
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCytotoxicity-guided isolation of elatostemanosides I–VI from Elatostema tenuicaudatum W. T. Wang and their cytotoxic activities†
Quoc-Dung Tran Huynh‡
 ab,
Thuy-Tien Thi Phan‡bc,
Ta-Wei Liud,
Truc-Ly Thi Duonge,
Su-Jung Hsudr,
Ching-Chuan Kuof,
Man-Hsiu Chud,
Yun-Han Wanga,
Thanh-Vu Nguyeng,
Yao-An Shenhij,
Yu-Jui Fankl,
Dang-Khoa Nguyenm,
Thanh-Hoa Vono and
Ching-Kuo Lee*adpq
ab,
Thuy-Tien Thi Phan‡bc,
Ta-Wei Liud,
Truc-Ly Thi Duonge,
Su-Jung Hsudr,
Ching-Chuan Kuof,
Man-Hsiu Chud,
Yun-Han Wanga,
Thanh-Vu Nguyeng,
Yao-An Shenhij,
Yu-Jui Fankl,
Dang-Khoa Nguyenm,
Thanh-Hoa Vono and
Ching-Kuo Lee*adpq
aPh. D. Program in Clinical Drug Development of Herbal Medicine, College of Pharmacy, Taipei Medical University, Taipei 11031, Taiwan. E-mail: cklee@tmu.edu.tw
bInstitute of Pharmaceutical Education and Research, Binh Duong University, Thu Dau Mot 820000, Binh Duong, Vietnam
cGraduate Institute of Biomedical Materials and Tissue Engineering, College of Biomedical Engineering, Taipei Medical University, Taipei 11031, Taiwan
dSchool of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei 11042, Taiwan
eFaculty of Traditional Medicine, Can Tho University of Medicine and Pharmacy, Can Tho 900000, Vietnam
fInstitute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Miaoli County 35053, Taiwan
gBiotechnology Center of Ho Chi Minh City, Ho Chi Minh City 700000, Vietnam
hDepartment of Pathology, School of Medicine, College of Medicine, Taipei Medical University, Taipei 110301, Taiwan
iGraduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, Taipei 110301, Taiwan
jInternational Master/Ph. D. Program in Medicine, College of Medicine, Taipei Medical University, Taipei 110301, Taiwan
kSchool of Biomedical Engineering, Taipei Medical University, Taipei 11031, Taiwan
lInternational PhD Program for Biomedical Engineering, Taipei Medical University, Taipei 110301, Taiwan
mFaculty of Pharmacy, Ton Duc Thang University, Ho Chi Minh 700000, Vietnam
nUniversity of Health Sciences, Vietnam National University Ho Chi Minh City, Ho Chi Minh 700000, Vietnam
oCenter for Discovery and Development of Healthcare Product, Vietnam National University Ho Chi Minh City, Ho Chi Minh 700000, Vietnam
pGraduate Institute of Pharmacognosy, College of Pharmacy, Taipei Medical University, Taipei 11042, Taiwan
qDepartment of Chemistry, Chung Yuan Christian University, Zhongli District, Taoyuan 32023, Taiwan
rInstitute of Fisheries Science, National Taiwan University, Taipei 106, Taiwan
First published on 4th April 2025
Abstract
Elatostema tenuicaudatum W. T. Wang, a medicinal plant traditionally utilized in herbal remedies, was explored for its cytotoxic properties. Bioassay-guided fractionation led to the discovery of six novel compounds, designated as elatostemanosides I–VI, with their structures elucidated through advanced spectroscopic methods and DP4+ analysis. Among these, compounds 2, 5, and 6 demonstrated moderate cytotoxicity against the human liver cancer cell line HepG2, exhibiting IC50 values of 18.2 ± 2.1, 32.1 ± 0.4, and 57.6 ± 1.3 µM, respectively. Notably, compound 6 also displayed significant activity against the human breast cancer cell line HCC1806, with an IC50 value of 35.4 ± 0.3 µM. Mechanistic studies revealed these compounds induced apoptosis by modulating the Bax/Bcl-2 ratio. Furthermore, structure–activity relationship (SAR) analysis underscored the importance of specific functional groups in mediating cytotoxic effects.
1. Introduction
Cancer is a leading cause of death worldwide, accounting for millions of cases and deaths each year.1 Among these, liver and breast cancers are particularly challenging to treat due to their complexity and drug resistance.2,3 To date, the number of chemotherapeutic drugs available for liver and breast cancers is still limited and often accompanied by significant side effects.4 Given these challenges, improving drug formulations to enhance the therapeutic efficacy of existing treatments is a crucial strategy. Advances in formulation strategies can optimize drug delivery, reduce side effects, and improve patient compliance.5–7 In addition, the discovery of new, effective, and safe therapeutic agents is crucial, especially from natural product resources.8 A variety of techniques are applied to search for potential candidates, with cytotoxicity screening being an essential initial step due to its effectiveness, simplicity, and cost- and time-efficiency.9Apoptosis, or programmed cell death, is a regulated process essential for cellular balance and eliminating damaged or malignant cells. It operates through pathways like the intrinsic (mitochondrial) pathway, which responds to intracellular stress and chemotherapeutic agents.10–12 Emerging drugs target these pathways to selectively kill cancer cells while sparing healthy ones, offering improved therapeutic outcomes.13,14 For studying apoptosis, flow cytometry is a powerful tool that offers high sensitivity and quantitative single-cell data.15 Using annexin V and propidium iodide (PI) staining, it enables precise classification of apoptotic stages, providing critical insights into apoptotic mechanisms and supporting anticancer drug discovery.16,17
One of the central regulators of the intrinsic pathway is the Bcl-2 family of proteins, which includes both pro-apoptotic (e.g., Bax, Bak) and anti-apoptotic (e.g., Bcl-2, Bcl-xL) members. The balance between these opposing factions determines the cell's fate. An increased Bax/Bcl-2 ratio favors mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c into the cytosol. Cytochrome c then binds to apoptotic protease activating factor-1 (Apaf-1), forming the apoptosome, which subsequently activates caspase-9. This activation triggers a caspase cascade culminating in the execution of apoptosis.18–21 Screening for cytotoxic agents based on their ability to modulate the Bax/Bcl-2 ratio has become a promising strategy in anticancer drug discovery. Compounds that upregulate pro-apoptotic proteins or inhibit anti-apoptotic ones can induce apoptosis selectively in cancer cells, providing a targeted approach to combat drug resistance and reduce side effects.22–24
Elatostema tenuicaudatum W. T. Wang, belonging to the Urticaceae family, is known both as an edible vegetable and an herbal medicine in the Vietnamese Northwest region. Besides being a delicious food, it is traditionally employed in ethnomedicine to treat diabetes, liver dysfunction, stomatitis, and cancer. A previous study isolated three dihydrophenanthrenes and assessed the anti-inflammatory activity of this plant.25 However, research on this plant remains limited. This study reports the cytotoxicity-guided isolation and the cytotoxic effects of compounds against human hepatocellular carcinoma (HepG2) and human triple-negative breast cancer (HCC1806) cell lines. Additionally, the structure–activity relationship (SAR) was investigated, alongside an analysis of the underlying mechanisms of action through flow cytometry and western blot techniques.
2. Results and discussion
2.1. Bioassay-guided isolation and structural elucidation
Fractions 1–11 were tested for cytotoxic effects at a concentration of 400 µg ml−1. The results indicated that fractions 5–8 exhibited cell viability below 70% (Fig. 1). As a result, these fractions were selected for further isolation, leading to the discovery of six new compounds 1–6. | ||
| Fig. 1 Cytotoxicity of fractions 1–11 (400 µg ml−1) and sorafenib (50 µM) against HepG2. | ||
The Polysaccharide Compositional Assay Kit was used to identify the sugar type by HPLC-MS or CE-DAD26,27 In the hydrolysates of all compounds 1–6, peaks corresponding to D-glucose-naphthylimidazole (NAIM) with a retention time of 4.92 minutes were observed by comparing to a reference standard. This confirmed the monosaccharide of compounds 1–6 as D-glucose.
Compound 1 was acquired as a white amorphous powder. It was assigned a formula form of C16H26O7 that was established based on peaks at m/z 353.15680 [M + Na]+, calcd for C16H26O7Na, m/z 375.16505 [M + HCOO]−, calcd for C17H27O9 in the HRESIMS. Its IR spectrum revealed the presence of hydroxy and conjugated carbonyl groups (3392 and 1694 cm−1, respectively). UV absorption at λmax 204, 226 and 290 nm were indicated in the UV spectrum. The 1H-NMR show one proton of aldehyde group signal at δH 9.35 (s); four olefinic protons at δH 6.67 (td, 7.3, 1.4), 6.11 (dd, 17.7, 11.0), 5.19 (dd, 11.0, 1.2) and 5.26 (dd, 17.7, 1.2); two tertiary methyl groups at δH 1.72 (s) and 1.38 (s) and anomer proton at δH 4.36 (d, 7.9). Combine 1H-NMR, 13C-NMR, DEPT and HSQC NMR data exhibited that compound 1 contains two CH3, four CH2, seven CH and three quaternary carbon groups. The chain bone was established based on 1D and 2D NMR signals as in Fig. 3. The linkage of glucose was identified according to the HMBC correlation from H-1′ to C-6 and the beta-orientation of sugar was also confirmed by J value equal to 7.9 Hz of H-1′ anomer. The Z configuration of C2–C3 double bond was established based on NOE correlation of H-1/H-3 and H-4/H-10 (Fig. 3). For the absolute configuration, linear correlation between experimental and calculated proton and carbon NMR values and DP4+ analysis was conducted to identify the configuration of the C-6 position.28–30 The results showed that 6S-1 and 6R-1 both have good correlation with correlation coefficient (R2) values of 0.9960 and 0.9965, respectively (Fig. 4). Furthermore, by DP4+ analysis, configuration 6S-1 acquired the match ratio values of 3.72% (uDP4+ – all data), 50.97% (sDP4+ – all data), 3.86% (DP4+ – all data) compared to configuration 6R-1 with match ratio values of 96.28% (uDP4+ – all data), 49.03% (sDP4+ – all data), 96.14% (DP4+ – all data) (ESI Tables S1–S3†). Therefore, the absolute configuration position of C-6 was identified as 6-R form, and their chemical structure was established as in Fig. 2, named elatostemanoside I. Compound 1 was a glycoside aldehyde form of the methiafolic acid family which was isolated from Jasminum hemsleyi.31
 | ||
| Fig. 2 Chemical structure of compounds 1–6. | ||
Compound 2 was isolated as a white amorphous powder. Its molecular formula of C17H22O8 was identified based on HRESIMS (m/z 355.13827 [M + H]+, calcd for C17H23O8, m/z 399.12857 [M + HCOO]− calcd for C18H23O10). The IR spectrum showed absorption bands corresponding to hydroxy (3388 cm−1) and conjugated carbonyl groups (1657 cm−1). The UV spectrum displayed absorption maxima at 208, 230, 273, and 307 nm. Based on 1D NMR spectrum, one tetrasubstituted benzene group with two olefinic protons at δH 6.90 (s, H-5), 7.68 (s, H-8); one methoxy group, one β-orientation proton anomer δH 4.94 (d, 7.2, H-Glc1) were explored. The O-sugar linkage was confirmed by the HMBC correlation of H-Glc1/C-6 and NOESY correlation of H-Glc1/H-5 and OMe linkage was also determined by HMBC signal of OMe/C-7 and NOESY signal of OMe/H-8. In addition, the position of C4a, C8a, C-1, C-4 were confirmed based on HMBC signal of H-5/C-4a, C-4, C-10a, H-8/C-1, C-4a. Therefore, the structure of 2 was shown in Fig. 2, named elatostemanoside II.
Compound 3 was obtained as a white amorphous powder. It showed the molecular formular of C20H30O8 which was deduced on the basis of the HRESIMS results at m/z 399.20109 [M + H]+, calcd for C20H31O8, m/z 397.18567 [M − H]−, calcd for C20H29O8. Its IR spectrum revealed the presence of hydroxy at 3360 cm−1 and UV spectrum showed the absorption maxima at 205, 222 and 280 nm. The 1H-NMR spectrum of 3 exhibited the presence of ABX system benzene rings [δH 6.68 (d, 2.1, H-2′), 7.07 (d, 8.2, H-5′), 6.60 (dd, 8.3, 2.1, H-6′)] and one anomer proton [δH 4.69 (d, 7.6, H-Glc1)]. The large coupling constant of H-Glc at 7.4 Hz also confirmed β-orientation position of the sugar. The 13C-NMR of compound 3 displayed 20 carbon signals, including six olefinic carbon signals [δC 140.2 (C-1′), 117.1(C-2′), 148.3 (C-3′), 144.8 (C-4′), 119.2 (C-5′) and 120.7 (C-6′)], 1 anomer carbon signal [δC 104.9 (C-Glc1′)], seven oxygenated carbon signals [δC 67.4 (C-3), 74.9 (C-Glc2), 77.7 (C-Glc3), 71.3 (C-Glc4), 78.3 (C-Glc5) and 62.4 (C-Glc6)]. Based on 1D, 2D NMR spectrum, the sugar signal (Glc-1 to Glc-6) and the aliphatic chain form and ABX system benzene ring were established (Fig. 3). The HMBC correlation from H-Glc1, H-2′, H-5′ to C-3′ confirmed the O-glycoside linkage of glucose sugar to C-3′. In addition, HMBC signals between H8/H-1′, H-2′, H-5′ and H7/H-1, H-2 were more important evidence for establishing the positions of atom on aliphatic part of 3 (Fig. 3). To determine the configuration at two positions of carbon C-1 and C-3, NOESY spectrum was assessed. The NOE correlation of H-1/H-5ax confirmed α-axial orientation of H-1. The β-equatorial orientation of H-3 was also established according to the NOE correlation of H-3/H-2ax,eq, H-4ax,eq (Fig. 3). Therefore, the structure of 3 was depicted on the Fig. 3, named elatostemanoside III.
 | ||
| Fig. 3 Key HMBC, NOESE and COSY correlations of compounds 1–6. | ||
 | ||
| Fig. 4 Linear correlations of the experimental 6S-1 and 6R-1 with the calculated 13C NMR chemical shifts for 1. | ||
Compound 4 was acquired as white amorphous powder. It possessed the molecular formular of C20H30O8 based on its HRESIMS results at m/z 399.20108 [M + H]+, calcd for C20H31O8, m/z 421.18258 [M + Na]+, calcd for C20H30O8Na, m/z 397.18570 [M − H]−, calcd for C20H29O8, m/z 443.19117 [M + HCOO]−, calcd for C21H30O10. Compound 4 had the same molecula formular and the similarity UV (λmax 205, 222 and 280 nm) and IR (3364 cm−1) spectrum to compound 3 that suggested to be an isomer of compound 3. This conclusion was be confirmed by analyzing its 1D and 2D NMR data and the major difference between them is the trisubstituted AMX system benzene instead of ABX in 3. The α-axial orientation of H-1, β-equatorial orientation of H-3 and β-glucose was determined similarly as those in 3 (Fig. 3). According to HMBC correlation of H-2′, H-6′, H-7, H-8/H-1′; H-4, H-6/H-5′, H-2, H-4, H-Glc1/H-3′, the position of substituted groups on the aromatic ring were established. From above the analysis, the structure of compound 4 was displayed in Fig. 2, named elatostemanoside IV.
Compound 5 was isolated as a white amorphous powder. The IR spectrum displayed a band at 3355 cm−1, indicating the presence of a hydroxy group, and the UV spectrum showed absorption maxima at 206, 226, and 282 nm. Its molecular formula of C20H30O8 was determined by the HRESIMS results at m/z 399.20109 [M + H]+, calcd for C20H31O8, m/z 421.18296 [M + Na]+, calcd for C20H30O8Na, m/z 397.18549 [M − H]−. It had same molecular formular with 3 and 4, that suggested they were isomers. Similarity to 3, NMR data 5 displayed three parts of the structure, one ABX aromatic ring, one β-orientation sugar and one aliphatic bone. In the HMBC spectrum, the correlation between H-2′, H-6′, H-7, H-8/C-1′ and H-2, H-5, H-7, H-8/C-1, H-2/C-8 confirmed the substituted positions at C-1 and C-1′, respectively. The O-glycoside linkage from sugar to C-3 was also established based on HMBC correlation from H-Glc1, H-2, H-4 to C-4 (Fig. 3). Besides, the NOE correlation of H-1/H-3, H-5 determined the β-axial orientation of H-3 and H-1 (Fig. 3). Therefore, the structure of 5 was showed in Fig. 2, named elatostemanoside V.
Compound 6 was acquired as white amorphous powder and have same formula molecular of C20H30O8 with 6 based on HRESIMS data m/z 399.20106 [M + H]+, calcd for C20H31O8, m/z 421.18293 [M + Na]+, calcd for C20H30O8Na, m/z 397.18562 [M − H]−, calcd for C20H29O8, m/z 795.37903 [2M − H]−, calcd for C40H59O16, m/z 443.19122 [M + HCOO]−, calcd for C21H31O10,. Additionally, the IR spectrum exhibited a band at 3358 cm−1 (hydroxy group). The UV spectrum showed λmax at 206, 226, and 282 nm. By analysis 1D and 2D NMR data, the planar structure was got as same as 5 (Fig. 3). This suggested they were stereoisomers. In the NOESY spectrum, the key correlation between H-1/H-5 confirmed the β-axial orientation of H-1 (Fig. 3). Furthermore, α-equatorial orientation of H-3 was also established based on NOE signal between H-3/H-4ax,eq, H-2ax,eq. Thus, the structure of 6 was depicted in Fig. 2, named elatostemanoside VI.
2.2. Cytotoxicity effect and structure–activity relationship (SAR) analysis
Among compounds 1–6, only compound 2 belongs to the hydroxynaphthalenone group, some members of which have been investigated for their cytotoxic effects on HepG2 cells. The results revealed that the compound with the lowest IC50 value was 88.23 µM.32 Additionally, to the best of our knowledge, no studies have been found on the effects of similar compounds on the remaining compounds with HepG2 or HCC1806 cells. Based on this evidence, combined with our experience, we selected concentrations of 100 µM for HepG2 and 40 µM for HCC1806 for the initial screening. Only compounds that showed the cytotoxic activity of more than 50% were continuously assessed IC50. On HepG2 model, compound 2 exhibited the strongest effect with IC50 value of 18.2 ± 2.1 µM, followed by 6 (IC50 = 32.1 ± 0.4 µM) and 5 (57.6 ± 1.3 µM). And on HCC1806 model, only compound 6 showed moderated cytotoxic activity with IC50 values of 35.4 ± 0.3 µM (Table 3). Furthermore, compound 2, 5, 6 were also tested cytotoxicity on normal cell fibroblast CCD-966SK. At concentration of 100 µM, all of them showed no toxicity with cell viability of 98.5%, 99.7% and 97.3%, respectively. These findings suggest that compounds 2, 5, and 6 exhibit selective cytotoxicity against cancer cells while demonstrating minimal toxicity toward normal fibroblast cells, even at high concentrations. This selective action highlights their potential as promising candidates for further investigation and development as anti-cancer agents.Compound 2 belongs to the hydroxynaphthalenon family which showed many bioactivities of potential neuroprotective, anti-cancer, anti-virus and anti-bacteria.32–38 For structure–activity relationship analysis (SAR), we used compounds a–g (Fig. 5) which were isolated in previous study32 and compound 2 in our study. Firstly, the presence of a bulky group at the C-4 position of the cyclohexanone ring was a crucial factor that significantly reduced cytotoxic effects. For instance, compound 2, which lacks a substituent at the C-4 position, exhibited an IC50 value of 18.20 µM. In comparison, compounds a and b, which share the same hydroxynaphthalenone core but have –OH groups at C-4, showed higher IC50 values of 56.9 µM and 67.9 µM, respectively. Compound c, similar in structure to compound b but with a bulkier –OCH3 group at C-4 instead of –OH, further increased the IC50 to 88.23 µM. Meanwhile, compounds d–g, which have the largest –O–Glc substituents at the C-4 position, replacing the –OH/−OCH3 or lack of substituents in compounds a, b, c, and 2, showed no activity. However, substituents on the benzene ring showed the opposite trend, where increased complexity led to higher cytotoxicity. For instance, the cytotoxicity of compound 2 (contained two substituents –OH and –O–Glc) was greater than that of compounds a (contained one substituent –OH) and b (no substituent). Additionally, the effect of the substituents on the cyclohexanone ring outweighed those on the benzene ring. For example, compounds d–g, containing an –O–Glc group conjugated to the C-4 position, showed no activity, regardless of the number or position of substituents on the benzene ring (Fig. 5). Final, compound 2, which exhibited the strongest cytotoxic activity against HepG2 cells, showed no effect on HCC1806 cells, indicating a selective effect on different cancer types.
 | ||
| Fig. 5 Cytotoxicity IC50 values of dihydrophenanthrenones on HepG2 cell model (NA: no activity, compounds a–g: data from previous study,32 2: compound 2) for SAR analysis. | ||
Compounds 3–6 have structures that contain a phenol ring conjugated to a cyclohexane ring by an ethane bridge. They are different by the position –OH and –O–Glc substituent on the phenol or cyclohexane ring. Notably, compounds 3, 4 with glycosidic substituent on phenol group do not show the cytotoxic effect, whereas compounds 5, 6 with glycosidic substituent on cyclohexan group. This suggested the position of glucose plays an essential role in the cytotoxicity effect. Besides, the cis and trans configuration of H-1 and H-3 have also an impact to the activity, that the trans showed the better effect than cis one. For example, compound 5 (cis position) and compound 6 (trans position) acquired the IC50 values of 57.6 ± 1.3 µM, 32.1 ± 0.4 µM respectively on HepG2. On HCC1806, compound 5 also showed the IC50 values of 35.4 ± 0.3 µM, whereas no activity on compound 6 (Fig. 6).
 | ||
| Fig. 6 Structure and cytotoxicity IC50 values for SAR analysis of compounds 3–6. | ||
By the cytotoxicity-guided technique, we successfully isolated three bioactive compounds from potential fractions. These compounds could be used as potential structures for the optimization and development of potential therapeutic agents for cancer treatment.
2.3. Cell apoptosis analysis
Following the assessment of their cytotoxicity effect on HepG2, HCC1806 cancer cells and non-cancerous cells, compounds 2, 5, and 6 were selected for further investigation due to their activity. To evaluate their potential to induce apoptosis, HepG2 cancer cells were treated with varying concentrations of the selected compounds for 72 hours. Subsequently, apoptotic cell percentages were quantified using a flow cytometry assay and the results were shown in Fig. 7. The data reveal a significant dose-dependent induction of apoptosis for all three compounds, with distinct variations in early and late apoptotic stages. Compound 2 exhibited a potent pro-apoptotic effect. At 10 µM, it induced apoptosis in 69.5% of HepG2 cells, with 59.6% in the early apoptotic stage and 17.1% in the late apoptotic stage. At 20 µM, the apoptotic effect was further enhanced, with 86.0% of cells undergoing apoptosis (66.7% early apoptosis and 19.3% late apoptosis). These findings indicate that compound 2 is highly effective in triggering apoptosis, particularly in the early stage. Compound 5 also demonstrated significant apoptotic activity. At a concentration of 40 µM, it caused apoptosis in 64.6% of cells, with 51.0% in early apoptosis and 15.8% in late apoptosis. Increasing the concentration to 60 µM resulted in an increase in total apoptosis to 71.6%, with 53.2% of cells in early apoptosis and 18.5% in late apoptosis. Among the three compounds, compound 6 induced the highest levels of apoptosis at comparable concentrations. At 20 µM, 71.9% of cells were apoptotic, with 53.7% in early apoptosis and 21.1% in late apoptosis. At 40 µM, the total apoptotic population rose to 84.7%, with 56.5% in early apoptosis and 28.2% in late apoptosis. These results suggest that compound 6 is particularly effective in inducing late apoptosis at higher concentrations. | ||
| Fig. 7 Apoptosis induction in HepG2 cancer cells by compounds 2, 5, and 6 after 72 hours of treatment (***P < 0.001 vs. control). | ||
2.4. Induction of apoptosis via modulation of the Bax/Bcl-2 ratio
The Bcl-2 protein family plays a pivotal role in regulating apoptosis, with the balance between pro-apoptotic and anti-apoptotic members determining cellular fate. Bax, a pro-apoptotic protein, promotes apoptosis by permeabilizing the mitochondrial outer membrane, leading to cytochrome c release and activation of the caspase cascade. The interaction between Bax and its anti-apoptotic counterpart, Bcl-2, is critical, as their relative levels influence whether a cell survives or undergoes apoptosis.39–41 Therefore, the Bax/Bcl-2 ratio is considered a more accurate predictor of apoptotic status than the absolute levels of either protein.42The effects of compounds 2, 5, and 6 on the Bax/Bcl-2 ratio in HepG2 cells were analyzed using western blot, as shown in Fig. 8. The results demonstrate a dose-dependent increase in the Bax/Bcl-2 ratio, indicating enhanced pro-apoptotic activity with increasing concentrations of the compounds. Compound 2 significantly increased the Bax/Bcl-2 ratio in a concentration-dependent manner. At 10 µM, the ratio increased to 1.43 compared to the control (1.0), and at 20 µM and 30 µM, the ratio further rose to 3.0 and 7.0, respectively. This steep increase highlights the strong pro-apoptotic effect of compound 2, particularly at higher concentrations. Similarly, compound 5 exhibited a pronounced effect on the Bax/Bcl-2 ratio. At 40 µM, the ratio increased to 1.5, and at 60 µM and 80 µM, it reached 3.2 and 6.4, respectively. These findings suggest that compound 5 effectively shifts the balance towards apoptosis by upregulating Bax or downregulating Bcl-2 expression. Compound 6 also demonstrated a dose-dependent increase in the Bax/Bcl-2 ratio, though to a lesser extent compared to compounds 2 and 5. At 20 µM, the ratio was 1.1, increasing to 2.6 at 40 µM and 3.4 at 60 µM. This moderate increase indicates that compound 6 induces apoptosis, albeit less efficiently than the other compounds at equivalent concentrations. These results also suggested the apoptosis activity of compound 2, 5 and 6 through the mitochondrial pathway. The Bax/Bcl-2 ratio is a critical determinant of the intrinsic apoptotic pathway, with higher ratios favoring mitochondrial membrane permeabilization and subsequent activation of caspases. The dose-dependent increases in the Bax/Bcl-2 ratio observed for compounds 2, 5 and 6 confirm their pro-apoptotic potential, with compound 2 showing the strongest effect, followed by compound 5 and compound 6. These results suggest that the compounds exert their effects through the modulation of pro- and anti-apoptotic proteins, effectively tipping the balance towards apoptosis. The differences in efficacy among the compounds highlight potential variations in their mechanisms of action or cellular uptake. These results also suggested that compounds 2, 5, 6 induced apoptosis through the mitochondrial pathway.43,44
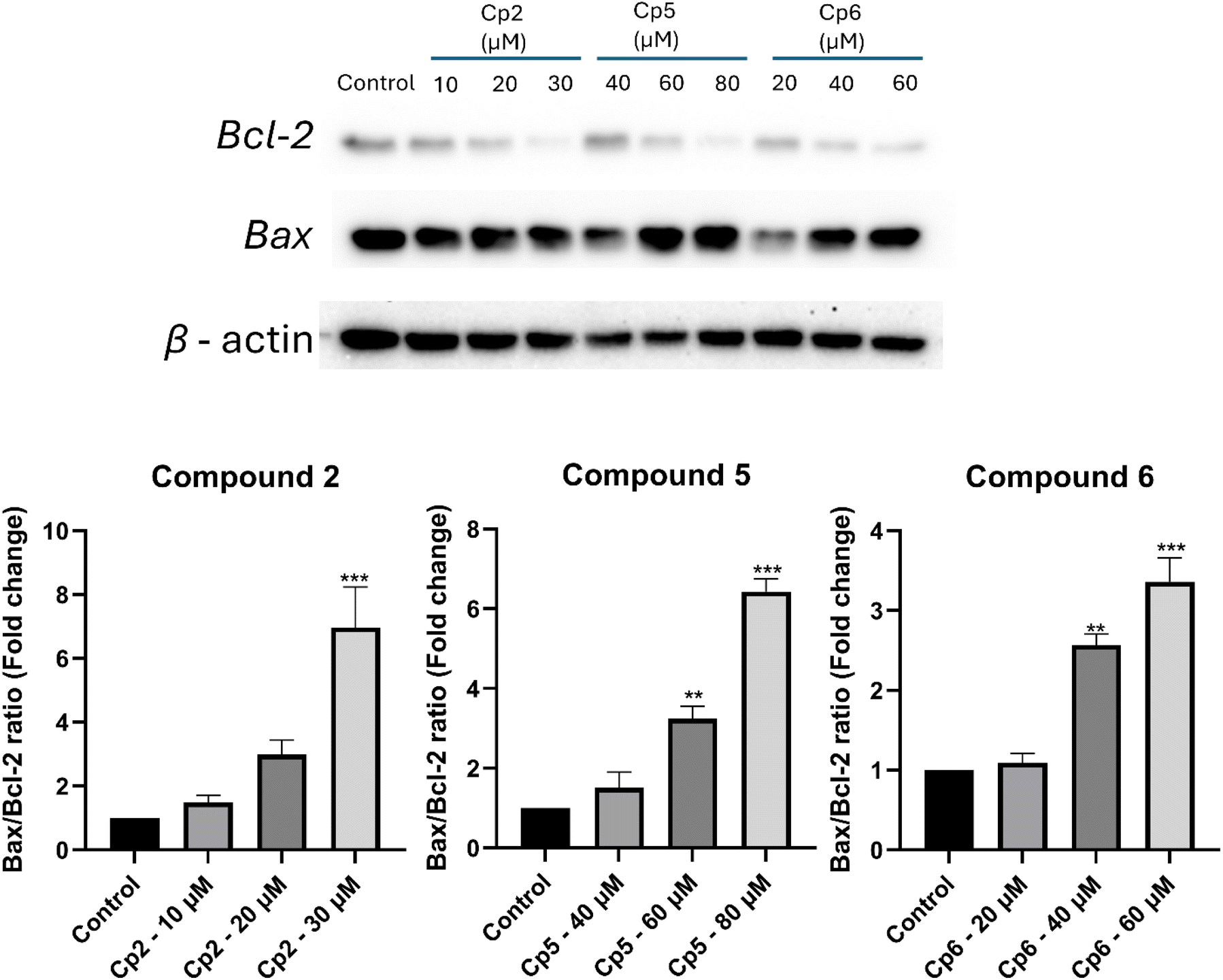 | ||
| Fig. 8 Effects of compound 2, 5, 6 on the Bax/Bcl-2 ratio HepG2 cells after treatment different concentrations: (A) presents representative results from western blot analysis, while (B) depicts the corresponding Bax/Bcl-2 ratio chart. The data are reported as mean ± standard deviation (SD) based on three independent experiments. Statistical significance is denoted by **, and ***, indicating P-values of <0.01, and <0.001 vs. control, respectively. | ||
3. Conclusion
In summary, six previously unreported glycosides (1–6) were isolated from Elatostema tenuicaudatum. Compounds 1 and 2 were identified as a menthiafolaldehyde glycoside and a dihydrophenanthrenone glycoside derivative, respectively. Compounds 3–6 featured a 6/2/6 structural framework, comprising a cyclohexane ring connected to a phenol ring via an ethane bridge. Their cytotoxic effects were also evaluated, and preliminary structure–activity relationships (SAR) were discussed. Notably, compound 2 exhibited the strongest cytotoxicity against HepG2 cells, while compound 6 showed the highest activity against HCC1806 cells. Compound 2, 5 and 6 also induce apoptosis in HepG2 model through the mitochondrial pathway by enhancing the Bax/Bcl-2 ratio.4. Material and methods
4.1. General experimental procedures
Amos GV-800S system (Amos instruments Corp., Taiwan) was used for extraction and concentrating. UV, IR, optical rotation, HRMS, NMR spectra were conducted on Thermo UV-vis Helios (USA), JASCO FT/IR 4100 (Japan), JASCO P-2000 polarimeter (Japan), Thermo Q-ExactiveTM Plus Hybrid Quadrupole Orbitrap (USA) and Agilent DD2 600 MHz system (USA), respectively. Hitachi L-7000 with DAD L-7455 detector system used Thermo Hypurity C18 column (250 × 4.6 mm, 5 µm) and Shimadzu 20-AR preparative HPLC system used Cosmosil C18 (250 × 20 mm, 5 µm) for preparative injection. Column chromatography was conducted on Isolera ONE MPLC (Uppsala, Sweden). The human liver cancer cell line HepG2 was obtained from the Bioresource Collection and Research Center (Taiwan) and the human breast cancer cell line HCC1808 was obtained from National Health Research Institutes (Taiwan), non-cancerous fibroblast (CCD-966SK) cell lines were sourced from Biotechnology Center of Ho Chi Minh City. Cytotoxicity experiments were assessed on Varioskan LUX spectrophotometer system (Thermo Fisher Scientific, Bremen, Germany). Flow cytometry was performed using the FITC Annexin V Apoptosis Detection Kit with PI (BioLegend) on an Invitrogen Attune™ NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific, Bremen, Germany). Antibodies against Bax, Bcl-2, and β-actin, along with secondary antibodies, were procured from Cell Signaling Technology for western blot analysis.4.2. Plant materials
E. tenuicaudatum aerial parts were collected from Hoa Binh province, Vietnam in 2020. The authentic name was confirmed by Dr Thanh-Hoa, Vo (University of Health Sciences – Vietnam National University Ho Chi Minh city). The plant voucher was stored at the Pharmacognosy laboratory, Taipei Medical University.4.3. Bioassay-guided isolation
A total of 117 g of E. tenuicaudatum extract was obtained from dried material, which had been extracted using 96% ethanol in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 ratio (2 kg of material to 40 liters of ethanol) via a reflux and concentration system. After removing the non-polar fraction through liquid–liquid partitioning with n-hexane, the aqueous layer was further separated into Fractions 1–11 using MPLC with a normal-phase column. This separation was achieved with a gradient mobile phase of n-hexane, ethyl acetate, and methanol. Fractions 1 to 11 were then screened for cytotoxicity in a HepG2 model, and those fractions showing cell viability below 70% were subsequently purified (Fig. 1). Fraction 5 was repeated preparative – HPLC on C18 column with mobile phase of CH3CN 18% and flow rate of 9 ml min−1 to get 3.2 mg of compound 1. Fraction 6 and fraction 7 was also applied onto preparative – HPLC (C18 column, CH3CN 24%, 9 ml min−1) to yield compound 3 (17.3 mg), compound 4 (9.7 mg), compound 5 (30.0 mg), compound 6 (40.0 mg). Fraction 8 was initially separated into Subfraction 8.1 to Subfraction 8.10 by C-18 MPLC with methanol–water (gradient). Then, Subfraction 8.4 was conducted on C18 column preparative-HPLC with CH3OH 28% in 8 ml min−1 to obtain 17.0 mg of compound 2 (Scheme 1).
:20 ratio (2 kg of material to 40 liters of ethanol) via a reflux and concentration system. After removing the non-polar fraction through liquid–liquid partitioning with n-hexane, the aqueous layer was further separated into Fractions 1–11 using MPLC with a normal-phase column. This separation was achieved with a gradient mobile phase of n-hexane, ethyl acetate, and methanol. Fractions 1 to 11 were then screened for cytotoxicity in a HepG2 model, and those fractions showing cell viability below 70% were subsequently purified (Fig. 1). Fraction 5 was repeated preparative – HPLC on C18 column with mobile phase of CH3CN 18% and flow rate of 9 ml min−1 to get 3.2 mg of compound 1. Fraction 6 and fraction 7 was also applied onto preparative – HPLC (C18 column, CH3CN 24%, 9 ml min−1) to yield compound 3 (17.3 mg), compound 4 (9.7 mg), compound 5 (30.0 mg), compound 6 (40.0 mg). Fraction 8 was initially separated into Subfraction 8.1 to Subfraction 8.10 by C-18 MPLC with methanol–water (gradient). Then, Subfraction 8.4 was conducted on C18 column preparative-HPLC with CH3OH 28% in 8 ml min−1 to obtain 17.0 mg of compound 2 (Scheme 1).
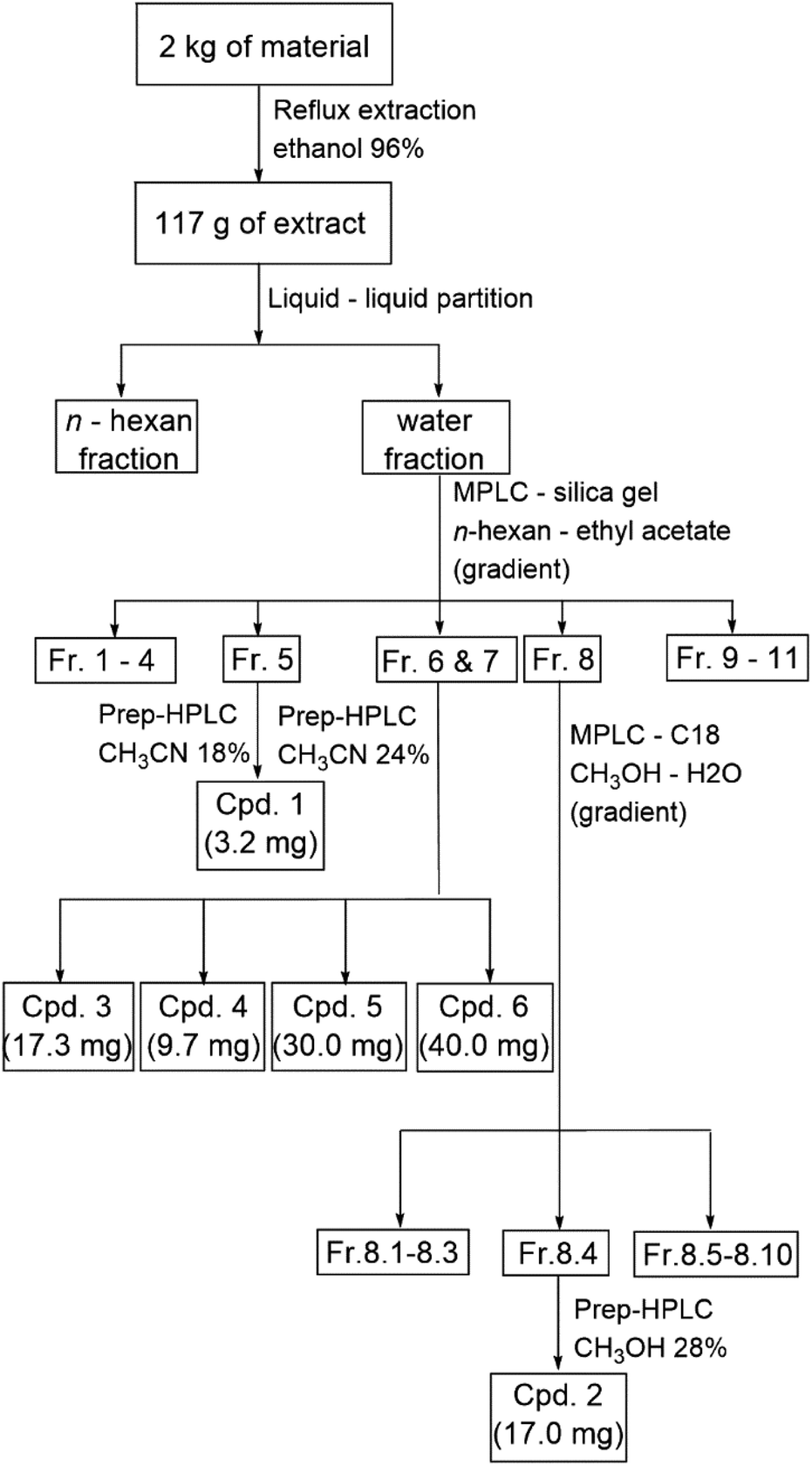 | ||
| Scheme 1 Extraction and isolation workflow of compounds 1–6 from Elatostema tenuicaudatum. | ||
| Position | 1 (CD3OD, 600 MHz) | Position | 2 (CD3OD, 600 MHz) | ||
|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | ||
| 1 | 197.3 | 9.35 (s) | 1 | 199.8 | |
| 2 | 140.3 | 2 | 39.4 | 2.58 (t, 6.0) | |
| 3 | 157.7 | 6.67 (td, 7.3, 1.4) | 3 | 24.7 | 24.7 (p, 6.0) |
| 4 | 24.8 | 2.53 (m) | 4 | 30.3 | 2.94 (p, 6.0) |
| 5 | 39.4 | 1.83 (m) | 4a | 126.7 | |
| 6 | 80.9 | 5 | 112.4 | 6.90 (s) | |
| 7 | 144.3 | 6.11 (dd, 17.7, 11.0) | 6 | 146.7 | |
| 8 | 115.3 | 5.19 (dd, 11.0, 1.2) | 7 | 156.0 | |
| 5.26 (dd, 17.7, 1.2) | |||||
| 9 | 9.1 | 1.72 (s) | 8 | 115.3 | 7.68 (s) |
| 10 | 24.1 | 1.38 (s) | 8a | 143.4 | |
| Glc-1 | 99.4 | 4.36 (d, 7.9) | O–Me | 56.7 | |
| Glc-2 | 75.1 | 3.17 (ovl) | Glc1 | 102.6 | 4.94 (d, 7.2) |
| Glc-3 | 78.3 | 3.34 (t, 9.3) | Glc2 | 74.9 | 3.46 (ovl) |
| Glc-4 | 71.8 | 3.28 (t, 9.3) | Glc3 | 77.9 | 3.49 (ovl) |
| Glc-5 | 77.7 | 3.17 (ovl) | Glc4 | 71.1 | 3.47 (ovl) |
| Glc-6 | 62.9 | 3.81 (dd, 11.8, 2.5) | Glc5 | 78.1 | 3.44 (ovl) |
| 3.63 (dd, 11.8, 5.7) | |||||
| Glc6 | 62.3 | 3.73 (dd, 12.2, 4.3) | |||
| 3.86 (dd, 12.2, 1.8) | |||||
| Position | 3 (CD3OD, 600 MHz) | 4 (CD3OD, 600 MHz) | 5 (CD3OD, 600 MHz) | 6 (CD3OD, 600 MHz) | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 32.4 | 1.75 (ovl) – ax | 32.2 | 1.75 (ovl) – ax | 37.2 | 1.32 (m) – eq | 32.6 | 1.79 (ovl) – ax |
| 2 | 40.5 | 1.28 (td, 11.0, 5.6) – ax | 40.5 | 1.28 (m) – αx | 41.7 | 1.00 (q, 11.9) – ax | 37.1 | 1.17 (1.17 (ddd, 13.6, 10.5, 2.7)) – ax |
| 1.75 (ovl) – eq | 1.75 (ovl) – eq | 2.13 (m) – eq | 1.95 (brd, 13.6) – eq | |||||
| 3 | 67.4 | 3.98 (m) – eq | 67.4 | 3.98 (m) – eq | 79.0 | 3.66 (ovl) | 75.1 | 4.05 (q, 3.7) – eq |
| 4 | 34.2 | 1.51 (ovl) – ax | 34.2 | 1.51 (ovl) – αx | 33.2 | 1.16 (1.16 (dtd, 13.2, 11.1, 3.4) – ax | 33.1 | 1.49 (ovl) – ax |
| 1.62 (ovl) – eq | 1.62 (ovl) – eq | 2.09 (brd, 11.1) – eq | 1.82 (ovl) – eq | |||||
| 5 | 21.1 | 1.50 (ovl) – ax | 21.1 | 1.51 (ovl) – ax | 25.1 | 1.24 (tt, 12.9, 3.4) – ax | 21.5 | 1.49 (ovl) – ax |
| 1.66 (ovl) – eq | 1.64 (ovl) – eq | 1.78 (dq, 12.9, 3.0) – eq | 1.72 (ovl) – eq | |||||
| 6 | 33.2 | 1.04 (m) – ax | 33.2 | 1.04 (m) – αx | 33.4 | 0.85 (m) – ax | 33.4 | 1.03 (m) – ax |
| 1.72 (ovl) – eq | 1.72 (ovl) – eq | 1.71 (brd, 13.5) – eq | 1.71 (ovl) – eq | |||||
| 7 | 39.7 | 1.47 (ovl) | 39.7 | 1.47 (ovl) | 40.4 | 1.49 (m) | 40.1 | 1.44 (ovl) |
| 8 | 33.7 | 2.51 (td, 7.5, 2.8) | 33.6 | 2.52 (td, 7.5, 2.6) | 33.5 | 2.48 (t, 7.9) | 33.7 | 2.46 (m) |
| 1′ | 140.2 | 136.1 | 135.7 | 136.1 | ||||
| 2′ | 117.1 | 6.68 (d, 2.1) | 119.1 | 7.03 (brs) | 116.5 | 6.60 (d, 2.1) | 116.5 | 6.61 (d, 2.1) |
| 3′ | 148.3 | 146.6 | 146.0 | 146.0 | ||||
| 4′ | 144.8 | 116.8 | 6.73 (brs) | 144.1 | 144.0 | |||
| 5′ | 119.2 | 7.07 (d, 8.2) | 146.2 | 116.3 | 6.65 (d, 8.1) | 116.3 | 6.65 (d, 8.0) | |
| 6′ | 120.7 | 6.60 (dd, 8.2, 2.1) | 124.5 | 6.73 (brs) | 120.6 | 6.48 (dd, 8.1, 2.1) | 120.6 | 6.48 (dd, 8.0, 2.1) |
| Glc1 | 104.9 | 4.69 (d, 7.6) | 104.6 | 4.73 (d, 7.5) | 102.4 | 4.39 (d, 7.8) | 102.6 | 4.32 (d, 7.8) |
| Glc2 | 74.9 | 3.47 (ovl) | 74.9 | 3.48 (ovl) | 75.2 | 3.14 (dd, 9.0, 7.8) | 75.2 | 3.18 (dd, 9.0, 7.8) |
| Glc3 | 77.7 | 3.46 (ovl) | 78.3 | 3.41 (m) | 78.1 | 3.35 (t, 9.0) | 78.2 | 3.37 (t, 9.0) |
| Glc4 | 71.3 | 3.40 (ovl) | 71.4 | 3.40 (m) | 71.7 | 3.27 (ovl) | 71.8 | 3.29 (ovl) |
| Glc5 | 78.3 | 3.39 (ovl) | 77.7 | 3.47 (ovl) | 77.9 | 3.27 (ovl) | 77.8 | 3.25 (ddd, 9.7, 5.5, 2.3) |
| Glc6 | 62.4 | 3.72 (dd, 12.0, 4.9) | 62.5 | 3.71 (dd, 12.1, 5.3) | 62.8 | 3.86 (dd, 11.9, 1.9) | 62.8 | 3.85 (dd, 11.8, 2.3) |
| 3.89 (dd, 12.0, 2.0) | 3.90 (dd, 12.1, 1.8) | 3.67 (ovl) | 3.66 (dd, 11.8, 5.5) | |||||
| Compounds | IC50 values on HepG2 (µM) | IC50 values on HCC1806 (µM) |
|---|---|---|
| 1 | >100 | >40 |
| 2 | 18.2 ± 2.1 | >40 |
| 3 | >100 | >40 |
| 4 | >100 | >40 |
| 5 | 57.6 ± 1.3 | >40 |
| 6 | 32.1 ± 0.4 | 35.4 ± 0.3 |
| Sorafenib | 12.3 ± 3.2 | — |
| Cisplatin | — | 8.3 ± 0.5 |
The spectroscopic data of compound 1–6 were showed in ESI Fig. S1–S59.†
4.4. Sugar identification
The sugar analysis was conducted by applying Polysaccharide Compositional Assay Kit of Sugar Light company (Taiwan and kit protocol). The UPLC-MS method utilized ACQUITY UPLC BEH C18 column (2.1 × 100 mm, 1.7 µm; Waters) and mobile phase consisting of two channels: channel A (0.1% formic acid in acetonitrile) and channel B (0.1% formic acid in water). The gradient program was as follows: at 0 and 6 minutes, channel A was at 7%, increasing to 13% at 8 minutes, 18% at 10 minutes, and reaching 50% at 10.5 minutes, maintaining this until 12 minutes, then returning to 7% by 12.1 minutes and remaining until 17 minutes. The flow rate was set at 0.3 µL min−1 with an injection volume of 10.0 µL. For mass spectrometry, electrospray ionization (ESI) was used in both positive ion modes. The spray voltages were 3.5 kV, while capillary and source heater temperatures were kept at 360 °C and 350 °C, respectively. The mass spectrometer executed at the SIM mode (m/z 319 for D-glucose-NAIM).4.5. NMR calculation
The chemical structure and initial conformers were generated using the Merck molecular force field (MMFF), followed by pre-optimization through the Semi-empirical (PM6) and Hartree–Fock (3-21G) methods in the Spartan 14 software (Wavefunction Inc., Irvine, CA, USA). Conformers with relative energies below 5 kcal mol−1 were then selected for further geometry optimization and frequency calculations using density functional theory (DFT) at the B3LYP/6-31G+(d,p) level in the gas phase, performed with Gaussian 09 W software. Next, NMR calculations were carried out using the GIAO method at the PCM/mpw1pw91/6-31+g(d,p) level, with theoretical NMR values determined based on Boltzmann-weighted averages. Finally, the computed proton and carbon NMR spectra were compared with experimental data, and the configuration was identified through DP4+ analysis.28–304.6. Cell viability assay
The cells were cultured in a complete medium composed of Dulbecco's Modified Eagle Medium (DMEM, Gibco), 10% Fetal Bovine Serum (FBS, Gibco), and 1% antibiotic–antimycotic BSA (Gibco). Once the cells reached 80% confluence, they were rinsed with PBS and detached using trypsin–EDTA (Gibco) at 37 °C for 5 minutes. The cells were then seeded into 96-well plates at a density of 3 × 104 cells per well (100 µL per well) and incubated for 24 hours at 37 °C with 5% CO2. Following this, 100 µL of fresh medium containing the test samples was added to replace the old medium. After 48 hours of incubation for HepG2 and 72 hours for HCC1806, cell viability was evaluated using the MTT assay according to standard protocols. Control cells were maintained under the same conditions for comparison. The absorbance readings were influenced by the solvent used in the sample, and cell viability was calculated using the formula: % viability = (total cells − viable cells)/total cells × 100. The compounds were screened at concentrations of 100 µM for HepG2 and 40 µM for HCC1806. Those that exhibited cytotoxicity exceeding 50% were further evaluated to determine their IC50 values. Sorafenib and cisplatin were used as positive control for HepG2 and HCC1806, respectively.4.7. Flow cytometry analysis
The effects of the tested compounds on cell cycle progression and apoptosis in HepG2 cells were evaluated using flow cytometry. HepG2 cells were seeded into 6-well plates at a density of 2 × 105 cells per well. For cell cycle analysis, the cells were treated with varying concentrations of the compounds for 72 hours. Staining was performed following the manufacturer's instructions. After treatment, the cells were collected, washed twice with cold BioLegend Cell Staining Buffer, and resuspended in Annexin V Binding Buffer at a concentration of 1.0 × 106 cells per ml. A 100 µL aliquot of the cell suspension was transferred into a 5 ml test tube. The cells were then incubated with 5 µL of FITC annexin V and 10 µL of propidium iodide (PI) solution for 15 minutes at room temperature in the dark. Following this, 400 µL of binding buffer was added to each sample. The samples were analyzed via flow cytometry within one hour, and cells positively stained with annexin V-FITC were identified as apoptotic. Data compensation and analysis were performed using FlowJo10 software.4.8. Western blot analysis
The effects of compounds 2, 5, and 6 on the Bax/Bcl-2 ratio in HepG2 cells were evaluated using western blot. HepG2 cells were cultured in 6-well plates at a density of 2 × 105 cells per well and treated with various concentrations of the compounds 72 hours. After treatment, the cells were harvested and lysed using lysis buffer supplemented with protease and phosphatase inhibitors. The protein concentration of the lysates was determined using the Bradford assay. Equal amounts of total protein (10 µg per sample) were separated on 12% SDS-PAGE gels and transferred to PVDF membranes. The membranes were blocked in 5% Bovine Serum Albumin (BSA) in Tris-buffered saline with 0.05% Tween-20 (TBS-T) for 1 hour at room temperature. Subsequently, the membranes were incubated overnight at 4 °C with primary antibodies against Bax (1:1000 dilution), Bcl-2 (1:1000 dilution), and β-actin (1:10000 dilution) as a loading control. After washing with TBS-T, the membranes were incubated with HRP-conjugated secondary antibodies (1:5000 dilution) for 2 hours at room temperature. The protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit, and the band intensities were normalized with β-actin quantified using ImageJ software.
Data availability
All relevant data are within the manuscript.Author contributions
Quoc-Dung Tran Huynh, Thuy-Tien Thi Phan: methodology, investigation, writing – original draft preparation, writing – review & editing. Truc-Ly Thi Duong, Su-Jung Hsu, Ching-Chuan Kuo, Thanh-Vu Nguyen, Yao-An Shen, Yu-Jui Fan: methodology, investigation. Ta-Wei Liu, Man-Hsiu Chu, Yun-Han Wang, Dang-Khoa Nguyen, Thanh-Hoa Vo: investigation, writing – reviewing and editing. Ching-Kuo Lee: resources, formal analysis, data curation, supervision, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Core Facility Center of Taipei Medical University and the Instrumentation Center of National Taiwan University, respectively, for the NMR and MS data acquisition. We are also grateful to Professor Tzong-Huei Lee – National Taiwan University, for the UV, IR, optical rotation spectra data. We greatly appreciate Professor Shu-Mei Lin from Food Science of National Chiayi University for providing HepG2 cells. This research was funded by the Ministry of Science and Technology (Taiwan), grant number MOST110-2320-B-038-039-MY3. Graphical abstract was created in BioRender. Huỳnh, D. (2025) https://BioRender.com/i61p560.Notes and references
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Cancer statistics, 2023, Ca-Cancer J. Clin., 2023, 73(1), 17–48, DOI:10.3322/caac.21763.
- J. K. Jallah, T. J. Dweh, A. Anjankar and O. Palma, A Review of the Advancements in Targeted Therapies for Breast Cancer, Cureus, 2023, e47847, DOI:10.7759/cureus.47847.
- F. Villarruel-Melquiades, M. E. Mendoza-Garrido, C. M. García-Cuellar, Y. Sánchez-Pérez, J. I. Pérez-Carreón and J. Camacho, Current and novel approaches in the pharmacological treatment of hepatocellular carcinoma, World J. Gastroenterol., 2023, 29(17), 2571–2599, DOI:10.3748/wjg.v29.i17.2571.
- U. Anand, A. Dey and A. K. S. Chandel, et al., Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2023, 10(4), 1367–1401, DOI:10.1016/j.gendis.2022.02.007.
- A. Saha, S. Mohapatra and P. Kurkute, et al., Targeted delivery of a novel peptide-docetaxel conjugate to MCF-7 cells through neuropilin-1 receptor: reduced toxicity and enhanced efficacy of docetaxel, RSC Adv., 2015, 5(112), 92596–92601, 10.1039/c5ra16741h.
- D. Bhunia, A. Saha, A. Adak, G. Das and S. Ghosh, A dual functional liposome specifically targets melanoma cells through integrin and ephrin receptors, RSC Adv., 2016, 6(114), 113487–113491, 10.1039/c6ra23864e.
- A. Saha, S. Mohapatra and G. Das, et al., Cancer Cell Specific Delivery of Photosystem I Through Integrin Targeted Liposome Shows Significant Anticancer Activity, ACS Appl. Mater. Interfaces, 2017, 9(1), 176–188, DOI:10.1021/acsami.6b13352.
- A. Naeem, P. Hu and M. Yang, et al., Natural Products as Anticancer Agents: Current Status and Future Perspectives, Molecules, 2022, 27(23), 8367, DOI:10.3390/molecules27238367.
- A. Setiawati, D. Candrasari, F. D. E. Setyajati, V. Prasetyo, D. Setyaningsih and Y. Hartini, Anticancer drug screening of natural products: in vitro cytotoxicity assays, techniques, and challenges, Asian Pac. J. Trop. Biomed., 2022, 12(7), 279–289, DOI:10.4103/2221-1691.350176.
- V. Singh, A. Khurana, U. Navik, P. Allawadhi, K. K. Bharani and R. Weiskirchen, Apoptosis and Pharmacological Therapies for Targeting Thereof for Cancer Therapeutics, Sci., 2022, 4(2), 15, DOI:10.3390/sci4020015.
- Y. Liu, T. Liu and T. Lei, et al., RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy, Int. J. Mol. Med., 2019, 44(3), 771–786, DOI:10.3892/ijmm.2019.4244.
- H. Kalkan, The Program Cell Death (Apoptosis) and the Therapy of Cancer, in Regulation and Dysfunction of Apoptosis, 2022, DOI:10.5772/intechopen.97289.
- B. A. Carneiro and W. S. El-Deiry, Targeting apoptosis in cancer therapy, Nat. Rev. Clin. Oncol., 2020, 17(7), 395–417, DOI:10.1038/s41571-020-0341-y.
- A. Saha, S. Mohapatra and P. Kurkute, et al., Interaction of Aβ peptide with tubulin causes an inhibition of tubulin polymerization and the apoptotic death of cancer cells, Chem. Commun., 2015, 51(12), 2249–2252, 10.1039/c4cc09390a.
- G. Banfalvi, Methods to detect apoptotic cell death, Apoptosis, 2017, 22(2), 306–323, DOI:10.1007/s10495-016-1333-3.
- C. M. Worsley, R. B. Veale and E. S. Mayne, Inducing apoptosis using chemical treatment and acidic pH, and detecting it using the Annexin V flow cytometric assay, PLoS One, 2022, 17, e0270599, DOI:10.1371/journal.pone.0270599.
- R. C. Wilkins, B. C. Kutzner, M. Truong, J. Sanchez-Dardon and J. R. N. McLean, Analysis of radiation-induced apoptosis in human lymphocytes: flow cytometry using annexin V and propidium iodide versus the neutral comet assay, Cytometry, 2002, 48(1), 14–19, DOI:10.1002/cyto.10098.
- P. E. Czabotar and A. J. Garcia-Saez, Mechanisms of BCL-2 family proteins in mitochondrial apoptosis, Nat. Rev. Mol. Cell Biol., 2023, 24(10), 732–748, DOI:10.1038/s41580-023-00629-4.
- P. E. Czabotar, G. Lessene, A. Strasser and J. M. Adams, Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy, Nat. Rev. Mol. Cell Biol., 2014, 15(1), 49–63, DOI:10.1038/nrm3722.
- A. Burlacu, Regulation of apoptosis by Bcl-2 family proteins, J. Cell. Mol. Med., 2003, 7(3), 249–257, DOI:10.1111/j.1582-4934.2003.tb00225.x.
- J. C. Martinou and R. J. Youle, Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics, Dev. Cell, 2011, 21(1), 92–101, DOI:10.1016/j.devcel.2011.06.017.
- L. Zhu, M. B. Han and Y. Gao, et al., Curcumin triggers apoptosis via upregulation of Bax/Bcl-2 ratio and caspase activation in SW872 human adipocytes, Mol. Med. Rep., 2015, 12(1), 1151–1156, DOI:10.3892/mmr.2015.3450.
- A. Daneshforouz, S. Nazemi, O. Gholami, M. Kafami and B. Amin, The cytotoxicity and apoptotic effects of verbascoside on breast cancer 4T1 cell line, BMC Pharmacol. Toxicol., 2021, 22(1), 72, DOI:10.1186/s40360-021-00540-8.
- J. Liu, C. Zhang and B. Jia, et al., Panax notoginseng saponins induce apoptosis in retinoblastoma Y79 cells via the PI3K/AKT signalling pathway, Exp. Eye Res., 2022, 216, 108954, DOI:10.1016/j.exer.2022.108954.
- D. D. Thien, T. D. Dai, N. H. Sa and N. T. Tam, New dihydrophenanthrenes from Elatostema tenuicaudatum, Nat. Prod. Res., 2024, 1–7, DOI:10.1080/14786419.2024.2359543.
- C. Y. Kuo, S. H. Wang and C. Lin, et al., Application of 2,3-naphthalenediamine in labeling natural carbohydrates for capillary electrophoresis, Molecules, 2012, 17(6), 7387–7400, DOI:10.3390/molecules17067387.
- T. H. Vo, Y. C. Lin and C. C. Liaw, et al., Triterpene glycosides and phenylpropane derivatives from Staurogyne concinnula possessing anti-angiogenic activity, Phytochemistry, 2021, 184, 112666, DOI:10.1016/j.phytochem.2021.112666.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts, J. Org. Chem., 2015, 80(24), 12526–12534, DOI:10.1021/acs.joc.5b02396.
- Y. R. Cho, S. Lee and H. Kim, et al., Pinuseldarone, a Clerodane-Type Diterpene from Pinus eldarica Needles and Phytochemicals as Novel Agents for Regulating Brown Adipogenesis and Thermogenesis, J. Nat. Prod., 2024, 87(1), 58–67, DOI:10.1021/acs.jnatprod.3c00779.
- B. R. Peng, N. B. A. Nguyen and L. Y. Chen, et al., Unprecedented anti-inflammatory biscembranoids with 3-hydroperoxy-3-methylcyclohex-1-ene moiety isolated from aquaculture Sarcophyton trocheliophorum, Aquac. Rep., 2023, 33, 101835, DOI:10.1016/j.aqrep.2023.101835.
- T. Tanahashi, A. Shimada and N. Nagakura, et al., Structure Elucidation of Six Acylated Iridoid Glucosides from Jasminum hemsleyi, Chem. Pharm. Bull., 1995, 43(5), 729–733, DOI:10.1248/cpb.43.729.
- Y. Zhou, B. Yang and Y. Jiang, et al., Studies on cytotoxic activity against HepG-2 cells of naphthoquinones from green walnut husks of Juglans mandshurica maxim, Molecules, 2015, 20(9), 15572–15588, DOI:10.3390/molecules200915572.
- V. Praveen Kumar, J. Renjitha and C. T. Fathimath Salfeena, et al., Antibacterial and antitubercular evaluation of dihydronaphthalenone-indole hybrid analogs, Chem. Biol. Drug Des., 2017, 90(5), 703–708, DOI:10.1111/cbdd.12990.
- L. I. U. Lijuan, Q. I. Fengqin and G. Xianfeng, Studies on the Cytotoxicity of Naphthoquinone Derivatives from the Fresh Rejuvenated Fruits of Juglans mandshurica, Chin. J. Mod. Appl. Pharm., 2010, 27(7), 574–577 Search PubMed . http://www.chinjmap.com/en/article/id/20100179.
- S. J. Nam, C. A. Kauffman, L. A. Paul, P. R. Jensen and W. Fenical, Actinoranone, a cytotoxic meroterpenoid of unprecedented structure from a marine adapted Streptomyces sp, Org. Lett., 2013, 15(21), 5400–5403, DOI:10.1021/ol402080s.
- Z. Yang, J. Ding, K. Ding, D. Chen, S. Cen, M. Ge and A. Phomonaphthalenone, A novel dihydronaphthalenone with anti-HIV activity from Phomopsis sp. HCCB04730, Phytochem. Lett., 2013, 6(2), 257–260, DOI:10.1016/j.phytol.2013.02.003.
- A. D. Kinghorn, N. R. Farnsworth and D. D. Soejarto, et al., Novel strategies for the discovery of plant-derived anticancer agents, Pure Appl. Chem., 1999, 71(9), 1611–1618, DOI:10.1351/pac199971091611.
- J. Li, L. Q. Li and H. P. Long, et al., Xylarinaps A–E, five pairs of naphthalenone derivatives with neuroprotective activities from Xylaria nigripes, Phytochemistry, 2021, 186, 112729, DOI:10.1016/j.phytochem.2021.112729.
- D. S. McClintock, M. T. Santore and V. Y. Lee, et al., Bcl-2 Family Members and Functional Electron Transport Chain Regulate Oxygen Deprivation-Induced Cell Death, Mol. Cell. Biol., 2002, 22(1), 94–104, DOI:10.1128/mcb.22.1.94-104.2002.
- D. Brenner and T. W. Mak, Mitochondrial cell death effectors, Curr. Opin. Cell Biol., 2009, 21(6), 871–877, DOI:10.1016/j.ceb.2009.09.004.
- R. J. Youle and A. Strasser, The BCL-2 protein family: opposing activities that mediate cell death, Nat. Rev. Mol. Cell Biol., 2008, 9(1), 47–59, DOI:10.1038/nrm2308.
- S. Cory and J. M. Adams, The BCL2 family: regulators of the cellular life-or-death switch, Nat. Rev. Cancer, 2002, 2(9) DOI:10.1038/nrc883.
- Q. Wang, L. Zhang and X. Yuan, et al., The relationship between the Bcl-2/Bax proteins and the mitochondria-mediated apoptosis pathway in the differentiation of adipose-derived stromal cells into neurons, PLoS One, 2016, 11(10), e0163327, DOI:10.1371/journal.pone.0163327.
- D. Ding, Y. R. Guo, R. L. Wu, W. Y. Qi and H. M. Xu, Two new isoquinoline alkaloids from Scolopendra subspinipes mutilans induce cell cycle arrest and apoptosis in human glioma cancer U87 cells, Fitoterapia, 2016, 110, 103–109, DOI:10.1016/j.fitote.2016.03.004.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra09007a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |