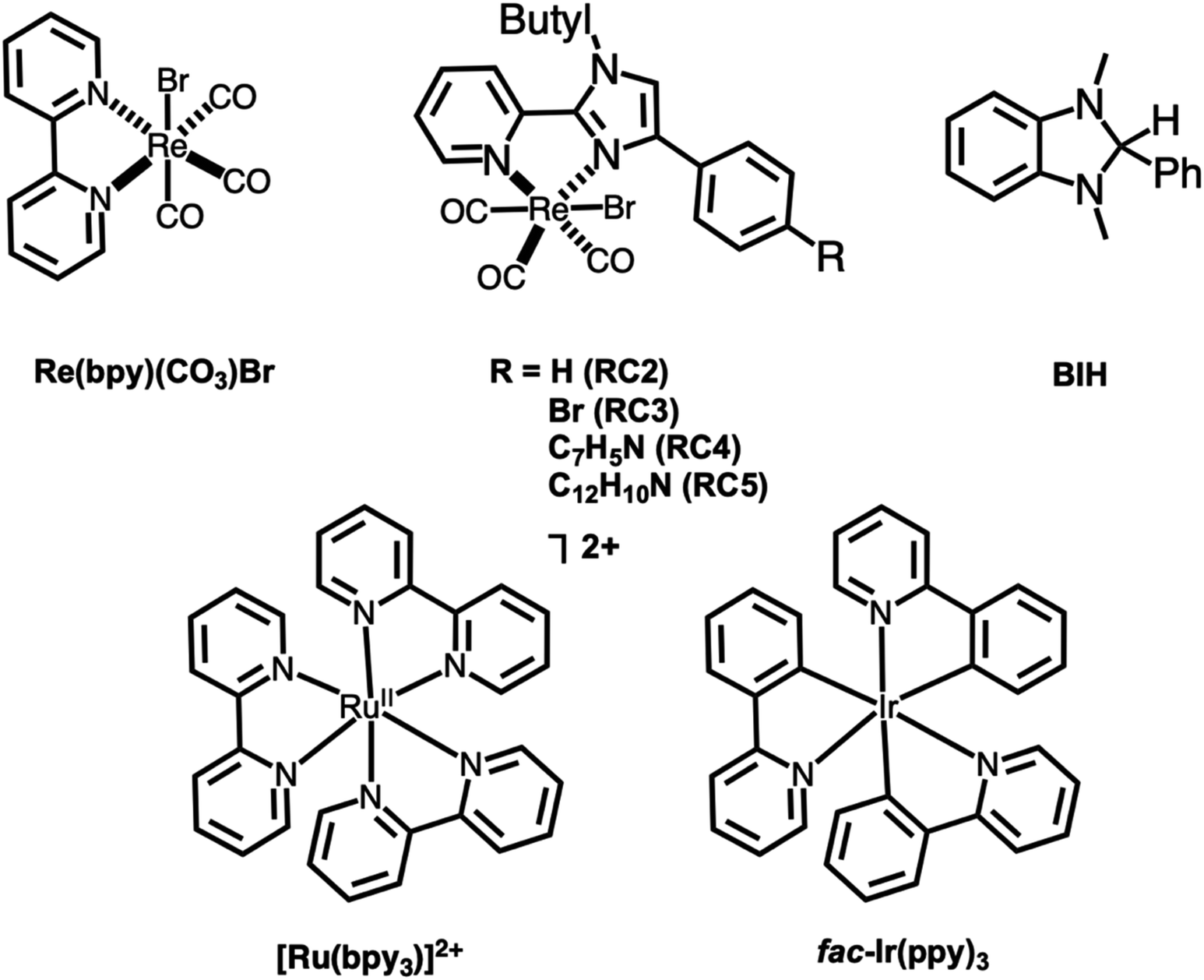
DOI:
10.1039/D5RA01561H
(Paper)
RSC Adv., 2025,
15, 12547-12556
Light-driven CO2 reduction with substituted imidazole-pyridine Re catalysts favoring formic acid production†
Received
5th March 2025
, Accepted 6th April 2025
First published on 22nd April 2025
Abstract
Removing carbon dioxide from the atmosphere is an attractive way to mitigate the greenhouse gas effect that contributes to climate change. A series of donor-pi (D-π), acceptor-pi (A-π), and π Re(I) pyridyl imidazole complexes have been synthesized and examined under photocatalytic conditions for the photocatalytic reduction of CO2. The catalytic activity of the complexes was further supported by cyclic voltammetry through the presence of a catalytic current under CO2 atmosphere. The D-π, π, and A-π complexes were studied to elucidate the effects of incorporating conjugated electron donating vs. withdrawing groups on the catalytic rates and product selectivity. The synthesized complexes were compared with Re(bpy)(CO)3Br (where bpy is 2,2′-bipyridine), the benchmark catalyst for this transformation. Remarkably, the complex with A-π pendant (RC4) outperformed the π (RC2–3) and D-π (RC5) complexes for the production of formic acid (HCO2H) in the presence of photosensitizer [Ru(bpy)3]2+ and sacrificial electron donor BIH (1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazoline). Among the investigated catalysts, RC4 with the A-π pendant showed the highest turnover number (TON) value of 844 for HCO2H production with 86% carbon selectivity. In stark contrast to the imidazole-pyridine based catalysts reported here that favor formic acid as a product, Re(bpy)(CO)3Br generated no formic acid under the same conditions. The imidazole-pyridine complexes also function as catalysts for CO2 reduction without an added photosensitizer, however, the TON values under self-sensitized conditions are poor.
Introduction
Currently, CO2 is the largest greenhouse gas emitted in the world.1 There is a considerable effort to convert this relatively untapped waste product into renewable fuels. While the thermodynamic reduction potentials of transforming CO2 to various products (CO, formic acid, methanol, methane, etc.) can be modest depending on the conditions,2 the kinetics are slow and a catalyst is required to lower activation barriers and direct selectivity for the desired product.3 Ideally, the energy required for catalysis is obtained directly from the sun or indirectly using solar photovoltaics to drive the electrochemistry. One reasonable goal for CO2 valorization is to selectively reduce CO2 to formic acid (HCO2H), which can be used directly in formic acid fuel cells4 or as a safe and easily transported H2 storage material.5,6 We note that CO is also a valuable product that can be transformed into hydrocarbon fuels via the Fischer–Tropsch process.7
The most well-studied mononuclear photocatalysts are derived from Lehn's original Re(bpy)(CO)3X (X = Cl, Br) catalyst,8–10 which has been modified extensively with the goal to improve the catalytic performance.11–14 Modification of the bidentate ligand has included introducing redox sites,15 ancillary Lewis acids,16 and other moieties to the second coordination sphere17 (proton relays,18 charged groups,19 hydrogen bond donors20,21) to facilitate proton transfer events or stabilize intermediates,8 change the local pH,22 or increase the onset of visible light absorption.23 Recently, Liang-Nian and coworkers reported an approach to increase the efficiency of the Re(bpy)-based photocatalyst by introducing chromophores onto the bipyridyl ligand, which yielded TONs as high as 1323 and ΦCO up to 55%.24 Another approach by Jurss and coworkers studied the effect of pendant light absorbing anthracene chromophores in mono- and di-nuclear anthracenyl Re(bpy) catalysts and found that especially in di-nuclear catalysts the ligand-based triplet state was highly influential.25 With these two works as a conceptual backdrop, we seek to improve the overall light absorption and increase the activity for Re-based photocatalysts. We hypothesized that incorporating a ligand design with an enhanced charge transfer absorption, either in a donor–acceptor (D–A) or acceptor–donor (A–D) configuration would provide a strongly red-shifted charge transfer absorption.26
Herein, we report the synthesis, characterization, and catalytic activity of a series of novel Re(I) imidazolylpyridine complexes (Fig. 1). We were able to confirm two of the metal complex structures through single crystal X-ray diffraction. The electrocatalytic and photocatalytic trials indicated that the complexes are able to reduce CO2 in sensitized photocatalytic reactions, and were modestly selective for HCO2H, which is rare for this class of Re photocatalysts.27–29
 |
| | Fig. 1 Structures of complexes studied herein. The photosensitizers used and sacrificial electron donor (BIH) are also shown. | |
Results and discussion
Catalyst synthesis
The synthesis of complexes RC2 and RC3 begin with an imidazole forming condensation between the alpha-brominated ketones 2, 5, or 10 and 2-amidinylpyridine to afford 3, 6, and 11 (Scheme 1).30 Imidazole alkylation with 1-bromobutane using NaH afforded 4, 7, and 12 in moderate yields. Alkylation of the imidazole unit was crucial to the success of the synthesis as the non-alkylated pyridyl-imidazole ligand was observed to chelate the Re metal center using either of the imidazole N atoms due to tautomerization. This led to mixtures of the two possible isomers that were not separable.31 Upon alkylation, however, only one isomer was observed to form, and Nuclear Overhauser Effect (NOESY) spectroscopy was performed to confirm which isomer was present after purification (Fig. S4†). Refluxing Re(CO)5Br in toluene with 4, 7, or 12 afforded catalysts RC2, RC3, or RC4. Catalyst RC5 was afforded with relative ease by Buchwald-coupling of diphenylamine with 7, followed by metalation with Re(CO)5Br in refluxing toluene. Suzuki-coupling of 3-cyanophenylboronic acid and 4-bromoacetophenone was performed to afford 9 which was then brominated at the ketone α carbon with NBS and p-toluenesulfonic acid to give 10. Detailed procedures for the synthesis of the ligands and their corresponding Re complexes is described in the ESI.†
 |
| | Scheme 1 Synthetic steps to prepare complexes RC2–RC5. Reaction conditions: (i) KHCO3, H2O, THF, amino(pyridin-2-yl)methaniminium chloride, reflux. Yields: 3: 70%; 6: 70%; 11: 75%. (ii) 1-Bromobutane, DMF, NaH, 80 °C. Yields: 4: 60%; 7: 77%; 12: 30%. (iii) Re(CO)5Br, toluene, reflux. Yields: 13: 55%; 14: 57%; 15: 23%; 16: 60%. (iv) Diphenyl amine, toluene, KOtBu, Pd(dba)2, tri-tert-butylphosphonium tetrafluoroborate, reflux, 76%. (v) 3-Cyanophenylboronic acid, Na2CO3, Pd(PPh3)4, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dioxane:water, 72%. (vi) p-TsOH, NBS, MeCN, reflux, 60%. :1 dioxane:water, 72%. (vi) p-TsOH, NBS, MeCN, reflux, 60%. | |
Crystallographic data
Suitable single crystals of RC2 and RC3 were obtained via vial-in-vial vapor diffusion using methylene chloride in the inner vial and diethyl ether in the outer vial after allowing the set-up to stand overnight (Fig. 2). Both crystals were bright yellow in color. Other instances of diimine pyridyl-imidazole rhenium crystals have been reported in the literature and a comparison of the selected bond lengths with a reference complex can be found below. The crystallographic data are summarized in Table 1. The Re1–Nimidazole and Re1–Npyridine bond lengths are slightly shorter (0.005 Angstroms) in RC2 than RC3, likely owing to the electron withdrawing nature of the bromine substituent lengthening the Re–N bonds in RC3. The Nimidazole–Re bond length in RC2 (2.181(2) Å) is longer than an analogous bond found in a benzimidazolylpyridine Re(I) complex (2.130(1) Å), indicating a weaker N–Re bond for the imidazole donor of RC2.32,33 Similarly, in a Re–pyridylimidazole complex with a 4-(N,N-dimethylamino)pyridine group instead of a bromine substituent, the Nimidazole–Re bond length is shorter as well (2.118 Å). This indicates that in RC2/RC3, the bond lengths are longer than those observed for related Re(azolylpyridine) complexes.
 |
| | Fig. 2 Crystal structures of RC2 (left) RC3 (right) omitting hydrogen atoms for clarity. | |
Table 1 Crystallographic data of catalysts RC2 and RC3
| |
RC2 |
RC3 |
| Empirical formula |
C21H19BrN3O3Re |
C21H18Br2N3O3Re |
| Formula weight |
627.50 |
706.40 |
| Crystal system |
Orthorhombic |
Monoclinic |
| Space group (no.) |
Pbca |
P21/c |
| a (Å) |
14.4135 (6) |
11.7322 (3) |
| b (Å) |
13.9657 (5) |
11.2485 (2) |
| c (Å) |
19.9434 (9) |
17.8809 (4) |
| α (deg) |
90 |
90 |
| β (deg) |
90 |
104.108 |
| γ (deg) |
90 |
90 |
| V (Å3) |
4014.5 (3) |
2288.56 |
| Z |
8 |
4 |
| F(000) (e) |
2400 |
1336 |
| μ (Mo-Kα) (mm−1) |
8.073 |
|
| Reflections collected |
43216 |
46047 |
| Independent reflections |
4593 (Rint = 0.0474) |
5254 (Rint = 0.0310) |
| Observed reflections (Fo ≥ 2σ(Fo)) |
43216 |
46047 |
| Refined parameters |
280 |
273 |
| Goodness-of-fit on F2 |
1.163 |
1.057 |
| Ra, Rwb (I ≥ 2σ(I)) |
0.0195, 0.0434 |
0.0149, 0.0292 |
| Ra, Rwb (all data) |
0.0212, 0.0440 |
0.0179, 0.0299 |
UV-Vis analysis
To characterize the light absorption properties of each complex, UV-visible absorption spectra were collected of the complexes dissolved in acetonitrile (MeCN) and are shown in Fig. 3. The optical properties of the complexes are summarized in Table 2. The data from these experiments revealed that the absorption onsets (λonset) of RC2–4 are blue-shifted compared to 1, in contrast to our hypothesis. We reason the blue shifts are due to the increased basicity of the imidazole donor in comparison to pyridine, in addition to the lower degree of conjugation in the imidazole ring. Complex RC5 shares a similar λonset to 1 at 425 nm. From the compounds, RC5 possesses the largest molar absorptivity coefficient while also possessing the most red-shifted MLCT absorption band in the visible light region. Interestingly, complexes RC2 and RC3 differ considerably in their molar absorptivity (Δε = 5853 M−1 cm−1) with the substitution of just one hydrogen for a bromine atom.
 |
| | Fig. 3 UV-Vis absorption spectra for catalysts 1–RC5 dissolved in MeCN. | |
Table 2 Optical properties of catalysts 1 and RC2–5a
| Catalyst |
λmax (nm) |
λonsetc (nm) |
ε (M−1 cm−1) |
EMLCT-GSd (eV) |
| Measured in acetonitrile. Indicates shoulder. Estimated from the intercept of the baseline and a tangent line on the UV-Vis spectrum on the low energy side of the absorption. Estimated from the onset of the low energy side of the absorption curve. The EMLCT-GS was converted from nm to eV using the equation 1240/EMLCT-GS nm = EMLCT-GS eV. |
| 1 |
292, 384b |
420 |
18951 |
2.95 |
| RC2 |
248, 337b |
370 |
3718 |
3.35 |
| RC3 |
246, 331b |
375 |
9571 |
3.31 |
| RC4 |
267, 336b |
380 |
19855 |
3.26 |
| RC5 |
306, 371b |
425 |
23381 |
2.92 |
Electrochemical analysis
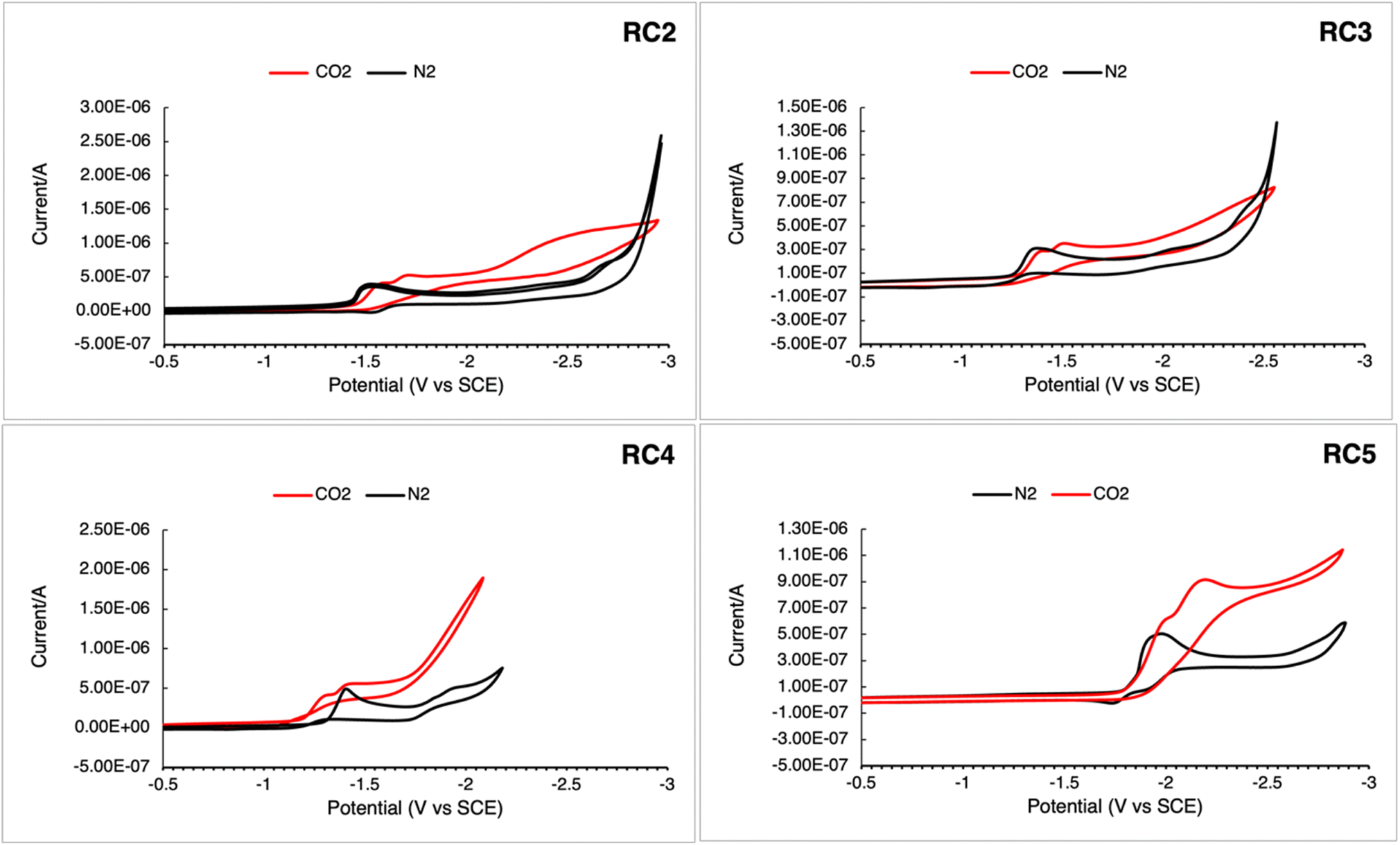
The redox properties of RC2–5 were measured using cyclic voltammetry (CV) to examine whether the complexes could serve as CO2 reduction catalysts under a CO2 atmosphere and shown in Fig. 4 and summarized in Table 3. When CO2 was sparged into each solution of dissolved complex, there was an increase in current associated with the first reduction potential, indicative of catalytic CO2 reduction activity. The increase in current for complexes RC2 and RC3 was also accompanied by shifts in the reduction wave to higher energy, while the current for RC5 was accompanied by a shift in the reduction wave to a lower energy.
 |
| | Fig. 4 Cyclic voltammograms of catalysts RC2–RC5. Spectra were recorded in dry MeCN, 0.1 M Bu4NPF6 in MeCN using a Pt pseudo-reference, Pt counter, and glassy carbon working electrode using a scan rate of 0.1 V s−1. All spectra were referenced to ferrocene (Fc) as an internal standard using the Fc+/Fc = 0.4 V couple vs. SCE.38 | |
Table 3 Electrochemical properties of catalysts RC2–RC5a
| Catalyst |
E(S+/S)b (V) |
E(S−/S)c (V) |
E(S*/S−)d (V) |
| Measured in dry MeCN. All values are reported vs. SCE and were measured by CV in 0.1 M Bu4NPF6 in MeCN using a Pt pseudo-reference, Pt counter, and a glassy carbon working electrode. Estimated as the onset of the oxidation peak due to irreversibility of the wave. Estimated as the onset of the reduction peak due to irreversibility of the wave. Estimated via the sum of the reduction potential and EMLCT-GS using the equation: E(S*/S−) = E(S−/S) + EMLCT-GS. |
| 1 (ref. 11) |
1.26 |
−1.20 |
1.33 |
| RC2 |
1.40 |
−1.35 |
2.00 |
| RC3 |
1.45 |
−1.30 |
2.01 |
| RC4 |
1.40 |
−1.35 |
1.91 |
| RC5 |
0.85 |
−1.55 |
1.59 |
| [Ru(bpy3)]2+ (ref. 34) |
1.29 |
−1.33 |
0.31 |
| fac-Ir(ppy)3 (ref. 34) |
0.73 |
−2.19 |
0.34 |
These measurements are also useful for determining whether the excited state reduction potentials of each complex are appropriately positioned for electron transfer from sacrificial electron donors like BIH or from photosensitizers (PS) such as Ir(ppy)3 or [Ru(bpy3)]2+. An energy level diagram showing relevant reduction potentials associated with the rhenium catalysts, the aforementioned photosensitizers, the standard reduction potential of CO2 to CO, and sacrificial electron donors BIH and TEA is given in Fig. 5. The reduction potentials of the dissolved complexes in nitrogen-sparged MeCN fall between −1.30 and −1.55 V vs. saturated calomel electrode (SCE), all of which are relatively higher in energy than the reduction potential of Re(bpy)(CO)3Br. The reduction potentials are also lower or very nearly lower in energy than both Ir(ppy)3 or [Ru(bpy3)]2+, which suggests that either could serve as a PS for the complexes. The excited state reduction potential of each complex falls between 1.59 and 2.01 V vs. SCE, which are all substantially lower than the oxidation potentials of both BIH and triethanolamine (TEOA) or triethylamine (TEA), which indicates that each can serve as an electron donor.
 |
| | Fig. 5 Energy level diagram illustrating of catalyst reduction potentials, calculated excited-state reduction potentials from the EMLCT-GS values, approximated range for the standard 2e−/2H+ reduction potential of CO2 to CO (and H2O) in acetonitrile based on the minimum and maximum pH range in the photocatalytic trials, and the oxidation potentials of BIH and TEA.35–37 | |
Photocatalytic CO2 reduction
The Re complexes were applied to the photocatalytic reduction of CO2 using anhydrous MeCN as the reaction solvent and a natural white light LED light source (λ > 400 nm) adjusted to 1 Sun intensity. Following initial optimization experiments (Table S2†) with catalyst RC2 and RC5, [Ru(bpy)3]2+ was selected as a PS due to its superior performance for photocatalytic CO2 reduction compared to Ir(ppy)3. A sacrificial electron donor BIH was used with TEOA where TEOA plays a secondary role as a base in deprotonating the BIH radical cation generated in the initial electron transfer process to the excited PS to limit back electron transfer.39 TEOA was chosen as the amine to pair with BIH as it gave better activity compared to another commonly used base, TEA. TEOA has been shown to react with CO2 to form a zwitterionic alkylcarbonate adduct that serves to capture CO2 in solution and facilitate proton transfer events.40 Gaseous products such as carbon monoxide (CO), methane (CH4), and hydrogen (H2) were detected and quantified by gas chromatography (GC). Formic acid (HCO2H) as a solution-phase product of the photocatalytic reactions was quantified by 1H NMR spectroscopy using ferrocene as an internal standard. The detailed procedure for quantifying HCO2H and a representative 1H NMR spectrum for this measurement is given in the ESI (Fig. S7†).
The results of photocatalytic CO2 reduction experiments are summarized and shown in Table 4 and Fig. 6. In the presence of PS [Ru(bpy)3]2+ and electron donor BIH, Re-complexes RC2–5 were found to be active in light-driven CO2 reduction with observed products HCO2H, CO, and H2. At a catalyst concentration of 1 μM, CO2 is primarily reduced to HCO2H as the dominant product, accompanied by H2 as a secondary byproduct from proton reduction. A smaller amount of CO was also formed. Interestingly, catalyst RC4, having resonance π-A group, exhibited the best catalytic performance for CO2 reduction with a turnover number (TON) value of 844 for HCO2H production and the highest selectivity for CO2 reduction products of 86% (Table 4, entry 1 and Fig. 6 (right)). Catalysts RC2 (TON for HCO2H = 280) and RC5 (TON for HCO2H = 275) with π-bridge and π-D group, respectively, showed lower catalytic activity overall than RC4, but similar activities and selectivities to one another with RC2 having a modestly higher carbon selectivity percentage (Table 4 and Fig. 6). Complex RC3 with Br-substituted π-bridge gave the second highest TON value for HCO2H of 593, which exceeds RC5 and the structurally similar complex RC2, but also produced the largest TON value for H2 at 362. This photocatalytic study establishes a clear correlation between the electron withdrawing π-A (–PhCN) and electron donating π-D (–NPh2) substituents attached to the imidazole ring. Among the catalysts investigated, the best performance was observed from the Re complex RC4 which contains a π-A group, followed by less electron withdrawing Br-substituted π-bridge RC3. Electronically neutral catalyst RC2 and π-D group substituted RC5 showed the lowest TON value for HCO2H generation. Notably, no HCO2H was observed for benchmark catalyst 1. A recent publication described that [Re(bpy)2(CO)2]+ also selectively formed HCO2H with a TON value of 428 from CO2 reduction, and they proposed a catalytic cycle similar to that of ruthenium catalyst [Ru(bpy)2(CO)2]2+.28 Doyle and coworkers have reported a photocatalyst, Re(CO)3(1-(1,10)phenanthroline-5-(4-nitro-naphthalimide))Cl, which is selective for HCO2H generation with a TON value of 533.41 We propose that the catalysts reported here also utilize the same catalytic cycle as that reported by Richeson and coworkers, which features a Re–H intermediate that undergoes CO2 insertion to give a Re–formato intermediate.28 Each of the imidazole-pyridine based catalysts reported here generate a mixture of HCO2H, CO, and H2 with HCO2H being the dominant product overall; likewise, TON values for H2 are greater than those of CO. The highest TON values for HCO2H production were obtained with catalysts bearing electron withdrawing substituents (RC3 and RC4) on the imidazole donor with HCO2H:H2 ratios of 5.7 (RC4) and 1.6 (RC3). The catalysts with more electron donating substituents (RC2 and RC5) slightly favor HCO2H, but with lower TON values and HCO2H:H2 ratios closer to 1. A rhenium-hydride species28 is expected to be a common intermediate responsible for the production of HCO2H and H2 during catalysis, both of which are formed in higher quantities than CO. The competition between HCO2H and H2 suggests that the hydricity of the putative Re–H is near 44 kcal mol−1 (i.e. the hydricity of formate in acetonitrile)42 and sensitive to the ligand substitutions. To the best of our knowledge, Re complex RC4 sets a new benchmark with the highest TON value for HCO2H production of 844 reported for rhenium-based CO2 reduction catalysts.
Table 4 Performance comparison of the catalysts in the photocatalytic reduction of CO2a
| Entry |
Catalyst |
HCO2H (TONb) |
CO (TONb) |
H2 (TONb) |
CSc (%) |
| Reactions were conducted in anhydrous MeCN with 1 μM catalyst/2 mL, 0.1 mM [Ru(bpy)3]2+, 50 mM BIH, and 5% (v/v) TEOA/MeCN. Turnover number (TON) values are reported at 72 h after catalysis was no longer occurring. Carbon-selective (CS) reduction percentage is calculated as CS% = [(CO TON + CH4 TON + HCO2H TON)/total observed product TON × 100]. Previously published; no HCO2H formation. |
| 1 |
RC4 |
844 |
88 |
148 |
86 |
| 2 |
RC3 |
593 |
126 |
362 |
67 |
| 3 |
RC2 |
280 |
150 |
224 |
66 |
| 4 |
RC5 |
275 |
113 |
266 |
59 |
| 5 |
1d |
0 |
436 |
222 |
67 |
 |
| | Fig. 6 CO formation over 72 h period (left) and the production of HCO2H, CO, and H2 after 72 h (right) in photocatalytic CO2 reduction reaction with catalysts RC2, RC3, RC4, RC5, and 1. All experiments were performed with 1 μM catalyst, 50 mM BIH, 0.1 mM [Ru(bpy)3]2+, and 5% (v/v) TEOA in anhydrous MeCN solution. The data points given are the average of two sample runs. | |
Under the same conditions, complex 1 outperformed catalyst RC2–5 regarding CO production (Fig. 6 and Table 4, entry 5). Comparing catalysts RC2–5, catalyst RC2 with a π-bridge on the imidazole ring showed comparatively better catalytic activity with a TON value of 150 for CO generation. In contrast, RC4 exhibited a relatively low TON value of 88 for CO evolution, strongly favoring HCO2H production. Toward H2 production, the highest activity (TONH2 = 362) was observed for complex RC3, followed by RC2 and RC5, with complex RC4 giving the lowest TON value for H2 (Table 4).
Catalytic performance of RC2–5 was also evaluated without using an external PS (Table S1†). Although the parent complex 1 is known to exhibit self-sensitized catalytic activity (TONCO = 55), only one of the Re complexes (RC5) absorbed visible light. Each of the catalysts exhibited very poor self-sensitized photocatalytic activity. Therefore, we conclude that catalysts RC2–RC5 required a PS for significant catalysis to occur.
The influence of catalyst concentration on TON values was also investigated using complexes RC4 and RC2 (Table 5). The TON values decreased dramatically when the catalyst concentration was changed from 1 μm to 10 and 100 μM. This is a typical result, as lowering the catalyst concentration has been shown to result in higher TON values owing to the catalytic sites being isolated from one another, therefore reducing deleterious side reactions.43,44
Table 5 Results of photocatalytic CO2 reduction with RC2 and RC4 at different catalyst concentrationsa
| [Catalyst] (μM) |
Catalyst |
HCO2H (TON) |
CO (TON) |
H2 (TON) |
CSb (%) |
| Maintained a constant irradiation time of 72 h and the same reaction conditions provided in Table 1. CS% = [(CO TON + CH4 TON + HCO2H TON)/total observed product TON × 100]. |
| 1 |
RC4 |
844 |
88 |
148 |
86 |
| RC2 |
280 |
150 |
224 |
66 |
| 10 |
RC4 |
53 |
16 |
18 |
79 |
| RC2 |
20 |
69 |
29 |
75 |
| 100 |
RC4 |
17 |
8 |
3 |
89 |
| RC2 |
8 |
6 |
5 |
74 |
The control experiments were performed using complex RC2 to determine the necessity of each component used in the photocatalytic CO2 reduction. The outcomes of these experiments are summarized in Table 6, where entry 1 is the result of our standard condition (all components present). Photocatalysis under N2 in the absence of CO2, or without the rhenium catalyst, or without irradiation (entries 5–7), resulted in negligible product formation, indicating the necessity of the substrate, the Re catalyst, and light, respectively. The impact of removing the PS (TONHCO2H with PS = 280 and without PS = 0, entries 1 and 2) and removing the electron donor BIH (TONHCO2H with BIH = 280 and without BIH = 10, entries 1 and 3) is significant. BIH is a better electron donor (more driving force for electron transfer) than TEOA, and thus, better catalytic activity is observed in the presence of BIH compared to using TEOA alone as the sacrificial electron donor. Interestingly, in the absence of TEOA, catalyst RC2 showed similar TON values for HCO2H compared to the experiment with TEOA (Table 6, entries 1 and 4). However, lower TON values for CO and higher values for H2 production are also observed without TEOA, indicating that the presence of TEOA strongly influences product selectivity.
Table 6 Summary of the photocatalytic control experiments with catalyst RC2a
| Entry |
Catalyst |
PSb |
e− donorc |
CO (TON) |
H2 (TON) |
HCO2H (TON) |
| Irradiation time was constant at 72 h. The activity under standard conditions is provided in entry 1. PS = [Ru(bpy)3]2+. When both components are listed both are present. Under nitrogen atmosphere. No irradiation. |
| 1 |
2 |
Yes |
BIH, TEOA |
150 |
224 |
280 |
| 2 |
2 |
No |
BIH, TEOA |
2 |
9 |
0 |
| 3 |
2 |
Yes |
TEOA |
16 |
14 |
10 |
| 4 |
2 |
Yes |
BIH |
45 |
388 |
228 |
| 5d |
2 |
Yes |
BIH, TEOA |
2 |
1 |
0 |
| 6 |
None |
Yes |
BIH, TEOA |
1 |
1 |
0 |
| 7e |
2 |
Yes |
BIH, TEOA |
0 |
0 |
0 |
In Table 7, catalysts RC2–5 are compared to recent Re(I) catalysts applied to light-driven CO2 reduction in the presence of a sacrificial electron donor. Müller et al.45 recently synthesized three rhenium tricarbonyl catalysts that produce TON values for CO as high as 125 without the presence of an external PS and using TEOA as the sacrificial electron donor. These excellent TONCO values were achieved by relatively simple modification to a 1,10-phenanthroline core by adding different heterocyclic substitutions at the 4 and 7 positions. Rotundo et al.46 also achieved similar TONCO values of 120 with ReI(CO)3 catalysts using BIH instead of TEOA while studying the effects of substituted 2,2′-bipyridine based ligands containing various functional groups in the second-coordination sphere. Liang and coworkers24 reported a self-sensitized rhenium catalyst utilizing pendant dipyrromethene-BF2 chromophores linked to a 2,2′-bipyridine core to achieve TONCO values as high as 1376 with BIH as the sacrificial electron donor. Although catalyst RC2 possesses similar TONCO values to Müller's and Rotundo's catalysts, their compounds function as effective self-sensitized photocatalysts while RC2 does not. With respect to formic acid production, RC4 in particular performs remarkably well compared to most Re catalysts that are generally known for their high selectivity for CO2-to-CO conversion. However, there are two recent rhenium catalysts that preferentially produce HCO2H. Spear et al.27 recently synthesized a rhenium catalyst supported by a phenanthroline nitro-naphthalimide ligand which selectively produced HCO2H in the absence of an external PS with a TON value as high as 533. Hameed et al.28 also achieve high selectivity and a HCO2H TON value of 428 utilizing the triflate salt of a bis(bipyridine) dicarbonyl rhenium complex which becomes less selective for HCO2H over the course of the reaction. Catalyst RC4 in comparison yields greater TON values for HCO2H, but with lower selectivity due to the formation of CO and H2, products that are not generated in large quantities or at all with the other two catalysts.
Table 7 Summary of the catalytic performance of selected rhenium catalysts applied to light-driven CO2 reduction
| Catalyst |
PS |
e− donor |
CO (TON) |
H2 (TON) |
HCO2H (TON) |
CSd (%) |
| NN = 1,10-phenanthroline having pyrrole. L = Ph-NH2 substituted 2,2′-bipyridine. BDP = 2,2′-bipyridine having dipyrromethene-BF2 chromophores. CS% = [(CO TON + HCO2H TON)/total observed product TON × 100]. |
| RC4 |
[Ru(bpy)3]2+ |
BIH, TEOA |
88 |
148 |
844 |
86 |
| RC4 |
No |
BIH, TEOA |
5 |
1 |
0 |
85 |
| fac-[Re(NN)a(CO)3Cl]45 |
No |
TEOA |
125 |
nd |
nd |
100 |
| fac-[ReCl(L)b(CO)3]46 |
No |
BIH |
120 |
nd |
nd |
100 |
| [Re(BDP)2c]24 |
[Ru(bpy)3]2+ |
BIH |
1376 |
nd |
nd |
100 |
| [Re(CO)3(1-(NN)a-5-(4-nitro-napthalimide))Cl]27 |
No |
BIH |
0.04 |
nd |
533 |
100 |
| cis-[Re(bpy)2(CO2)]+OTf−(1+OTf−)28 |
[Ru(bpy)3]2+ |
BIH, TEOA |
nd |
38 |
428 |
92 |
Conclusions
Carbon dioxide valorization to fuels or fuel precursors is an attractive strategy to reduce the overall use of non-renewable carbon fuel sources. Here, we synthesized four novel imidazolyl-pyridine Re complexes that were applied to the photocatalytic reduction of CO2 and compared to benchmark catalyst Re(bpy)(CO)3Br, 1. By replacing one of the pyridyl rings for a substituted imidazole group containing electron donating or accepting moieties, we were able to tune the electronic properties and light absorption of the complexes. Of note is that each of the imidazolyl complexes favored HCO2H production, which is unusual for Re(CO)3 based catalysts which overwhelming mediate the conversion of CO2-to-CO with high selectivity. Catalysts RC3 and RC4 strongly favored HCO2H generation, with RC4 being the most active and selective catalyst overall with an overall carbon selectivity (CS) of 86%. The CS percentage of RC3 is 67% due to a relatively large TON value for H2 evolution. Catalysts RC2 and RC5 also favor HCO2H as the dominant CO2 reduction product but make comparably higher amounts of CO and H2 and thus have lower carbon selectivity of 66 and 59%, respectively. In sharp contrast, benchmark catalyst 1 does not produce HCO2H under the same conditions. The production of HCO2H is rare for rhenium-based catalysts for CO2 reduction with only a few examples known in the literature. Catalyst RC4 gave a TON value for HCO2H as high as 844 at 1 μM catalyst concentration, further demonstrating the high stability of this catalyst under photochemical conditions.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2428908 and 2428909 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. I. S. and J. W. J. thank the National Science Foundation for generous funding through a CAREER award (CHE-1848478).
References
- U. S. E. P. Agency, Overview of Greenhouse Gases, https://www.epa.gov/ghgemissions/overview-greenhouse-gases.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels, Chem. Soc. Rev., 2009, 38(1), 89–99 RSC.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Electrocatalytic carbon dioxide reduction: from fundamental principles to catalyst design, Mater. Today Adv., 2020, 7, 100074 CrossRef.
- Z. Ma, U. Legrand, E. Pahija, J. R. Tavares and D. C. Boffito, From CO2 to Formic Acid Fuel Cells, Ind. Eng. Chem. Res., 2021, 60(2), 803–815 CrossRef CAS.
- F. Joó, Breakthroughs in Hydrogen Storage—Formic Acid as a Sustainable Storage Material for Hydrogen, ChemSusChem, 2008, 1(10), 805–808 CrossRef PubMed.
- J. Eppinger and K.-W. Huang, Formic Acid as a Hydrogen Energy Carrier, ACS Energy Lett., 2017, 2(1), 188–195 CrossRef CAS.
- H. Jahangiri, J. Bennett, P. Mahjoubi, K. Wilson and S. Gu, A review of advanced catalyst development for Fischer–Tropsch synthesis of hydrocarbons from biomass derived syn-gas, Catal. Sci. Technol., 2014, 4(8), 2210–2229 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–Co2+ combinations as homogeneous catalysts, J. Chem. Soc. Chem. Commun., 1983,(9), 536–538 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine), J. Chem. Soc. Chem. Commun., 1984,(6), 328–330 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2′-Bipyridine)tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts, Helv. Chim. Acta, 1986, 69(8), 1990–2012 CrossRef CAS.
- L. Kearney, M. P. Brandon, A. Coleman, A. M. Chippindale, F. Hartl, R. Lalrempuia, M. Pižl and M. T. Pryce, Ligand-Structure Effects on N-Heterocyclic Carbene Rhenium Photo- and Electrocatalysts of CO2 Reduction, Molecules, 2023, 28(10), 4149 CrossRef CAS PubMed.
- S. Sinha, E. K. Berdichevsky and J. J. Warren, Electrocatalytic CO2 reduction using rhenium(I) complexes with modified 2-(2′-pyridyl)imidazole ligands, Inorg. Chim. Acta, 2017, 460, 63–68 CrossRef CAS.
- H. Y. V. Ching, X. Wang, M. He, N. Perujo Holland, R. Guillot, C. Slim, S. Griveau, H. C. Bertrand, C. Policar, F. Bedioui and M. Fontecave, Rhenium Complexes Based on 2-Pyridyl-1,2,3-triazole Ligands: A New Class of CO2 Reduction Catalysts, Inorg. Chem., 2017, 56(5), 2966–2976 CrossRef CAS.
- L. Rotundo, E. Azzi, A. Deagostino, C. Garino, L. Nencini, E. Priola, P. Quagliotto, R. Rocca, R. Gobetto and C. Nervi, Electronic Effects of Substituents on fac-M(bpy-R)(CO)3 (M = Mn, Re) Complexes for Homogeneous CO2 Electroreduction, Front. Chem., 2019, 7, 417 CrossRef CAS.
- R. Stichauer, A. Helmers, J. Bremer, M. Rohdenburg, A. Wark, E. Lork and M. Vogt, Rhenium(I) Triscarbonyl Complexes with Redox-Active Amino- and Iminopyridine Ligands: Metal–Ligand Cooperation as Trigger for the Reversible Binding of CO2 via a Dearmomatization/Rearomatization Reaction Sequence, Organometallics, 2017, 36(4), 839–848 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Homogeneous CO Hydrogenation: Ligand Effects on the Lewis Acid-Assisted Reductive Coupling of Carbon Monoxide, Organometallics, 2010, 29(20), 4499–4516 CrossRef CAS.
- S. S. Roy, K. Talukdar and J. W. Jurss, Electro- and Photochemical Reduction of CO2 by Molecular Manganese Catalysts: Exploring the Positional Effect of Second-Sphere Hydrogen-Bond Donors, ChemSusChem, 2021, 14(2), 662–670 CrossRef CAS.
- A. Wilting, T. Stolper, R. A. Mata and I. Siewert, Dinuclear Rhenium Complex with a Proton Responsive Ligand as a Redox Catalyst for the Electrochemical CO2 Reduction, Inorg. Chem., 2017, 56(7), 4176–4185 CrossRef CAS PubMed.
- S. Sahu, P. L. Cheung, C. W. Machan, S. A. Chabolla, C. P. Kubiak and N. C. Gianneschi, Charged Macromolecular Rhenium Bipyridine Catalysts with Tunable CO2 Reduction Potentials, Chem.–Eur. J., 2017, 23(36), 8619–8622 CrossRef CAS PubMed.
- J. O. Taylor, G. Neri, L. Banerji, A. J. Cowan and F. Hartl, Strong Impact of Intramolecular Hydrogen Bonding on the Cathodic Path of [Re(3,3′-dihydroxy-2,2′-bipyridine)(CO)3Cl] and Catalytic Reduction of Carbon Dioxide, Inorg. Chem., 2020, 59(8), 5564–5578 CrossRef CAS PubMed.
- A. N. Hellman, R. Haiges and S. C. Marinescu, Rhenium bipyridine catalysts with hydrogen bonding pendant amines for CO2 reduction, Dalton Trans., 2019, 48(38), 14251–14255 RSC.
- L. Rotundo, C. Garino, E. Priola, D. Sassone, H. Rao, B. Ma, M. Robert, J. Fiedler, R. Gobetto and C. Nervi, Electrochemical and Photochemical Reduction of CO2 Catalyzed by Re(I) Complexes Carrying Local Proton Sources, Organometallics, 2019, 38(6), 1351–1360 CrossRef CAS.
- L.-Q. Qiu, K.-H. Chen, Z.-W. Yang and L.-N. He, A rhenium catalyst with bifunctional pyrene groups boosts natural light-driven CO2 reduction, Green Chem., 2020, 22(24), 8614–8622 RSC.
- L.-Q. Qiu, Z.-W. Yang, X. Yao, X.-Y. Li and L.-N. He, Highly Robust Rhenium(I) Bipyridyl Complexes Containing Dipyrromethene-BF2 Chromophores for Visible Light-Driven CO2 Reduction, ChemSusChem, 2022, 15(14), e202200337 CrossRef CAS.
- N. P. Liyanage, W. Yang, S. Guertin, S. Sinha Roy, C. A. Carpenter, R. E. Adams, R. H. Schmehl, J. H. Delcamp and J. W. Jurss, Photochemical CO2 reduction with mononuclear and dinuclear rhenium catalysts bearing a pendant anthracene chromophore, Chem. Commun., 2019, 55(7), 993–996 RSC.
- M. Rémond, Z. Zheng, E. Jeanneau, C. Andraud, Y. Bretonnière and S. Redon, 4,5,5-Trimethyl-2,5-dihydrofuran-Based Electron-Withdrawing Groups for NIR-Emitting Push–Pull Dipolar Fluorophores, J. Org. Chem., 2019, 84(16), 9965–9974 CrossRef PubMed.
- A. Spear, R. L. Schuarca, J. Q. Bond, T. M. Korter, J. Zubieta and R. P. Doyle, Photocatalytic turnover of CO2 under visible light by [Re(CO)3(1-(1,10) phenanthroline-5-(4-nitro-naphthalimide))Cl] in tandem with the sacrificial donor BIH, RSC Adv., 2022, 12(9), 5080–5084 RSC.
- Y. Hameed, P. Berro, B. Gabidullin and D. Richeson, An integrated Re(I) photocatalyst/sensitizer that activates the formation of formic acid from reduction of CO2, Chem. Commun., 2019, 55(74), 11041–11044 RSC.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Photocatalytic CO2 Reduction to Formic Acid Using a Ru(II)–Re(I) Supramolecular Complex in an Aqueous Solution, Inorg. Chem., 2015, 54(4), 1800–1807 CrossRef CAS PubMed.
- K. Hirano, S. Urban, C. Wang and F. Glorius, A Modular Synthesis of Highly Substituted Imidazolium Salts, Org. Lett., 2009, 11(4), 1019–1022 CrossRef CAS.
- E. Van Den Berge and R. Robiette, Development of a Regioselective N-Methylation of (Benz)imidazoles Providing the More Sterically Hindered Isomer, J. Org. Chem., 2013, 78(23), 12220–12223 CrossRef CAS PubMed.
- K. Wang, L. Huang, L. Gao, L. Jin and C. Huang, Synthesis, Crystal Structure, and Photoelectric Properties of Re(CO)3ClL (L = 2-(1-Ethylbenzimidazol-2-yl)pyridine), Inorg. Chem., 2002, 41(13), 3353–3358 CrossRef CAS PubMed.
- P. Cantero-López, Y. Hidalgo-Rosa, Z. Sandoval-Olivares, J. Santoyo-Flores, P. Mella, L. Arrué, C. Zúñiga, R. Arratia-Pérez and D. Páez-Hernández, The role of zero-field splitting and π-stacking interaction of different nitrogen-donor ligands on the optical properties of luminescent rhenium tricarbonyl complexes, New J. Chem., 2021, 45(25), 11192–11201 RSC.
- S. K. Pagire, N. Kumagai and M. Shibasaki, The Different Faces of [Ru(bpy)3Cl2] and fac[Ir(ppy)3] Photocatalysts: Redox Potential Controlled Synthesis of Sulfonylated Fluorenes and Pyrroloindoles from Unactivated Olefins and Sulfonyl Chlorides, Org. Lett., 2020, 22(20), 7853–7858 CrossRef CAS PubMed.
- C. Costentin and J. M. Saveant, Homogeneous Molecular Catalysis of Electrochemical Reactions: Catalyst Benchmarking and Optimization Strategies, J. Am. Chem. Soc., 2017, 139(24), 8245–8250 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion, Acc. Chem. Res., 2015, 48(12), 2996–3006 CrossRef CAS PubMed.
- C. Costentin, G. Passard and J. M. Saveant, Benchmarking of homogeneous electrocatalysts: overpotential, turnover frequency, limiting turnover number, J. Am. Chem. Soc., 2015, 137(16), 5461–5467 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96(2), 877–910 CrossRef CAS PubMed.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Architecture of Supramolecular Metal Complexes for Photocatalytic CO2 Reduction: Ruthenium–Rhenium Bi- and Tetranuclear Complexes, Inorg. Chem., 2005, 44(7), 2326–2336 CrossRef CAS PubMed.
- R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szalda and E. Fujita, Unexpected Roles of Triethanolamine in the Photochemical Reduction of CO2 to Formate by Ruthenium Complexes, J. Am. Chem. Soc., 2020, 142(5), 2413–2428 CrossRef CAS PubMed.
- D. R. Case, A. Spear, A. F. Henwood, M. Nanao, S. Dampf, T. M. Korter, T. Gunnlaugsson, J. Zubieta and R. P. Doyle, [Re(CO)3(5-PAN)Cl], a rhenium(I) naphthalimide complex for the visible light photocatalytic reduction of CO2, Dalton Trans., 2021, 50(10), 3479–3486 RSC.
- D. L. DuBois and D. E. Berning, Hydricity of transition-metal hydrides and its role in CO2 reduction, Appl. Organomet. Chem., 2000, 14(12), 860–862 CrossRef CAS.
- H. Shirley, X. Su, H. Sanjanwala, K. Talukdar, J. W. Jurss and J. H. Delcamp, Durable Solar-Powered Systems with Ni-Catalysts for Conversion of CO2 or CO to CH4, J. Am. Chem. Soc., 2019, 141(16), 6617–6622 CrossRef CAS PubMed.
- E. G. Ha, J. A. Chang, S. M. Byun, C. Pac, D. M. Jang, J. Park and S. O. Kang, High-turnover visible-light photoreduction of CO2 by a Re(I) complex stabilized on dye-sensitized TiO2, Chem. Commun., 2014, 50(34), 4462–4464 RSC.
- A. V. Müller, L. A. Faustino, K. T. de Oliveira, A. O. T. Patrocinio and A. S. Polo, Visible-Light-Driven Photocatalytic CO2 Reduction by Re(I) Photocatalysts with N-Heterocyclic Substituents, ACS Catal., 2023, 13(1), 633–646 CrossRef.
- L. Rotundo, D. C. Grills, R. Gobetto, E. Priola, C. Nervi, D. E. Polyansky and E. Fujita, Photochemical CO2 Reduction Using Rhenium(I) Tricarbonyl Complexes with Bipyridyl-Type Ligands with and without Second Coordination Sphere Effects, ChemPhotoChem, 2021, 5(6), 526–537 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Sean Parkin
b,
Sean Parkin