
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling PPARβ/δ nuclear receptor agonists via a drug-repurposing approach: HTVS-based ligand identification, molecular dynamics, pharmacokinetics, and in vitro anti-steatotic validation
Sumit Kumar Mandal†
a,
Mohammed Muzaffar-Ur-Rehman†b,
Sonakshi Puria,
Banoth Karan Kumarb,
Pankaj Kumar Sharma a,
Murugesan Sankaranarayananb and
P. R. Deepa*a
a,
Murugesan Sankaranarayananb and
P. R. Deepa*a
aBiochemistry and Enzyme Biotechnology Laboratory, Department of Biological Sciences, Birla Institute of Technology and Science Pilani, Pilani Campus, Pilani-333 031, Rajasthan, India. E-mail: p20200425@pilani.bits-pilani.ac.in; pankajsharma@pilani.bits-pilani.ac.in; deepa@pilani.bits-pilani.ac.in
bMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Pilani-333 031, Rajasthan, India. E-mail: p20210457@pilani.bits-pilani.ac.in; p20180042@pilani.bits-pilani.ac.in; murugesan@pilani.bits-pilani.ac.in
First published on 4th April 2025
Abstract
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors with a crucial regulatory role in carbohydrate and lipid metabolism and are emerging druggable targets in “metabolic syndrome” (MetS) and cancers. However, there is a need to identify ligands that can activate specific PPAR subtypes, particularly PPARβ/δ, which is less studied compared with other PPAR isoforms (α and γ). Herein, using the drug-repurposing approach, the ZINC database of clinically approved drugs was screened to target the PPARβ/δ receptor through high-throughput-virtual-screening, followed by molecular docking and molecular dynamics (MD) simulation. The top-scoring ligands were subjected to drug-likeness analysis. The hit molecule was tested in an in vitro model of NAFLD (non-alcoholic fatty liver disease). The top five ligands with strong binding affinity towards PPARβ/δ were canagliflozin > empagliflozin > lumacaftor > eprosartan > dapagliflozin. RMSD/RMSF analysis demonstrated stable protein–ligand complexation (PLC) by the top-scoring ligands with PPARβ/δ. In silico ADMET prediction analysis revealed favorable pharmacokinetic profiles of these top five ligands. Canagliflozin showed significant (P < 0.001) dose-dependent decrease in lipid accumulation and the associated oxidative stress-inflammatory response, suggesting its promising anti-steatotic potential. These outcomes pave the way for further validation and development of PPAR activity-modulating therapeutics.
Introduction
Peroxisome proliferator-activated receptors (PPARs) are a type of nuclear receptors that regulate target gene expression upon binding with ligands.1 They are subdivided into three isoforms, namely, PPARα, PPARβ/δ and PPARγ, based on their diverse functions and differential expressions in various tissues and cell types, including the heart, fat tissue, liver and skeletal muscles.2The activation mechanism is quite similar among the three PPAR isoforms, yet they show some differences. For example, PPARα is activated by fatty acids, while PPARγ is activated by thiazolidinedione drugs and is reported to be regulated by a natural product, chelerythrine, resulting in an anti-diabetic effect.3,4 PPARβ/δ, alternatively, has broad ligand specificity, including for fatty acids, eicosanoids, and ligands such as leukotriene B4 and prostacyclin.5 When these ligands bind to PPARs' ligand binding domain, a conformational change is induced, enabling the PPARs to interact with co-activators and co-repressors. Once activated, the PPARs bind to specific DNA sequences, known as PPAR response elements (PPREs), within the promotor region of the target gene, resulting in the transactivation of the gene.6 Transrepression is another PPAR mechanism that occurs without a ligand and is involved in the anti-inflammatory effects of PPARs.7
The multi-faceted regulation of lipid metabolism and associated energy mechanisms that have been reported2,8 are compiled in Fig. 1. It shows that activation of PPARβ/δ inhibits the STAT3/SOCS3 pathway and improves insulin sensitivity in adipose tissue.9 A recent study has shown that treatment with leptin increases the expression of FGF21 in visceral adipose tissue by activating PPARβ/δ.10 GW501516, a PPARβ/δ agonist, lowers triglyceride levels in hepatic tissue by restoring hepatic AMPK levels via activation of the PGC-1/α-lipin-1/PPARα pathway in a rat model.11,12 Similarly, GW501516 reduces multiple clinical features of metabolic syndrome by increasing the oxidation of fatty acids in skeletal muscles.13 Metabolic syndrome represents a complement of related disorders that include hyperlipidemia, diabetes mellitus, hypertension, and cardiovascular disorders, which may have the hepatic complication of non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH).
 | ||
| Fig. 1 The crucial role of PPAR-β/δ in the regulation of lipid metabolism. FA: fatty acids, TCA: tricarboxylic acid, FATP: fatty acid transport protein; FABP: fatty acid binding protein; CPT1A: carnitine palmitoyl transferase I; RXR: retinoid X receptor; ATP: adenosine triphosphate; acetyl-CoA: acetyl coenzyme A; NFκB: nuclear factor kappa B; IL1, 6, 10: interleukin 1, 6, 10; FGF21: fibroblast growth factor 21; GSIS: glucose-stimulated insulin secretion; GLP-1: glucagon-like peptide 1. | ||
Studies have also shown that lipolytic functions of PPARβ/δ, which have a potentially beneficial effect on fatty liver disease, are mediated by activating the autophagy pathway and attenuating endoplasmic reticulum stress in obese mice.14,15 Other beneficial effects reported with respect to PPARβ/δ agonists are mitigation of oxidative stress,16 inflammation17,18 and mitochondrial biogenesis,19 which make it a promising target for identifying PPARβ/δ agonists that could potentially treat metabolic disorders.20 One of the risks of metabolic syndrome is the development of cancers. In this context, it is important to note that PPARβ/δ has also been evaluated for its potential anti-cancer effects.16,21–23
Recently, using a machine learning approach, newer PPARβ/δ agonists were reported.24 However, there are limited reports on PPARβ/δ agonists, which prompted the present study. Herein, we screened a database with clinically approved drugs as potential pharmacological candidates using high-throughput virtual screening (HTVS) technique, molecular docking/dynamics simulations and pharmacokinetic profiling. This was followed by biochemical testing of a hit-ligand agonist molecule in an in vitro steatotic cell culture model of NAFLD.
Experimental
Materials and methods
Computational studies were conducted using the Maestro module within Schrödinger software (v13.3, Schrodinger LLC, NY, USA). Molecular dynamics simulations were carried out utilizing the Desmond module of Schrödinger software, specifically developed by D. E. Shaw (ver. 6.5). Simulations were performed on a Tyrone workstation running on a Linux platform (Ubuntu 22.04 LTS), equipped with 160 GB of RAM and an 11 GB NVIDIA graphics card (GeForce RTX 208 Ti).HepG2 cells were sourced from the National Centre for Cell Science (NCCS – Pune, India). The minimum essential medium (MEM) was procured from Gibco Life Technologies (Carlsbad, USA), while fetal bovine serum (FBS), oleic acid, and Oil Red O (ORO) powder were obtained from HiMedia (Mumbai, India). All other chemicals were purchased from Sigma-Aldrich (Schnelldorf, Germany) and were of the highest analytical grade.
Protein preparation
The protein structures of PPARβ/δ (PDB ID: 3GZ9) were retrieved from the RCSB protein databank (PDB). This protein crystal structure includes a co-crystal ligand, which is reported to be a full agonist of PPARβ/δ and partial agonist of PPARγ.25,26Prior to docking, proteins must be prepared since their crystal structures might have issues including improper bonding orders, missing side chains, loops, and atoms. The Protein Preparation Wizard was used to create the crystal structure of the two proteins, and the purpose of this process was to examine the protein structures and confirm the assignment of bonds and binding orders, the insertion of hydrogens, the identification of disulfide bonds, the completion of any missing loops or side chains, and the rectification of any incorrectly labelled components. Moreover, the OPLS-2005 force field was used to minimize crystal protein structures. Heavy atoms in the structures were constrained to relieve torsional tension using a harmonic potential of 25 kcal mol−1 throughout the procedure, whereas hydrogen was left un-constrained.27
Ligand preparation
The ligand library of US FDA approved drugs was obtained in SDF format from the approved drug library ZINC database, which comprises 1615 drugs. The ligands were prepared using the LigPrep module of the Schrodinger program for computational studies. Desalting was performed using Epik v4.7, which is based on Hammett and Taft approaches, to achieve the biologically appropriate pH 7 after charge neutralization. PROPKA was used to modify the protonation states of the polar residues. A maximum of one stereoisomer was produced for each compound. In order to produce a low energy conformer, energy minimization of the compounds was carried out utilizing the OPLS-2005 force field.Receptor grid generation
The grid box was generated using the receptor's native ligand pose. The receptor grid was generated using receptor-grid generation wizard of the Maestro module of the software. The docked ligands were similar in size compared with native ligands with respect to the grid box, which was the centroid of the native ligands of protein receptors. Using the OPLS-2005 force field, the protein's atoms were fixed within the default van der Waals scaling factor and partial charge cut-off values of 1 and 0.25. The grid box's dimensions were set to 10 × 10 × 10 Å with the co-crystal co-ordinates (x = 34.58, y = 70.56, and z = −2.71) and a grid spacing of 1 Å distance.28Virtual screening and molecular docking
The molecular docking process for FDA-approved drugs from the ZINC29 database was conducted using the glide module of Schrödinger, LLC, located in New York, USA. Initially, the HTVS tool was employed to determine whether the ligands were capable of binding to the active site of PPARβ/δ. The ligands that passed this screening were then subjected to further refinement using the standard precision (SP) mode, followed by the extra precision (XP) mode. The HTVS filter output was used as input for the SP filter, and the ligands that successfully passed the SP filter were subsequently subjected to XP docking. Each screening mode utilizes the same scoring function to minimize false positives, but they differ in terms of speed and accuracy. The docking analyses were visualized using the Maestro module of the software. The docking binding energy was utilized to calculate the docking binding affinity of the compounds towards the active site of PPARβ/δ using the following relationship:ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKd. lnKd. |
In the equation, ΔG represents the changes in docking binding energy, T represents temperature, R represents the Boltzmann gas constant (R = 1.987 cal mol−1 K−1), and Kd represents dissociation constant. This equation enables the estimation of binding affinity based on the docking binding energy.30
Validation of the docking protocol
The selected protein structures were re-docked with their co-crystal ligands at their active site using the XP docking mode. The generated conformations were superimposed against the native pose to determine their root mean square deviation (RMSD). A deviation value of ≤2.0 Å validates the docking methodology can be used for docking of the test ligands.MM-GBSA studies
Molecular mechanics with generalised Born and surface area solvation (MM-GBSA) is an efficient method to calculate the absolute free energy of a protein–ligand complex.31 It is the summation of gas-phase energy, solvation energy and entropic contributions. To carry out this study, the docked protein and the ligand's docked conformation were used, and calculations were carried out in the Prime module of Schrodinger software.32 The solvation model was dielectric surface generalized Born (VSGB) continuum in the presence of the OPLS4 force field.33Molecular dynamics (MD) simulation studies
The protein–ligand complexes were prepared, optimized, and minimized using the Protein Preparation Wizard of the OPLS 2005 force field. To assess the conformational stability of the protein–ligand complex interactions, a molecular dynamics (MD) simulation was performed using the Desmond module of Schrodinger. The simulation was run for 100 nanoseconds (ns) and employed the OPLS-2005 force field. The protein–ligand complex was placed at the center of an orthorhombic cubic box with dimensions of 10 Å × 10 Å × 10 Å. The simulation included explicit TIP3P water molecules and buffers between the protein and box edges. In order to neutralize the system, counter ions (such as Na+ or Cl−) were added. Four Cl− ions and 0.15 M NaCl salt concentration were used, and the box volume was minimized using the System Builder wizard in Maestro software.27,34–36The analysis focused on the ligand molecules that showed significant results. The prepared system underwent energy minimization for 100 picoseconds (ps). The final production run consisted of a 100 ns MD simulation conducted under constant temperature and pressure (NPT ensemble) with a relaxation period of 1 ps and a temperature of 300 K. The Nose–Hoover chain coupling technique was used for temperature control. Bonding interactions were calculated with a time step of 2 fs using the reversible reference system propagator algorithm (RESPA) integrator.37,38 The trajectories obtained from the simulation were examined for the root mean square deviation (RMSD) and root mean square fluctuation (RMSF) of the protein–ligand complex, as well as the RMSF of the ligand itself.
In silico ADMET prediction analysis
After the HTVS and docking of 1615 FDA approved molecules with PPARβ/δ, the pKCSM tool was used to evaluate the ADMET (absorption, distribution, metabolism, elimination, and toxicity) of the top five molecules with the highest docking scores to determine their significant pharmacokinetic features. The ligands with optimal drug-likeness and bioavailability profiles were selected to carry out molecular dynamics studies. The following ADMET parameters were included in the report: absorption: Caco-2 permeability, water solubility, human intestinal absorption, P-glycoprotein substrate, P-glycoprotein I and II inhibitors, and skin permeability; distribution: steady state volume of distribution (VDss), fraction unbound, blood–brain barrier (BBB) permeability, and central nervous system (CNS) permeability; metabolism: cytochrome P450 inhibitors, CYP2D, AMES toxicity, maximum tolerated dosage, hepatotoxicity, and skin sensitization.39Canagliflozin, identified as the top scoring ligand, was experimentally evaluated to evaluate its effectiveness in reducing steatosis in a cell culture model of non-alcoholic fatty liver disease (NAFLD). The subsequent sections detail the experimental confirmation of the pharmacological efficacy of canagliflozin.
MTT assay for cell viability
HepG2 cells were acquired from the National Centre for Cell Science in Pune, India, and cultured in T-25 cm2 cell culture flasks. HepG2 cells are a widely accepted and valuable model for studying hepatic steatosis owing to their retention of key hepatocyte functions, including lipid metabolism and accumulation. Furthermore, their established use in steatosis research provides a pool of comparative data, and their ease of culture and reproducibility make them a reliable in vitro model.40 Using this cellular model of NAFLD, we examined the anti-steatotic efficacy of the top-hit ligand, canagliflozin. The cells were maintained at 37 °C with 5% CO2 in MEM containing 4.5 g L−1 glucose and 110 mg L−1 sodium pyruvate, supplemented with 10% bovine calf serum (HiMedia). The cell cultures were passaged every 2–3 days, typically when they reached 80% confluence. Cell viability was assessed by their ability to metabolize the tetrazolium salt MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (SRL Chemicals, Mumbai, India).To prepare canagliflozin for experiments, it was initially dissolved in dimethyl sulphoxide (DMSO) to prepare a stock solution (SS) with a concentration of 1 mM. Working concentrations of canagliflozin, ranging from 5–100 μM, were then prepared from the stock solution, ensuring that the final concentration of DMSO in the test dilutions did not exceed 1%. Confluent monolayers of HepG2 cells were grown in 96-well plates for 24 hours. These cells were treated with different concentrations of canagliflozin in triplicate and incubated at 37 °C in a CO2 environment for 24 hours. After the 24 hour treatment period, the supernatants were aspirated from the wells, and 100 μL of MTT solution (2 mg mL−1) was added to each well. The plates were then incubated for 3 hours at 37 °C, followed by the addition of 100 μL DMSO to dissolve the formazan crystals. Optical density was measured at 570 nm using a multi-well spectrophotometer (Multiskan, Thermo Fisher Scientific, Waltham, MA). The safe dose concentration, often considered the concentration at which 80% of cells remain viable, was determined using non-linear regression curve analysis within GraphPad Prism statistical software.27
ORO staining
To test the anti-steatotic effect of the top-agonist canagliflozin (10, 15, and 20 μM), it was compared with the standard drug saroglitazar (10 μM).41 To prepare an ORO solution, 0.5 grams of ORO was diluted in 100 mL of isopropanol to make a stock solution. A working dilution was then prepared by mixing 6 parts of the ORO stock with 4 parts of deionized water. For cell staining, the cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature. After removing paraformaldehyde, the cells were washed with PBS. Subsequently, the freshly prepared ORO working solution was added to the cells, and the cells were kept undisturbed for 15 minutes. The cells were then rinsed with water and immediately examined under a light microscope. To quantify the accumulation of neutral lipids, the stained lipid droplets were dissolved using 100 μL of 100% isopropanol for 5 minutes at room temperature, and the absorbance was measured at 492 nm.27Lipid peroxidation (LPO) assay
To assess LPO levels, HepG2 cells were seeded in 6 cm plates at a density of 1 × 105 cells per well. Subsequently, the cells were exposed to different concentrations of canagliflozin (10, 15, and 20 μM) and saroglitazar (10 μM) for 24 hours. Following the treatment, the control and treated cells were collected and sonicated; then, the total protein content was determined using the Bradford method. LPO levels in HepG2 cells were quantified through the thiobarbituric acid-reactive substance (TBARS) assay.Following treatment, the cells were treated with lysis buffer, homogenized, and centrifuged at 13000×g for 15 minutes at 4 °C for the lipid peroxidation assay using a modified protocol. The resulting supernatant was collected for subsequent analysis. The cell lysates from each experimental group were adjusted to contain an equal quantity of protein (1 mg mL−1) and were treated with 250 μL of 10% trichloroacetic acid (TCA). Afterward, these lysates were mixed with 375 μL of 1% (w/v) thiobarbituric acid (TBA) under acidic conditions. This solution was heated in a boiling water bath for 15 minutes, promoting the formation of a pink-colored adduct. The absorbance of this resultant adduct was measured spectrophotometrically at 530 nm using the Multiskan FC (Thermo Scientific, DE). The values obtained were expressed as micromoles (μM) of malondialdehyde per milligram (mg) of protein.28
Statistical analysis
In vitro experiments were conducted in a minimum of two to three separate trials, with each trial performed in triplicate sets. Statistical analysis was carried out using one-way ANOVA to compare the various experimental groups. Significant differences among values were denoted by P ≤ 0.05 (*), P ≤ 0.01 (**), and P ≤ 0.001 (***).Results and discussion
Molecular docking analysis
A total of 1615 compounds were docked into the active site of PPARβ/δ. Using a step-by-step procedure, compounds were primarily screened using HTVS, yielding a top scoring hit, which was then screened using the SP mode. The drugs that failed to bind in the SP mode were excluded, and the remaining drugs were finally docked using an XP mode. The top scoring five ligands were shortlisted. which show the highest affinity towards the PPARβ/δ target protein (Table 1).| S. no | Ligands | Drug name | PPARβ/δ (kcal mol−1) | MM-GBSA (kcal mol−1) |
|---|---|---|---|---|
| 1 | ZINC000043207238 | Canagliflozin | −13.907 | −73.80 |
| 2 | ZINC000036520252 | Empagliflozin | −12.756 | −77.09 |
| 3 | ZINC000064033452 | Lumacaftor | −12.509 | −58.09 |
| 4 | ZINC000029319828 | Eprosartan | −11.944 | −54.77 |
| 5 | ZINC000003819138 | Dapagliflozin | −11.86 | −71.22 |
| 6 | Co-crystal ligand | (2,3-Dimethyl-4-{[2-(prop-2-yn-1-yloxy)-4-{[4-(trifluoromethyl)phenoxy]methyl}phenyl]sulfanyl}phenoxy)acetic acid | −12.74 | −96.96 |
Table 2 summarizes the docking interactions and bond distances of the top five ligands against PPARβ/δ. Canagliflozin, a sodium-glucose transport protein sub-type 2 inhibitor used for the treatment of type 2 diabetes, exhibited significant docking scores against PPARβ/δ (−13.907 kcal mol−1). When canagliflozin was docked against PPARβ/δ, it formed four hydrogen bond interactions with Thr288, Thr289, His449 and Phe282. Empagliflozin, a sodium-glucose co-transporter 2 inhibitor used to improve glycemic control in adults with type 2 diabetes mellitus, showed a similar interaction pattern. It formed hydrogen bonds with Thr288 and halogen bonds with Thr288, His449, Lys367. Lumacaftor, used for the treatment of cystic fibrosis, interacted with PPAR β/δ through hydrogen bonds with His449 and formed one halogen bond with His449. Eprosartan, an angiotensin II receptor inhibitor used for the treatment of high blood pressure, formed two aromatic bond interactions with Cys285 and Phe327. It also exhibited one halogen bond interaction with Thr289. Dapagliflozin, a sodium-glucose transport protein sub-type 2 inhibitor used in type 2 diabetes treatment, formed hydrogen bonds with Thr289 and one aromatic H-bond with Phe327 (Fig. 2). Overall, the docking interactions and bond distances of the top five drugs with different PPARβ/δ molecules are summarized in Table 2.
| Ligands | PPARβ/δ | ||
|---|---|---|---|
| Residues | Type of interactions | Distance (Å) | |
| a Abbreviations: PPARβ/δ = peroxisome proliferator activated receptor β/δ; kcal = kilocalorie; mol = mole. | |||
| ZINC000043207238 (canagliflozin) | THR288 | Halogen bond | 1.93 |
| THR289 | H-Bond | 1.81 | |
| HIS449 | Halogen bond | 2.00 | |
| HIS449 | Halogen bond | 2.50 | |
| PHE282 | Halogen bond | 2.58 | |
| PHE282 | Halogen bond | 2.12 | |
| PHE282 | Halogen bond | 2.00 | |
| PHE282 | Halogen bond | 2.44 | |
| ZINC000036520252 (empagliflozin) | THR288 | H-Bond | 2.12 |
| THR288 | Halogen bond | 3.06 | |
| HIS449 | Halogen bond | 5.71 | |
| LYS367 | Halogen bond | 6.68 | |
| ZINC000064033452 (lumacaftor) | HIS449 | H-Bond | 1.92 |
| HID449 | Halogen bond | 3.73 | |
| ZINC000029319828 (eprosartan) | CYS285 | Aromatic H-bond | 2.13 |
| PHE327 | Aromatic H-bond | 2.43 | |
| THR289 | Halogen bond | 2.07 | |
| ZINC000003819138 (dapagliflozin) | PHE327 | Aromatic H-bond | 3.64 |
| THR289 | H-Bond | 2.74 | |
| Co-crystal ligand | THR288 | H-Bond | 2.00 |
| TYR473 | H-Bond | 1.82 | |
| HIS449 | H-Bond | 1.80 | |
| HIS449 | Pi–Pi | 4.47 | |
| THR288 | H-Bond | 2.00 | |
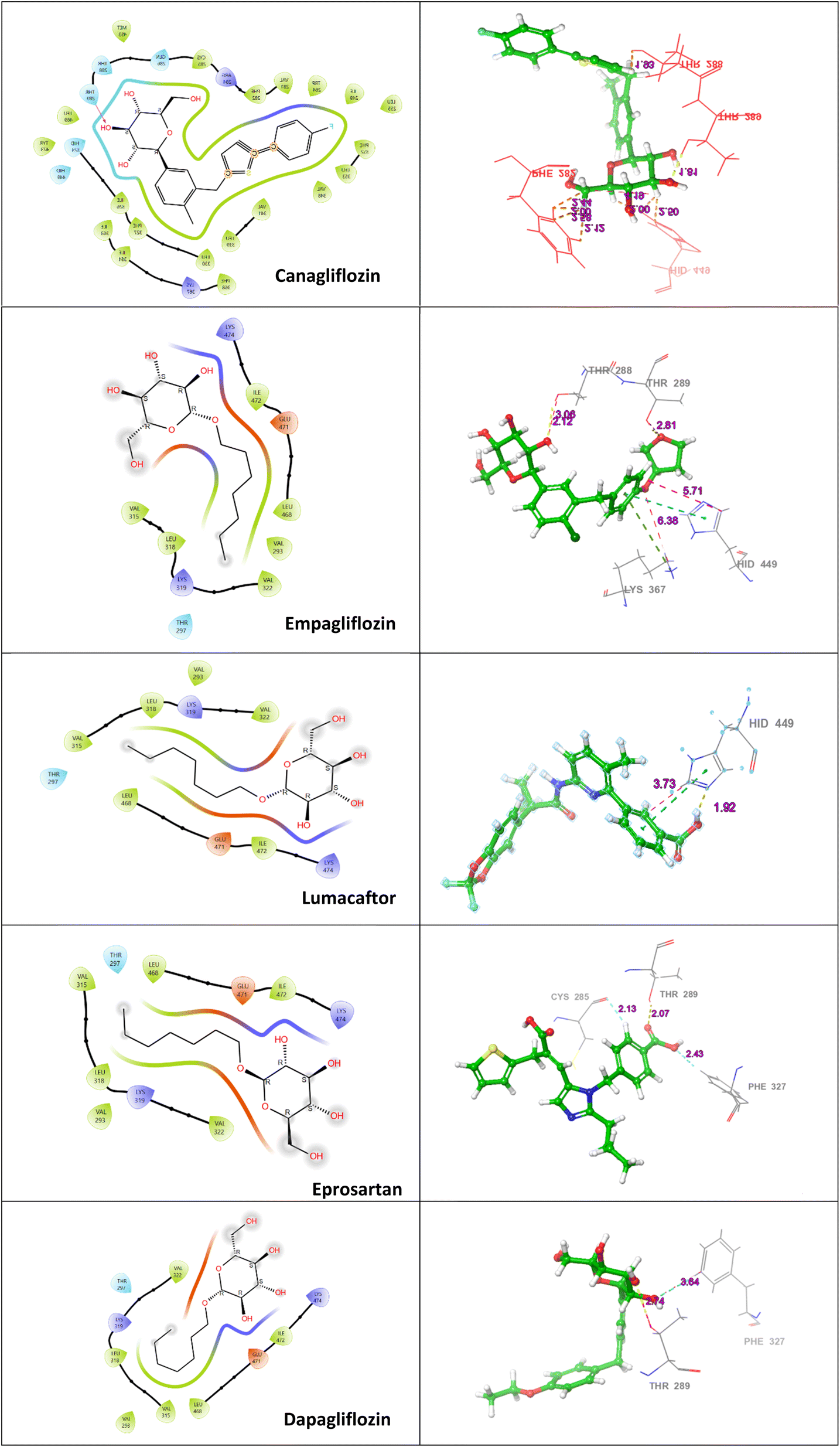 | ||
| Fig. 2 Representation of 2D and 3D docked poses of canagliflozin, empagliflozin, lumacaftor, eprosartan, and dapagliflozin in the active site of PPARβ/δ (PDB: 3GZ9). The active site residues are represented by coloured discs, while interaction bonds are depicted as dashed lines. | ||
Molecular dynamics simulation studies
Molecular dynamics results of the co-crystal and the hit molecule (ZINC000043207238) have shown a stable RMSD plot. In the case of the co-crystal ligand, the docked complex exhibits three H-bond interactions. One H bond was formed via interaction of the oxygen atom between the rings A and B with Thr288. The other two interactions were due to interaction of carboxylic terminal with His449 and Tyr473. In the initial stage of the dynamics, the interaction between the ligand and Thr288 disappeared, resulting in an RMSD of 1.2 Å. During the entire simulation, there were very few deviations in the ligand within the range of 2.0 Å (from 1.2 Å to 3.2 Å) (Fig. 3(i)). The residues interacting with the ligand had fewer fluctuations between 0.6 Å and 1.8 Å, as can be seen in the RMSF plot (Fig. 3(ii)), indicating stable protein in the presence of a co-crystal ligand. The major contributing residues in the interactions include His449 (41% H bonds and 40% pi–pi stacking) and Tyr473 (43% H bonds) (Fig. 4(i)).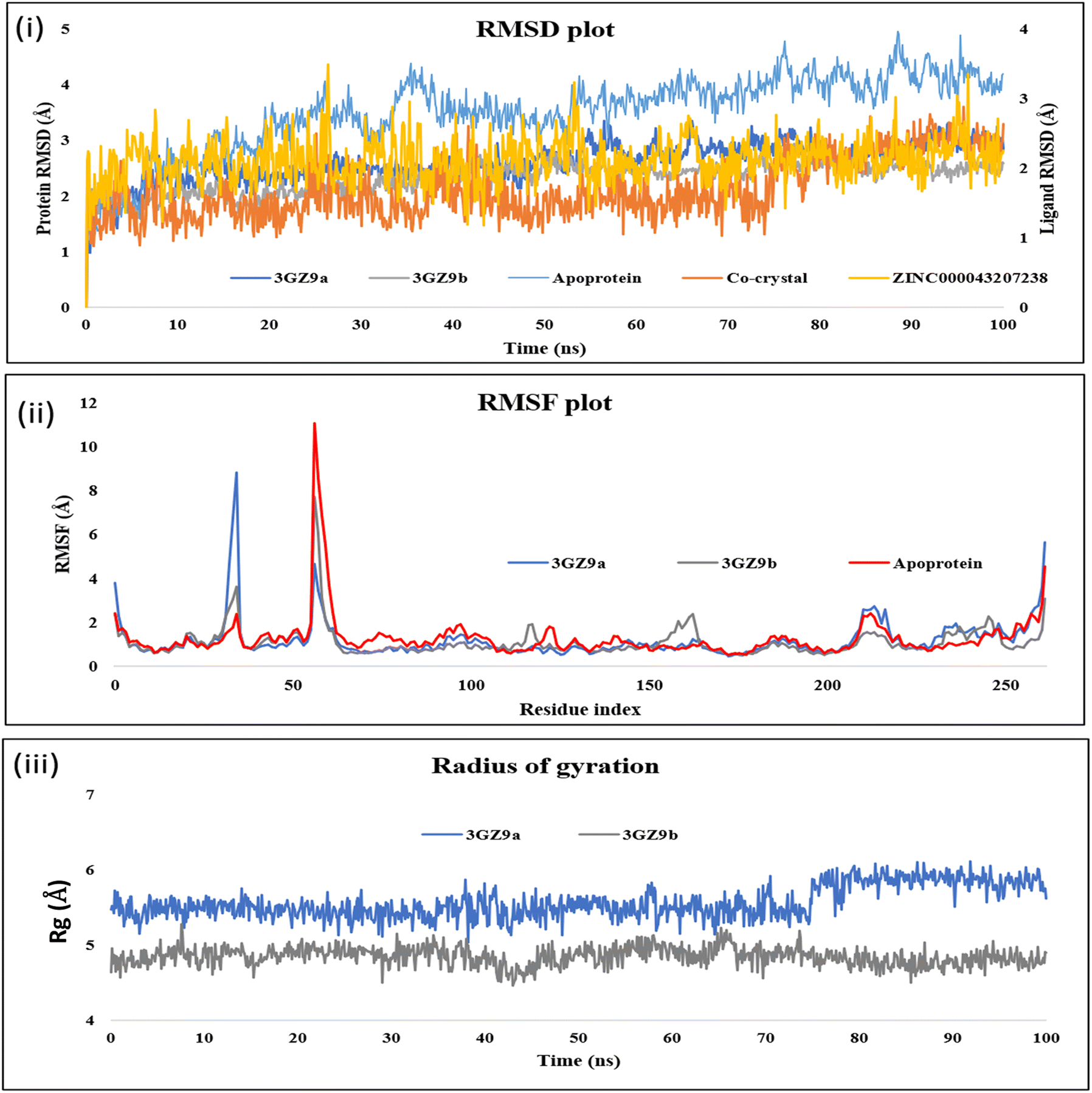 | ||
| Fig. 3 (i) RMSD plot depicting the deviation of the protein and ligands during the simulation. 3GZ9a indicates the protein with the hit compound, while 3GZ9b indicates the protein with the co-crystal ligand, orange line indicates the co-crystal ligand, and yellow indicates the hit compound. (ii) RMSF plot depicting fluctuations of the protein residues during simulation. 3GZ9a indicates the protein with respect to the co-crystal ligand and 3GZ9b with respect to the hit compound. (iii) Depiction of the radius of gyration (Rg) graph of the top-scoring ligand (canagliflozin) complexed with 3GZ9 and co-crystal ligand complexed with 3GZ9 during 100 ns MD Simulation. 3GZ9a indicates the protein with respect to the co-crystal ligand and 3GZ9b with respect to the hit compound. | ||
 | ||
| Fig. 4 (i) Analysis of amino acid residue interactions during molecular dynamics simulation. 2D interaction diagram of co-crystal representing the (a) docked pose (b) residual contribution during the simulation and (c) histogram graph and percentage of interaction fraction wherein green indicates an H bond, purple indicates a hydrophobic bond, pink indicates an ionic bond, and blue indicates water bridges. (ii) Analysis of amino acid residue interactions during molecular dynamics simulation. 2D interaction diagram of ZINC000043207238 (canagliflozin) representing the (a) docked pose (b) residual contribution during the simulation, and (c) histogram graph and percentage of interaction fraction, wherein green indicates an H bond, purple indicates an hydrophobic bond, pink indicates an ionic bond, and blue indicates water bridges. | ||
The radius of gyration (Rg), which determines the compactness of the protein, shows very less deviation between 5.1 Å and 6.0 Å (Fig. 3(iii)). In the case of the hit compound, the docked complex had only one H-bond interaction with Gln286 with a distance of 1.81 Å. After initiating dynamics, the bond between the 5th hydroxy group of glucopyranose of the hit molecule and Gln286 disappeared, and the interaction of His323 with the 3rd hydroxy group was seen, resulting in a change in conformation of the docked ligand, thereby increasing the RMSD to 2.4 Å (Fig. 3). Furthermore, new interactions were observed with Leu339, Val341, Lys367, His449 and Tyr473 until 40 ns. After 55 ns, two additional residues, Gln286 and His323, were observed to form strong interactions. Among these, residues that contributed to direct interactions with the ligand include Gln286, Tyr473, and His449, with 24%, 11%, and 36%, respectively. The residue His323 contributed to water-mediated interactions (11%). In addition, other residues such as Leu339, Val341, and Lys367 formed water-mediated interactions, and their contributions were less than 10% (Fig. 4(ii)). The RMSF plot (Fig. 3(ii)) also shows very low fluctuations of the residues under interaction with the ligand (<2.0 Å). The deviations in the Rg was <1.5 Å (between 4.0–5.25 Å) (Fig. 3(iii)), and the overall deviations in the RMSD plot were not more than 2.0 Å (1.5–3.2 Å). This indicates that the ligand is stable in the active site pocket and is retained within the pocket till the end of the simulation.
For comparison, the PPARβ/δ protein in its apo-form was subjected to dynamics studies for 100 ns. As depicted in Fig. 3(i), the RMSD of the apo-protein has a deviation ranging from 2.0 Å to 5.0 Å, while the protein when complexed with the co-crystal or the hit compound has a lower deviation (2.0–3.0 Å). This indicates that the presence of ligands in the active site of the protein resulted in stability, as evident from the decreased protein deviation or residual fluctuation observed in the respective RMSD and RMSF plots (Fig. 3(i and ii). We also observed most of the interactions with the co-crystal ligand are due to the acetic acid group, while the hit compound engages via hydroxyl groups on its pyranose rings. The co-crystal and the hit compound showed similar behaviour in their dynamics and interaction pattern, which suggested that the hit compound (canagliflozin) could have agonistic behaviour similar to that of the co-crystal ligand, which encouraged us to further carry out in vitro investigation.
In silico ADMET prediction and drug-likeness analysis
The top five agonists for the protein target of PPARβ/δ were chosen. All the ligands showed better docking scores against PPARβ/δ protein targets. Further testing was performed on these five ligands for in silico drug-likeness prediction and ADMET analysis. These top scored ligands met the requirements for polarity, hydrophobicity, and lipophilicity. This study aids in the selection of the finest molecules with drug-like properties and polarities that are biologically permeable.Table 3 presents a summary of the predicted ADMET characteristic results for the top five PPARβ/δ agonists. Based on the comparison between the marginal value and the actual value, ADMET results were interpreted as high Caco-2 permeability predicted value >0.90, intestinal absorption less than 30% is considered to be poorly absorbed, human VDss is low if it is less than 0.71 L kg−1 and high if it is more than 2.81 L kg−1, BBB permeability logBB >0.3 is considered to cross BBB, and logBB 1 are poorly distributed. Compounds with a logPS of >2 and <3 have the ability to permeate the central nervous system (CNS). The favourable pharmacokinetic profile of the top scoring ligands, as revealed by the above ADMET analysis, is yet another point for considering these ligands as potential PPARβ/δ agonist drug candidates.
| Parameters | Absorption | Distribution | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Water solubility (logmol L−1) |
Caco2 permeability (logcm s−1) |
Intestinal absorption (% human) | Skin permeability (logKp) |
P-Glycoprotein substrate | P-Glycoprotein inhibitor | VDss (human) (log L kg−1) | Fraction unbound (human) (Fu) | BBB permeability (logBB) |
CNS permeability (logPS) |
|
| Canagliflozin | −4.21 | 0.85 | 0.85 | −2.7 | Yes | Yes | −0.71 | 0.08 | −0.99 | −3.02 |
| Empagliflozin | −3.56 | −0.02 | 55.87 | −2.7 | Yes | Yes | −0.38 | 0.07 | −1.10 | −3.51 |
| Lumacaftor | −3.18 | 0.63 | 78.71 | −2.7 | Yes | No | −1.46 | 0.03 | −0.47 | −2.81 |
| Eprosartan | −2.89 | 0.67 | 57.17 | −2.7 | Yes | No | −0.72 | 0.14 | −0.87 | −2.47 |
| Dapagliflozin | −3.43 | 0.94 | 55.86 | −2.7 | Yes | Yes | −0.33 | 0.08 | −0.99 | −3.34 |
| Parameters | Metabolism | Excretion | Toxicity | Lipinski's rule of five | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP substrate | CYP inhibitors | Total clearance (log mL min−1 kg−1) | Renal OCT2 substrate | AMES toxicity | Hepatotoxicity | Skin sensitization | Molecular weight | logP |
Rotatable bonds | Acceptors | Donors | Surface area | |
| Canagliflozin | No | No | 0.035 | No | No | No | No | 444.52 | 2.97 | 5 | 6 | 4 | 183.56 |
| Empagliflozin | No | No | 0.40 | No | No | No | No | 450.91 | 1.613 | 6 | 7 | 4 | 185.31 |
| Lumacaftor | No | No | −0.10 | No | No | Yes | No | 452.41 | 4.75 | 5 | 5 | 2 | 186.53 |
| Eprosartan | No | No | 0.67 | No | No | Yes | No | 424.52 | 4.74 | 10 | 5 | 2 | 178.70 |
| Dapagliflozin | No | No | 0.19 | No | No | No | No | 408.88 | 1.84 | 6 | 6 | 4 | 168.47 |
The ADMET analysis of canagliflozin in our study is in resonance with other research reports. Studies have shown that canagliflozin is rapidly and completely absorbed from the gastrointestinal tract, with a peak plasma concentration reached within 1–2 hours after oral administration.42 Food does not significantly affect its absorption. Canagliflozin is highly protein-bound (99%) in the plasma, mainly to albumin. It has a large volume of distribution, indicating that it gets distributed extensively to tissues.43 Canagliflozin is primarily metabolized in the liver via glucuronidation, a phase II metabolic pathway. The major enzymes involved are UGT1A9 and UGT2B4, whereas CYP3A4, a phase I enzyme, plays a minor role in its metabolism.44 Canagliflozin is mainly excreted in the feces (about 50%) as an unchanged drug and metabolite. About 33% is excreted in the urine as metabolites. The terminal half-life of canagliflozin is about 10–13 hours.45 Overall, the outcomes of ADMET analysis of canagliflozin in the present study and from other reports indicate that it has favorable pharmacokinetic properties, which contribute to its efficacy and safety profile in the treatment of type 2 diabetes and potentially other metabolic disorders.
Canagliflozin suppresses intracellular lipid accumulation in HepG2 cells
Before assessing the anti-steatotic impact of canagliflozin, we determined its safe dosage by examining its dose-dependent effects on HepG2 cell viability using the MTT assay. Subsequently, we employed non-cytotoxic doses of canagliflozin (10, 15, and 20 μM) for further in vitro investigations (Fig. 5a). | ||
| Fig. 5 Inhibitory effects of canagliflozin on lipid accumulation in the in vitro model of NAFLD. (a) Canagliflozin's inhibitory effects on lipid accumulation in an in vitro model of NAFLD were evaluated by assessing dosage-dependent impacts on HepG2 cells after 24 hours of treatment using the MTT assay. Results are presented as the mean ± S.D. of a minimum of three experiments conducted in triplicate. (b) Photomicrographs at 10× magnification illustrated intense Oil Red O (ORO) staining in the untreated steatotic NAFLD group (i), contrasted by reduced lipid staining in the canagliflozin-treated groups (ii–v). White arrows indicate lipid accumulation stained with ORO. (c) Quantification of ORO staining revealed a dose-dependent inhibition of lipid accumulation (steatosis). Values are expressed as the mean ± S.D. of a minimum of three experiments. (d) The reduction in malondialdehyde (MDA) levels in the treated groups confirmed the anti-NAFLD protection conferred by canagliflozin. Values are expressed as the mean ± S.D. of a minimum of three experiments conducted in triplicate. Symbols *, **, and *** denote statistical significance at P ≤ 0.05, 0.01, and 0.001, respectively. | ||
To induce cellular lipid deposits in HepG2 hepatocytes, we supplemented them with free fatty acids (oleic acid) for 48 hours. Canagliflozin, identified as the top hit, demonstrated a concentration-dependent reduction in lipid accumulation in steatotic cells following a 24 hour treatment, which was evaluated using ORO staining (Fig. 5a and b). This anti-steatotic effect was comparable to that observed with the positive control saroglitazar (10 μM). Specifically, neutral lipids such as triglycerides, stained with ORO, were quantified spectrophotometrically at 492 nm. Fig. 5c illustrates a consistent reduction in triglyceride (TG) accumulation as the concentration of canagliflozin increased (p < 0.05). The doses of 10, 15, and 20 μM resulted in lipid accumulation reductions in the steatotic cells by 29.03%, 37.57%, and 48.45%, respectively, while saroglitazar, the reference anti-steatotic drug control, induced a 33.39% reduction.
Lipotoxicity induced by steatosis resulted in oxidative stress in the hepatocytes, evident from an increase in malondialdehyde levels in the disease (control) group (Fig. 5d). This oxidative stress-induced lipid peroxidation was attenuated in the cells treated with canagliflozin. In this study, we observed a reduction in lipid peroxidation by 27.99%, 33.50%, and 46.10% when treated with 10, 15, and 20 μM of canagliflozin, respectively. This is similar to the oxidative reduction observed when cells were treated with the reference drug saroglitazar at 10 μM.
Conclusion
Using a high throughput virtual screening and molecular docking approach, a list of top-scoring ligands with potential as PPARβ/δ receptor-agonists were identified in this study. These ligands can be experimentally validated in metabolic syndrome and cancers, where the nuclear receptor PPARβ/δ plays a significant mechanistic role. The top-hit molecule (canagliflozin), in the present study, showed favourable protein–ligand complex stability, pharmacokinetic properties, and pharmacological efficacy in controlling hepatic fat accumulation and associated oxidative stress in NAFLD/NASH.Abbreviations
| PPAR | Peroxisome proliferator-activated receptors |
| PPREs | Peroxisome proliferator-activated receptor response elements |
| FDA | Food and drug administration |
| RXR | Retinoid X receptor |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| HTVS | High throughput virtual screening |
| MD | Molecular dynamics |
| PDB | Protein Data Bank |
| OPLS-2005 | Optimized potentials for liquid simulations 2005 |
| SP | Standard precision |
| XP | Extra precision |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| Rg | Radius of gyration |
| MM-GBSA | Molecular mechanics with generalised Born and surface area solvation |
| RESPA | Reversible reference system propagator algorithms |
| VSGB | Variable dielectric generalized Born |
Data availability
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.Author contributions
P. R. Deepa (PRD), Sankaranarayanan Murugesan (SM): Conceptualization; Sumit Kumar Mandal (SKM), Mohammed Muzaffar-Ur-Rehman (MMR), Sonakshi Puri (SP), Banoth Karan Kumar (BKK): methodology & experimentation; SKM, SP, BKK, MMR: data curation, software; SKM, SP, MMR, & BKK: writing and original draft preparation; SKM, SP, BKK, MMR, SM: software and data validation; PRD, SM, PKS: visualization, investigation, supervision; SKM, SP, MMR, PRD, SM, PKS: writing – reviewing and editing.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The research grant from the Indian Council of Medical Research (52/13/2022-BIO/BMS), Govt. of India, New Delhi, is acknowledged. SKM, SP and MMR acknowledged the Institute Fellowship from BITS Pilani, and BKK is grateful to the Ministry of Tribal Affairs, Government of India, for the research fellowship. SM acknowledges the research support from the Department of Biotechnology (Indo-Spain Bilateral Programme), Govt. of India, New Delhi.References
- J. N. Feige, L. Gelman, L. Michalik, B. Desvergne and W. Wahli, Prog. Lipid Res., 2006, 45, 120–159 CAS.
- B. Grygiel-Górniak, Nutr. J., 2014, 13, 1–10 Search PubMed.
- V. M. W. Gee, F. S. L. Wong, L. Ramachandran, G. Sethi, A. P. Kumar and C. W. Yap, J. Comput.-Aided Mol. Des., 2014, 28, 1143–1151 CAS.
- W. Zheng, L. Qiu, R. Wang, X. Feng, Y. Han, Y. Zhu, D. Chen, Y. Liu, L. Jin and Y. Li, Sci. Rep., 2015, 5, 12222 CAS.
- L. Han, W. J. Shen, S. Bittner, F. B. Kraemer and S. Azhar, Future Cardiol., 2017, 13, 279–296 CAS.
- S. Tyagi, P. Gupta, A. Saini, C. Kaushal and S. Sharma, J. Adv. Pharm. Technol. Res., 2011, 2, 236–240 CAS.
- A. Fougerat, A. Montagner, N. Loiseau, H. Guillou and W. Wahli, Cells, 2020, 9, 1638 CAS.
- J.-C. Fruchart, B. Staels and P. Duriez, Curr. Atheroscler. Rep., 2001, 3, 83–92 CrossRef CAS PubMed.
- L. Serrano-Marco, R. Rodríguez-Calvo, I. El Kochairi, X. Palomer, L. Michalik, W. Wahli and M. Vázquez-Carrera, Diabetes, 2011, 60, 1990–1999 CrossRef CAS PubMed.
- L. Mazuecos, C. Pintado, B. Rubio, E. Guisantes-Batán, A. Andrés and N. Gallardo, Int. J. Mol. Sci., 2021, 22(9), 4624 CrossRef CAS PubMed.
- E. Barroso, R. Rodríguez-Calvo, L. Serrano-Marco, A. M. Astudillo, J. Balsinde, X. Palomer and M. Vázquez-Carrera, Endocrinology, 2011, 152, 1848–1859 CrossRef CAS PubMed.
- S. Li, Y. Zhi, W. Mu, M. Li and G. Lv, Eur. J. Pharmacol., 2023, 176300 Search PubMed.
- D. Sprecher, T. Johnson, E. Olson, S. Hirschberg, A. Liu, Z. Fang, P. Hegde, D. Richards, L. Sarov-blat, J. C. Strum, S. Basu, J. Cheeseman, B. A. Fielding, S. M. Humphreys, T. Danoff, N. R. Moore, P. Murgatroyd, S. O. Rahilly, P. Sutton, T. Willson, D. Hassall, K. N. Frayn and F. Karpe, Diabetes, 2008, 57(2), 332–339 CrossRef PubMed.
- L. Tong, L. Wang, S. Yao, L. Jin, J. Yang, Y. Zhang, G. Ning and Z. Zhang, Cell Death Dis., 2022, 151, 113127 Search PubMed.
- F. M. Silva-Veiga, T. L. Rachid, L. de Oliveira, F. Graus-Nunes, C. A. Mandarim-de-Lacerda and V. Souza-Mello, Mol. Cell. Endocrinol., 2018, 474, 227–237 CrossRef CAS PubMed.
- S. Du, N. Wagner and K. Wagner, PPAR Res., 2020, 2020(1), 3608315 Search PubMed.
- Y. Tobita, T. Arima, Y. Nakano, M. Uchiyama, A. Shimizu and H. Takahashi, Int. J. Mol. Sci., 2020, 21, 1–13 Search PubMed.
- W. Shan, P. S. Palkar, I. A. Murray, E. I. Mcdevitt, M. J. Kennett, B. H. Kang, H. C. Isom, G. H. Perdew, F. J. Gonzalez and J. M. Peters, Toxicol. Sci., 2008, 105, 418–428 CrossRef CAS PubMed.
- M. Webb, D. P. Sideris and M. Biddle, Bioorg. Med. Chem. Lett., 2019, 29, 1270–1277 CrossRef CAS PubMed.
- L. Xiao and N. Wang, J. Mol. Cell. Cardiol., 2022, 169, 1–9 CrossRef CAS PubMed.
- X. Wang, G. Wang, Y. Shi, L. Sun, R. Gorczynski, Y.-J. Li, Z. Xu and D. E. Spaner, Oncogenesis, 2016, 5, e232 CrossRef CAS PubMed.
- T. T. L. Wu, H. S. Niu, L. J. Chen, J. T. Cheng and Y. C. Tong, Eur. J. Pharmacol., 2016, 775, 35–42 CAS.
- N. Wagner and K. D. Wagner, Cells, 2022, 11(15), 2432 CAS.
- B. Da’adoosh, D. Marcus, A. Rayan, F. King, J. Che and A. Goldblum, Sci. Rep., 2019, 9, 1106 CrossRef PubMed.
- R. V Connors, Z. Wang, M. Harrison, A. Zhang, M. Wanska, S. Hiscock, B. Fox, M. Dore, M. Labelle and A. Sudom, Bioorg. Med. Chem. Lett., 2009, 19, 3550–3554 Search PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CAS.
- S. K. Mandal, B. K. Kumar, P. K. Sharma, S. Murugesan and P. R. Deepa, Comput. Biol. Med., 2022, 147, 105796 CAS.
- S. K. Mandal, S. Puri, B. K. Kumar, M. Muzaffar-Ur-Rehman, P. K. Sharma, M. Sankaranarayanan and P. R. Deepa, Mol. Diversity, 2024, 28, 1423–1438 CAS.
- D. E. Shaw, J. P. Grossman, J. A. Bank, B. Batson, J. A. Butts, J. C. Chao, M. M. Deneroff, R. O. Dror, A. Even, C. H. Fenton, A. Forte, J. Gagliardo, G. Gill, B. Greskamp, C. R. Ho, D. J. Ierardi, L. Iserovich, J. S. Kuskin, R. H. Larson, T. Layman, L. S. Lee, A. K. Lerer, C. Li, D. Killebrew, K. M. Mackenzie, S. Y. H. Mok, M. A. Moraes, R. Mueller, L. J. Nociolo, J. L. Peticolas, T. Quan, D. Ramot, J. K. Salmon, D. P. Scarpazza, U. Ben Schafer, N. Siddique, C. W. Snyder, J. Spengler, P. T. P. Tang, M. Theobald, H. Toma, B. Towles, B. Vitale, S. C. Wang and C. Young, in International Conference for High Performance Computing, Networking, Storage and Analysis, SC, IEEE, 2014, vol. 2015, pp. 41–53 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CAS.
- S. Genheden, O. Kuhn, P. Mikulskis, D. Hoffmann and U. Ryde, J. Chem. Inf. Model., 2012, 52, 2079–2088 CrossRef CAS PubMed.
- C. Mulakala and V. N. Viswanadhan, J. Mol. Graphics Modell., 2013, 46, 41–51 CAS.
- Y. Sixto-López, M. Bello and J. Correa-Basurto, J. Comput.-Aided Mol. Des., 2020, 34, 857–878 Search PubMed.
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, SC'06, 2006, pp. 84 Search PubMed.
- M. P. Jacobson, R. A. Friesner, Z. Xiang and B. Honig, J. Mol. Biol., 2002, 320, 597–608 CrossRef CAS PubMed.
- V. Kumar, J. K. Dhanjal, S. C. Kaul, R. Wadhwa and D. Sundar, J. Biomol. Struct. Dyn., 2020, 39, 1–13 Search PubMed.
- M. Ikeguchi, J. Comput. Chem., 2004, 25, 529–541 CrossRef CAS PubMed.
- S. J. Stuart, R. Zhou and B. J. Berne, J. Chem. Phys., 1996, 105, 1426–1436 CrossRef CAS.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- F. A. Müller and S. J. Sturla, Curr. Opin. Toxicol., 2019, 16, 9–16 CrossRef.
- M. R. Jain, S. R. Giri, B. Bhoi, C. Trivedi, A. Rath, R. Rathod, R. Ranvir, S. Kadam, H. Patel and P. Swain, Liver Int., 2018, 38, 1084–1094 CrossRef CAS PubMed.
- D. Devineni and D. Polidori, Clin. Pharmacokinet., 2015, 54, 1027–1041 CrossRef CAS PubMed.
- E. Hoeben, W. De Winter, M. Neyens, D. Devineni, A. Vermeulen and A. Dunne, Clin. Pharmacokinet., 2016, 55, 209–223 CrossRef CAS PubMed.
- P. Kaur, B. S. Behera, S. Singh and A. Munshi, Eur. J. Pharmacol., 2021, 904, 174169 CrossRef CAS.
- D. Devineni, J. Murphy, S. Wang, H. Stieltjes, P. Rothenberg, E. Scheers and R. N. V. S. Mamidi, Clin. Pharmacol. Drug Dev., 2015, 4, 295–304 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |