
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, molecular docking, and in vitro activity of a novel angiotensin-converting enzyme 2 inhibitor, LMS1007: a potential molecule in Covid-19 and cancer treatments†
Loai Saadah *a,
Ghina'a Abu Deiabb,
Qosay Al-Balasc and
Iman Bashetide
*a,
Ghina'a Abu Deiabb,
Qosay Al-Balasc and
Iman Bashetide
aFaculty of Pharmacy, Applied Science Private University, 11937 Amman, Jordan. E-mail: l_saadah@asu.edu.jo; Tel: +962798222044
bFaculty of Pharmacy, Yarmouk University, 21163 Irbid, Jordan. E-mail: ghinaadeiab@gmail.com; Tel: +962791101303
cFaculty of Pharmacy, Jordan University for Science & Technology, 22110 Irbid, Jordan. E-mail: qabalas@just.edu.jo; Tel: +962776337216
dFaculty of Pharmacy, Jadara University, 21110 Irbid, Jordan. E-mail: i.basheti@jadara.edu.jo; Tel: +962797708060
eFaculty of Medicine and Health, School of Pharmacy, The University of Sydney, 2006 NSW, Australia
First published on 8th May 2025
Abstract
Angiotensin-converting enzyme 2 (ACE2) is a validated commonly studied in the pathology of several diseases, including novel coronavirus and breast cancer. Herein, we report the synthesis, molecular docking, and validation of a novel ACE2 inhibitor that was previously discovered by our team based on diverse scaffolds of other ACE2 inhibitors and carnosine. The synthesized 4-subsitituted imidazole compound, namely, LMS1007, was characterized through 1H-NMR, LC-MS, and SFC. LMS1007 was then tested in vitro with ACE2 and viral spike protein–ACE2 inhibitor kits and was found to be approximately 100 times more potent as an ACE2 inhibitor than carnosine. However, it was less potent than the standard ACE2 inhibitor. In the same concentration range of the standard drug for ACE2 inhibition, LMS1007 demonstrated similar inhibitory effects on the interaction of the viral spike protein with ACE2. LMS1007 had an inhibitory concentration of 50% (IC50) at a concentration of 2.3 mM in all kits. LMS1007, similar to carnosine in breast cancer cell lines, exhibited potential inhibitory effects on the ACE2-mediated host uptake of Covid-19. Thus, a thorough review and discussion are provided on the role of ACE2 as an attractive target for the development of new drugs for Covid-19 treatment.
Introduction
Angiotensin-converting enzyme 2 (ACE2) has emerged as a pivotal molecular mediator in diverse pathological processes, notably including Covid-19 and breast cancer.1 The host's ACE2 receptor facilitates viral entry by binding to the viral spike glycoprotein, thereby enabling viral internalization into host cells.2 Interestingly, the modulation of ACE2 activity presents a viable therapeutic strategy to disrupt this critical viral–host interaction. For example, ACE2 downregulation has a protective effect against severe acute respiratory syndrome coronavirus 2 (SARS CoV-2) in cystic fibrosis patients.3 Additionally, allosteric changes that are induced by heparin facilitate ACE2–spike protein binding and viral entry.4Carnosine, a naturally occurring supplement, stands out in virtual screening and in silico studies as a potential ACE2 inhibitor, underpinning its broad-spectrum pharmacological efficacy (Fig. 1A).5 One of our research teams substantiated this hypothesis in an experimental seminal study.6 Therein, carnosine had shown both ACE2 and triple negative breast cancer (TNBC) cell line proliferation inhibition at the same concentration range. The IC50 value of carnosine for ACE2 activity inhibition and the antiproliferative effect was in the range of 100–300 mM. This constituted the first time that ACE2 inhibition and the antiproliferative effect were presented as a coupled effect. However, it is not clear whether ACE2 inhibition would have positive implications for SARS CoV-2 replication as it did for TNBC cell line proliferation. In this same study, MCF-7 proliferation increased but non-significantly in the 100–300 mM range. This contrasts with two other research groups that have shown that the antiproliferative effect of carnosine is associated with an IC50 value of about 68 mM.7 In this range,8 it has been shown that carnosine reduces the breast cancer tumor size in mice. Moreover, emerging research shows that various drugs interacting with the ACE2/Ang1-7/Mas and ACE/AngII/AT1 axes affect their anticancer properties.9 Therefore, further studies in this area may help elaborate on the role of ACE2 inhibition in various diseases and cell lines.
 | ||
| Fig. 1 Structures of (A) Carnosine, (B) ORE1001, and (C) LMS1007. | ||
Collaterally, an international patent has been previously registered by our research group for 25 novel substituted imidazole-based compounds that have been computationally predicted as potential ACE2 inhibitors.10 The compounds have been designed and studied in silico, resulting in a lower Gibbs free energy when docked with ACE2. The results indicated that the compounds have higher affinity and ACE2 inhibition than carnosine and ORE1001 (also known as MLN-4760), another standard ACE2 inhibitor (Fig. 1B). Pharmacokinetic predictor tools have shown that some of these compounds have drug-like properties. Therefore, in this review, we report the detailed docking, chemical modeling, chemical synthesis, characterization, and experimental validation of one of the compounds, namely LMS1007 (Fig. 1C). For the complete structures of all 25 compounds that were docked, please refer to our international patent.10
The present review aims to achieve four main objectives. First, a comprehensive molecular docking and chemical modeling of the novel compounds are presented to identify the most promising candidate for synthesis. Second, LMS1007 has been synthesized and characterized. Third, experimental validation of the LMS1007 inhibitory activity against ACE2 and viral spike protein–ACE2 binding has been reported. Finally, the most current literature on the topic is discussed to further emphasize the need for more research on ACE2 in Covid-19, as well as other diseases. The research hypotheses, derived from prior studies, posit the following:
LMS1007 will exhibit superior or more potent ACE2 inhibitory activity compared to carnosine.
LMS1007 will effectively inhibit ACE2 and its interaction with the Covid-19 spike protein within the same concentration range.
Methods
Detailed docking and chemical modeling
The minimization steps were as follows; first, the protein was solvated with water to mimic its natural environment using the “solvation” protocol. An explicit periodic boundary was selected to solve the protein with the periodic boundary conditions. Then, counterions were added to help keep the system neutral with a desirable value. The second step involved the minimization stage, which was successfully accomplished via three steps by avoiding any deformed structures of the protein. (1) The protein backbone and side chains were fixed by applying harmonic strain within the first minimization. Only the water molecules and the hydrogens of the protein were allowed to move. Smart minimizer was applied, as the starting structure is expected to be poor. Within it, the steepest descend protocol was applied to a gradient RMS of 3, and then a conjugate gradient protocol was applied to reach an acceptable energy minimized structure. (2) The backbone of the protein was fixed, and the side chains with the rest of the system were allowed to move and resonate with the same smart minimizer protocol. (3) The system was allowed to completely move without restrictions by using the same smart minimizer protocol. Finally, the water molecules, ions and counterions were removed from the system. The protein was ready for the subsequent steps of ligand docking.
Experimental chemistry
Synthesis of benzyl D-histidinate, 2.
 To a solution of compound 1 (100.0 g, 644.0 mmol) in 250.0 mL BnOH, SOCl2 (60.8 mL, 837.9 mmol) was added dropwise at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (dichloromethane/methanol = 5/1, reactant Rf = 0.08) until a new spot of compound 2 appeared. The reaction mixture was concentrated under reduced pressure, resulting in the crude product (230.5 g) as a white solid. Compound 2 was used in the next step without further purification. 1H NMR: (400 MHz, DMSO-d6) δ = 8.39 (s, 1H), 7.32–7.31 (m, 4H), 7.25–7.21 (m, 2H), 4.49 (s, 2H), 4.07 (t, J = 6.4 Hz, 1H), 3.21–3.09 (m, 2H).
To a solution of compound 1 (100.0 g, 644.0 mmol) in 250.0 mL BnOH, SOCl2 (60.8 mL, 837.9 mmol) was added dropwise at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (dichloromethane/methanol = 5/1, reactant Rf = 0.08) until a new spot of compound 2 appeared. The reaction mixture was concentrated under reduced pressure, resulting in the crude product (230.5 g) as a white solid. Compound 2 was used in the next step without further purification. 1H NMR: (400 MHz, DMSO-d6) δ = 8.39 (s, 1H), 7.32–7.31 (m, 4H), 7.25–7.21 (m, 2H), 4.49 (s, 2H), 4.07 (t, J = 6.4 Hz, 1H), 3.21–3.09 (m, 2H).
Synthesis of tert-butyl (R)-4-(3-(benzyloxy)-2-((tert-butoxycarbonyl)amino)-3-oxopropyl)-1H-imidazole-1-carboxylate, 3.
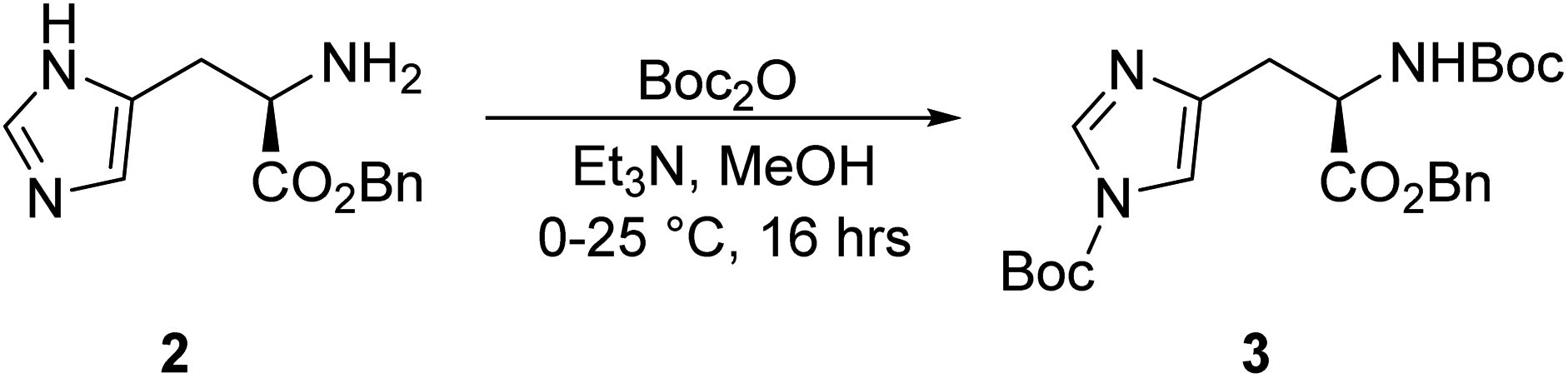 To a solution of compound 2 (230.5 g, 939.0 mmol) in 4600.0 mL methanol, Et3N (183.0 mL, 1.32 mol) and Boc2O (539.0 mL, 2.35 mol) were added dropwise at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.11, product Rf = 0.66) until compound 2 was completely consumed, and three new spots with lower polarity were detected. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified using column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in the crude product of compound 3 (22.5 g, 50.5 mmol) as a white solid.
To a solution of compound 2 (230.5 g, 939.0 mmol) in 4600.0 mL methanol, Et3N (183.0 mL, 1.32 mol) and Boc2O (539.0 mL, 2.35 mol) were added dropwise at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.11, product Rf = 0.66) until compound 2 was completely consumed, and three new spots with lower polarity were detected. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified using column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in the crude product of compound 3 (22.5 g, 50.5 mmol) as a white solid.
Synthesis of benzyl (tert-butoxycarbonyl)-D-histidinate, 4.
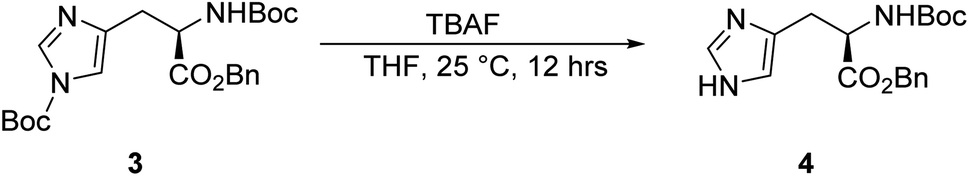 To a solution of compound 3 (22.5 g, 50.5 mmol) in 225.0 mL tetrahydrofuran (THF) was added TBAF (27.7 mL, 101.0 mmol) dropwise at 25 °C. Then, the mixture was stirred at 25 °C for 16 h. LC-MS showed that compound 3 was completely consumed, resulting in compound 4. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in compound 4 (12.8 g, 37.0 mmol, 23.7% yield) as a yellow solid. 1HNMR: (400 MHz, CDCl3) δ = 7.45 (s, 1H), 7.30–7.23 (m, 5H), 6.72 (s, 1H), 5.71 (br s, 1H), 4.46 (br s, 1H), 3.02 (br d, J = 4.1 Hz, 2H), 2.76–2.71 (m, 1H), 1.36 (s, 9H).
To a solution of compound 3 (22.5 g, 50.5 mmol) in 225.0 mL tetrahydrofuran (THF) was added TBAF (27.7 mL, 101.0 mmol) dropwise at 25 °C. Then, the mixture was stirred at 25 °C for 16 h. LC-MS showed that compound 3 was completely consumed, resulting in compound 4. The reaction mixture was concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in compound 4 (12.8 g, 37.0 mmol, 23.7% yield) as a yellow solid. 1HNMR: (400 MHz, CDCl3) δ = 7.45 (s, 1H), 7.30–7.23 (m, 5H), 6.72 (s, 1H), 5.71 (br s, 1H), 4.46 (br s, 1H), 3.02 (br d, J = 4.1 Hz, 2H), 2.76–2.71 (m, 1H), 1.36 (s, 9H).
Synthesis of benzyl Nα-(tert-butoxycarbonyl)-Nτ-(3,5-dichlorobenzyl)-D-histidinate, 6.
 To a solution of compound 4 (10.0 g, 28.9 mmol) in 100.0 mL MeCN, compound 5 (11.3 g, 57.9 mmol) and K2CO3 (24.0 g, 173.0 mmol) were added at 25 °C. The resulting mixture was stirred at the same temperature for 16 h. LC-MS showed that compound 4 was completely consumed and ∼13% desired mass was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1) to result compound 6 (4.0 g, 7.9 mmol, 27.3% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ = 7.34 (s, 1H), 7.24–7.19 (m, 6H), 6.87 (s, 2H), 6.24 (br s, 1H), 5.87 (br d, J = 7.6 Hz, 1H), 5.13 (br d, J = 12.3 Hz, 1H), 4.96 (br d, J = 12.2 Hz, 1H), 4.82 (br d, J = 6.6 Hz, 2H), 4.53 (br d, J = 3.2 Hz, 1H), 3.00–2.92 (m, 2H), 1.35 (s, 9H).
To a solution of compound 4 (10.0 g, 28.9 mmol) in 100.0 mL MeCN, compound 5 (11.3 g, 57.9 mmol) and K2CO3 (24.0 g, 173.0 mmol) were added at 25 °C. The resulting mixture was stirred at the same temperature for 16 h. LC-MS showed that compound 4 was completely consumed and ∼13% desired mass was detected. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1) to result compound 6 (4.0 g, 7.9 mmol, 27.3% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ = 7.34 (s, 1H), 7.24–7.19 (m, 6H), 6.87 (s, 2H), 6.24 (br s, 1H), 5.87 (br d, J = 7.6 Hz, 1H), 5.13 (br d, J = 12.3 Hz, 1H), 4.96 (br d, J = 12.2 Hz, 1H), 4.82 (br d, J = 6.6 Hz, 2H), 4.53 (br d, J = 3.2 Hz, 1H), 3.00–2.92 (m, 2H), 1.35 (s, 9H).
Synthesis of benzyl Nτ-(3,5-dichlorobenzyl)-D-histidinate, 7.
 A solution of compound 6 (5.0 g, 9.9 mmol) in HCl/dioxane (4 M, 57.0 mL) was stirred at 25 °C for 2 h. LC-MS showed that compound 6 was completely consumed. The reaction mixture was concentrated under reduced pressure, diluted with 50.0 mL aqueous Na2CO3 and extracted with 100.0 mL dichloromethane (DCM, 50.0 mL × 2). The combined organic layers were washed with 100.0 mL aqueous NaCl (50.0 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure, resulting in the crude product. The crude product was used in the next step without further purification. Compound 7 (4.1 g, crude) was obtained as a yellow solid.
A solution of compound 6 (5.0 g, 9.9 mmol) in HCl/dioxane (4 M, 57.0 mL) was stirred at 25 °C for 2 h. LC-MS showed that compound 6 was completely consumed. The reaction mixture was concentrated under reduced pressure, diluted with 50.0 mL aqueous Na2CO3 and extracted with 100.0 mL dichloromethane (DCM, 50.0 mL × 2). The combined organic layers were washed with 100.0 mL aqueous NaCl (50.0 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure, resulting in the crude product. The crude product was used in the next step without further purification. Compound 7 (4.1 g, crude) was obtained as a yellow solid.
Synthesis of benzyl Nα-((R)-2-(benzyloxy)-1-cyclopentyl-2-oxoethyl)-Nτ-(3,5-dichlorobenzyl)-D-histidinate, 9.
 To a solution of compound 7 (2.0 g, 5.0 mmol) in 20.0 mL DCM, compound 8 (2.30 g, 9.9 mmol) and AcOH (28.3 μL, 494.0 μmol) were added at 25 °C. After 1 h, NaB(OAc)3H (3.2 g, 14.8 mmol) was slowly added. The resulting mixture was stirred at 25 °C for 16 h. LC-MS showed that compound 7 was completely consumed. Then, the mixture was adjusted to pH ∼9 with 5.0 mL aqueous NaHCO3, and the mixture was extracted with 5.0 mL ethyl acetate (2.0 mL × 2). The combined organic layers were washed with 4.0 mL brine (2.0 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1) to give the racemate product (0.8 g, 1.3 mmol, 26.0% yield) as a white solid. The racemic mixture was further separated by SFC (condition: column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [CO2-IPA (0.1% NH3H2O)]; B%: 0%, isocratic elution mode) to give the chiral desired compound (9, 0.23 g, 1.3 mmol) as a yellow oil.
To a solution of compound 7 (2.0 g, 5.0 mmol) in 20.0 mL DCM, compound 8 (2.30 g, 9.9 mmol) and AcOH (28.3 μL, 494.0 μmol) were added at 25 °C. After 1 h, NaB(OAc)3H (3.2 g, 14.8 mmol) was slowly added. The resulting mixture was stirred at 25 °C for 16 h. LC-MS showed that compound 7 was completely consumed. Then, the mixture was adjusted to pH ∼9 with 5.0 mL aqueous NaHCO3, and the mixture was extracted with 5.0 mL ethyl acetate (2.0 mL × 2). The combined organic layers were washed with 4.0 mL brine (2.0 mL × 2), dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1) to give the racemate product (0.8 g, 1.3 mmol, 26.0% yield) as a white solid. The racemic mixture was further separated by SFC (condition: column: DAICEL CHIRALPAK AD (250 mm × 30 mm, 10 μm); mobile phase: [CO2-IPA (0.1% NH3H2O)]; B%: 0%, isocratic elution mode) to give the chiral desired compound (9, 0.23 g, 1.3 mmol) as a yellow oil.
Synthesis of LMS1007.
 To a solution of compound 9 (0.23 g, 435.0 μmol) in 2.0 mL THF was added HCl (3 M, 1.5 mL, 10.0 eq.). The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 1/1, reactant Rf = 0.40, product Rf = 0.21) until compound 9 was completely consumed. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 100 × 40 mm × 3 μm; mobile phase: [H2O (0.04% HCl)–ACN]; gradient: 15–35% B over 18.0 min). Compound LMS1007 (23.9 mg, 53.2 μmol, 12.24% yield, 97.9% purity) was obtained as a white solid. 1HNMR: (400 MHz, methanol-d4) δ = 8.94 (s, 1H), 7.55 (t, J = 1.8 Hz, 1H), 7.52 (s, 1H), 7.43 (d, J = 1.8 Hz, 2H), 5.41 (s, 2H), 3.94 (t, J = 6.3 Hz, 1H), 3.47 (br d, J = 6.9 Hz, 1H), 3.29–3.24 (m, 2H), 2.29–2.24 (m, 1H), 1.80–1.72 (m, 2H), 1.63–1.57 (m, 4H), 1.41–1.34 (m, 2H). LMS1007 was also checked by LC-MS and SFC. LMS1007 was stored at −20 °C until use. LMS1007 was prepared by dissolving different concentrations with different solvents. Amounts (≤10 mg) of LMS1007 were dissolved in water or DMSO (11%), similar to our previous work on carnosine.
To a solution of compound 9 (0.23 g, 435.0 μmol) in 2.0 mL THF was added HCl (3 M, 1.5 mL, 10.0 eq.). The mixture was stirred at 25 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 1/1, reactant Rf = 0.40, product Rf = 0.21) until compound 9 was completely consumed. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-HPLC (column: Phenomenex Luna C18 100 × 40 mm × 3 μm; mobile phase: [H2O (0.04% HCl)–ACN]; gradient: 15–35% B over 18.0 min). Compound LMS1007 (23.9 mg, 53.2 μmol, 12.24% yield, 97.9% purity) was obtained as a white solid. 1HNMR: (400 MHz, methanol-d4) δ = 8.94 (s, 1H), 7.55 (t, J = 1.8 Hz, 1H), 7.52 (s, 1H), 7.43 (d, J = 1.8 Hz, 2H), 5.41 (s, 2H), 3.94 (t, J = 6.3 Hz, 1H), 3.47 (br d, J = 6.9 Hz, 1H), 3.29–3.24 (m, 2H), 2.29–2.24 (m, 1H), 1.80–1.72 (m, 2H), 1.63–1.57 (m, 4H), 1.41–1.34 (m, 2H). LMS1007 was also checked by LC-MS and SFC. LMS1007 was stored at −20 °C until use. LMS1007 was prepared by dissolving different concentrations with different solvents. Amounts (≤10 mg) of LMS1007 were dissolved in water or DMSO (11%), similar to our previous work on carnosine.
Synthesis of 1,3-dichloro-5-(chloromethyl)benzene, 5.
 A mixture of compound 10 (10.0 g, 56.5 mmol) in 80.0 mL SOCl2 was degassed, purged 3 times with N2, and stirred at 60 °C for 12 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 1/1, reactant Rf = 0.30, product Rf = 0.69) until compound 10 was completely consumed. The reaction mixture was concentrated under reduced pressure to give a residue. The crude product was used in the next step without further purification. Compound 5 (8 g, crude) was obtained as a yellow solid.
A mixture of compound 10 (10.0 g, 56.5 mmol) in 80.0 mL SOCl2 was degassed, purged 3 times with N2, and stirred at 60 °C for 12 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 1/1, reactant Rf = 0.30, product Rf = 0.69) until compound 10 was completely consumed. The reaction mixture was concentrated under reduced pressure to give a residue. The crude product was used in the next step without further purification. Compound 5 (8 g, crude) was obtained as a yellow solid.
Synthesis of benzyl 2-cyclopentylacetate, 12.
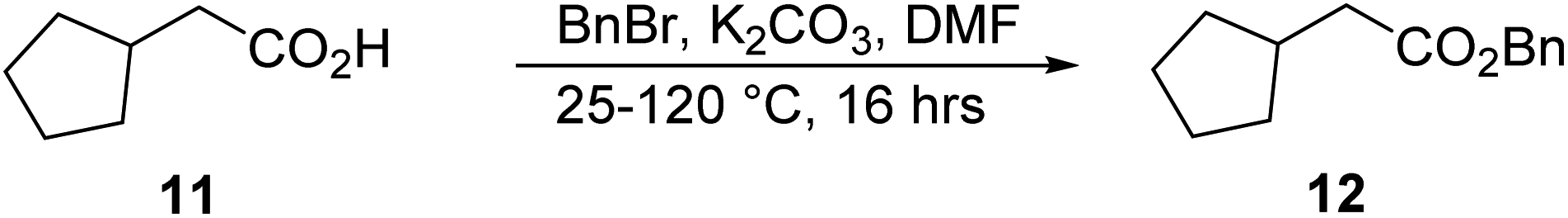 A mixture of compound 11 (19.6 mL, 156.0 mmol), BnBr (18.5 mL, 156.0 mmol), and K2CO3 (32.4 g, 234.1 mmol) were dissolved in 160.0 mL dimethylformamide (DMF) at 25 °C. Then, the mixture was stirred at 120 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.31, product Rf = 0.65) until compound 11 was completely consumed, and two new spots with lower polarity were detected. After completion, the reaction mixture was quenched with 100.0 mL of saturated aqueous NH4Cl solution and extracted with 200.0 mL ethyl acetate (100.0 mL × 2). The combined organic layers were washed with 100.0 mL brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in compound 12 (23.0 g, 105.4 mmol, 67.5% yield) as a yellow oil. 1H NMR: (400 MHz, CDCl3) δ = 7.42–7.33 (m, 5H), 5.15 (s, 2H), 2.41–2.39 (m, 2H), 2.29 (td, J = 7.6, 15.3 Hz, 1H), 1.86–1.84 (m, 2H), 1.65–1.58 (m, 4H), 1.21–1.19 (m, 2H).
A mixture of compound 11 (19.6 mL, 156.0 mmol), BnBr (18.5 mL, 156.0 mmol), and K2CO3 (32.4 g, 234.1 mmol) were dissolved in 160.0 mL dimethylformamide (DMF) at 25 °C. Then, the mixture was stirred at 120 °C for 16 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.31, product Rf = 0.65) until compound 11 was completely consumed, and two new spots with lower polarity were detected. After completion, the reaction mixture was quenched with 100.0 mL of saturated aqueous NH4Cl solution and extracted with 200.0 mL ethyl acetate (100.0 mL × 2). The combined organic layers were washed with 100.0 mL brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 10/1), resulting in compound 12 (23.0 g, 105.4 mmol, 67.5% yield) as a yellow oil. 1H NMR: (400 MHz, CDCl3) δ = 7.42–7.33 (m, 5H), 5.15 (s, 2H), 2.41–2.39 (m, 2H), 2.29 (td, J = 7.6, 15.3 Hz, 1H), 1.86–1.84 (m, 2H), 1.65–1.58 (m, 4H), 1.21–1.19 (m, 2H).
Synthesis of benzyl 2-cyclopentyl-2-hydroxyacetate, 14.
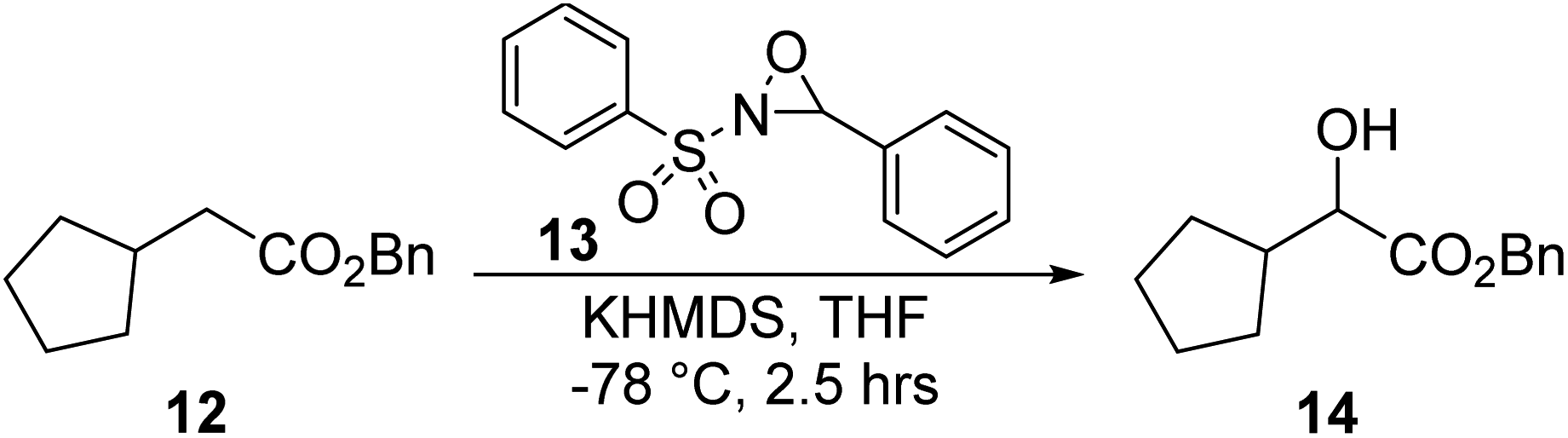 To a mixture of KHMDS (1 M, 126.0 mL, 1.2 eq.) in 100.0 mL THF, a solution of compound 12 (23.0 g, 105.4 mmol) in 100.0 mL THF was added dropwise at −78 °C. After the mixture was stirred for 30 min, a solution of compound 13 (33.0 g, 126.0 mmol) in 100.0 mL THF was added dropwise over 1 h. The reaction mixture was stirred at −78 °C for 1 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.75, product Rf = 0.30) until compound 12 was completely consumed. The reaction mixture was quenched with 200.0 mL aqueous NH4Cl and extracted with 200.0 mL diethyl ether (100.0 mL × 2). The combined organic layers were washed with 100.0 mL aqueous NaCl, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 50/1 to 10/1), resulting in compound 14 (7.8 g, 33.2 mmol, 31.5% yield) as a yellow oil.
To a mixture of KHMDS (1 M, 126.0 mL, 1.2 eq.) in 100.0 mL THF, a solution of compound 12 (23.0 g, 105.4 mmol) in 100.0 mL THF was added dropwise at −78 °C. After the mixture was stirred for 30 min, a solution of compound 13 (33.0 g, 126.0 mmol) in 100.0 mL THF was added dropwise over 1 h. The reaction mixture was stirred at −78 °C for 1 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 3/1, reactant Rf = 0.75, product Rf = 0.30) until compound 12 was completely consumed. The reaction mixture was quenched with 200.0 mL aqueous NH4Cl and extracted with 200.0 mL diethyl ether (100.0 mL × 2). The combined organic layers were washed with 100.0 mL aqueous NaCl, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 50/1 to 10/1), resulting in compound 14 (7.8 g, 33.2 mmol, 31.5% yield) as a yellow oil.
Synthesis of benzyl 2-cyclopentyl-2-oxoacetate, 8.
 A mixture of compound 14 (7.8 g, 33.2 mmol, 1.0 eq.) and Dess–Martin (22.5 g, 53.2 mmol, 16.5 mL, 1.6 eq.) in 78.0 mL DCM was degassed, purged 3 times with N2 gas, and stirred at 25 °C for 12 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 8/1, reactant Rf = 0.33, product Rf = 0.53) until compound 14 was completely consumed and two new spots were formed. The reaction mixture was quenched with 100.0 mL of 10% sodium thiosulfate at 20 °C and extracted with 100.0 mL DCM (50.0 mL × 2). The combined organic layers were washed with 50.0 mL aqueous NaCl, and subsequently concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 1/1) to give compound 8 (4.8 g, 20.6 mmol, 62.0% yield) as a yellow oil. 1H NMR: (400 MHz, CDCl3) δ = 7.31–7.27 (m, 5H), 5.20 (s, 2H), 3.45–3.37 (m, 1H), 1.80–1.74 (m, 4H), 1.57–1.59 (m, 4H).
A mixture of compound 14 (7.8 g, 33.2 mmol, 1.0 eq.) and Dess–Martin (22.5 g, 53.2 mmol, 16.5 mL, 1.6 eq.) in 78.0 mL DCM was degassed, purged 3 times with N2 gas, and stirred at 25 °C for 12 h. The reaction was monitored by TLC (petroleum ether/ethyl acetate = 8/1, reactant Rf = 0.33, product Rf = 0.53) until compound 14 was completely consumed and two new spots were formed. The reaction mixture was quenched with 100.0 mL of 10% sodium thiosulfate at 20 °C and extracted with 100.0 mL DCM (50.0 mL × 2). The combined organic layers were washed with 50.0 mL aqueous NaCl, and subsequently concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 100/1 to 1/1) to give compound 8 (4.8 g, 20.6 mmol, 62.0% yield) as a yellow oil. 1H NMR: (400 MHz, CDCl3) δ = 7.31–7.27 (m, 5H), 5.20 (s, 2H), 3.45–3.37 (m, 1H), 1.80–1.74 (m, 4H), 1.57–1.59 (m, 4H).
Experimental validation of activities and hypotheses
LMS1007, the standard kit inhibitor, and DMSO were tested in triplicate/quadruplicate sets at up to four dilutions. For LMS1007, the tested concentrations included 23.0 nM, 230.0 nM, 2.3.0 μM, 23.0 μM, 230.0 μM, 2.3 mM, and 23.0 mM. The standard kit inhibitor concentrations included 6.25 nM, 12.5 nM, 25.0 nM, and 50.0 nM. For DMSO, there were up to four concentrations: 0.0625%, 0.125%, 0.25%, and 0.5%. Subsequently, the activity percentage was calculated by dividing the slope of the fluorescence line for each measurement by the slope of the matching fluorescence line slope for enzyme control. Finally, the inhibition percentage was calculated for each concentration as (1 − % activity).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dilution of the LMS1007 was prepared in the provided assay buffer. 50.0 μL of the LMS1007 dilution was added to the designated wells, while 50.0 μL of the vehicle control was added to the control wells. The plate was incubated at room temperature for 30 minutes on a shaker.
:10 dilution of the LMS1007 was prepared in the provided assay buffer. 50.0 μL of the LMS1007 dilution was added to the designated wells, while 50.0 μL of the vehicle control was added to the control wells. The plate was incubated at room temperature for 30 minutes on a shaker.50.0 μL of ACE2 inhibitor screening reagent (His-tagged ACE2 protein) was added to all wells.
The plate was incubated at room temperature for 1 hour on a shaker. Wells were washed three times with wash buffer to remove the unbound ACE2 protein. 100.0 μL of Anti-His-HRP conjugate was added to each well. The plate was incubated for 1 hour at room temperature on a shaker.
Wells were washed three times with wash buffer. 100.0 μL of TMB substrate solution was added to each well and incubated in the dark at room temperature for 10 minutes. The reaction was stopped by adding 50.0 μL of stop solution to each well. The absorbance was immediately measured at 450 nm using a microplate reader. The percent inhibition was calculated by comparing the absorbance of wells with LMS1007 to the vehicle control wells using a similar method to the ACE2 inhibitor kit. The IC50 value for LMS1007 was determined by non-linear regression analysis using similar curve fitting as well. Vehicle control wells ensured that any inhibition observed was specific to the test compound and not the assay conditions.
:10 dilution of LMS1007 was prepared in the provided assay buffer. 50.0 μL of the LMS1007 dilution was added to the designated wells, while 50.0 μL of the vehicle control was added to the control wells. The plate was incubated at room temperature for 30 minutes on a shaker. 50.0 μL of ACE2 protein was added to all wells. The plate was incubated at room temperature for 1 hour on a shaker set to 300 rpm. Wells were washed three times with 300.0 μL of wash buffer to remove the unbound ACE2.100.0 μL of HRP-conjugate was added to all wells. Then, the plate was incubated at room temperature on a shaker, followed by washing with 300.0 μL of wash buffer. 100.0 μL of the TMB substrate solution was added to each well. The wells were incubated in the dark at room temperature for 10 minutes. 50.0 μL of stop solution was added to each well to terminate the reaction. Finally, the absorbance was immediately measured at 450 nm using a microplate reader.
Percent inhibition was calculated by comparing the absorbance of wells with LMS1007 to the vehicle control wells using a similar method to the previous two kits. The IC50 value for LMS1007 was determined by non-linear regression analysis using similar curve fitting as well. Vehicle control wells ensured that any inhibition observed was specific to the test compound and not the assay conditions.
Review search strategy
The National Library of Medicine (NLM) secondary database of biomedical and life science literature (PubMed, accessed July 14th, 2024) was queried using the term (Angiotensin Converting Enzyme 2 [Title]). The filters that were used included publications in the last year, in English language, and in humans. Retrieved studies were thoroughly screened with focus on Covid-19 related papers. Our objective for the review was to summarize the current knowledge on ACE2's role in Covid-19 disease. Specifically, to determine the biological and mechanistic relations of ACE2 and Covid-19, presence of inhibitors or activators of ACE2, and the experimental proofs of biological activity of these drugs in Covid-19.Results
Detailed docking and molecular modeling
803 kcal mol−1 and the minimization criteria of a gradient of 0.1 was satisfied. | ||
| Fig. 2 (A) The ACE2 (solid blue colour) interacts with spike protein (red), and the three active spheres are assigned. (B) ACE2 receptor with its three active sites assigned at the interface. | ||
 | ||
| Fig. 3 The spike receptor with its three active sites assigned to the interface. | ||
To start with, the ACE2 protein with the first active site (9.9 Å) showed no poses of any ligand, which could be explained by the inability of any ligand to bind to the active site at this position. This could be due to many reasons, such as inadequacy of the active site for binding the suggested ligands and the presence of repulsive forces that prevent proper binding. The second active site allowed for the binding of 70 ligands out of 260, with the highest scoring compound being the S,S-diastereomer of LMS1004 with a LibDock score value of 106.0. The third active site was the best of the three to have ligands bound to it, in which all 260 compounds had returned with docking scores. The highest score was related to the S,S,S-diastereomer of LMS1023, with a score of 105.3, while the S,S,R-diastereomer of LMS1025 was in the second rank with a score of 104.8. It can be pointed out from Fig. 4, panels A and B, that one of the carboxylic acid groups of the S,S,S-diastereomer of LMS1023 performs ionic interaction with the amino acids LYS353, while the other carboxylic acid shows unfavorable repulsive interactions with GLU35. The terminal hydrophobic side chain does not have any hydrophobic interactions. Similarly, the central imidazole ring does not have any interactions. The benzene ring interacts with the active site with hydrophobic interactions, while the fluorine atoms in it interact with LYS68.
 | ||
| Fig. 4 (A) 3D view of compound LMS1023SSS bound in its active site with a surrounding surface showing the H-donor (purple) and H-acceptor areas (green). (B) 2D view of LMS1023SSS within the active site, showing its interactions with the amino acids at the ACE2 surface. (C) 3D view of compound LMS004RS bound in its active site with a surrounding surface showing the H-donor (purple) and H-acceptor areas (green). (D) 2D view of LMS1004RS within the active site, showing its interactions with the amino acids at the spike surface. | ||
Then, the spike protein (9.9 Å) was employed for docking the assigned ligands and the results were like those of the ACE2 active site (9.9 Å) with zero compounds managing to bind to the surface of the protein. Subsequently, the 9.95 Å active site was docked with the same parameters that were used in every docking trial, and only 27 ligands were able to dock within this area. The highest compound, the R,R-diastereomer of LMS008 scored only 80.5. The third active site behaved in the same way as the ACE2 surface, and achieved the highest number of compounds to fit within it. Still, only 38 compounds have managed to bind to this site, where the R,S-diastereomer of LMS1004 scored the highest with a LibDock score of 97.3. The interaction plot of the R,S-diastereomer of LMS1004 can be seen in Fig. 4, panels C and D. The two carboxylic acid groups participated in binding with the active site. However, their interactions are limited to H-donor and H-acceptor. As there are no ionizable counterparts, they perform ion–dipole interactions with ASN501 and H-bonding with TYR449 and ALN498. In addition, a hydrophobic interaction is detected at the hydrophobic aliphatic cyclopropyl chain of the compound. Methoxy groups at the benzene ring have not shown any participation with the receptor.
Chemistry of LMS1007
The synthesis of LMS1007 started with the protection of the carboxylic acid functionality in D-histidine (1), resulting in compound 2 (Fig. 5). Further protection of the amino group was achieved with a Boc group to afford compound 3. The protection of the imidazole ring nitrogen was not aimed so it was deprotected in the next step using TBAF, resulting in compound 4. The imidazole ring was then functionalized with compound 5, which was synthesized from its commercially available corresponding alcohol derivative (10) to give compound 6. Having compound 6, the amino group deprotection was achieved using HCl/dioxane. The resulting compound (7) was functionalized with compound 8 that was obtained in three steps, as described later. The reaction resulted in a racemic mixture of the product for the new chiral center, and was then further purified by SFC to result in the required diastereomer. Finally, deprotection of the benzyl groups resulted in LMS1007. LMS1007 and the synthesized intermediates were confirmed by 1H-NMR and LC-MS. (All actual spectra files and graphs are available upon request). | ||
| Fig. 5 The total synthesis of LMS1007. | ||
Compound 8 was smoothly obtained from the commercially available 2-cyclopentyl acetic acid (11, Scheme 1). The carboxylic acid group was first protected with a benzyl group to result in compound 12, which was functionalized on the α-carbon with a hydroxyl group using compound 13 and a base to afford compound 14. Compound 14 was then converted to compound 8 by a reduction reaction.
 | ||
| Scheme 1 The total synthesis of LMS1007. | ||
Experimental validation of activities and hypotheses
LMS1007 ACE2 inhibition
LMS1007 inhibited ACE2 at doses in the range of 0.23 mM to 23.00 mM with IC50 of 2.27 mM. Computation of the inhibition percentage for each concentration was accomplished by calculating the ratio for the slopes of two lines. In the numerator, the slope was derived from the meticulously fitted linear equation characterizing fluorescence across five temporal points for each concentration. The denominator was the slope of the commensurate average slope associated with the enzyme control (DMSO), as delineated in Table 1. However, the inhibition exhibited a tempered character when juxtaposed with a standard inhibitor. The standard inhibitor demonstrated a more potent ACE2 inhibitory effect within a markedly lower and narrower concentration range ranging from 6.25 nM to 50 nM. On the other hand, the ACE2 inhibition was noticed at a maximum of 23 mM and a minimum of 0.23 mM. Fig. 6 panel A shows the triplicate inhibition% of both drugs at the tested concentrations. It is evident from the graph that LMS1007 at 23 mM produces a similar ACE2 inhibition% to the standard kit inhibitor at 50 nM. Similarly, the 2.3 mM of LMS1007 produces a similar effect to the standard kit inhibitor at 25 nM, and so on for the lower concentrations.| Test compound | Time point in minutes | ||||
|---|---|---|---|---|---|
| 2 | 17 | 32 | 47 | 62 | |
| a Measurement sample size (n) = 3 for each time point, x is the time point and y is the fluorescence reading; actual slopes and intercepts were divided by 1000 and rounded to the nearest integer.b % inhibition was calculated from activity%, which was the fraction of the trendline slope to the corresponding enzyme control dilution slope.c DMSO is the control. | |||||
| LMS1007(mM) | |||||
| 23 | 299 ± 22 | 365 ± 27 | 427 ± 33 | 496 ± 38 | 560 ± 43 |
| Slope = 4.4% inhibition = 96% | |||||
| 2.3 | 3403 ± 520 | 4129 ± 633 | 4862 ± 762 | 5586 ± 874 | 6314 ± 993 |
| Slope = 48.5% inhibition = 50% | |||||
| 0.23 | 6478 ± 329 | 7863 ± 394 | 9249 ± 468 | 10635 ± 544 | 12022 ± 624 |
| Slope = 92.4% inhibition = 6% | |||||
| 0.023 | 6711 ± 208 | 8143 ± 208 | 9577 ± 218 | 11014 ± 235 | 12446 ± 256 |
| Slope = 95.6% inhibition = 2% | |||||
| 0.0023 | 8324 ± 261 | 9754 ± 263 | 11204 ± 262 | 12656 ± 261 | 14108 ± 261 |
| Slope = 96.5% inhibition = 1% | |||||
| 0.00023 | 5997 ± 545 | 7459 ± 508 | 8921 ± 475 | 10382 ± 442 | 11844 ± 410 |
| Slope = 97.4% inhibition = 0% | |||||
|
|||||
| Kit inhibitor (nM) | |||||
| 50 | 972 ± 187 | 833 ± 116 | 952 ± 31 | 1027 ± 140 | 1072 ± 130 |
| Slope = 2.6% inhibition = 100% | |||||
| 25 | 1524 ± 302 | 2109 ± 153 | 2087 ± 419 | 2380 ± 337 | 2595 ± 223 |
| Slope = 16.1% inhibition = 50% | |||||
| 12.5 | 1651 ± 243 | 2131 ± 145 | 2388 ± 117 | 2934 ± 117 | 3176 ± 338 |
| Slope = 25.7% inhibition = 19% | |||||
| 6.25 | 1875 ± 95 | 2608 ± 210 | 3097 ± 69 | 3833 ± 648 | 3987 ± 271 |
| Slope = 36.3% inhibition = 0% | |||||
|
|||||
| Enzyme/DMSO controlc | |||||
| Enzyme controla | 1876 ± 238 | 2407 ± 319 | 2836 ± 401 | 3363 ± 401 | 3792 ± 438 |
| Slope = 31.9% inhibition = 0% | |||||
| Enzyme controlb | 6542 ± 169 | 8557 ± 310 | 9918 ± 199 | 10863 ± 620 | 12727 ± 81 |
| Slope = 97.8% inhibition = 0% | |||||
 | ||
| Fig. 6 Mean inhibition% of ACE2 at concentrations ranging from 0.23 μM to 23 mM for LMS1007 and from 6.25 nm to 50 nM for the standard kit inhibitor. | ||
LMS1007 and viral spike–ACE2 inhibition kits
In the range of concentrations in Fig. 6 panel B, LMS1007 displays similar inhibition on the SARS CoV-2 spike protein and ACE2 interaction with about the same IC50 of 2.3 mM. As a result, the ACE2 active site may either directly or indirectly be involved in the viral spike interaction with ACE2. However, the ACE2–spike S1 kit shows static inhibition across the whole range of concentrations. IC50 in this third kit is impossible to calculate.Review and discussion
The ACE2 enzyme plays a key enzymatic role under a variety of conditions. Readers should note that the ACE2 role is ubiquitous, just like the entire renin angiotensin aldosterone systems (RAAS) that it belongs to. Most notable are the applications for ACE2 in cardiovascular and related diseases. For example, candesartan has been reported to reduce renal ubiquitination, and lysosomal degradation in male mice.12 Growing evidence suggests that ubiquitination is the main cellular mechanism by which the host regulates the ACE2 levels in tissues, including in Covid-19 patients.13 To elaborate a bit more on this emerging concept, ACE2 ubiquitination contributes to hypertension, but offers a new druggable target for elevated blood pressure.14 In the latest research by Elgazzaz et al.,14 the full mechanisms are reported. Therefore, according to Wang et al., candesartan resulted in an upregulation of ACE2 in mice, and hence gender-differential, reno-protective and antihypertensive effect.12 The upregulation-mediated ACE2 renoprotection has been confirmed in another rat model as well.15 In contrast, ablation of ACE2 in rats followed by Lactobacillus paracasei, which were genetically engineered for the production of ACE2, has resulted in a female-rat specific reduction in the blood pressure.16 Clinically, this renoprotection has been confirmed in 777 patients, where patients with higher ACE2 in plasma and urine had a lower progression of diabetic kidney disease and albuminuria.17 It was suggested that the benefit of ACE2 upregulation extends to pulmonary hypertension.18 Moreover, genetic polymorphisms in ACE2 may account for a greater probability of arterial hypertension and cardiovascular disease in systemic sclerosis patients.19 Logically, these cardiovascular findings would generate various hypotheses to test the ACE2 role in heart failure. In fact, a study on 103 myocardial tissue samples sourced from patients in various heart failure stages have shown that ACE2 significantly increased and decreased, respectively, in cardiomyocytes and non-cardiomyocytes from heart failure stages A to C (P < 0.001).20 These changes in ACE2 were associated with left ventricular remodeling in these histopathological samples. Kellum et al. have confirmed that these cardiac effects are especially derived from the loss of ACE2 in pericytes, suggesting that these cells are both the target of SARS CoV-2 cardiac infection and elicitor of arrhythmogenesis.21 This active and promising research on ACE2 has triggered researchers to develop innovative models for the identification of ACE2 as a promising marker in varied pathologies, including biosensors for viral spike–ACE2.22–24 Now, these various research articles, although they may seem confusing, point to one fact that ACE2 modulation is worth investigating, as both ACE2 inhibitors and activators may help fine-tune a variety of pathologies. Cancer is another broad group of diseases, where ACE2 seems to have important pathological roles. For example, researchers have found that ACE2 overexpression in the liver impacts the risk of fibrosis and hepatocarcinoma by inhibiting Hepatic Stellate Cell (HSC) autophagy.25 This is mediated through the AMP-activated protein kinase (AMPK)/mammalian target of the rapamycin (mTOR) pathway. On this basis, ACE2 inhibitors, blockers, or modulators could have implications in the development and progression of liver cancer. Saadah et al. found that ACE2 modulation is a promising target, especially in TNBC.6 One sole study about Kawasaki disease has shown that ACE2 was higher and correlated with lower platelet levels in this population.26 Excluding the four SARS CoV-2 studies cited in this paragraph, we have a total of ten in Fig. 6 diseases other than SARS CoV-2. However, our query of NLM-PubMed, which returned a total of 47 articles, showed that most studies (remaining 37, 79%) were related to SARS CoV-2 and Covid-19.Taking the Covid-19 articles, there has been only one study that found no correlation of plasma ACE2 and that it might not be a good surrogate marker of lung ACE2.28 However, this study has provided no dynamic assessment of the ACE2 level and activity in these different groups. Indeed, it has also found divergent changes in these levels by age, gender, and antihypertensive treatment. Additionally, other researchers have found that the wild type ACE2 has higher plasma ACE2 levels compared to other variants, and this dimension was missing in the previous study by Xie et al.29 This soluble ACE2 has an important role and was protective against the aggressive Delta SARS CoV-2 for extended durations post infection.30 Moreover, the current research direction in this area has pushed towards developing 3D-lung models that incorporate the ACE2 expression in the cells due to the consensus that this molecule is pivotal in Covid-19 lung infections.31 Although soluble ACE2 levels are similar in SARS CoV-2 positive and negative patients, the expression levels indicated by micro RNA (miRNA), miR-141-3p, are different.32 This means that the virus-positive patients may, in fact, respond by downregulating/upregulating ACE2. Guo et al. confirmed that miRNA is downregulated initially in SARS CoV-2-infected patients, and then upregulated later in the disease cycle.33 A similar finding in ACE2 research in breast cancer has been described.16,34–36 There, the less aggressive breast cancer cells have an initially marked reduction in the ACE2 levels compared to the normal tissues, followed by a rise in the ACE2 expression in TNBC cell lines. Moreover, higher ACE2 transcription is positively correlated with the transmission of SARS CoV-2 among hospital-admitted patients, but not healthcare providers during Covid-19 outbreaks.37 Furthermore, the expression of ACE2 and risk of ocular SARS CoV-2 infection are higher in smokers than non-smokers.38 In smokers, it has been also shown that washing eyes with water reduces ACE2 and SARS CoV-2 ocular infections. This concept of expression regulation is further presented in both patients with colon tissues affected by cancer and with unaffected tissues in response to SARS CoV-2 infection.39 At other healthcare front lines, more soluble ACE2 in the brain seem to increase the risk for Alzheimer disease and other Central Nervous System (CNS) pathologies even though its relation to SARS CoV-2 CNS infection was unclear.40 Moreover, pityriasis rosea-like lesions and other cutaneous complications have been less and of mitigated severity in Covid-19 patients with higher ACE2 expression.41 As an extension to increased expression, multiple studies have shown that this was coupled with increased ACE2 peptidase activity.42,43 Indeed, one of these studies has found that the increased ACE2 peptidase activity is specific to SARS CoV-2 among all other coronaviruses.43
The current work offers a novel approach in determining the potential active sites. These were suggested based on protein surfaces analysis based on measuring distances between the proteins. Therefore, a comparative study with the active site in others work will not be possible or applicable. Studies on the ACE2 docking are based on the defined active site that has a zinc atom, and this active site is not fully involved in the spike–ACE2 recognition. Mechanistically, the previous reports had some of the amino acids involved in the spike Receptor Binding Domain (RBD) interaction with the host ACE2, similar to our docking and chemical modeling analysis.44,45 For example, the amino acid ASN501 has been constantly found to form H-bonding with TYR449 and ALN498 similar to the current analysis. Interrupting the RBD-ACE2 interactions subsequently inhibit various host protein interactions, which then lead to protection against SARS CoV-2 entry into host cells.46 Additionally, SARS CoV-2 is a dynamic disease with new variants like XBB.1.5 and XBB.1.16, having novel RBD that can evade the host antibodies against the virus.47 On the other hand, ACE2 is a highly glycated enzyme and the glycans at the C-terminus can influence its binding with viral spike.48 In contrast de-glycosylation at other sites can improve the spike RBD-ACE2 binding.49 This variation in the effect of de-glycosylation on viral spike RBD-ACE2 binding is perfectly in line with research that also found that SARS CoV-2 activates ACE2 in a glycosylation-independent manner.42 Researchers have also shown that ACE2 activation is linked to its hinge-bending region, while upregulation involves various signaling pathways, transcription factors, and epigenetic modulators.49 On the other hand, Mei et al. have suggested that the dual-targeting of ACE2/Neuropilin-1 (NRP1) could be a promising broad-spectrum target for treating SARS-CoV-2 emerging variants of coronavirus.50 Accordingly, it has been recommended that future studies explore novel roles of ACE2 activators/inhibitors or up-/down-regulators in disease treatments to translate discoveries in this field to bedside applications.
Before exploring the discoveries made on the ACE2 modulators, we will cover a few more elements on this protein herein. Genetically, the G/G genotype of ACE2, rs2106809, resulted in patients that had more severe Covid-19 symptoms and adverse disease outcomes.51 So, ACE2 genetics play a role too. Additionally, in mice, the manipulation of these genes has enabled some groups to produce new strains of mice with higher ACE2 expression and lower severity of Covid-19 mortality compared to the original mouse models.52 Other groups have shown that Covid-19 vaccine recipients and convalescent patients had more ACE2 autoantibodies.53 Collectively, these studies and the previous ones are consistent in increasing the demand for testing all types of ACE2 modulators on SARS CoV-2 infection and progression and in other related disease pathways.
In terms of finding ACE2 modulators and confirming the therapeutic potential in SARS CoV-2 and other related diseases, there seems to be more value to ACE2 inhibition than activation. For example, Melhem et al. have previously shown that carnosine is a mild ACE2 inhibitor in the range of 100–300 mM concentration. This is the same range where carnosine demonstrates broad spectrum anticancer effects, including in breast malignancies.6 Rebelo and co-workers were able to use an ACE2 decoy, namely NL-CVX1, that mimics ACE2 and thus inhibits the spike protein recognition by the host ACE2 to interrupt the viral spread.54 Yet, in their study, the decoy-exposed patients still made SARS CoV-2 antibodies and memory T cells. Most other investigations have focused on preventing the spike–ACE2 interaction. For example, Pashameah and colleagues have discovered raffinose sulfate inhibitory effects on spike–ACE2 with no effect for the drug on ACE2 peptidase activity.55 This inhibitory effect is coupled with an improved tolerability compared to other sulfated polysaccharides, e.g., heparin and heparan. Luteolin also has inhibitory effects on spike–ACE2 binding.56 As a result, luteolin has been shown to be associated with less platelet aggregation and thrombosis. This link between ACE2 and platelet spread and aggregation in Covid-19 patients has been confirmed by another group.57 In another study, natural herbal medicine Eunkyo-san diminishes the levels of both inflammatory cytokines and ACE2, and may therefore help in mitigating the cytokine storm of Covid-19.58 Biscoumarin glycosides from P. trimera roots hold potential as natural ACE-2 inhibitors for preventing SARS-CoV-2 infection both in silico and in vitro.59 One of the very first drugs that were repurposed for Covid-19 is hydroxychloroquine, and findings showed that only the (S)-(+) enantiomer inhibits ACE2 in comparison to the (R)-(−) enantiomer.60 However, data have shown that ursodeoxycholic acid prevents both the Delta and Omicron variants of SARS CoV-2 infection in hamster models.61 More ACE2 inhibitors are on the way as well, with Harman et al. proposing the most potent ACE2 inhibitors in vitro, a group of constrained bicyclic peptides, as candidates for SARS CoV-2 infection management.62 Studies conducted by our research teams, including the current one, have shown a clear match of ACE2 inhibition and in vitro effects in two diseases, triple negative cell line proliferation and SARS CoV-2–host interaction. Current work adds further evidence that ACE2 inhibition at the active site indirectly interfere with ACE2–spike interaction in a manner independent of the spike S1 unit responsible for this coupling by the two proteins. Therefore, future studies must further elaborate on the possible mechanisms of how this ACE2 inhibition possibly induces conformational changes that prevent the host–viral proteins interaction. These results weigh on adding more research efforts into exploring the role of ACE2 in these and other diseases. More potent ACE2 and potent direct ACE2–spike S1 inhibitors must be tested as well. Among the various inhibitors, ORE-1001 (aka MLN-4760) is the most potent. However, clinical applications of this drug were challenged owing to the diverse effects that it had. The current clinical status of this molecule involves the use radiofluorine derivatives for PET imaging and crystallography to further understand the role it has in human disease.63 Thus, the current research may help to fully elicit the complexity of the ACE2 role, hence facilitating the development of selective and disease-specific ACE2 inhibitors.
Conclusion
The scientific drug discovery community is progressively working toward the development of ACE2 or ACE2–spike inhibitors. However, despite the limited in vitro and in vivo proofs of this concept, a full picture of the inhibition of ACE2 is still missing, in addition to its role in preventing SARS CoV-2 infection. The current analysis has added a few pieces to the puzzle. LMS1007, the novel mild ACE2 inhibitor, was 100 times more potent than carnosine. Both showed activity on two different diseases in their same respective range of ACE2 inhibitions. Carnosine was effective against TNBC at an IC50 of about 230 mM.6 Meanwhile, LMS1007 was effective in inhibiting SARS CoV-2 spike ACE2 interaction at an IC50 of 2.3 mM. Future experiments should scale up the validation of ACE2 as a target for drug therapy in Covid-19 and TNBC. More research is needed on the myriads of effects for the ACE2 inhibition in the mesh of diseases affected by this novel and key enzyme. These diseases include a variety of cancers, neurodegenerative, autoimmune, and infectious diseases. Both carnosine and LMS1007 are potential ACE2 inhibitor leads for anti-cancer treatment and for combating Covid-19.Abbreviations
| ACE2 | Angiotensin converting enzyme 2 |
| Covid-19 | Novel coronavirus disease of 2019 |
| SARS CoV-2 | Severe acute respiratory syndrome corona virus 2 |
| SBDD | Structure-based drug docking |
| TNBC | Triple negative breast cancer |
Data availability
Datasets are provided as a ESI EXCEL file.† Spectra files and graphs of LMS1007 are available as a ESI file.† All other intermediate compound spectrum files and graphs are available from the corresponding author upon reasonable request.Author contributions
All the authors contributed to the conceptualization and writing of this research. LS performed the initial design, in silico testing, preliminary docking, secured funds, registered a patent, and finally experimentally validated LMS1007. GA proposed the synthetic routes, managed the synthesis and characterization of LMS1007, and contributed to the writing and analysis of the data. QA carried out the detailed docking and chemical modeling, prepared all the figures pertaining to the specific amino acids at the target sites, and contributed to the writing and analysis of the data. IB supervised the project and coordinated all efforts and contributed to the writing and analysis of the data.Conflicts of interest
The authors have no competing interests to disclose.Acknowledgements
L. S. secured funding for this project from Applied Science Private University # DRGS-2021-2022-6. The authors would like to thank Applied Science Private University for their kind funding for this research with the grant number DRGS-2021-2022-6.References
- H. Vatansev, C. Kadiyoran, M. Cumhur Cure and E. Cure, COVID-19 infection can cause chemotherapy resistance development in patients with breast cancer and tamoxifen may cause susceptibility to COVID-19 infection, Med. Hypotheses, 2020, 143, 110091, DOI:10.1016/j.mehy.2020.110091.
- C. F. Yang, C. C. Liao and H. W. Hsu, et al., Human ACE2 protein is a molecular switch controlling the mode of SARS-CoV-2 transmission, J. Biomed. Sci., 2023, 30(1), 87, DOI:10.1186/s12929-023-00980-w.
- V. Bezzerri, V. Gentili and M. Api, et al., SARS-CoV-2 viral entry and replication is impaired in Cystic Fibrosis airways due to ACE2 downregulation, Nat. Commun., 2023, 14(1), 132, DOI:10.1038/s41467-023-35862-0.
- S. J. L. Petitjean, S. Eeckhout, M. Delguste, Q. Zhang, K. Durlet and D. Alsteens, Heparin-Induced Allosteric Changes in SARS-CoV-2 Spike Protein Facilitate ACE2 Binding and Viral Entry, Nano Lett., 2023, 23(24), 11678–11684, DOI:10.1021/acs.nanolett.3c03550.
- L. M. Saadah, G. I. A. Deiab, Q. Al-Balas and I. A. Basheti, Carnosine to Combat Novel Coronavirus (nCoV): Molecular Docking and Modeling to Cocrystallized Host Angiotensin-Converting Enzyme 2 (ACE2) and Viral Spike Protein, Molecules, 2020, 25(23), 5605, DOI:10.3390/molecules25235605.
- S. A. Melhem, L. M. Saadah, Z. S. Attallah, I. A. Mansi, S. H. Hamed and W. H. Talib, Deciphering angiotensin converting enzyme 2 (ACE2) inhibition dynamics: Carnosine's modulatory role in breast cancer proliferation - A clinical sciences perspective, Heliyon, 2024, 10(19), e38685, DOI:10.1016/j.heliyon.2024.e38685.
- P. M. E. Gaafar, N. S. El-Salamouni, R. M. Farid, H. A. Hazzah, M. W. Helmy and O. Y. Abdallah, Pegylated liquisomes: A novel combined passive targeting nanoplatform of L-carnosine for breast cancer, Int. J. Pharm., 2021, 602, 120666, DOI:10.1016/j.ijpharm.2021.120666.
- R. M. Farid, P. M. E. Gaafar, H. A. Hazzah, M. W. Helmy and O. Y. Abdallah, Chemotherapeutic potential of L-carnosine from stimuli-responsive magnetic nanoparticles against breast cancer model, Nanomedicine, 2020, 15(9), 891–911, DOI:10.2217/nnm-2019-0428.
- R. Fatehi, M. Nouraei, M. Panahiyan, M. Rashedinia and N. Firouzabadi, Modulation of ACE2/Ang1-7/Mas and ACE/AngII/AT1 axes affects anticancer properties of sertraline in MCF-7 breast cancer cells, Biochem. Biophys. Rep., 2024, 38, 101738, DOI:10.1016/j.bbrep.2024.101738.
- L. Saadah, G. Abu Deiab, I. Basheti and Q. Al-Balas, Compounds and Pharmaceutical Compositions for Treating Coronavirus Disease and Methods of Preparation Thereof, WO2023238167, Worldwide, 2023. C07.
- J. Lan, J. Ge and J. Yu, et al., Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature, 2020, 581(7807), 215–220, DOI:10.1038/s41586-020-2180-5.
- P. Wang, Z. Ren and W. Wang, et al., Candesartan upregulates angiotensin-converting enzyme 2 in kidneys of male animals by decreased ubiquitination, FASEB J., 2024, 38(6), e23537, DOI:10.1096/fj.202302707R.
- M. Elgazzaz, C. M. Filipeanu and E. Lazartigues, Angiotensin-Converting Enzyme 2 Posttranslational Modifications and Implications for Hypertension and SARS-CoV-2: 2023 Lewis K. Dahl Memorial Lecture, Hypertension, 2024, 81(7), 1438–1449, DOI:10.1161/HYPERTENSIONAHA.124.22067.
- M. Elgazzaz, N. Lakkappa and C. Berdasco, et al., UBR1 Promotes Sex-Dependent ACE2 Ubiquitination in Hypertension, Hypertension, 2025, 82(1), 84–95, DOI:10.1161/HYPERTENSIONAHA.124.23196.
- A. M. Sanad, F. Qadri and E. Popova, et al., Transgenic angiotensin-converting enzyme 2 overexpression in the rat vasculature protects kidneys from ageing-induced injury, Kidney Int., 2023, 104(2), 293–304, DOI:10.1016/j.kint.2023.04.007.
- X. Mei, B. Mell and S. Aryal, et al., Genetically engineered Lactobacillus paracasei rescues colonic angiotensin converting enzyme 2 (ACE2) and attenuates hypertension in female Ace2 knock out rats, Pharmacol. Res., 2023, 196, 106920, DOI:10.1016/j.phrs.2023.106920.
- A. Ueno, Y. Onishi and K. Mise, et al., Plasma angiotensin-converting enzyme 2 (ACE2) is a marker for renal outcome of diabetic kidney disease (DKD) (U-CARE study 3), BMJ Open Diabetes Res. Care., 2024, 12(3), e004237, DOI:10.1136/bmjdrc-2024-004237.
- K. A. Papavassiliou, V. A. Gogou and A. G. Papavassiliou, Angiotensin-Converting Enzyme 2 (ACE2) Signaling in Pulmonary Arterial Hypertension: Underpinning Mechanisms and Potential Targeting Strategies, Int. J. Mol. Sci., 2023, 24(24), 17441, DOI:10.3390/ijms242417441.
- B. Miziołek, C. Kruszniewska-Rajs and J. Gola, et al., Correlation between angiotensin-converting-enzyme 2 gene polymorphisms and systemic sclerosis, Clin. Exp. Rheumatol., 2023, 41(8), 1652–1658, DOI:10.55563/clinexprheumatol/utmjnq.
- V. Siratavičiūtė, D. Pangonytė and L. Utkienė, et al., Myocardial Angiotensin-Converting Enzyme 2 Protein Expression in Ischemic Heart Failure, Int. J. Mol. Sci., 2023, 24(24), 17145, DOI:10.3390/ijms242417145.
- C. L. Kellum, L. G. Kirkland and T. K. Nelson, et al., Sympathetic remodeling and altered angiotensin-converting enzyme 2 localization occur in patients with cardiac disease but are not exacerbated by severe COVID-19, Auton. Neurosci., 2024, 251, 103134, DOI:10.1016/j.autneu.2023.103134.
- Y. Liu, Y. He, G. Zhang, J. Yang and Y. Li, Multifunctional Self-Signaling nanoMIP and Its Application for a Washing-Free Assay of Human Angiotensin-Converting Enzyme 2, Anal. Chem., 2024, 96(19), 7602–7608, DOI:10.1021/acs.analchem.4c00501.
- S. Karthikeyan, A. Sundaramoorthy and S. Kandasamy, et al., A biophysical approach of tyrphostin AG879 binding information in: bovine serum albumin, human ErbB2, c-RAF1 kinase, SARS-CoV-2 main protease and angiotensin-converting enzyme 2, J. Biomol. Struct. Dyn., 2024, 42(3), 1455–1468, DOI:10.1080/07391102.2023.2204368.
- (a) Y. Yang, J. Cui, D. Luo, et al., Rapid Detection of SARS-CoV-2 Variants Using an Angiotensin-Converting Enzyme 2-Based Surface-Enhanced Raman Spectroscopy Sensor Enhanced by CoVari Deep Learning Algorithms. ACS Sens.. 9(6):3158–3169. doi: DOI:10.1021/acssensors.4c00488; (b) J. Xie, Q. F. Huang and Z. Zhang, et al., Angiotensin-converting enzyme 2 in human plasma and lung tissue, Blood Press., 2023, 32(1), 6–15, DOI:10.1080/08037051.2022.2154745.
- M. Barone, Angiotensin-converting enzyme 2 and AMPK/mTOR pathway in the treatment of liver fibrosis: Should we consider further implications?, World J. Gastroenterol., 2024, 30(18), 2391–2396, DOI:10.3748/wjg.v30.i18.2391.
- Y. Gan, Y. Feng and X. Zhou, et al., Serum levels of angiotensin-converting enzyme 2 in children with Kawasaki disease, Clin. Exp. Med., 2023, 23(4), 1325–1330, DOI:10.1007/s10238-022-00933-x.
- N. Naderi and M. Rahimzadeh, The Role of Soluble ACE2 as a Prognostic Marker in Severe COVID-19: A Brief Meta-Analysis, Endocr., Metab. Immune Disord.:Drug Targets, 2023, 23(1), 70–76, DOI:10.2174/1871530322666220623121922.
- J. Xie, Q. F. Huang and Z. Zhang, et al., Angiotensin-converting enzyme 2 in human plasma and lung tissue, Blood Press., 2023, 32(1), 6–15, DOI:10.1080/08037051.2022.2154745.
- R. Ahmed, A. A. Saba and A. Paul, et al., Intronic Variants of the Angiotensin-Converting Enzyme 2 Gene Modulate Plasma ACE2 Levels and Possibly Confer Protection against Severe COVID-19, BioMed Res. Int., 2023, 2023, 5705076, DOI:10.1155/2023/5705076.
- C. Cianfarini, L. Hassler and J. Wysocki, et al., Soluble Angiotensin-Converting Enzyme 2 Protein Improves Survival and Lowers Viral Titers in Lethal Mouse Model of Severe Acute Respiratory Syndrome Coronavirus Type 2 Infection with the Delta Variant, Cells, 2024, 13(3), 203, DOI:10.3390/cells13030203.
- J. F. Dos Santos, E. M. Dos Reis, F. V. Berti, G. Colla, J. Koepp and V. A. Nunes, CALU-3 lung cells three-dimensionally assembled onto CellFate® matrix present angiotensin-converting enzyme-2 activity, Biotechnol. Bioeng., 2023, 120(12), 3602–3611, DOI:10.1002/bit.28552.
- E. Kakavandi, K. Sadeghi and M. Shayestehpour, et al., Evaluation of angiotensin converting enzyme 2 (ACE2), angiotensin II (Ang II), miR-141-3p, and miR-421 levels in SARS-CoV-2 patients: a case-control study, BMC Infect. Dis., 2024, 24(1), 429, DOI:10.1186/s12879-024-09310-3.
- N. Guo, X. Li and Z. Han, et al., Expression of angiotensin converting enzyme 2 mRNA in different SARS-CoV-2 infection states and times, J. Biomol. Struct. Dyn., 2023, 41(20), 11255–11261, DOI:10.1080/07391102.2022.2160821.
- H. S. Parmar, A. Nayak, P. K. Gavel, H. C. Jha, S. Bhagwat and R. Sharma, Cross Talk between COVID-19 and Breast Cancer, Curr. Cancer Drug Targets, 2021, 21(7), 575–600, DOI:10.2174/1568009621666210216102236.
- V. K. Bhari, D. Kumar, S. Kumar and R. Mishra, SARS-CoV-2 cell receptor gene ACE2 -mediated immunomodulation in breast cancer subtypes, Biochem. Biophys. Rep., 2020, 24, 100844, DOI:10.1016/j.bbrep.2020.100844.
- M. G. Nair, J. S. Prabhu and S. Ts, High expression of ACE2 in HER2 subtype of breast cancer is a marker of poor prognosis, Cancer Treat Res Commun., 2021, 27, 100321, DOI:10.1016/j.ctarc.2021.100321.
- A. M. Nikiforuk, K. S. Kuchinski and K. Short, et al., Nasopharyngeal angiotensin converting enzyme 2 (ACE2) expression as a risk-factor for SARS-CoV-2 transmission in concurrent hospital associated outbreaks, BMC Infect. Dis., 2024, 24(1), 262, DOI:10.1186/s12879-024-09067-9.
- H. Fujishima, H. Yazu, E. Shimizu, N. Okada and K. Fukagawa, Eye Washing Downregulated Angiotensin-Converting Enzyme 2 in Conjunctival Tissue Samples from Smokers, Int. J. Mol. Sci., 2023, 24(24), 17526, DOI:10.3390/ijms242417526.
- M. A. Alotaibi, T. M. I. Al-Hazani and M. A. Alwaili, et al., SARS-CoV-2 virus associated angiotensin converting enzyme 2 expression modulation in colorectal cancer: Insights from mRNA and protein analysis COVID-19 associated (ACE2) expression in colorectal cancer, Microb. Pathog., 2023, 185, 106389, DOI:10.1016/j.micpath.2023.106389.
- L. Reveret, M. Leclerc and V. Emond, et al., Higher angiotensin-converting enzyme 2 (ACE2) levels in the brain of individuals with Alzheimer's disease, Acta Neuropathol. Commun., 2023, 11, 173 CrossRef PubMed.
- A. G. Abdou, M. Fayed and A. G. A. Farag, Immunohistochemical Expression of Angiotensin-Converting Enzyme 2 in the Skin of Patients Affected by COVID-19, Am. J. Dermatopathol., 2024, 46(1), 1–13, DOI:10.1097/DAD.0000000000002498.
- X. A. Mendiola-Salazar, M. A. Munguía-Laguna, M. Franco, A. Cano-Martínez, J. Santamaría Sosa and R. Bautista-Pérez, SARS-CoV-2 Spike Protein Enhances Carboxypeptidase Activity of Angiotensin-Converting Enzyme 2, Int. J. Mol. Sci., 2024, 25(11), 6276, DOI:10.3390/ijms25116276.
- J. Bejoy, C. I. Williams and H. J. Cole, et al., Effects of spike proteins on angiotensin converting enzyme 2 (ACE2), Arch. Biochem. Biophys., 2023, 748, 109769, DOI:10.1016/j.abb.2023.109769.
- I. Biskupek and A. Gieldon, Two-Stage Recognition Mechanism of the SARS-CoV-2 Receptor-Binding Domain to Angiotensin-Converting Enzyme-2 (ACE2), Int. J. Mol. Sci., 2024, 25(1), 679, DOI:10.3390/ijms25010679.
- C. Peng, X. Lv, Z. Zhang, J. Lin and D. Li, The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2, Molecules, 2024, 29(8), 1875, DOI:10.3390/molecules29081875.
- S. Karthikeyan, A. Sundaramoorthy and S. Kandasamy, et al., A biophysical approach of tyrphostin AG879 binding information in: bovine serum albumin, human ErbB2, c-RAF1 kinase, SARS-CoV-2 main protease and angiotensin-converting enzyme 2, J. Biomol. Struct. Dyn., 2024, 42(3), 1455–1468, DOI:10.1080/07391102.2023.2204368.
- T. Sharma, B. Gerstman and P. Chapagain, Distinctive Features of the XBB.1.5 and XBB.1.16 Spike Protein Receptor-Binding Domains and Their Roles in Conformational Changes and Angiotensin-Converting Enzyme 2 Binding, Int. J. Mol. Sci., 2023, 24(16), 12586, DOI:10.3390/ijms241612586.
- L. Wei, Y. Chen and X. Feng, et al., Elucidation of N-/O-glycosylation and site-specific mapping of sialic acid linkage isomers of SARS-CoV-2 human receptor angiotensin-converting enzyme 2, Analyst, 2023, 148(20), 5002–5011, 10.1039/d3an01079a.
- Y. Zhou, C. Tan and R. Zenobi, Rapid Profiling of the Glycosylation Effects on the Binding of SARS-CoV-2 Spike Protein to Angiotensin-Converting Enzyme 2 Using MALDI-MS with High Mass Detection, Anal. Chem., 2024, 96(5), 1898–1905, DOI:10.1021/acs.analchem.3c03930.
- S. Mei, Y. Zou and S. Jiang, et al., Highly potent dual-targeting angiotensin-converting enzyme 2 (ACE2) and Neuropilin-1 (NRP1) peptides: A promising broad-spectrum therapeutic strategy against SARS-CoV-2 infection, Eur. J. Med. Chem., 2024, 263, 115908, DOI:10.1016/j.ejmech.2023.115908.
- H. Mohammadi-Berenjestanaki, E. Mohammadali and M. Khasayesi, et al., Association between angiotensin-converting enzyme-2 gene polymorphism (rs2106809) with severity and outcome of COVID-19 infection, Mol. Biol. Rep., 2023, 50(8), 6669–6679, DOI:10.1007/s11033-023-08493-3.
- C. M. Bradshaw, T. Georgieva and T. N. Tankersley, et al., Cutting Edge: Characterization of Low Copy Number Human Angiotensin-Converting Enzyme 2-Transgenic Mice as an Improved Model of SARS-CoV-2 Infection, J. Immunol., 2024, 212(4), 523–528, DOI:10.4049/jimmunol.2300591.
- J. Y. H. Tsoi, J. Cai and J. Situ, et al., Autoantibodies against angiotensin-converting enzyme 2 (ACE2) after COVID-19 infection or vaccination, J. Med. Virol., 2023, 95(12), e29313, DOI:10.1002/jmv.29313.
- M. Rebelo, C. Tang and A. R. Coelho, et al., De Novo Human Angiotensin-Converting Enzyme 2 Decoy NL-CVX1 Protects Mice From Severe Disease After Severe Acute Respiratory Syndrome Coronavirus 2 Infection, J. Infect. Dis., 2023, 228(6), 723–733, DOI:10.1093/infdis/jiad135.
- R. A. Pashameah, R. Soltane and A. M. Sayed, Discovery of raffinose sulfate as a potential SARS CoV-2 inhibitor via blocking its binding with angiotensin converting enzyme 2, Int. J. Biol. Macromol., 2023, 248, 125818, DOI:10.1016/j.ijbiomac.2023.125818.
- J. Zhu, H. Yan and M. Shi, et al., Luteolin inhibits spike protein of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) binding to angiotensin-converting enzyme 2, Phytother. Res., 2023, 37(8), 3508–3521, DOI:10.1002/ptr.7826.
- H. A. Waggiallah, M. M. Eltayeb, A. M. Hjazi and Y. M. Elmosaad, High expression of angiotensin-converting enzyme 2 receptor (ACE-2), transmembrane protease serine (TMPRSS), and P-selectin in platelets lead to thrombosis formation in COVID-19 patients, Eur. Rev. Med. Pharmacol. Sci., 2024, 28(5), 1847–1856, DOI:10.26355/eurrev_202403_35598.
- H. Y. Kim, K. M. Jeong and S. H. Kim, et al., Modulating effect of Eunkyo-san on expression of inflammatory cytokines and angiotensin-converting enzyme 2 in human mast cells, In Vitro Cell. Dev. Biol.: Anim., 2024, 60(2), 195–208, DOI:10.1007/s11626-024-00847-w.
- N. X. Ha, T. T. Huong and P. N. Khanh, et al., In vitro and in Silico Study of New Biscoumarin Glycosides from Paramignya trimera against Angiotensin-Converting Enzyme 2 (ACE-2) for Preventing SARS-CoV-2 Infection, Chem. Pharm. Bull., 2024, 72(6), 574–583, DOI:10.1248/cpb.c23-00844.
- F. Aiello, F. Balzano, G. Uccello Barretta, I. D'Acquarica, G. Mazzoccanti and I. Agranat, Chiral distinction between hydroxychloroquine enantiomers in binding to angiotensin-converting enzyme 2, the forward receptor of SARS-CoV-2, J. Pharm. Biomed. Anal., 2024, 237, 115770, DOI:10.1016/j.jpba.2023.115770.
- K. Lee, Y. Na and M. Kim, et al., Ursodeoxycholic acid may protect from severe acute respiratory syndrome coronavirus 2 Omicron variant by reducing angiotensin-converting enzyme 2, Pharmacol. Res. Perspect., 2024, 12(2), e1194, DOI:10.1002/prp2.1194.
- M. A. J. Harman, S. J. Stanway and H. Scott, et al., Structure-Guided Chemical Optimization of Bicyclic Peptide (Bicycle) Inhibitors of Angiotensin-Converting Enzyme 2, J. Med. Chem., 2023, 66(14), 9881–9893, DOI:10.1021/acs.jmedchem.3c00710.
- J. Wang, D. Beyer and C. Vaccarin, et al., Development of radiofluorinated MLN-4760 derivatives for PET imaging of the SARS-CoV-2 entry receptor ACE2, Eur. J. Nucl. Med. Mol. Imaging, 2024, 52(1), 9–21, DOI:10.1007/s00259-024-06831-6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01134e |
| This journal is © The Royal Society of Chemistry 2025 |