
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-free regioselective synthesis of spiro-oxazolidines through [3 + 2] annulation reactions of azadienes with haloalcohols†
Amol T. Savekar‡
 ,
Sonali M. Vitnor‡,
Vishal B. Karande and
Suresh B. Waghmode*
,
Sonali M. Vitnor‡,
Vishal B. Karande and
Suresh B. Waghmode*
Department of Chemistry, Savitribai Phule Pune University (Formerly University of Pune), Ganeshkhind, Pune-411007, India. E-mail: suresh.waghmode@unipune.ac.in; suresh.waghmode@gmail.com
First published on 4th April 2025
Abstract
The transition-metal-free regioselective [3 + 2] annulation of azadienes with haloalcohols for the preparation of highly functionalized spiro-oxazolidines is experimentally simple and proceeds under mild conditions. The metal-free protocols have more significance than the metal-catalyzed ones when the toxicity associated with the metal catalyst is considered. This transformation features a broad substrate scope, good yields, and excellent regioselectivity. Moreover, large-scale synthesis and representative transformations of spiro-oxazolidines were carried out to provide additional evidence on the practicality of this approach.
Introduction
Spirocyclic skeletons are privileged structural moieties in many biologically active natural products owing to their inherent three-dimensional architecture.1 Among the useful classes of oxygen- and nitrogen-based heterocycles, oxazolidine is a significant motif present in various natural products and pharmaceuticals (as shown in Fig. 1), and it is used as a chiral auxiliary and chiral ligand in diverse asymmetric transformations.2,3 In particular, spiro-oxazolidines, with heteroatom-substituted quaternary stereocenters, are valuable building blocks to construct medicinally relevant compounds.4 | ||
| Fig. 1 Representative examples of bioactive oxazolidine scaffolds. | ||
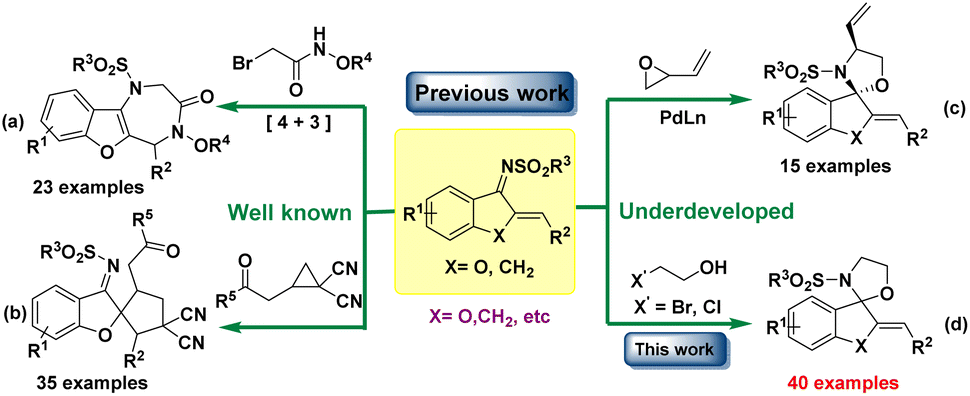
Since the existence of heteroatoms can bring new synthetic and biological values, the focus has recently been shifted to utilizing azadienes (1 and 4) to prepare heterocycles containing multi-heteroatoms.5 Azadiene contains exocyclic alkylidene and an imine unit, which acts as a four-atom synthon in various Michael additions and subsequent cascade reactions, delivering diverse cyclic compounds through [4 + n] cycloaddition reactions due to the driving force for aromatization.6 Recently, Zhao's group reported [4 + 3] annulation through 1,4 addition of α-bromohydroxamates to azadienes to access benzofuran-fused seven-membered heterocycles (Scheme 1a).7 In contrast to well-developed [4 + n] cyclizations through 1,4-addition, which are applicable only for the synthesis of aromatized heterocycles, these azadienes can also serve as two-atom synthons to undergo [2 + n] annulation to synthesize spirocyclic motifs.8 Very recently, Liu's group developed a methodology for the synthesis of spirocyclopentanone through [3 + 2] cycloaddition of cyclopropanes with azadienes (Scheme 1b).9 Except a few reports, the synthesis of spiro-oxazolidine scaffolds has not been developed yet.10 The 1,2-addition reactions are comparatively less developed; Trost's group have recently demonstrated Pd-catalyzed [3 + 2] spiroannulation from azadienes and vinyl epoxides (Scheme 1c).11 These reported methods involved noble metal-catalyzed protocol, high cost, multistep cascade reactions, drastic reaction conditions, and limited substrate scope, adding to their drawbacks. The quest to explore alternatives to transition metal catalysts is mainly attributed to the toxicity inherent in such systems, especially when it comes to synthesizing heterocycles of biological relevance. Hence, protocols that proceed under transition-metal-free conditions are always desirable among the scientific community.12
 | ||
| Scheme 1 Annulation reactions of azadienes with various reaction partners. | ||
However, to our knowledge, the regioselective transition-metal-free [3 + 2] spiroannulation of azadienes has not been studied yet. Therefore, it is highly desirable to synthesize spiro-oxazolidines under transition-metal-free and ambient reaction conditions. Herein, we describe the development of an approach for the regioselective synthesis of spiro-oxazolidines under ambient reaction conditions.
Recently, haloalcohols (Br, Cl, and I) and their homologs were explored and developed as Michael donors for [3 + 2]/[4 + 2] annulation reactions with various Michael acceptors for the synthesis of an important class of heterocyclic compounds.13 Based on this background and as a continuation of our interest in bioactive-fused polycyclic structure synthesis,14 we carried out [3 + 2] annulation of azadienes with haloalcohols under milder reaction conditions to obtain the corresponding spiro-oxazolidines, which are constituents of various natural products and pharmaceuticals (Scheme 1d).
Results and discussion
The investigation started by evaluating the reaction of benzofuran-derived azadienes 1a (1.0 equiv.), and 2-bromoethanol 2a (1.5 equiv.) with, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (2.5 equiv.) as a base in MeCN at room temperature (i.e., 25 °C) for 3 h, and the desired (Z)-2-benzylidene-3′-tosyl-2H-spiro[benzofuran-3,2′-oxazolidine] product 3a was generated in 28% yield (Table 1; entry 1). After extensive screening of various bases such as K3PO4, K2CO3, Et3N, NaH, Cs2CO3, and t-BuOK, surprisingly it was found that Cs2CO3 provided the highest 81% yield in 3 h (entries 2–7). Similarly, organic solvents were also studied, namely 1,4-dioxane, DMF, DMSO, THF, dichloromethane (DCM), toluene, ethyl acetate (EtOAc), and acetone; among the studied solvents, acetone (83%, yield) proved to be the best (entries 8–15). Furthermore, variation in the stoichiometry of 2-bromoethanol did not show promising results (entries 16 and 17). A lower yield was observed when the base loading decreased or increased (entries; 18 and 19).
|
|||||
|---|---|---|---|---|---|
| Sr. no. | 2a (equiv.) | Base | Solvent | Time (h) | Yieldb (%) |
| a All reactions were performed with 1a (0.27 mmol, 1.0 equiv.), 2a (0.40 mmol, 1.5 equiv.), and base (0.67 mmol, 2.5 equiv.) in solvent (2.0 mL) at room temperature (rt) under N2.b Isolated yields.c Cs2CO3 (0.53 mmol, 2.0 equiv.).d Cs2CO3 (0.80 mmol, 3.0 equiv.), nr = no reaction. | |||||
| 1 | 1.5 | DBU | MeCN | 3 | 28 |
| 2 | 1.5 | K3PO4 | MeCN | 6 | 78 |
| 3 | 1.5 | K2CO3 | MeCN | 12 | 52 |
| 4 | 1.5 | Et3N | MeCN | 12 | Trace |
| 5 | 1.5 | NaH | MeCN | 12 | 40 |
| 6 | 1.5 | Cs2CO3 | MeCN | 3 | 81 |
| 7 | 1.5 | t-BuOK | MeCN | 12 | 40 |
| 8 | 1.5 | Cs2CO3 | 1,4-Dioxane | 12 | 10 |
| 9 | 1.5 | Cs2CO3 | DMF | 6 | 72 |
| 10 | 1.5 | Cs2CO3 | DMSO | 12 | Trace |
| 11 | 1.5 | Cs2CO3 | THF | 12 | 5 |
| 12 | 1.5 | Cs2CO3 | DCM | 12 | nr |
| 13 | 1.5 | Cs2CO3 | Toluene | 12 | nr |
| 14 | 1.5 | Cs2CO3 | EtOAc | 2 | 30 |
| 15 | 1.5 | Cs2CO3 | Acetone | 2 | 83 |
| 16 | 1 | Cs2CO3 | Acetone | 2 | 75 |
| 17 | 2 | Cs2CO3 | Acetone | 2 | 83 |
| 18c | 1.5 | Cs2CO3 | Acetone | 2 | 73 |
| 19d | 1.5 | Cs2CO3 | Acetone | 2 | 81 |

After optimizing the reaction conditions, we began exploring the scope of the [3 + 2] annulation reaction (Scheme 2). When various N-protecting groups such as N-SO2Ph, N-SO2PhBr, N-Ns, N-SO2PhCF3, and N-Ms containing azadienes were used as substrates, the corresponding target products 3b–3f were obtained in good yields (63–76%). We observed that spirocyclization was facilitated by 2-chloroethanol and 2-iodoethanol provided the product with lower yields (20–31%). Various substitutions at ortho, meta, and para positions on the phenyl of azadienes provided products in good to high yields (3g–3n). The electron-withdrawing groups 4-NO2 and 3-CF3, on azadienes, gave superior yield (3i–3k, 80–83%) to electron-donating groups 4-OMe and 4-Me (3g–3h, 60–68%). Further, the mono-substituted halogens (–F, –Cl, and –Br) on the phenyl ring were well tolerated to produce corresponding products 3l–3n in good to excellent yields (64–80%), which offers new possibilities for the cross-coupling type of manipulations. The sterically hindered 9-anthryl and 2-naphthyl substituted azadienes delivered products 3o–3p in 48% and 83% yields, respectively. The azadiene bearing a heterocyclic 2-thiophenyl group was converted into the expected product 3q in 57% yield. The reaction also extended to t-butyl substituents on the azadiene yielding product 3r in 54% yield. Furthermore, the azadiene-bearing substituent at the benzofuryl ring was readily converted to afford the corresponding product 3s in high yields (80%). Unfortunately, the reaction of azadienes with 3-bromopropan-1-ol did not give our desired products (3t) under optimized reaction conditions.
 | ||
| Scheme 2 Substrate scope for the synthesis of spiro-oxazolidine derivatives.a,b aAll reactions were performed with 1 (0.27 mmol, 1.0 equiv.), 2 (0.40 mmol, 1.5 equiv.), and Cs2CO3 (0.67 mmol, 2.5 equiv.) in 2.0 mL acetone at room temperature for 2 h. bIsolated yields. c2-Chloroethanol and t-BuOK. d2-Iodoethanol and t-BuOK. | ||
Further, we expanded this methodology to indanone-derived azadienes 4 and 2-bromoethanol 2a as model substrates under standard reaction conditions. As shown in Table 2, in the presence of Cs2CO3 in acetone at room temperature, the desired product (Z)-2-benzylidene-3′-tosyl-2,3-dihydrospiro[indene-1,2′-oxazolidine] 5a was obtained in 61% yield after 3 h (Table 2, entry 1). Replacing Cs2CO3 with inorganic bases such as K3PO4, K2CO3, NaH, and organic bases DBU, Et3N, and t-BuOK effectively mediated this strategy except K2CO3 and Et3N, while Cs2CO3 was the optimal one (entries 2–7). The yields were significantly reduced when we performed the reaction in other solvents, namely MeCN, 1,4-dioxane, DMF, DMSO, THF, DCM, toluene, and EtOAc (entries 11–15); among the studied solvents, MeCN (63%, yield) proved to be the best.
|
||||
|---|---|---|---|---|
| Sr. no. | Base | Solvent | Time (h) | Yieldb (%) |
| a All reactions were performed with 4a (0.27 mmol, 1.0 equiv.), 2a (0.40 mmol, 1.5 equiv.), and base (0.67 mmol, 2.5 equiv.) in solvent (2.0 mL) at room temperature (rt) under N2.b Isolated yields. nr = no reaction. | ||||
| 1 | Cs2CO3 | Acetone | 3 | 61 |
| 2 | K3PO4 | Acetone | 3 | 59 |
| 3 | K2CO3 | Acetone | 12 | nr |
| 4 | Et3N | Acetone | 12 | nr |
| 5 | NaH | Acetone | 3 | 45 |
| 6 | DBU | Acetone | 3 | 21 |
| 7 | t-BuOK | Acetone | 5 | 59 |
| 8 | Cs2CO3 | MeCN | 2 | 63 |
| 9 | Cs2CO3 | 1,4-Dioxane | 3 | 53 |
| 10 | Cs2CO3 | DMF | 6 | 43 |
| 11 | Cs2CO3 | DMSO | 12 | nr |
| 12 | Cs2CO3 | THF | 6 | Trace |
| 13 | Cs2CO3 | DCM | 12 | nr |
| 15 | Cs2CO3 | Toluene | 12 | nr |
| 16 | Cs2CO3 | EtOAc | 6 | 29 |
The substrate scope was subsequently investigated with the optimal reaction conditions in hand (Table 2, entry 8). As shown in Scheme 3, various N-protecting groups containing azadienes converted into the desired products 5b–5e in moderate to good yields (62–76%). In our substrate scope generalization studies, 2-chloroethanol and 2-iodoethanol provided products with good yields (35–42%). An examination of the substituent effects on the phenyl ring of azadiene showed that electron-withdrawing (–NO2, –CF3, 5g, 5h, 72–77%) groups were superior to electron-donating (–CH3, 5f, 62%) groups. Mono- and di-substituted halogens (–F, –Cl, and –Br) were well tolerated to produce corresponding products 5i–5o in good to high yields (66–77%). Azadiene bearing 2-thiophenyl, 2-naphthyl, and 2-furanyl groups were converted into the corresponding products 5p–5r (60–74%). Halogen-substituted indanyl ring afforded the corresponding products 5s–5u in good yields (67–75%).
 | ||
| Scheme 3 Substrate scope for the synthesis of spiro-oxazolidine derivatives.a,b aAll reactions were performed with 4 (0.27 mmol, 1.0 equiv.), 2 (0.40 mmol, 1.5 equiv.), and base (0.67 mmol, 2.5 equiv.) in solvent (2.0 mL) at room temperature (rt) under N2. bIsolated yields. c2-Chloroethanol and K3PO4. d2-Iodoethanol and Cs2CO3. | ||
With the optimized reaction conditions, we explored its generality for isatin-derived N-Boc ketimine 6a with 2-bromoethanol 2a (Scheme 4). It is worth noting that isatin-derived N-Boc ketimine (6a) also tolerated the [3 + 2] annulation reaction and gave the corresponding oxospiro[indoline-3,2′-oxazolidine] 7a in good yield (56%). This provides an alternative to the convenient assembly of spiro-oxazolidines from another perspective.
 | ||
| Scheme 4 Synthesis of spiro-oxazolidines derivatives from isatin-derived ketimines. | ||
Based on literature reports, a plausible reaction mechanism has been proposed to explain the cascade 1,2-addition followed by the spiro-cyclization that leads to the formation of product (Scheme 5). Initially, the base abstracts the alcoholic proton of 2a, and then in situ generated oxoanion attacks on imine of azadiene 1a via direct addition to deliver intermediate A. Then, displacement of bromine takes place to form the desired product (3a).
 | ||
| Scheme 5 Plausible reaction mechanism for the synthesis of spiro-oxazolidines. | ||
To demonstrate the synthetic potential of this transformation, we investigated the scaled-up preparation of 3a and 5a under the standard reaction conditions (Scheme 6a and b). The reaction of 1.0 g of 1a and 4a proceeded smoothly, to deliver products 3a and 5a with yields of 75% and 58% in 3 h, respectively. Furthermore, to investigate the potential utility and enrich the spirocyclic compound's molecular complexity, we carried out the derivatization of the product 3a (Scheme 7). When m-CPBA was used for the epoxidation of 3a in DCM, epoxide 8 was obtained with excellent yields and diastereoselectivity (91%, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) in 12 h (Scheme 7a). Various epoxides are valuable building blocks in chemical synthesis and such structural motifs are present in biologically active molecules.15 Hydrogenation of 3a readily generated product 9 in good yields with moderate diastereoselectivity (Scheme 7b).
:1 dr) in 12 h (Scheme 7a). Various epoxides are valuable building blocks in chemical synthesis and such structural motifs are present in biologically active molecules.15 Hydrogenation of 3a readily generated product 9 in good yields with moderate diastereoselectivity (Scheme 7b).
 | ||
| Scheme 6 Gram-scale synthesis. | ||
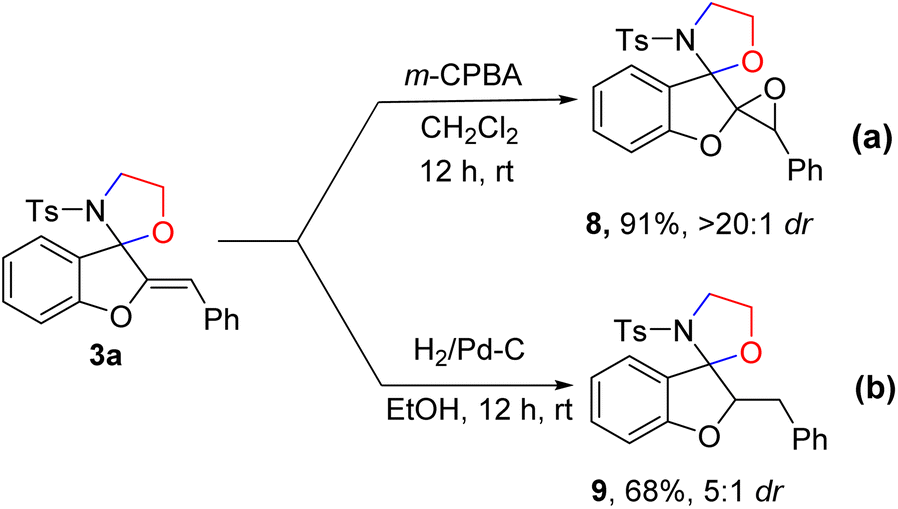 | ||
| Scheme 7 Synthetic transformation of product 3a. | ||
Conclusions
In summary, we have developed a highly regioselective and chemoselective [3 + 2] spiroannulative transformation of azadienes. The reaction was carried out in one pot without transition metals and under mild reaction conditions. This method demonstrated good substrate generality, and was successfully applied to large-scale synthesis. Product derivations further confirmed the applicability of this method. However, this feeding sequence-controlled divergent reaction was limited to a special type of substrate.Data availability
The data supporting this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
ATS thanks UGC New Delhi for the research fellowship (JUNE-18-144311), and SMV thanks NF-OBC (NBCFDC Ref. No. 231610106961). SBW and VBK thank SERB-SURE project SUR/2022/000608 for financial support. They also thank CIF SPPU, Pune, for the spectral analysis facility.References
-
(a) C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514–5533 CAS
; (b) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed
-
(a) T. Fukuyama, L. Li, A. A. Laird and R. K. Frank, J. Am. Chem. Soc., 1987, 109, 1587–1589 CrossRef CAS
-
(a) C. Agami and F. Couty, Eur. J. Org. Chem., 2004, 677–685 CrossRef CAS
-
(a) J. D. Scott and R. M. Williams, J. Am. Chem. Soc., 2002, 124, 2951–2956 CrossRef CAS PubMed
-
(a) Q. Deng and X. Meng, Chem.–Asian J., 2020, 15, 2838–2853 CrossRef CAS PubMed
-
(a) J. Chen and Y. Huang, Org. Lett., 2017, 19, 5609–5612 CrossRef CAS PubMed
- Q. Y. Fang and L. M. Zhao, Chem. Commun., 2020, 56, 14079–14082 RSC
-
(a) Q. Y. Fang, M. H. Yi, X. X. Wu and L.-M. Zhao, Org. Lett., 2020, 22, 5266–5270 CrossRef CAS PubMed
- S. Li, Z.-H. Dong, S.-Y. Dan, M.-J. Zheng, T. Long, J. Zhan, Q. Zhou, W.-D. Chu and Q. Z. Liu, Org. Chem. Front., 2024, 11, 2905–2910 RSC
-
(a) T. Rajasekaran, G. Karthik, B. Sridhar and B. V Subba Reddy, Org. Lett., 2013, 15, 1512–1515 CrossRef CAS PubMed
-
(a) B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2021, 60, 5806–5810 CrossRef CAS PubMed
-
(a) V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957–11986 RSC
-
(a) M. K. Ghorai, D. Shukla and K. Das, J. Org. Chem., 2009, 74, 7013–7022 CrossRef CAS PubMed
-
(a) R. A. Gaikwad, S. B. Kamble and S. B. Waghmode, Chem.–Asian J., 2022, 17, e202200931 CrossRef CAS PubMed
-
(a) M. González-Pérez, R. Gómez-Bombarelli, J. Arenas-Valgañón, M. T. Pérez-Prior, M. P. García-Santos, E. Calle and J. Casado, Chem. Res. Toxicol., 2012, 25, 2755–2762 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01423a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |