(4) |
17.8492(7) |
| α (°) |
90 |
73.1880(2) |
90 |
104.192(2) |
90 |
110.5390(4) |
| β (°) |
90 |
88.4520(2) |
90 |
107.674(2) |
95.0490(2) |
94.03430(4) |
| γ (°) |
90 |
82.9820(2) |
90 |
111.408(3) |
90 |
116.6932(5) |
| V (Å3) |
3032.70(4) |
812.73(4) |
3655.20(8) |
772.33(4) |
3246.41(11) |
3872.45(3) |
| Z |
8 |
2 |
8 |
1 |
4 |
4 |
| Dcalc (g cm−3) |
1.320 |
1.372 |
1.402 |
1.428 |
1.500 |
1.429 |
| F (000) |
1264 |
348 |
1600 |
343 |
1500 |
1708 |
| μ (mm−1) |
0.683 |
2.172 |
2.011 |
1.378 |
2.849 |
2.463 |
| Max/min transmission |
1.000/0.195 |
0.932/0.848 |
1.000/0.665 |
1.000/0.357 |
0.893/0.893 |
0.894/0.733 |
| Refl. measured |
87929 |
9431 |
17398 |
19558 |
22281 |
144207 |
| Refl. unique (Rint) |
3221(0.0401) |
3231(0.0247) |
6961(0.0342) |
3044(0.0479) |
5115(0.0413) |
15597(0.0368) |
| Data/restraints/parameters |
3221/0/295 |
3231/0/220 |
6961/0/633 |
3044/0/214 |
5115/0/442 |
15597/0/1267 |
| Completeness |
1.000 |
0.994 |
0.999 |
0.999 |
1.000 |
0.999 |
| Largest diff. peak & hole (Δρ/e Å−3) |
0.151/−0.188 |
0.154/−0.261 |
0.186/−0.272 |
0.270/−0.462 |
0.405/−0.697 |
0.351/−0.430 |
| R1/wR2 [I > 2σ(I)]a |
0.0341/0.0852 |
0.0343/0.0893 |
0.0323/0.0845 |
0.0352/0.0877 |
0.0441/0.1069 |
0.0307/0.0914 |
| R1/wR2 (all reflect.)a |
0.0353/0.0862 |
0.0425/0.0934 |
0.0347/0.0863 |
0.0372/0.0896 |
0.0518/0.1104 |
0.0326/0.0929 |
| Goodness-of-fit on F2b |
1.062 |
1.054 |
1.057 |
1.086 |
1.084 |
1.061 |
| CCDC number |
2425911 |
2425912 |
2425913 |
2425914 |
2425915 |
2425916 |
Computational method
The Gaussian 0963 software was used for the computational procedures. The molecular structures for 1, 2 and 3 were used to design the initial gas phase geometry for DFT optimization using the functional B3LYP or M06 and the basis sets 6-31G(d) or SDD (Fig. S4‡).64,65 The UV-vis. spectra and excited-state properties were calculated by TD-DFT with different arrangements of the functionals (e.g., B3LYP, cam-B3LYP, M06 and M06-2X) and the basis sets (e.g., 6-31G(d), SDD and SVP), respectively (Fig. 1 and S7–S9‡). The spectra were further calculated using the mixed basis sets LANL2DZ (for copper) and 6-31G(d) (for other atoms) with B3LYP and M06 functionals, respectively. The spectra thus obtained from different arrangements are almost similar, with a small change in the positions of the bands, and match well with the experimental spectra (Fig. 1 and S7–S9‡). These outcomes validate the legitimacy and consistency of the simulation protocol. The polarization continuum model (PCM) using CHCl3 as the solvent and 72 excited states (roots) was used for the computations. The spectra were constructed with SpecDis soft66 using a Gaussian band shape with exponential half-width, σ = 0.33 eV.
Antibacterial activity study
The agar-well diffusion method30,33,34 was used to measure the antibacterial activity of the Schiff bases (HL1, HL2, HL3) and complexes (1, 2, 3) against Escherichia coli (gram −ve, ATCC 11303) and Staphylococcus aureus (gram +ve, ATCC 9144). In Mueller–Hinton Broth, bacterial colonies were cultivated at 120 rpm and 37 ± 2 °C. On the agar plates, sterile micropipette tips were used to make 5–7 mm diameter wells. The complex solution in DMSO (ca. 250–300 μg mL−1) was placed onto the wells with the same solvent mixture as a blank (a −ve control) and showed no or very little inhibition. A dose (ca. 30 μg) of a chloramphenicol disc (standard) was used as a +ve control. The agar plates were kept at 4 °C for 3 hours to prevent any diffusion or penetration of the complex. The diameter of the inhibitory zone was measured after 20 hours of incubation on agar plates.
Data availability
Data will be made available upon request.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors acknowledge the financial assistance provided by the Alexander von Humboldt Foundation (AvH), Germany under the Research Group Linkage Program (2020–2023). We acknowledge WMSRC at Jahangirnagar University, Bangladesh for conducting 1H/13C NMR experiment. Our sincere thanks to Professor M. M. Islam, Department of Chemistry, Dhaka University, Bangladesh for CV run. We thank to computecanada.ca (https://ccdb.computecanada.ca/) for computational resources.
References
- G. Crisponi, V. M. Nurchi, D. Fanni, C. Gerosa, S. Nemolato and G. Faa, Coord. Chem. Rev., 2010, 254, 876–889, DOI:10.1016/j.ccr.2009.12.018.
- J. A. Drewry and P. T. Gunning, Coord. Chem. Rev., 2011, 255, 459–472, DOI:10.1016/j.ccr.2010.10.018.
- W. J. Lian, X. T. Wang, C. Z. Xie, H. Tian, X. Q. Song, H. T. Pan, X. Qiao and J. Y. Xu, Dalton Trans., 2016, 45(22), 9073–9087, 10.1039/C6DT00461J.
-
(a) K. Y. Djoko, P. S. Donnelly and A. G. McEwan, Metallomics, 2014, 6(12), 2250–2259, 10.1039/c4mt00226a;
(b) S. D. Oladipo and R. C. Luckay, New J. Chem., 2024, 48(30), 13276–13288, 10.1039/D4NJ01621A.
- L. M. Balsa, V. Ferretti, M. Sottile, P. Nunes, J. C. Pessoa, I. Correia and I. E. León, Dalton Trans., 2024, 53(7), 3039–3051, 10.1039/D3DT02964F.
- C. K. Pal, S. Mahato, M. Joshi, S. Paul, A. R. Choudhury and B. Biswas, Inorg. Chim. Acta, 2020, 506, 119541, DOI:10.1016/j.ica.2020.119541.
- N. Assadawi, M. Ferderer, N. Kusi-Appauh, H. Yu, C. T. Dillon, R. Sluyter, C. Richardson and S. F. Ralph, Dalton Trans., 2024, 53(30), 12720–12739, 10.1039/D4DT00448E.
- S. Kumar, A. Arora, V. K. Maikhuri, A. Chaudhary, R. Kumar, V. S. Parmar, B. K. Singh and D. Mathur, RSC Adv., 2024, 14(24), 17102–17139, 10.1039/D4RA00590B.
- R. Morales-Guevara, J. A. Fuentes, D. Paez-Hernández and A. Carreño, RSC Adv., 2021, 11(59), 37181–37193, 10.1039/D1RA90173G.
- N. Arora, P. Dhiman, S. Kumar, G. Singh and V. Monga, Bioorg. Chem., 2020, 97, 103668, DOI:10.1016/j.bioorg.2020.103668.
- B. Bieszczad, D. Garbicz, D. Trzybiński, D. Mielecki, K. Woźniak, E. Grzesiuk and A. Mieczkowski, Molecules, 2020, 25(4), 906, DOI:10.3390/molecules25040906.
- D. Iguchi, R. Erra-Balsells and S. M. Bonesi, Photochem. Photobiol. Sci., 2016, 15, 105–116, 10.1039/c5pp00349k.
- G. Spighi, M. A. Gaveau, J. M. Mestdagh, L. Poisson and B. Soep, Phys. Chem. Chem. Phys., 2014, 16(20), 9610–9618, 10.1039/C4CP00423J.
- C. Lei, W. Yang, Z. Lin, Y. Tao, R. Ye, Y. Jiang, Y. Chen and B. Zhou, RSC Adv., 2024, 14(28), 20339–20350, 10.1039/D4RA02797C.
- B. Zhang, C. Chen, J. Tian, S. L. Gao, Y. Yang, X. Li and J. Zhang, New J. Chem., 2024, 48, 20399–20409, 10.1039/D4NJ03860F.
- C. Pigot, G. Noirbent, T. T. Bui, S. Péralta, S. Duval, D. Gigmes, M. Nechab and F. Dumur, New J. Chem., 2021, 45(13), 5808–5821, 10.1039/D1NJ00275A.
- P. Subbaraj, A. Ramu, N. Raman and J. Dharmaraja, J. Saudi Chem. Soc., 2015, 19(2), 207–216, DOI:10.1016/j.jscs.2014.05.002.
- A. F. Mkrtchyan, L. A. Hayriyan, A. J. Karapetyan, A. S. Tovmasyan, A. H. Tsaturyan, V. N. Khrustalev, V. I. Maleev and A. S. aghyan, New J. Chem., 2020, 44(28), 11927–11932, 10.1039/D0NJ02072A.
- Q. Wang, X. Zhang and X. Fan, Org. Biomol. Chem., 2018, 16(41), 7737–7747, 10.1039/C8OB01846D.
- K. Surana, B. Chaudhary, M. Diwaker and S. Sharma, Med. Chem. Commun., 2018, 9(11), 1803–1817, 10.1039/C8MD00300A.
- D. Pagnozzi, N. Pala, G. Biosa, R. Dallocchio, A. Dessì, P. K. Singh, D. Rogolino, A. Di Fiore, G. De Simone, C. T. Supuran and M. Sechi, ACS Med. Chem. Lett., 2022, 13(2), 271–277, DOI:10.1021/acsmedchemlett.1c00644.
- N. G. Tsierkezos, J. Solution Chem., 2007, 36, 1301–1310, DOI:10.1007/s10953-007-9188-4.
- N. G. Tsierkezos and U. Ritter, Phys. Chem. Liq., 2011, 49, 729–742, DOI:10.1080/00319104.2010.508039.
- W. R. Fawcett and M. Fedurco, J. Phys. Chem., 1993, 97, 7075–7080, DOI:10.1021/j100129a025.
- K. Kohring, J. Wiesner, M. Altenkämper, J. Sakowski, K. Silber, A. Hillebrecht, P. Haebel, H. M. Dahse, R. Ortmann, H. Jomaa, G. Klebe and M. Schlitzer, ChemMedChem, 2008, 3, 1217–1231, DOI:10.1002/cmdc.200800043.
- M. I. Esteva, K. Kettler, C. Maidana, L. Fichera, A. M. Ruiz, E. J. Bontempi, B. Andersson, H. M. Dahse, P. Haebel, R. Ortmann, G. Klebe and M. Schlitzer, J. Med. Chem., 2005, 48, 7186–7191, DOI:10.1021/jm050456x.
- J. Ren, P. P. Chamberlain, A. Stamp, S. A. Short, K. L. Weaver, K. R. Romines, R. Hazen, A. Freeman, R. G. Ferris, C. W. Andrews, L. Boone, J. H. Chan and D. K. Stammers, J. Med. Chem., 2008, 51, 5000–5008, DOI:10.1021/jm8004493.
- T. J. Khoo, M. K. bin Break, K. A. Crouse, M. I. M. Tahir, A. M. Ali, A. R. Cowley, D. J. Watkin and M. T. H. Tarafder, Inorg. Chim. Acta, 2014, 413, 68–76, DOI:10.1016/j.ica.2014.01.001.
- Y. D. Kurt, B. Ülküseven, S. Güner and Y. Köseoğlu, Transition Met. Chem., 2007, 32(4), 494–500, DOI:10.1007/s11243-007-0196-4.
- S. Kumar and M. Choudhary, New J. Chem., 2022, 46, 4911–4926, 10.1039/D2NJ00283C.
- M. Aslam, I. Anis, N. Afza, A. Nelofar and S. Yousuf, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2011, 67(12), o3442–o3443, DOI:10.1107/s1600536811048690.
- M. Enamullah, I. Haque, A. K. Resma, D. Woschko and C. Janiak, Molecules, 2023, 28(1), 172, DOI:10.3390/molecules28010172.
- T. I. Tonny, I. Haque, M. S. Abdullah, B. K. Sidhu, D. E. Herbert and M. Enamullah, J. Coord. Chem., 2024, 77, 2487–2507, DOI:10.1080/00958972.2024.2428323.
- M. Enamullah, T. Aziz, I. Haque, A. Mohabbat, A. Kacperkiewicz, D. E. Herbert and C. Janiak, J. Mol. Struct., 2024, 1312, 138509, DOI:10.1016/j.molstruc.2024.138509.
- M. Enamullah, A. Mim, I. Haque, B. K. Sidhu, A. Kacperkiewicz and D. E. Herbert, New J. Chem., 2023, 47, 21804–21814, 10.1039/D3NJ04915A.
- M. Enamullah, A. K. M. Royhan Uddin, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva, A.-C. Chamayou and C. Janiak, Dalton Trans., 2014, 43(8), 3313–3329, 10.1039/C3DT52871E.
- I. Haque, M. Enamullah, A. K. Resma, N. T. Jhumur, D. Woschko, A. Mohabbat, J. v. Leusen, P. Kögerler and C. Janiak, Chem. – Asian J., 2024, 19, e202400915, DOI:10.1002/asia.202400915.
- M. Enamullah, M. A. Hossain, M. K. Islam, D. Woschko and C. Janiak, Dalton Trans., 2021, 50, 9236–9249, 10.1039/D1DT01671G.
- N. Kordestani, H. A. Rudbari, G. Bruno, S. Rosario, J. D. Braun, D. E. Herbert, O. Blacque, I. Correia, M. A.-M. Zaman, M. M. Bindu, C. Janiak and M. Enamullah, Dalton Trans., 2020, 49, 8247–8264, 10.1039/D0DT01649G.
- A. Mim, M. Enamullah, I. Haque, A. Mohabbat and C. Janiak, J. Mol. Struct., 2023, 1291, 135669, DOI:10.1016/j.molstruc.2023.135669.
- T. Akitsu and Y. Einaga, Polyhedron, 2005, 24, 2933–2943, DOI:10.1016/j.poly.2005.06.018.
- J. M. Fernandez-G, C. Ausbun-Valdés, E. E. Gonzaléz-Guerrero and R. A. Toscano, Z. Anorg. Allg. Chem., 2007, 633, 1251–1256, DOI:10.1002/zaac.200700130.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B:Struct. Sci., 2004, 60(6), 627–668, DOI:10.1107/S0108768104020300.
-
(a) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392, 10.1039/B203191B;
(b) J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816, 10.1039/B704980C.
-
(a) M. Enamullah, M. A. Islam, A.-C. Kautz and C. Janiak, J. Coord. Chem., 2018, 71, 2557–2568, DOI:10.1080/00958972.2018.1506111;
(b) M. Enamullah, M. Al-M. Zaman, M. M. Bindu, M. K. Islam and M. A. Islam, J. Mol. Struct., 2020, 1201, 127207, DOI:10.1016/j.molstruc.2019.127207;
(c) M. Enamullah, B. A. Joy and M. K. Islam, J. Mol. Struct., 2019, 1175, 56–64, DOI:10.1016/j.molstruc.2018.07.068.
- A. Zianna, G. Psomas, A. Hatzidimitriou and M. Lalia-Kantouri, RSC Adv., 2015, 5, 37495–37511, 10.1039/C4RA16484A.
- M. Enamullah, M. A. Islam, B. A. Joy and G. J. Reiß, Inorg. Chim. Acta, 2016, 453, 202–209, DOI:10.1016/j.ica.2016.08.013.
- S. M. Abdallah, G. G. Mohamed, M. A. Zayed and M. S. A. El-Ela, Spectrochim. Acta, Part A, 2009, 73, 833–840, DOI:10.1016/j.saa.2009.04.005.
- S. Djebbar-Sid, O. Benali-Baitich and J. P. Deloume, Polyhedron, 1997, 16, 2175–2182, DOI:10.1016/S0277-5387(96)00555-4.
- M. Enamullah, M. A. Quddus, M. M. Rahman and T. E. Burrow, J. Mol. Struct., 2017, 1130, 765–774, DOI:10.1016/j.molstruc.2016.11.002.
- A. F. Orchard, in Magnetochemistry, Oxford University Press, Oxford, 2003 Search PubMed.
- A. Saadati, H. A. Rudbari, M. Aryaeifar, O. Blacque, I. Correia, M. K. Islam, D. Woschko, T. H. H. Sohi, C. Janiak and M. Enamullah, CrystEngComm, 2023, 25, 365–377, 10.1039/D2CE01311H.
- G. Pescitelli, S. Lüdeke, A. C. Chamayou, M. Marolt, V. Justus, M. Ǵorecki, L. Arrico, L. Di Bari, M. A. Islam, I. Gruber, M. Enamullah and C. Janiak, Inorg. Chem., 2018, 57, 13397–13408, DOI:10.1021/acs.inorgchem.8b01932.
- M. Enamullah, I. Haque, A. Mim, M. K. Islam, B. K. Sidhu, D. E. Herbert, D. Woschko and C. Janiak, J. Mol. Struct., 2023, 1292, 136078, DOI:10.1016/j.molstruc.2023.136078.
- A. Sahraei, H. Kargar, M. Hakimi and M. N. Tahir, J. Mol. Struct., 2017, 1149, 576–584, DOI:10.1016/j.molstruc.2017.08.022.
- A. Sahraei, H. Kargar, M. Hakimi and M. N. Tahir, Transition Met. Chem., 2017, 42(6), 483–489, DOI:10.1007/s11243-017-0152-x.
- H. Kargar, A. A. Ardakani, M. N. Tahir, M. Ashfaq and K. S. Munawar, J. Mol. Struct., 2021, 1229, 129842, DOI:10.1016/j.molstruc.2020.129842.
- CrysAlisPro, Rigaku Oxford Diffraction, Release 1.171.40.103a; Agilent Technologies Ltd, Yarnton, UK, 2014, available online: https://scholar.google.com/scholar_lookup?title=Rigaku+Oxford+Diffraction%26author=CrysAlisPro%26publication_year=2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- K. Brandenburg, Diamond (Version 4.5), in Crystal and Molecular Structure Visualization, Crystal Impact, K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2009–2022 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnemberg, D. Williams-Young, F. Ding, F. LIpparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. llam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision A.03), Wallingford CT, 2016 Search PubMed.
- A. Kumar, A. Rajput, P. Kaur, I. Verma, R. D. Erande, S. Javed, J. Kłak, S. F. Alrebei, A. J. Mota, E. Colacio and H. Arora, Dalton Trans., 2023, 52, 7225–7238, 10.1039/D3DT00604B.
- P. Agarwal, A. Kumar, I. Verma, R. D. Erande, J. Kłak, A. J. Mota, H. Arora and A. Rajput, New J. Chem., 2021, 45, 1203–1215, 10.1039/D0NJ00484G.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pescitelli, SpecDis Version 1.71, Berlin, Germany, 2017 Search PubMed.
Footnotes |
| † Dedicated in memory of Professor ABP Lever, Department of Chemistry, York University, Toronto, Canada. |
| ‡ Electronic supplementary information (ESI) available. CCDC [2425911–2425916]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra01982f |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Imdadul Haque
*ab,
Imdadul Haque