DOI:
10.1039/D5RA01525A
(Paper)
RSC Adv., 2025,
15, 15131-15137
Influence of the calcination conditions of the support on the activity of ruthenium-encapsulated porous hollow silica sphere catalysts for hydrogenation of carbon dioxide into formic acid
Received
4th March 2025
, Accepted 23rd April 2025
First published on 8th May 2025
Abstract
The present study investigated the influence of the calcination conditions of porous hollow silica spheres on the activity of a ruthenium-encapsulated porous hollow silica sphere catalyst for hydrogenation of carbon dioxide into formic acid. The hollow spheres were prepared at various calcination temperatures in air or in an argon flow. The amount of residual carbon content in the ruthenium-encapsulated hollow silica sphere catalysts increased with a decrease in the calcination temperature of the hollow silica sphere supports in air. Energy dispersive X-ray spectroscopy (EDS) and thermogravimetric (TG) analyses revealed that cetyltrimethylammonium bromide (CTAB) preferentially decomposed at calcination temperatures of up to 673 K, and most of the CTAB and carbon templates decomposed with the collapse of the hollow sphere catalyst particles in the catalysts calcined at 873 K. Moreover, the highest amounts of residual CTAB and carbon templates were found in the catalysts calcined in the argon flow. Differential thermal analysis (DTA), transmission electron microscopy (TEM), nitrogen sorption and X-ray diffraction (XRD) measurements showed that active ruthenium species were highly dispersed in the hollow spheres calcined in air, while a small amount of active ruthenium species with low dispersion were supported on the hollow spheres calcined in the argon flow. The catalyst calcined at 473 K exhibited the highest turnover number (TON) for formic acid formation (350 mol-HCOOH per mol-Ru), suggesting that the catalysts exhibited high activity not only owing to the high dispersion of the active species but also owing to the effective conduction of reaction heat by residual carbon species originating from CTAB in the nanospaces of the hollow spheres' shells.
Introduction
Anthropogenic consumption of materials has led to emission and accumulation of enormous amount of harmful compounds, such as greenhouse gases and ozone depletion compounds, in the global environment. Several compounds have induced extraordinary phenomena in nature, such as climate change and damage to ecosystems. Carbon dioxide has been regarded as a harmful compound affecting the global climate as a greenhouse gas.1–3 To reduce excess amount of this compound, various technologies have been developed.4–7 Among these technologies, the catalytic conversion of CO2 is not only expected to reduce the amount of the compound but could also allow obtaining useful chemicals if appropriate reactants and/or catalysts are chosen for its conversion.8–15 Hydrogenation reactions have been intensely developed and are considered potential processes for industrial use.11,13,14 Formic acid can be synthesized via a hydrogenation process under relatively mild reaction conditions, while the reverse reaction of formic acid to produce hydrogen can also proceed under mild conditions.15–21 For the features, formic acid has been regarded as a hydrogen-storage material and basic compound to produce various chemicals. Liquid-phase processes have been reported as effective processes, and homogeneous catalysts have been used for promoting these processes.22–27 Alternatively, heterogeneous catalysts have also been developed for the reaction because of their attributes such as reusability and recoverability. Various heterogeneous catalysts, including noble metals as active species, have been reported as highly active catalysts for the reaction.28–34 Our research group originally developed fine particle catalysts via a solvothermal process from metal salt solutions using alcohol solvents.28,30 In the samples, ruthenium fine particle catalysts were obtained through the solvothermal process in methanol solution and exhibited high activity for formic acid synthesis from supercritical carbon dioxide using mixed solvents with ethanol and deionized water.28 However, a significant dissolution of the active species in the reaction solution was observed under the conditions with a high amount of water solvent, resulting in a drastic decrease in the catalytic activity. Based on this result, our research group investigated the potential for encapsulation of the fine ruthenium particle catalyst in porous hollow silica spheres and assessed its effect on the activity and durability of the ruthenium-based catalysts. The ruthenium-encapsulated hollow silica sphere catalysts exhibited high activity and durability for the hydrogenation of supercritical carbon dioxide into formic acid even in pure deionized water solvent.35,36 Otherwise, the hydrogenation reaction into formic acid exothermically proceeds via the reaction formula described below;37
| CO2 + H2 ⇄ HCOOH, ΔG0 = −4 kJ mol−1 (in aqueous solution) |
This suggests that the reaction could be promoted by removing the reaction heat to effectively shift the reaction equilibrium for formic acid formation based on Le Chatelier's principle.
In the present study, we investigated the influence of co-encapsulated carbon-based materials on the activity of ruthenium-encapsulated hollow silica spheres. It has been reported that carbon-based materials possess varied thermal conductivity depending on factors such as their crystallinity and functional groups, and their conductivity could reportedly reach up to one order of magnitude higher than that of highly thermal conductive metals.38–41 In the present study, hollow silica spheres were prepared with spherical carbon templates fabricated from aqueous glucose solution and a cation surfactant template, namely cetyltrimethylammonium bromide (CTAB), which was used to form the porous shell structure of the hollow spheres. Various states of carbon-based materials could be expected to be prepared in the hollow spheres from these templates. Therefore, for controlling the states of the carbon-based materials formed from these templates, we investigated the calcination conditions to obtain hollow silica spheres and the influence of the conditions on the activity of a ruthenium-encapsulated hollow silica sphere catalyst for the hydrogenation of carbon dioxide into formic acid.
Experimental section
Catalyst preparation
For the fabrication of ruthenium-encapsulated hollow silica spheres, spherical carbon particles were prepared following a previous study and were used as templates.35 First, glucose (16.05 g, FUJIFILM Wako Chem. Co., ≧98.0%) was dissolved in a mixed solvent comprising 100 mL of deionized water and 3 mL of ethyl alcohol (FUJIFILM Wako Chem. Co., ≧99.5%) in a Teflon-lined stainless autoclave. The solution was heated at 373 K for 1 h following its hydrothermal treatment at 443 K for 9 h. The resulting suspension was filtrated and dried at 373 K for 2 h to obtain the template particles. Hollow silica spheres were prepared by coating silica on the templates via a sol–gel-based method following calcination processes to remove the carbonous components. The prepared templates (0.2000 g) were mixed with a surfactant, i.e., cetyltrimethylammonium bromide (CTAB, 0.0930 g, Kanto Chem. Co. Ltd, ≧96.0%), and L(+)-arginine (0.0872 g, FUJIFILM Wako Chem. Co., ≧98.0%) in a mixed solution comprising 15 mL of methyl alcohol and 2 mL of deionized water. After stirring the solution at 333 K for 30 min, tetraethoxysilane (0.56 mL, FUJIFILM Wako Chem. Co., ≧98.0%) was introduced into the solution, and then the solution was stirred at 333 K for 2 h. The resulting suspension was centrifuged (6000 rpm, 5 min) and washed with ethyl alcohol three times, and the obtained powder was then dried and calcined at 473–873 K for 3 h in air or in an argon flow (50 mL min−1). Precursors of the ruthenium-encapsulated hollow silica spheres were prepared via an immersion process in 15 mL of methyl alcohol solution comprising ruthenium chloride hydrate (RuCl3·nH2O (n ≈ 1.87), FUJIFILM Wako Chem. Co., ≧85.0%) and the hollow silica spheres degassed at 423 K for 6 h under vacuum conditions. After stirring at 298 K for 3 h, the suspension was solvothermally treated in a Teflon-lined stainless autoclave at 423 K for 10 h followed by centrifugation (8000 rpm, 10 min) and drying to obtain ruthenium-encapsulated hollow silica sphere catalysts.
Characterizations
The morphology of the ruthenium-encapsulated hollow silica sphere catalysts was analysed using a FE2000 field emission transmission electron microscopy instrument (FE-TEM, Hitachi). Compositional analyses of the catalysts were performed with a Brucker XFrash Mini SVE instrument for performing the energy dispersive X-ray spectroscopy (EDS) analyses. The thermochemical behaviours of the samples were examined using a Shimadzu DTG-60 thermogravimetric-differential thermal analysis system (TG-DTA), at a heating rate of 20 K min−1 in air up to 1273 K. The physicochemical properties of the hollow silica sphere supports were analysed by nitrogen sorption measurements with an ASAP2010 MC at 77 K system (Micrometrics). Powder X-ray diffraction (PXRD) patterns of the catalysts were recorded using a MultiFlex X-ray diffractometer (Rigaku) with Cu Kα radiation (λ = 0.15406 nm) operating at 30 kV and 16 mA to identify the crystalline phases, including for the catalysts.
Evaluation of the catalytic activity
The hydrogenation reaction of carbon dioxide was carried out in a 120 mL stainless steel autoclave with a magnetic stirrer (Taiatsu Techno). The catalyst was put into the autoclave with 5 mL of triethylamine and 17 mL of deionized water. The autoclave was then heated to 393 K, and the reactor was then pressurized to 5 MPa with H2 followed by the introduction of carbon dioxide from a cooled (268 K) reservoir by a high-pressure liquid chromatography pump up to 13.0 MPa total pressure, at which point the reaction was regarded to have started and was then maintained for 1 h. After the reaction, ethyl acetate (2 mL) was added to the mixture as an internal standard for the quantitative analysis of the product, and the liquid mixture from the autoclave was analysed using a Shimadzu GC 8A gas chromatograph equipped with 15% TSG-1 on a SHINCARBON A column (Shimadzu, 2 m × 3 mm) and a thermal conductivity detector. The yield was evaluated in terms of the turnover number (TON) of formic acid, which is the number of moles of formic acid produced per mole of ruthenium. The experimental error of the TON was within ca 7.3% in the present study.
Results and discussion
Morphological analysis of the hollow silica spheres prepared under various calcination conditions was conducted by TEM measurements. Fig. 1 displays the TEM images of the samples calcined at the temperatures ranging from 473 to 873 K in air. The samples calcined at 473 and 673 K consisted of homogeneous spherical particles with diameters of ca 500 and 850 nm. In the particles calcined at 673 K, hollow voids with shell thicknesses of ca 50 nm were observed. The result indicates that the spherical carbon templates remained intact at 473 K, but decomposed at 673 K to form hollow voids with expansion of the particle size. Otherwise, the sample calcined at 873 K included collapsed hollow spherical particles, indicating that a large amount of CTAB and/or the spherical templates had decomposed and the shell of the hollow sphere particles collapsed at this calcination temperature. Otherwise, fine particles surrounded by white circles were observed for all the samples, as shown in Fig. 1, and the particle sizes in the samples calcined at 473, 673, and 873 K were ca 5.0, 7.5, and 7.5 nm, respectively. In the sample calcined at 873 K, integration of the particles was observed, as shown in the white square. The results suggest that the active ruthenium particles were well-dispersed in the samples calcined at 473 and 673 K, while the active species were partially aggregated in the sample calcined at 873 K. In order to confirm the amount of residual carbonous compounds and encapsulated active ruthenium species, the compositions of the catalysts were evaluated from the results of the EDX analysis, as shown in Table 1. For the samples calcined in air, all the samples included almost the same ruthenium contents. Meanwhile the carbon contents of the samples calcined at 473 and 673 K were almost at the same level, while the carbon content of the sample calcined at 873 K was significantly lower. This result indicates that most of CTAB and/or the templates decomposed at around 873 K. Under the atmosphere with a very low oxygen concentration, carbonous compounds are difficult to decompose even at high temperature and thus a large amount of residual carbon species may be included in the calcined sample. In order to observe the influence of the atmosphere during the calcination process on the residual compounds, the composition of the sample calcined in an argon flow at 673 K was obtained and is listed in Table 1 for comparison. It can be seen that the sample calcined in argon flow possessed the highest carbon content among all the samples, indicating that most of CTAB and the spherical carbon templates remained in the sample after the calcination process. In addition, the amount of active ruthenium species was much lower than the other samples calcined in air, suggesting that the carbonous compounds formed from CTAB plugged in the nanopores in the shell of the hollow spheres, resulting in a relatively low amount of encapsulated active ruthenium species in the nanopores compared with the samples calcined in air. For a more detailed analysis of the residual carbonous compounds, the thermochemical properties of the samples calcined in air and in argon flow were analysed by TG-DTA, as shown in Fig. 2. Drastic weight losses centred at around 655 K were observed in the TG curves of the samples calcined at 473 and 673 K, and all the samples exhibited almost the same losses, as shown in Fig. 2A. In this figure, the weight loss of pristine CTAB is also shown, and the thermogravimetric behaviour was almost the same as those of the samples calcined at 473 and 673 K. This indicates that many parts of the CTAB and the templates remained in the sample throughout the calcination processes up to 673 K, while the residual surfactant mainly decomposed during the TG measurements of the catalysts calcined at 473 and 673 K. Otherwise, the thermogravimetric behaviour of the sample calcined at 873 K in air was significantly different from those of the other samples and pristine CTAB, and the weight loss in the same temperature range was significantly low. This indicates that most of the CTAB in the sample decomposed during the calcination process at 873 K, while the residual carbon templates mainly decomposed during the TG measurements. Fig. 2B displays the curves from the DTA of the catalysts conducted in parallel with the TGA. In this figure, a major exothermic peak centred at around 650 K could be observed in the curves of the catalysts calcined at 473 and 673 K in air. In this figure, the curves of the carbon templates and pristine CTAB are shown, and only the curve of the pristine CTAB included two endothermic peaks, indicating that the exothermic peak of the catalysts calcined at 473 and 673 K in air could not be assigned to the residual carbon templates and CTAB. It has previously been reported that the exothermic peak assigned to the oxidation of the metallic ruthenium of supported catalysts could be observed at around 650 K,42 which is similar to the exothermic peak of the catalysts calcined at 473 and 673 K in air in the present study. These results indicate that the metallic ruthenium species in the catalysts calcined at 473 and 673 K in air was oxidized at around these temperatures. The peak area in the curve of the catalyst calcined at 873 K in air was significantly smaller compared with the curves of the catalysts calcined at 473 and 673 K, suggesting that metallic ruthenium species with large particle sizes were gradually oxidized, thereby displaying a broad peak. Otherwise, the exothermic peak in the curve of the catalyst calcined in argon flow observed at around 573 K was relatively low compared with the other catalysts. This suggests that the active species of the catalyst calcined in air, especially those calcined at 473 and 673 K in air, were encapsulated in nanopores in the hollow sphere shells, while those of the catalyst calcined in argon flow were dispersed on the surface of the hollow spheres due to the plugging of the nanopores by the residual surfactant. To compare the state of the active ruthenium species in the samples at various calcination temperatures and atmospheres, powder XRD measurements of the catalysts were also conducted, as shown in Fig. 3. As shown in the figure, all the profiles included peaks at 2θ = ca 23.0°, 38.0°, and 43.5° assigned to amorphous silica, the metallic Ru(100) and Ru(101) planes (JCPDS card no. 06-0663), respectively.35,36,43,44 A broad peak was observed at 2θ = 43.5° in all the profiles of the catalysts calcined in air, assigned to the metallic ruthenium, with a significantly low intensity in the profiles of the catalysts calcined at 473 and 673 K in air. The result indicates that highly dispersed active ruthenium species were encapsulated in the nanopores of the hollow silica spheres calcined up to 673 K. The peak intensity of the catalyst calcined at 873 K in air was higher than those of the catalysts calcined at 473 and 673 K air, indicating that the dispersion of the active ruthenium species of the catalyst calcined at 873 K was slightly lower than that of the catalyst calcined at 473 and 673 K in air. Otherwise, the profile of the catalyst calcined at 673 K in the argon flow included two diffraction peaks at 2θ = 38.0° and 43.5° with relatively high intensities, suggesting that the active ruthenium species had partially aggregated during the preparation step of the active species due to the plugging of the pores in the hollow sphere shells with the residual surfactant, leading to the active species being dispersed only on the surface of the hollow spheres.
 |
| | Fig. 1 TEM images of the ruthenium-encapsulated porous hollow silica sphere catalysts calcined at (a) 473, (b) 673, and (c) 873 K in air. | |
Table 1 Compositions of the ruthenium-encapsulated porous hollow silica sphere catalysts prepared at various calcination temperatures evaluated through EDX analyses
| Calcination temp. [K] |
Atmosphere |
Si [at%] |
C [at%] |
Ru [at%] |
| 473 |
Air |
14.9 |
79.7 |
5.4 |
| 673 |
Air |
17.7 |
76.3 |
6.1 |
| 673 |
Argon |
6.8 |
91.2 |
2.0 |
| 873 |
Air |
68.3 |
25.7 |
6.0 |
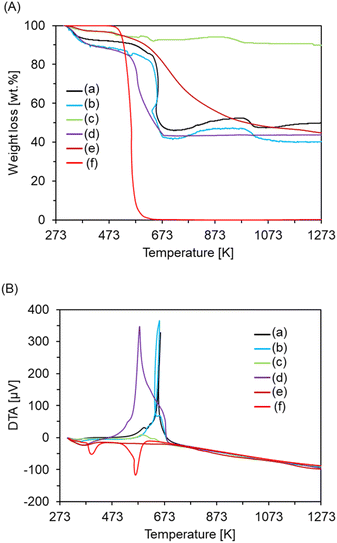 |
| | Fig. 2 (A) TG and (B) DTA curves of ruthenium-encapsulated porous hollow silica sphere catalysts calcined at (a) 473, (b) 673, (c) 873 K in air and (d) 673 K in an argon flow; (e) spherical carbon templates; and (f) pristine CTAB. Conditions: 20 K min−1 in air. | |
 |
| | Fig. 3 Powder XRD profiles of ruthenium-encapsulated porous hollow silica spheres calcined at (a) 473, (b) 673, (c) 873 K in air and (d) 673 K in an argon flow. | |
To identify the textural properties of the catalysts, we evaluated various physicochemical properties of the catalysts calcined in air and argon atmospheres. Fig. 4 displays the nitrogen sorption isotherms (A) and pore-size distributions (B) of the catalysts under various calcination conditions. The samples calcined at 673 and 873 K in air exhibited a significantly higher amount of nitrogen sorption than the samples calcined at 473 K in air and 673 K in argon flow. The isotherms of the samples calcined at 673 and 873 K in air were classified as being type IV isotherms according to the IUPAC classification system for physisorption isotherms with hysteresis loops in the relative pressure range of 0.45–1.0, and the samples displayed a type H3 loop.45 Over P/P0 ≈ 0.9, a drastic increase in the nitrogen sorption amount was observed in their isotherms, which was associated with the interparticle spaces formed by the hollow spherical particles. From the analyses of the pore-size distributions, as shown in Fig. 4B, a negligible amount of pore sizes was observed for the sample calcined at 673 K in the argon flow, while a small peak centred at ca 2.3 nm was observed in the pore-size distribution of the sample calcined at 473 K in air, suggesting that the two samples possessed small amounts of pores due to the high amounts of residual carbon templates and CTAB. Meanwhile, pores up to ca 5 nm were mainly included in the sample calcinated at 673 K in air, with a smaller amount of pores in the range of 5–100 nm, indicating that a large amount of CTAB had decomposed via calcination at this temperature. With increasing the calcination temperature up to 873 K, the number of nanopores up to 5 nm decreased, while the number of large pores in the range of ca 12–100 nm increased, suggesting a partial collapse of the hollow spherical particles. Table 2 lists various textural properties of the catalysts calcined under various conditions. Compared with the catalysts calcined at 473 K in air and 673 K in the argon flow, the specific surface areas and pore volumes of the catalyst calcined at 673 and 873 K in air were significantly higher, indicating that the amounts of residual carbon templates and CTAB were significantly low in the catalyst calcined at 673 and 873 K in air compared with those in the catalysts calcined at 473 K in air and 673 K in the argon flow. From those and the results in Table 1, the nitrogen sorption amount, specific surface area, and pore volume of the sample calcined at 473 K in air were found to be significantly lower than those of the sample calcined at 673 K in air, despite the amount of active ruthenium species being almost the same. This suggests that the sample calcined at 473 K included a higher amount of residual carbon species originating from the CTAB than the sample calcined at 673 K, and both the active ruthenium species and the residual carbon species were encapsulated in almost all the nanospaces in the shells of the hollow silica spheres. Otherwise, the ruthenium content in the sample calcined at 673 K in the argon flow was relatively low accompanied with a relatively low nitrogen sorption, specific surface area, and pore volume, indicating that a significant amount of the residual carbon species originating from the CTAB was encapsulated in many parts of the nanospaces in the shells of the hollow silica spheres.
 |
| | Fig. 4 (A) Nitrogen sorption isotherms and (B) pore-size distributions of ruthenium-encapsulated porous hollow silica spheres calcined at (a) 473, (b) 673, and (c) 873 K in air and (d) 673 K in an argon flow. | |
Table 2 Textural properties of the ruthenium-encapsulated porous hollow silica sphere catalyst under various calcination conditions
| Calcination temp. [K] |
Atmosphere |
Surface area [m2 g−1] |
Pore volumea [cm3 g−1] |
| Calculated from the desorption branch using the BJH method. |
| 473 |
Air |
23.4 |
0.0449 |
| 673 |
Air |
483.3 |
0.2654 |
| 673 |
Argon |
8.6 |
0.0200 |
| 873 |
Air |
296.9 |
0.2842 |
Fig. 5 shows the dependence of the TON for the synthesis of formic acid on the calcination temperature. From the results, it could be seen that the TON of the catalysts calcined in air increased with decreasing the calcination temperature, and the TON of the catalyst calcined in the argon flow was lower compared with the catalyst calcined in air. The results of the EDX analysis listed in Table 1 and the nitrogen sorption measurements in Fig. 4 suggest that a certain amount of active ruthenium species and residual carbon species originating from CTAB were encapsulated in the nanospaces of the hollow spheres. Consequently, the sample with an appropriate amount of encapsulated active ruthenium species accompanied with encapsulated residual carbon species in the nanospaces exhibited high activity for the hydrogenation of carbon dioxide into formic acid probably because of the high dispersion of the active species and thermal conductivity from the appropriate amount of the residual carbon species in the nanospaces.
 |
| | Fig. 5 Dependence of the activity for hydrogenation of carbon dioxide over ruthenium-encapsulated porous hollow silica sphere catalysts on calcination temperature. | |
Conclusions
This work investigated the influence of the calcination conditions of porous hollow silica spheres on the activity of ruthenium-based catalysts encapsulated in the hollow spheres. From the TEM images of the shells of the hollow spheres obtained via calcination in air over 473 K that parts of the hollow spherical particles had collapsed. From the TG-DTA, EDX, and nitrogen sorption analyses, it was revealed that many parts of the residual carbon templates and/or CTAB still remained in the samples obtained via calcination processes up to 673 K, and that CTAB preferentially decomposed to form nanopores below 5 nm in the hollow spheres. The result of the nitrogen sorption measurements also exhibited that the sample calcined at 873 K had a high number of pores in the range of 12–100 nm compared with the other samples, while a negligible amount of pores was observed in the sample calcined at 673 K in an argon flow. This suggested that the particles of the sample calcined at 873 K in air partially had collapsed, while most of the carbon templates and CTAB remained in the sample calcined at 673 K in the argon flow. The dispersion of the active ruthenium species increased with decreasing the calcination temperature in air, and both the dispersion and amount of the active species were relatively low in the sample calcined in the argon flow compared with those of the samples calcined in air, as indicated by the result of the XRD measurements. This suggested that the samples possessing large amount of nanopores in the shells of the hollow spheres from the calcination processes used to remove most of CTAB included highly dispersed active ruthenium species in the catalysts. The TONs for the synthesis of formic acid over the catalysts calcined at 473 and 673 K in air were significantly higher compared with the catalysts calcined at 873 K in air and 673 K in the argon flow. Consequently, the catalysts that included both a high amount of nanopores for encapsulation of the active ruthenium species and a certain amount of residual carbon species originating from the CTAB, which were probably useful for controlling the radiation or adsorption of the reaction heat, exhibited high activity for the hydrogenation of carbon dioxide into formic acid.
Data availability
All the data supporting this article have been included in the main manuscript.
Author contributions
Syntheses, characterizations, and activity tests by M. Kawaguchi and R. Takeda; work led by T. Umegaki and Y. Kojima; data analyses and manuscript writing by T. Umegaki; manuscript validation by all the authors.
Conflicts of interest
There are no conflicts to declare.
References
- T. R. Anderson, E. Hawkins and P. D. Jones, Endeavour, 2016, 40, 178 CrossRef PubMed
 .
. - R. J. Fleming, Environ. Earth Sci., 2018, 77, 262 CrossRef .
- M. Filonchyk, M. P. Peterson, L. Zhang, V. Hurynovich and Y. He, Sci. Total Environ., 2024, 935, 173359 CrossRef CAS .
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557 CrossRef CAS .
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87 CrossRef CAS .
- H. Ma, Y. Tong, X. Wang and H. Wang, RSC Sustainability, 2024, 2, 2731 RSC .
- K. S. Kappagantula, Y. Jiang, F. Pierobon, M. R. E. Rabby, J. Ramos, Y. Ni, A. Nittala, J. King, E. Nickerson, N. C. Nelson, W. Joo, R. John, J. C. Linehan, R. N. Aranzazu, S. K. Nune and D. J. Heldebrant, Green Chem., 2025, 27, 2392–2403 RSC .
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580 RSC .
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg and A. Bardow, Chem. Rev., 2018, 118, 434 CrossRef CAS PubMed .
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937 CrossRef CAS .
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed .
- K. M. G. Langie, K. Tak, C. Kim, H. W. Lee, K. Park, D. Kim, W. Jung, C. W. Lee, H.-S. Oh, D. K. Lee, J. H. Koh, B. K. Min, D. H. Won and U. Lee, Nat. Commun., 2022, 13, 7482 CrossRef CAS .
- D. N. Gorbunov, M. V. Nenasheva, M. V. Terenina, Y. S. Kardasheva, S. V. Kardashev, E. R. Naranov, A. L. Bugaev, A. V. Soldatov, A. L. Maximov and E. A. Karakhanov, Petrol. Chem., 2022, 62, 1 CrossRef CAS .
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245 CrossRef CAS .
- L. Zuo, Y. Deng, L. Chen, T. He, J. Yang and J. Zhang, ACS Catal., 2024, 14, 16795 CrossRef CAS .
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijin, Chem. Rev., 2017, 117, 9804 CrossRef PubMed .
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631 CrossRef CAS PubMed .
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247 RSC .
- H. Li, B. Peng, S. Lv, Q. Zhou, Z. Yan, X. Luan, X. Liu, C. Niu, Y. Liu, J. Hou, Z. Wang, Y. Chen, B. Yan, Z. Tang, C. Hou, K. Q. Y. Wu and R. Xu, Carbon Capture Sci. Technol., 2024, 13, 100322 CrossRef CAS .
- K. Park, H. Park, H. Yoon, K. R. Lee, S. Ahn, C. Kim, U. Lee, K. -D Jung and S. Yoon, Catal. Sci. Technol., 2024, 14, 5811 RSC .
- Y. Ma, R. Xu, X. Wu, Y. Wu, L. Zhaom, G. Wang, F. Li and Z. Shi, Top. Curr. Chem., 2025, 383, 2 CrossRef CAS .
- P. Munshi, A. D. Main, J. C. Linehan, C.-C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963 CrossRef CAS .
- J. Elek, L. Nádasdi, G. Papp, G. Laurenczy and F. Joó, Appl. Catal., A, 2003, 255, 59 CrossRef CAS .
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702 CrossRef CAS .
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168 CrossRef CAS PubMed .
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274 CrossRef CAS .
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526 CrossRef CAS .
- T. Umegaki, Y. Enomoto and Y. Kojima, Catal. Sci. Technol., 2016, 6, 409 RSC .
- S. K. Kabra, E. Turpeinen, M. Huuhtanen, R. L. Keiski and G. D. Yadav, Chem. Eng. J., 2016, 285, 625 CrossRef CAS .
- T. Umegaki, Y. Satomi and Y. Kojima, J. Jpn. Inst. Energy, 2017, 96, 487 CrossRef CAS .
- W. Zhang, S. Wang, Y. Zhao and X. Ma, Fuel Process. Technol., 2018, 178, 98 CrossRef CAS .
- M. S. Maru, S. Ram, R. S. Shukla and N.-U. H. Khan, Mol. Catal., 2018, 446, 23 CrossRef CAS .
- Q. Sun, X. Fu, R. Si, C.-H. Wang and N. Yan, ChemCatChem, 2019, 11, 5093 CrossRef CAS .
- Q. Sun, B. W. J. Chen, N. Wang, Q. He, A. Chang, C.-M. Yang, H. Asakura, T. Tanaka, M. J. Hülsey, C.-H. Wang, J. Yu and N. Yan, Angew. Chem., Int. Ed., 2020, 59, 20183 CrossRef CAS PubMed .
- T. Umegaki, E. Nagakubo, K. Saeki and Y. Kojima, Mater. Adv., 2023, 4, 6353 RSC .
- T. Umegaki, K. Saeki and Y. Kojima, Can. J. Chem. Eng., 2025 DOI:10.1002/cjce.25589 .
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231 CrossRef CAS .
- A. A. Balandin, Nat. Mater., 2011, 10, 569 CrossRef CAS .
- B. Kumanek and D. Janas, J. Mater. Sci., 2019, 54, 7397 CrossRef CAS .
- X. Guo, S. Cheng, W. Cai, Y. Zhang and X.-a. Zhang, Mater. Design, 2021, 209, 109936 CrossRef CAS .
- Q. Zhang, J. Peng, H. Xiong, S. Jiang, W. Li, X. Fu, S. Shang, J. Xu and G. He, Appl. Catal. B Environ., 2025, 362, 124759 CrossRef CAS .
- L. Jinxiang, Y. Lixin, G. Shiuying, H. Lijuan, T. Renyuan and L. Dongbai, Thermochim. Acta, 1988, 123, 121 CrossRef .
- V. Srivastava, Catal. Lett., 2016, 146, 2630 CrossRef CAS .
- J. Cored, A. García-Ortiz, S. Iborra, M. J. Climent, L. Liu, C.-H. Chuang, T.-S. Chan, C. Escudero, P. Concepción and A. Corma, J. Am. Chem. Soc., 2019, 141, 19304 CrossRef CAS .
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201 CrossRef .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Mahiro Kawaguchi,
Rintaro Takeda and
Yoshiyuki Kojima
*,
Mahiro Kawaguchi,
Rintaro Takeda and
Yoshiyuki Kojima