DOI:
10.1039/D5RA01322D
(Paper)
RSC Adv., 2025,
15, 13799-13811
Acid-modified Cu–Ce/HZSM-5 adsorbent removes trace phosphorus impurities from recycled hydrogen during polysilicon production†
Received
24th February 2025
, Accepted 18th April 2025
First published on 29th April 2025
Abstract
In this study, an acid-modified Cu–Ce/HZSM-5 bimetallic adsorbent was developed for the removal of trace PH3 impurities from simulated recycled hydrogen in a chemical vapor deposition furnace for polysilicon production. A systematic examination was subsequently conducted to examine the effects of acidic substance type and concentration, and the addition of Ce on the adsorbent's ability to remove PH3. The results showed that the adsorbent, when treated with 13% HNO3 and an appropriate level of Ce (nCe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nCu = 1:40), exhibited superior adsorption performance, achieving a PH3 breakthrough adsorption capacity of 135.2 mg g−1. After three regeneration cycles, the sorbent achieved optimal performance. Further investigation indicated that the improved efficiency of this sorbent in removing PH3 was primarily due to the formation of Cu2(OH)3NO3, an increase in microporous volume, the enhanced distribution of metallic oxides, a higher quantity of reactive oxygen species on the surface, an increased concentration of acidic sites, and the exposure of additional reactive species (Cu2(OH)3NO3, CuO). Moreover, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) characterization indicated that the deactivation of the adsorbent was mainly caused by the continuous consumption of Cu2(OH)3NO3 and CuO, along with the accumulation of reaction products (P2O5, Cu3P, Cu (PO3)2, and Cu2P2O7) on the adsorbent surface.
:nCu = 1:40), exhibited superior adsorption performance, achieving a PH3 breakthrough adsorption capacity of 135.2 mg g−1. After three regeneration cycles, the sorbent achieved optimal performance. Further investigation indicated that the improved efficiency of this sorbent in removing PH3 was primarily due to the formation of Cu2(OH)3NO3, an increase in microporous volume, the enhanced distribution of metallic oxides, a higher quantity of reactive oxygen species on the surface, an increased concentration of acidic sites, and the exposure of additional reactive species (Cu2(OH)3NO3, CuO). Moreover, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) characterization indicated that the deactivation of the adsorbent was mainly caused by the continuous consumption of Cu2(OH)3NO3 and CuO, along with the accumulation of reaction products (P2O5, Cu3P, Cu (PO3)2, and Cu2P2O7) on the adsorbent surface.
1 Introduction
As a major producer of polysilicon, China generates toxic gases such as PH3 during production. If PH3-containing waste gases are released directly into the atmosphere, they can pose severe environmental and public health risks.1 In addition, PH3 is recognized as highly hazardous due to its high biotoxicity and its detrimental effects on industrial processes, including equipment corrosion and product quality degradation.2 In the Siemens process for polysilicon production, recycling PH3-contaminated hydrogen back into the chemical vapor deposition furnace can lead to participation in the reaction, ultimately causing PH3 accumulation during tail gas recycling and deposition in the polysilicon, thus reducing product quality.3,4 Therefore, developing an adsorbent to efficiently remove PH3 from hydrogen is of significant industrial importance. This study focused on purifying recycled hydrogen and maintaining PH3 levels below critical thresholds.
Traditional methods for removing PH3 can be categorized into wet and dry processes. The wet method involves oxidizing PH3 impurities using solutions such as potassium permanganate or sodium hypochlorite–potassium hydroxide, converting them into phosphorous oxides that are easily absorbed. However, this process introduces significant amounts of water into the treated gas, which reacts with SiHCl3, necessitating additional dehydration steps if solution absorption is employed.5,6 The dry methods mainly include combustion, catalytic thermal decomposition, and adsorption techniques. Among these, materials such as activated carbon and molecular sieves are widely used due to their cost-effectiveness and practicality for removing trace PH3 contaminants from various gaseous substances.7–10 Historically, removing PH3 from silane relied on dry adsorption using activated carbon or zeolite at extremely low temperatures, but without a systematic analysis of the adsorption mechanism.11,12 Recent research into chemical absorption has focused on the incorporation of active substances such as Cu2+, Zn2+, and Fe3+ into the adsorptive materials. This approach has been shown to significantly enhance the transformation of PH3 into different substances, improving its removal efficiency. Notably, compared with other metal ions, Cu2+ has been identified as a particularly effective active species,13–15 with several Cu-based adsorbents, including 30Cu@TiO2, Cu–S-AC, and 3DCuO/C, demonstrating excellent PH3 removal capacities.16–18 These methods are best suited for situations where the gas stream contains a relatively high concentration of PH3. However, adjustments may be needed for handling trace amounts of PH3 in polysilicon tail gas systems. Our previous research provides valuable insights into addressing this challenge.6,19,20 Luo et al. synthesized a highly efficient PH3 adsorbent using Ce doping and found that the Ce1/20–Cu23/TiO2 adsorbent exhibited excellent PH3 removal performance with a saturation capacity of 149.14 mg g−1.21 Therefore, Ce doping enhanced CuO dispersion, surface oxygen adsorption, and lowered the reduction temperature. Among the above-mentioned methods, the adsorption–oxidation method has shown the most promise, with significantly higher reactivity for reducing gases and mild reaction conditions.
Selecting appropriate support is crucial for developing efficient adsorbents. Solid adsorbents without support exhibit limited specific surface areas and pore capacities, with a tendency to agglomerate during both synthesis and PH3 removal. To address these limitations, numerous studies have focused on impregnating metallic oxides into mesoporous silica substrates with large surface areas. HZSM-5, with strong acidity, a large specific surface area, a unique three-dimensional structure, and the ability to withstand high temperatures above 500 °C for extended periods, has been frequently used as a support material for adsorption and impurity removal.13,22 Notably, Cu supported on HZSM-5 has shown promise for PH3 removal from chemical vapor deposition furnaces, especially in applications such as the production of electronic-grade polysilicon. Studies have shown that the effectiveness of sorbents in removing PH3 is directly correlated to the state of active species and their dispersion. By changing the state of the active species and improving their dispersion, the functionality of these sorbents or catalysts may be significantly enhanced. Effective methods for achieving this include second metal doping23,24 and acid modification.25,26 Song et al. used HNO3 modification to introduce oxygen-containing functional groups such as NO2− and NO3− onto the surface of SBA-15, causing the active species to migrate from the interior to the surface, thereby improving the adsorption–oxidation activity of the sorbent.27 Li et al. found that hydrogen nitrate-modified Cu/SBA-15 exhibited the best PH3 removal performance, with a capacity of 104.84 mg g−1.26 Doping with a secondary metal has been shown to improve the dispersion of active species and enhance their oxidative capacity, thereby enhancing the adsorption–oxidation activity of the adsorbent.28 Therefore, to enhance the oxidative performance of active species, it is critical to introduce suitable oxygen-rich functional groups into the sorbent and dope secondary metals to form strong interactions with active species.
Despite these advancements, no studies have explored the use of acid modification on Cu–Ce/HZSM-5 or evaluated its effectiveness in PH3 removal for polysilicon production. The effects of acid modification and Cu–Ce mixed oxides on the adsorption–oxidation of PH3 in polysilicon reduction tail gas remain unclear. Therefore, in this study, we conducted a systematic investigation by modifying the adsorbent with different types of acids, followed by Ce doping as a secondary metal for further modification. We further examined the use of Cu–Ce mixed metal oxides supported on HZSM-5 for PH3 removal. This comprehensive investigation focused on evaluating the adsorbent's capacity to capture PH3, both in its original form and after modification. This process is expected to play a critical role in selecting and advancing PH3-adsorbing materials for use in polysilicon manufacturing.
2 Experimental section
2.1 Preparation and regeneration method of the sorbents
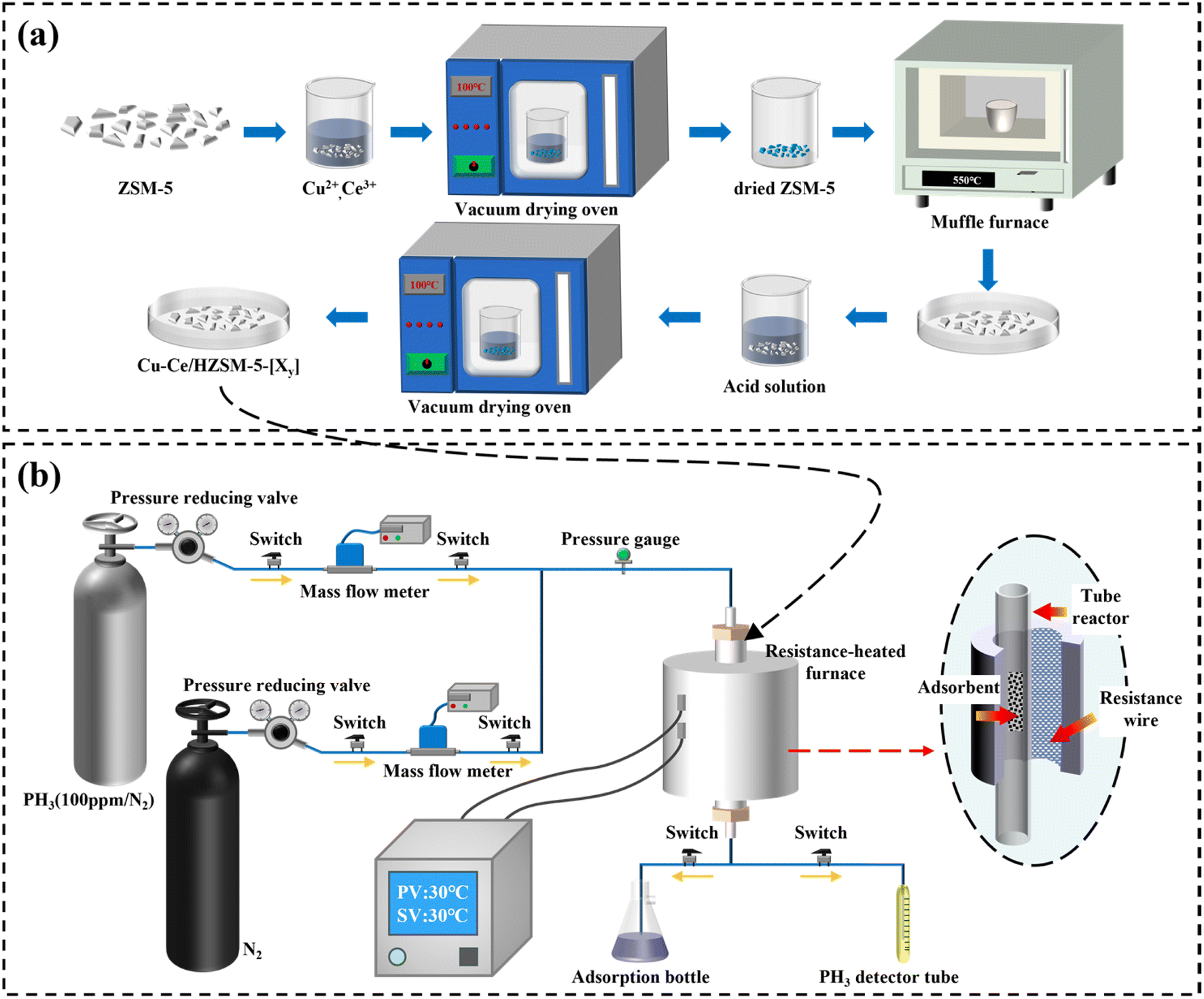
The adsorbent used in this study was synthesized via a wet impregnation technique, as illustrated in Fig. 1(a). The various acids used for modification and the HZSM-5 powder (Si:Al = 25) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd., while nitrate salts were obtained from Shanghai Macklin Biochemical Technology Co., Ltd. The specific preparation process for the adsorbent was as follows: precise amounts of Cu(NO3)2 and Ce(NO3)3·6H2O were weighed and dissolved in deionized water in a beaker, followed by ultrasonic treatment for 2 min to ensure complete dissolution. Next, 2 g of HZSM-5 support was added to the solution, and the mixture was agitated for approximately 5 h using a magnetic stirrer to ensure complete integration of the support with the solution. The resulting mixture was then transferred to a vacuum oven and dried at 100 °C overnight. Then, the dried solid was subjected to calcination at 550 °C for 5 h in a muffle furnace, ensuring the transformation of Cu (NO3)2 and Ce (NO3)3·6H2O into their respective oxides, CuO and CeO2. After calcination, the powder was immersed in various acidic solutions of different concentrations, followed by a final drying step overnight at 100 °C, resulting in the final adsorptive material. The restructured Cu–Ce/HZSM-5 composites were denoted as CumCen/HZSM-5-[Xy], where m represents the weight percentage of Cu relative to the HZSM-5 support (wt%), n signifies the molar ratio of Ce to Cu, X indicates the type of acid used (B: none, N:HNO3, S:H2SO4, Cl:HCl, P:H3PO4), and y specifies the weight concentration of the acid in purified water (wt%, with y values of 5, 10, 13, or 15). The Cu loading, acid impregnation concentration, and Ce addition amount were initially determined based on the relevant literature and subsequently adjusted based on the experimental results to establish an appropriate range for the experiments.21,26–28 In addition, the deactivated adsorbent was regenerated in a muffle furnace at 450 °C for 240 min.
 |
| | Fig. 1 (a) Preparation process of Cu–Ce/HZSM-5-[Xy]; (b) adsorption–oxidation performance test device. | |
2.2 Sorbent characterization
The X-ray diffraction (XRD) profiles were obtained using a diffractometer (D8-Focus, Bruker, Germany) with Cu Kα radiation. The diffraction angles ranged from 10° to 80°, with a scanning speed of 10° min−1. The resulting data were analyzed using Jade software (version 9.0). Fourier transform infrared (FT-IR) spectroscopy was performed using an infrared (IR) spectrophotometer (Nicolet iS50, FEI, USA), with a total of 32 scans conducted in the wavenumber range of 400–4000 cm−1 and a resolution of 4 cm−1. Isotherms depicting the adsorption and desorption behavior of the adsorbent materials were recorded by an automated gas sorption analyzer (ASAP 2460, Micromeritics, USA) at 77 K. Prior to analysis, the materials were outgassed at 473 K for 7 h to ensure accurate results. The pore size distribution and volumes of the micropores and mesopores were determined using the Horvath–Kawazoe (HK) and Barrett–Joyner–Halenda (BJH) methods. The topography and elemental composition of the adsorptive materials were analyzed by scanning electron microscopy (SEM, Hitachi S-4800, Japan) coupled with energy-dispersive X-ray spectroscopy (EDS). To investigate the elemental chemical states on the adsorbent surface, X-ray photoelectron spectroscopy (XPS) was conducted using a K-Alpha system (Thermo Fisher Scientific, USA). The spectral peaks were calibrated by Avantage software, using the carbon 1s peak at 284.8 eV as the reference. The quantity and strength of the acidic sites were measured by a chemisorption analyzer (BELCat II, Microtrac, Japan). Approximately 100 mg of the sample was heated to 120 °C under a helium stream (30–50 mL min−1) and maintained at this temperature for 60 min. The temperature was then reduced to 50 °C, and the sample was exposed to a 10% NH3/He mixture (30–50 mL min−1) for 60 min, followed by flushing with helium. The sample was subsequently heated to 700 °C in a helium stream, and the desorbed species were identified by a thermal conductivity detector (TCD). The reduction capabilities of the adsorbent were further investigated by hydrogen-temperature programmed reduction (H2-TPR) with a chemisorption analyzer (AutoChem II 2920, Micromeritics, USA). Approximately 100 mg of the sample was placed in a U-shaped quartz reactor and initially heated to 100 °C in a helium atmosphere for 60 min. The temperature then gradually increased to 500 °C at a rate of 10 °C min−1 in a hydrogen–argon gas mixture, and the desorbed gases were monitored using TCD.
2.3 Dephosphorization experiment
Fig. 1(b) illustrates the setup for PH3 adsorption and the removal experiment. The adsorption process was carried out in a quartz column with a diameter of 14 mm, containing 0.2 g of the adsorbent. Initially, the column was heated to 150 °C and exposed to a nitrogen flow at 60 mL min−1 for 2 h to remove any atmospheric or moisture-related impurities from the adsorbent and the column. After this pre-conditioning step, the heat was turned off, and the column was allowed to cool to ambient temperature (30 °C). Once the column had cooled, nitrogen flow was stopped, and a mixture of 100 ppm PH3 was introduced into the adsorption column at a flow rate of 80 mL min−1. Because the PH3 concentration during the polycrystalline silicon reduction process typically ranged from 50 to 200 ppm, this concentration was chosen to simulate the PH3 levels in a polycrystalline silicon reduction furnace. The PH3 concentration at the outlet of the reactor was monitored every 30 min using a PH3-specific detection tube. This detection tube utilized chemical absorption, containing a reagent that reacted with PH3. As gas passed through the tube, it reacted with the reagent, causing a color change. The degree of color change was proportional to the concentration of PH3, allowing for accurate and quantitative measurement of its concentration. The adsorption process was considered to have reached breakthrough when the removal efficiency fell below 60%. At this point, the valves for the N2 and PH3 gas mixture were closed, and the deactivated sorbent was collected and stored in a sealed bottle. Each experiment was repeated twice to ensure the reliability of the collected data. The adsorption capacity at the point of 60% efficiency in removing PH3 was referred to as the breakthrough capacity. The corresponding calculations for this value are provided in eqn (1) and (2):| |
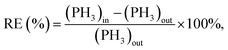 | (1) |
| |
 | (2) |
where (PH3)in represents the concentration of PH3 in the incoming gas, (PH3)out is the concentration of PH3 in the exiting gas, m denotes the mass of the fresh adsorbent (0.2 g), t indicates the duration of the reaction (min), and Q represents the volume of the gas flow, which was recorded at 80 mL min−1.
3 Results and discussion
3.1 The effect of different Cu loadings
The variation in active species loading was found to significantly influence the adsorption–oxidation activity of the adsorbent for PH3.29 Therefore, a comprehensive investigation was conducted to determine the optimal incorporation level of active species (Cu) by evaluating the sorptive–oxidative efficiency of sorbents with different Cu contents. The experimental results are presented in Fig. 2(a) and (b), indicating that all sorbents exhibited a gradual decline in performance over time, eventually reaching deactivation. Notably, the breakthrough capacity for adsorption significantly increased as the Cu loading increased from 5% to 30%. However, when the Cu loading further increased to 40%, the breakthrough adsorption capacity started to decrease. Compared with Cu30/HZSM-5-[B], a decrease in PH3 uptake performance was observed for Cu40/HZSM-5-[B], with the capacity dropping from 70.4 to 64.8 mg g−1 (Fig. 2(b)), representing an overall reduction of approximately 7.9%.
 |
| | Fig. 2 Effect of different Cu loading amounts on PH3 removal performance; (a) PH3 breakthrough curve; (b) adsorption capacity; (c) XRD patterns (adsorption conditions: PH3 (100 ppm)/N2, temperature (30 °C), gas inlet flow rate (24000 mL min−1)). | |
Fluctuations in Cu loading significantly impacted the adsorbent's structure,30 influencing its effectiveness, as demonstrated by the XRD and BET analyses. Fig. 2(c) presents the XRD patterns of the blank HZSM-5 and HZSM-5 samples loaded with different Cu contents. A distinct set of diffraction peaks corresponding to HZSM-5 was clearly observed in the 2θ range of 10–34°, indicating that the introduction of Cu did not disrupt the crystalline framework of HZSM-5.31 In all Cu-loaded samples, the characteristic diffraction patterns of CuO were visible at 2θ angles of 35.56°, 38.84°, 48.94°, 58.52°, and 61.58° (PDF#80-0076), with a noticeable increase in peak intensity as the Cu content increased. This suggested that, through calcination, copper nitrate was completely converted into CuO, which became the active species of the adsorbent. For Cu40/HZSM-5-[B], the peak intensity of CuO was significantly higher compared with Cu30/HZSM-5-[B], implying that excess CuO could accumulate on the surface of Cu40/HZSM-5-[B], a phenomenon known to weaken bonding between the Cu species on the surface of the adsorbent, thereby reducing the adsorptive capacity.32 Experimental results supported this conclusion, as increasing Cu loading to 40% led to a noticeable decrease in the breakthrough adsorption capacity compared with 30% Cu loading (Fig. 2(a) and (b)).
BET analysis was conducted to further investigate the effects of varying Cu concentrations on the structure of the adsorbent, with the results shown in Fig. S1.† The N2 adsorption–desorption curves of the adsorbents with different Cu loadings, including blank HZSM-5, exhibited a type IV isotherm. Notably, the presence of hysteresis within a similar range of relative pressures (P/P0 = 0.5–1.0) indicated that incorporating Cu species into the HZSM-5 framework did not significantly disrupt its pore structure.33 These findings supported the use of HZSM-5 as an adsorbent support and suggested that its adsorption and removal performance could be enhanced through various modification methods. Furthermore, after adjusting the Cu content, both the specific surface area and overall porosity of the adsorbents decreased, with the degree of reduction varying inconsistently (Table S1†). This decrease could potentially hinder the diffusion of PH3 molecules within the material, negatively affecting PH3 adsorption and oxidation. However, the experimental results showed that despite these changes in physical properties, the performance of PH3 adsorption and oxidation remained largely unaffected. Notably, as the Cu loading increased from 5% to 30%, the adsorption performance continued to improve. This improvement attributed to the increased content of active species, which enhanced oxidation activity. Therefore, although the decrease in surface area and pore volume suggested a potential negative impact, the increased availability of active species counteracted this effect, resulting in an overall improvement in performance. This observation was consistent with findings reported in related research,34 leading to the selection of Cu30/HZSM-5-[B] for further comprehensive investigation.
3.2 The efficacy of adsorbents treated with various acids
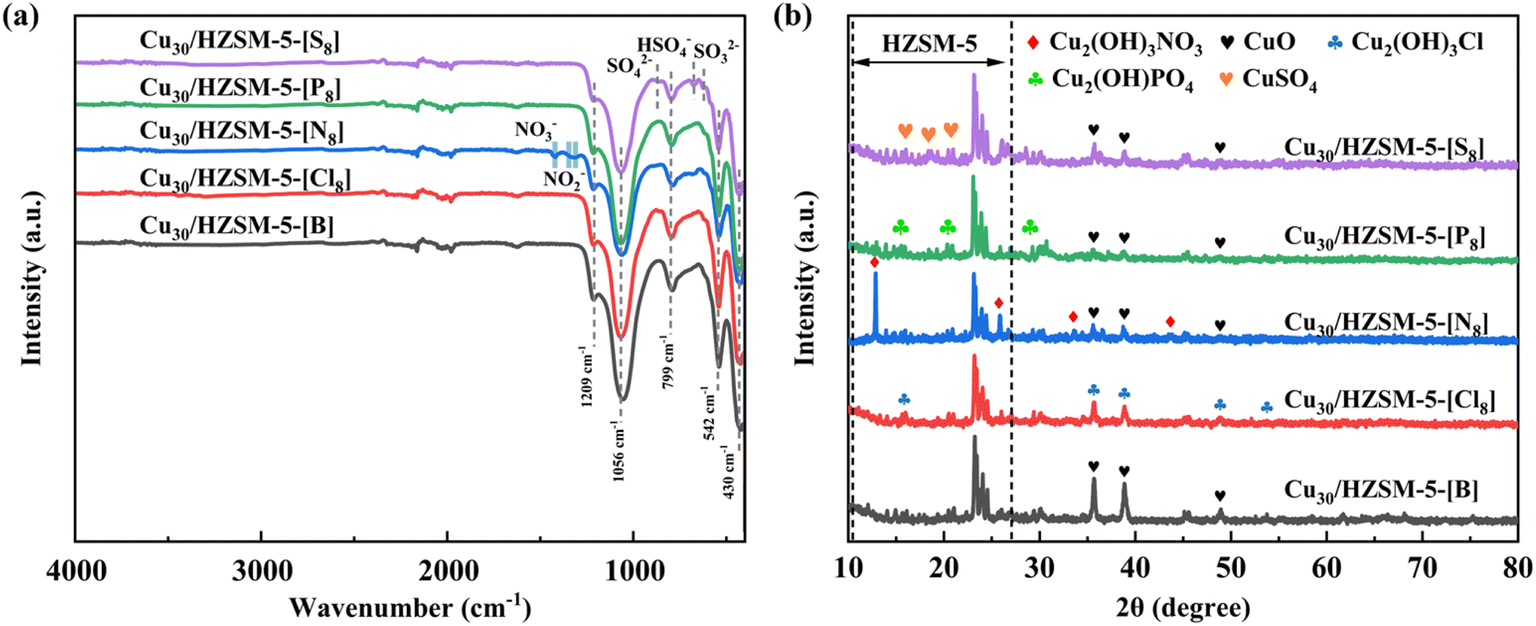
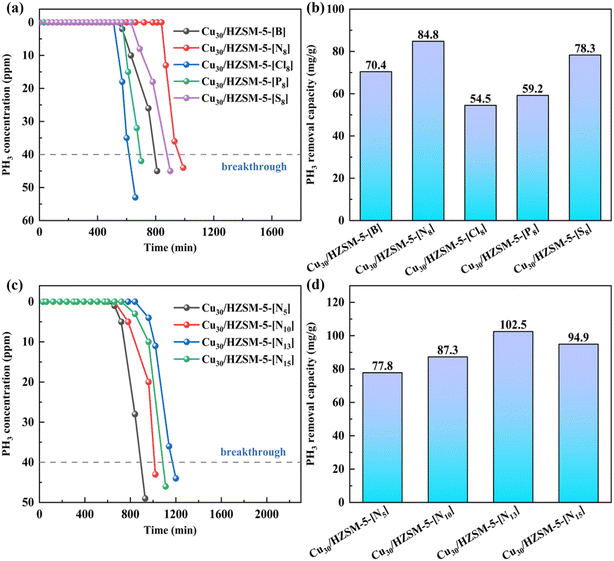
To further enhance the adsorption–oxidation activity of the adsorbent for PH3, in this study, we modified the adsorbents with different types of acids and evaluated their impact on PH3 removal performance. The experimental results are shown in Fig. 3(a) and (b), indicating that the effect of different types of acids on the adsorption performance of the adsorbents follows the order: Cu30/HZSM-5-[N8] > Cu30/HZSM-5-[S8] > Cu30/HZSM-5-[P8] > Cu30/HZSM-5-[Cl8] > Cu30/HZSM-5-[B]. The hydrogen nitrate-treated sample (Cu30/HZSM-5-[N8]) demonstrated superior PH3 removal performance, with a breakthrough time of 990 min and an adsorption capacity of 84.8 mg g−1, representing a 16.9% increase compared with the untreated sample, which had a capacity of 70.4 mg g−1. This improvement could be attributed to hydrogen nitrate, which facilitated PH3 removal by providing sufficient NO3− and NO2− groups, as shown in eqn (3).26 These groups could oxidize PH3 into P or PO43−, thereby enhancing the oxidative capacity of the adsorbent. In addition, NO3− and NO2− exhibited strong chemical adsorption of PH3, which further contributed to its effective removal.27 As a result, the inclusion of nitrate (NO3−) and nitrite (NO2−) ions improved both the adsorption and oxidation capacity of the adsorbent for PH3. Compared with the unmodified adsorbents, adsorbents modified with hydrogen nitrate and sulfuric acid exhibited better performance. By contrast, modifications with hydrochloric acid and phosphoric acid negatively impacted the adsorption capabilities of the adsorbent. Notably, the adsorbent modified with hydrogen nitrate (Cu30/HZSM-5-[N8]) was the most effective for PH3 adsorption and oxidation. Compared with adsorbents modified with other acids, hydrogen nitrate yielded the best results, establishing Cu30/HZSM-5-[N8] as the top-performing material in this study. FT-IR, XRD, and SEM characterization were used to analyze the type and quantity of functional groups, active species, and their distribution on the adsorbent surfaces following acid treatment. The analysis results are presented in Fig. 4(a) and (b) and 7(c–g):| | |
PH3 + NO2−/NO3− → NH2− + NH2− + P/PO43−.
| (3) |
 |
| | Fig. 3 PH3 breakthrough curves and adsorption capacities of adsorbents with different acid types (a and b) and different hydrogen nitrate impregnation concentrations (c and d) (adsorption conditions: PH3 (100 ppm)/N2, temperature (30 °C), gas inlet flow rate (24000 mL min−1)). | |
 |
| | Fig. 4 (a) FT-IR spectroscopy, and (b) XRD profiles for different adsorbent materials. | |
Fig. 4(a) presents the FT-IR spectral analysis results for the specimens modified with various acidic substances, indicating that all samples exhibited characteristic absorption peaks at 799, 1056, and 1209 cm−1, with noticeable changes in intensity after acid modification. These peaks were attributed to the symmetric oscillations of the outer bonds connecting the tetrahedral structures,35 resonances within the (Si, Al)O4 tetrahedral network of the HZSM-5 material, and the stretching vibrations of the outer bonds between the tetrahedra.36 In addition, the acid-modified adsorbent samples showed the formation of new functional groups. For example, the sample modified with hydrogen nitrate exhibited characteristic absorption peaks at 1432, 1342, and 1320 cm−1, corresponding to the stretching vibrations of the NO3− and NO2− groups.26,27 Similarly, the specimen treated with sulfuric acid displayed absorption peaks at 869, 677, and 625 cm−1, which were attributed to the stretching vibrations of the SO42−, HSO4−, and SO32− groups, respectively. These results suggested that different types of acids could react with copper ions to form corresponding copper salts, which were deposited on the adsorbent surface, thereby altering the original physical properties of the material. According to the experimental results and infrared analysis, the adsorption and oxidation capacity of the adsorbent were closely linked to the types of functional groups formed, with these functional groups playing a crucial role in generating relevant reactive species.
XRD analysis was performed to verify the changes in surface compound varieties of the modified adsorbents resulting from treatment with different acids, and the results are presented in Fig. 4(b). We observed that all samples exhibited the characteristic diffraction peaks of HZSM-5, except for the adsorbent modified with hydrochloric acid, where CuO was completely converted to basic copper chloride; the characteristic diffraction peaks corresponding to CuO (2θ = 35.72°, 38.87°, and 48.93°) were observed in the blank sample and the other acid-modified samples.30 In addition, the specimen treated with hydrogen nitrate contained new diffraction peaks at 2θ = 12.83°, 25.81°, 33.58°, and 43.73°, corresponding to Cu2(OH)3NO3 (PDF# 45-0592), a compound that could transform into CuO under certain conditions. For the adsorbent modified with sulfuric acid, a series of peaks in the 2θ = 15–20° were associated with the presence of copper sulfate. Similarly, the characteristic diffraction peaks of copper salts were detected in samples modified with hydrochloric acid and phosphoric acid. Based on the differences observed in the experimental results (Fig. 3(a) and (b)), we inferred that these salts affected the adsorption and removal capacity of the adsorbent materials for PH3, with Cu2(OH)3NO3 exhibiting more effective performance compared with the other substances. These findings were consistent with the FT-IR results, reinforcing the notion that the efficiency of the adsorbent was largely influenced by the specific form of copper salt produced through acid modification. Based on these experimental results, the adsorbent with the best PH3 removal performance (Cu30/HZSM-5-[N8]) was selected for further investigation.
3.3 Effect of hydrogen nitrate concentration
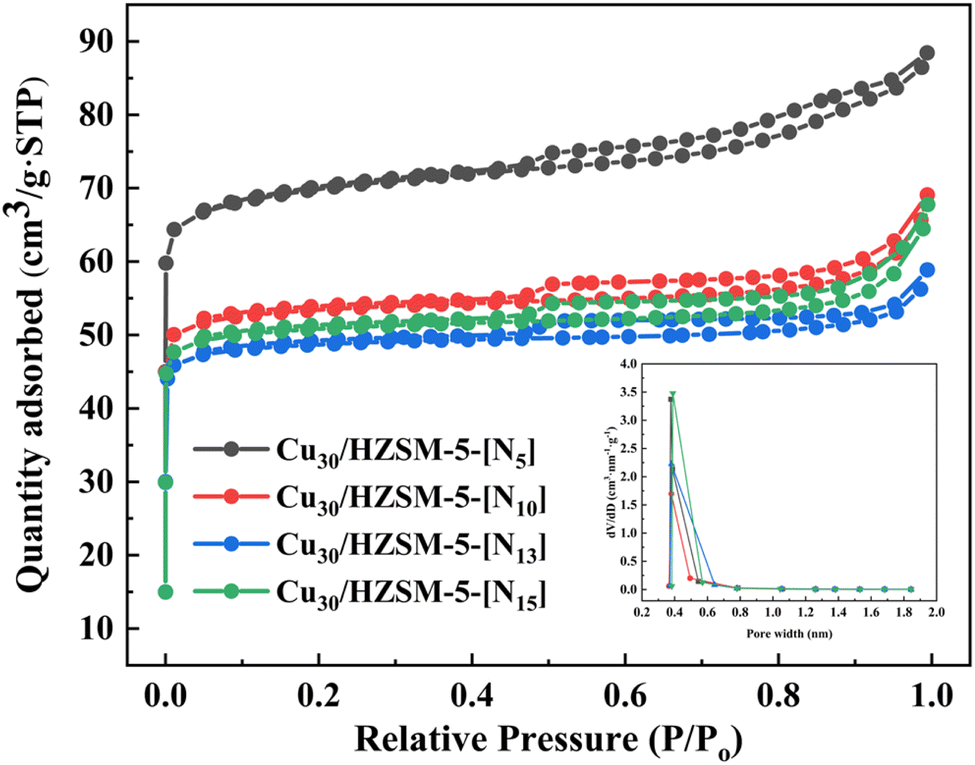
Previous research found that acid concentration significantly influenced the adsorption performance of adsorbents.37 To determine the optimal acid modification level, in this study, we conducted tests on PH3 adsorption efficiency using hydrogen nitrate-modified adsorbents with varying concentrations. In addition, BET analysis was performed to assess the changes in the physical properties of adsorbents modified with different hydrogen nitrate concentrations. The results of these tests and analyses are presented in Fig. 3(c) and (d) and 5. The experimental results indicated that PH3 removal efficiency varied with hydrogen nitrate concentration. At low hydrogen nitrate concentrations, no significant changes in adsorption performance were observed. However, at high concentrations, the adsorption capacity sharply decreased, exhibiting a volcanic distribution trend. Notably, compared with 10% and 15% hydrogen nitrate concentrations, the adsorbent modified with 13% hydrogen nitrate demonstrated superior adsorption performance (Fig. 3(c) and (d)), with a fresh adsorbent breakthrough capacity of 102.5 mg g−1. These findings suggested that the optimal hydrogen nitrate concentration significantly enhanced the adsorptive capacity of the material for PH3 removal. Furthermore, varying hydrogen nitrate concentrations had a notable impact on the adsorbent's pore structure, which subsequently affected its adsorption efficiency. To better understand these effects, BET characterization was conducted on adsorbent samples modified with different hydrogen nitrate concentrations, with the results presented in Fig. 5 and Table 1.
 |
| | Fig. 5 N2 adsorption/desorption isotherms of different types of adsorbents. | |
Table 1 Physical properties of different samples
| Sample |
S (m2 g−1) |
Vtotal (cm3 g−1) |
Dp (nm) |
| Cu30/HZSM-5-[N5] |
277 |
0.14 |
1.97 |
| Cu30/HZSM-5-[N10] |
216 |
0.11 |
1.98 |
| Cu30/HZSM-5-[N13] |
198 |
0.09 |
1.84 |
| Cu30/HZSM-5-[N15] |
206 |
0.11 |
2.04 |
Fig. 5 displays the nitrogen adsorption–desorption isotherms and pore size distributions for samples modified with different hydrogen nitrate concentrations. According to the nomenclature and classification outlined by IUPAC, all adsorbents exhibited nitrogen adsorption–desorption isotherms corresponding to type IV, similar to HZSM-5 (Fig. S1†). This indicated that modification with hydrogen nitrate did not compromise the intrinsic porous structure of HZSM-5. Furthermore, as shown in Fig. 5, despite increasing concentrations of hydrogen nitrate, the pore size distribution primarily remained within the micropore range of 0–2 nm. Table 1 presents the data on specific surface areas, total pore volumes, and mean pore diameters for adsorbents treated with varying hydrogen nitrate concentrations, as determined by BET analysis. The data revealed a trend where both the specific surface area and total pore volume progressively decreased with higher hydrogen nitrate concentrations. However, at an impregnation level of 15%, both the specific surface area and total pore volume experienced an increase. According to the experimental results (Fig. 3(c) and (d)), the specimen treated with 13% hydrogen nitrate exhibited the smallest specific surface area, total pore volume, and mean pore size, but demonstrated superior PH3 absorption efficiency. This phenomenon could be attributed to the enhanced chemical activity induced by the hydrogen nitrate modification process. Acid treatment likely led to the formation of additional active sites on the adsorbent surface, which possibly compensated for the reduction in surface area. These newly formed active sites, including acidic or oxygenated functional groups, significantly enhanced the interaction between the adsorbent and PH3 molecules, leading to improved adsorption performance. Therefore, although the physical properties of the surface area decreased, the chemical activity of the material was enhanced, resulting in better adsorption behavior. Similar findings have also been reported in other comparative studies.26,38
3.4 The effect of Ce-doping
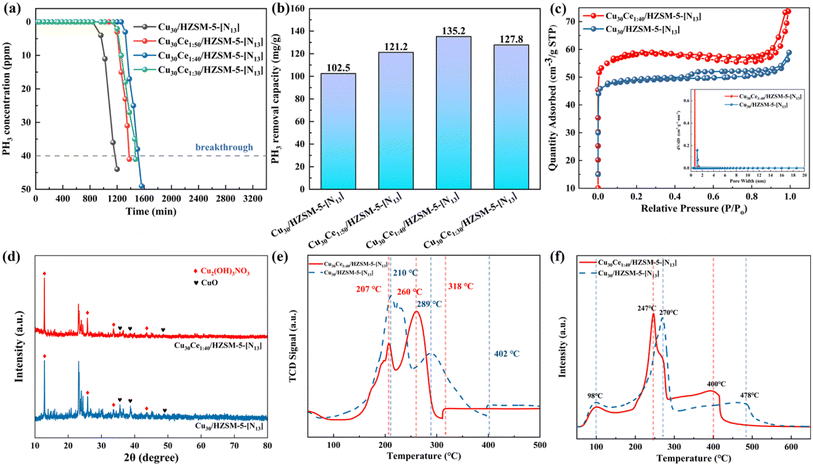
According to previous research, hydrogen nitrate impregnation significantly enhanced the adsorption–oxidation activity of the adsorbent by introducing oxygen-containing functional groups and changing the state of the active species (CuO). However, this impregnation treatment could lead to pore blockage in the support, resulting in a substantial decrease in the specific surface area and total pore volume. This reduction could hinder the dispersion of the active species and the diffusion of PH3 molecules within the pores. To address this issue, a secondary metal doping method was employed. Numerous studies have shown that adsorbents doped with rare earth element Ce as the secondary metal will exhibit better control over PH3 adsorption–oxidation activity compared with other metals, such as Fe, Zn, and La.39,40 Based on these findings, in this study, we used Ce as a secondary metal to further investigate its impact on the PH3 removal performance of the adsorbent. Sophisticated analytical methods were employed to examine the changes in the adsorbent's physical properties before and after Ce incorporation, providing insights into how Ce addition affected the physicochemical properties of the adsorbent, which in turn influenced its adsorption activity for PH3. A graphical representation of the PH3 breakthrough profiles and the corresponding adsorption capacities for different Ce concentrations in the adsorbents is shown in Fig. 6(a) and (b). Notably, the adsorbent without Ce experienced breakthrough at 1200 min, with a breakthrough adsorption capacity of 102.5 mg g−1. However, with the incorporation of Ce, a significant enhancement in the adsorbent's adsorption capacity was observed. As the Ce content continued to increase, the adsorption performance began to decline. This was mainly due to excessive CeO2 covering the adsorbent surface, which concealed active sites and obstructed PH3 diffusion within the pores, leading to decreased activity. Under conditions where the cerium-to-copper ratio was set to 1:40, the adsorbent demonstrated optimal efficacy, with a breakthrough capacity of 135.2 mg g−1, which was approximately 24% higher than that of the Ce-free adsorbent (102.5 mg g−1). Table S3† details the PH3 absorption capacities reported in various studies for different adsorbents. Clearly, the Cu30Ce1:40/HZSM-5-[N13] adsorbent demonstrated the highest PH3 breakthrough capacity under air conditions.
 |
| | Fig. 6 Effect of different Ce doping concentrations on PH3 removal: (a) breakthrough curves for PH3, (b) PH3 capacity, (c) BET patterns, (d) XRD patterns, (e) H2-TPR profiles, and (f) NH3–TPD curves of the materials before and after Ce incorporation (adsorption conditions: PH3 (100 ppm)/N2, temperature (30 °C), gas inlet flow rate (24000 mL min−1)). | |
To examine the changes in the physical and chemical properties of the adsorbent materials before and after Ce incorporation, we selected undoped Cu30/HZSM-5-[N13] and Ce-doped Cu30Ce1:40/HZSM-5-[N13] variants for analysis using BET, XRD, H2-TPR, and NH3-TPD techniques, with the results shown in Fig. 6(c–f). The BET characterization results in Fig. 6(c) revealed that the N2 adsorption/desorption curves for both adsorbents were very similar, indicating that Ce doping did not disrupt the pore structure of the adsorbents. After Ce doping, the pore size distribution of the adsorbent remained primarily within the micropore range, with no significant changes observed. Furthermore, Table 2 shows that after Ce incorporation, the adsorbent's specific surface area, total pore volume, and mean pore size increased, providing more adsorption sites and enhancing its adsorption activity. The SEM results also supported this finding, demonstrating that Ce doping resulted in a noticeably coarser and more uneven surface texture, with an abundance of irregularly shaped particles (Fig. 7(d–h)). This increased textural roughness and particle inconsistency contributed to a larger specific surface area and greater overall pore capacity, providing more sites for adsorption and thereby enhancing the adsorbent's capacity for PH3 adsorption and oxidation.
Table 2 Physical properties of the sorbent before and after Ce doping
| Sample |
S (m2 g−1) |
Vtotal (cm3 g−1) |
Dp (nm) |
| Cu30Ce1:40/HZSM-5-[N13] |
228 |
0.12 |
2.08 |
| Cu30/HZSM-5-[N13] |
198 |
0.09 |
1.84 |
 |
| | Fig. 7 (a–h) SEM micrographs; (i–l) EDS mapping images of HZSM-5, Cu30/HZSM-5-[B], Cu30/HZSM-5-[N13], and Cu30Ce1:40/HZSM-5-[N13] showing the elemental distribution of Cu. | |
To assess the changes in phase composition of the adsorbents before and after Ce doping, XRD analysis was performed on the Cu30/HZSM-5-[N13] and Cu30Ce1:40/HZSM-5-[N13] adsorbents, with the results presented in Fig. 6(d). The XRD patterns of the two samples were remarkably similar in terms of key peaks, with the intensities serving as the primary differences. Notably, the characteristic peaks for HZSM-5 were detectable between 10° and 30°, suggesting that the incorporation of Ce did not compromise the integrity of the HZSM-5 matrix. In addition, the diffraction peaks for Cu2(OH)3NO3 and CuO remained intact, which was crucial for the modification of the adsorbent using Ce as a secondary doping metal. The incorporation of Ce resulted in a slight reduction in the sharpness of the Cu2(OH)3NO3 and CuO diffraction peaks, likely due to the improved dispersion of these compounds following doping. Furthermore, no distinct peaks corresponding to CeO2 or Ce-related compounds were observed in the spectra of the Ce-doped adsorbent samples, suggesting that Ce was present either in a non-crystalline state or was finely dispersed as small particles on the HZSM-5 substrate.
H2-TPR analysis has been commonly used to assess the reduction behavior of sorbents and identify various Cu species following impregnation.41 In this study, this technique was employed to explore how Ce doping affected the reducibility of the sorbent.28 As shown in Fig. 6(e), for the Cu30/HZSM-5-[N13] sample, two reduction peaks were observed at 210 °C and 289 °C, corresponding to the reduction of small CuO particles and larger CuO particles, respectively. The peak at 402 °C was associated with the reduction of CuO crystals. By contrast, the Cu30Ce1:40/HZSM-5-[N13] sample exhibited a main reduction peak around 260 °C, indicative of the reduction of larger CuO particle aggregates.31 Additional peaks near 207 °C and 318 °C reflected the transformation of small CuO aggregates with minimal interaction with CeO2 or Cu2+ ions, as well as the conversion of CuO crystals to Cu. Notably, the Cu30Ce1:40/HZSM-5-[N13] sample had a lower reduction peak temperature compared with Cu30/HZSM-5-[N13], which could be attributed to Ce doping increasing the adsorbent's ability to store oxygen and enhancing the presence of reactive oxygen species on the surface,42,43 thereby facilitating the PH3 adsorption and oxidation process.
Acidic sites are considered crucial for determining the surface acidity of adsorbents, significantly influencing their reactivity with target gases.44 In this work, the concentration and strength of these acidic sites were assessed by NH3–TPD analysis, with the results shown in Fig. 6(f). The NH3–TPD curves revealed three distinct desorption peaks: a low-temperature peak around 98 °C, corresponding to NH3 loosely bound to weak Lewis acid sites; a higher-temperature peak near 250 °C, indicative of NH3 bound more firmly to strong Lewis acid sites, likely associated with isolated Cu2+ ions; and a third peak above 400 °C, representing the release of ammonia from Brønsted acid sites.45 Notably, for the Cu30Ce1:40/HZSM-5-[N13] samples, peaks 2 and 3 exhibited slight shifts to lower temperatures, along with an increase in their peak areas. This suggested that Ce incorporation reduced the strength of both Lewis and Brønsted acidic sites while enhancing the adsorbent's ability to adsorb substances. According to these results, we concluded that Ce doping improved the creation of more acidic sites, thereby increasing PH3 removal efficiency.
Fig. 7(a–l) presents the SEM and Cu elemental mapping images of the various sorbents. The HZSM-5 sample displayed numerous small, uniformly shaped particles with smooth surfaces (Fig. 7(a–e)), which were typical for HZSM-5 crystals.46 After Cu loading, the morphology of Cu30/HZSM-5-[B] showed minimal change (Fig. 7(b–f)), despite the increase in particle size, which was presumably attributed to the agglomeration of copper species (CuO) on the substrate.47 By contrast, hydrogen nitrate impregnation significantly changed the morphology of Cu30/HZSM-5-[N13] (Fig. 7(c–g)), resulting in extensive surface agglomeration. However, Ce doping mitigated this aggregation, resulting in Cu30Ce1:40/HZSM-5-[N13] with numerous irregularly shaped particles and a significantly rougher surface (Fig. 7(d–h)). This structure likely came from the strong interaction between copper oxide and CeO2, which formed more pores on the sorbent surface and facilitated PH3 molecule adsorption. The elemental mapping images (Fig. 7(i–l)) further revealed that Cu30/HZSM-5-[B] exhibited significant clustering of Cu species on its surface, a phenomenon that could impede the PH3 adsorption–oxidation process by forming phosphates that blocked internal active sites. However, the Ce-doped adsorbents showed no obvious accumulation of Cu species (Cu2(OH)3NO3 and CuO), indicating that these treatments enhanced the exposure and uniform dispersion of Cu species. This improved dispersion was likely a key factor behind the significantly enhanced adsorption performance of Cu30Ce1:40/HZSM-5-[N13] compared with Cu30/HZSM-5-[B]. This unique structure, combined with the BET findings, was believed to enhance the specific surface area and microporosity, which in turn improved the adsorption of PH3.
3.5 Deactivation mechanism
To identify the primary reaction products on the surface of the Cu30Ce1:40/HZSM-5-[N13] sample adsorbent and investigate its deactivation mechanism, fresh and deactivated adsorbents were characterized using various techniques. According to the characterization results, plausible hypotheses regarding the reaction pathway and factors contributing to deactivation were proposed. As illustrated in Fig. 7(d–h) and 8, significant microstructural differences of the Cu30Ce1:40/HZSM-5-[N13] sample were observed before and after deactivation. Following its deactivation, the adsorbent exhibited a significant clustering effect on its surface, likely due to various interactions occurring on the Cu30Ce1:40/HZSM-5-[N13] surface during the PH3 adsorption process. EDS analysis confirmed a homogeneous distribution of P, O, Ce, and Cu across the surface of the deactivated Cu30Ce1:40/HZSM-5-[N13] specimen (Fig. 8(c)). The uniform distribution of phosphorus indicated the effective adsorption of PH3, with the reaction products evenly distributed across the adsorbent surface.
 |
| | Fig. 8 (a and b) SEM micrographs of the deactivated adsorbent; (c) EDS mapping image of the deactivated adsorbent showing the elemental distribution of O, P, Cu, and Ce. | |
Fig. 9(a) presents the XRD patterns of the fresh and deactivated adsorbents. As shown in Fig. 9(a), the diffraction peak intensities of Cu2(OH)3NO3 and CuO significantly decreased after deactivation (PDF# 45-0592, 48-1548), indicating that Cu2(OH)3NO3 and CuO were continuously consumed as active species during PH3 adsorption–oxidation until they were fully transformed into other by-products. Newly emerged diffraction signals at 2θ = 20.86° and 30.22° were attributed to P2O5 (PDF# 87-0592), while the signals at 2θ = 26.98°, 28.54°, 32.79°, 34.32°, 35.72°, and 42.41° were indicative of copper phosphates (Cu(PO3)2 and Cu2P2O7) (PDF# 29-0572, 79-2075). In addition, the peaks at 2θ = 37.52°, 38.71°, 40.88°, 45.08°, and 45.54° corresponded to cuprous phosphide (Cu3P) (PDF# 71-2261). These products were consistent with the literature findings related to PH3 adsorption and removal.13,28 Therefore, according to the XRD characterization results, we concluded that during the PH3 removal process, P2O5, Cu3P, and various copper phosphates formed. As the reaction continued, these products accumulated, ultimately leading to adsorbent deactivation.
 |
| | Fig. 9 (a) XRD patterns and XPS results of (b) O 1s spectra, (c) P 2p spectra, and (d) Cu 2p spectra of fresh and used Cu30Ce1:40/HZSM-5-[N13] adsorbents. | |
XPS examination provided additional insights into the changes in surface species before and after adsorbent deactivation, as shown in Fig. 9(b–d). Fig. 9(b) presents the O 1s XPS spectra of Cu30Ce1:40/HZSM-5-[N13] before and after deactivation. As shown in Fig. 9(b), the O 1 s spectrum of the fresh sample displayed two prominent peaks, 530.9 ± 0.3 eV (lattice oxygen, Oα) and 532.6 ± 0.3 eV (adsorbed oxygen, Oβ), while the spectrum of the deactivated sample exhibited three distinct peaks at 530.9 ± 0.3 eV (lattice oxygen, Oα), 532.6 ± 0.3 eV (adsorbed oxygen, Oβ), and 534.6 ± 0.3 eV (adsorbed water, Oγ).48 Notably, compared with the fresh adsorbent, the lattice oxygen content significantly decreased in the deactivated adsorbent, and a hydroxyl peak (Oγ) corresponding to H2O molecules appeared. Because the gas itself did not contain H2O, the appearance of the hydroxyl peak (Oγ) in the deactivated adsorbent suggested that H2O was generated during the PH3 adsorption–oxidation process. According to these results, most of the active species (Cu2(OH)3NO3 and CuO) were converted to Cu3P, and by-products such as H2O also formed. As shown in Fig. 9(c), the P 2p XPS spectrum of the fresh Cu30Ce1:40/HZSM-5-[N13] contained no signal peaks, indicating the absence of phosphorus species in the fresh adsorbent, consistent with the actual situation. In the P 2p spectrum of the deactivated adsorbent, phosphorus species were detected on the surface, and the peaks at 129.3 ± 0.3 and 134.3 ± 0.3 eV corresponded to P3− (Cu3P) and P5+ (P2O5, H3PO4, or phosphate) signatures, respectively, consistent with findings reported in previous studies.49,50 Fig. 9(d) presents the Cu 2p XPS spectra of the fresh and deactivated adsorbents. For the fresh adsorbent, the peaks at 934.9 ± 0.3 eV corresponded to Cu2(OH)3NO3 and CuO, while for the deactivated adsorbent, new peaks at 932.6 ± 0.3 eV corresponded to Cu+ (Cu3P/Cu2O),51,52 in accordance with the Cu 2p binding energies. Notably, the Cu2+ peaks, originating from Cu2(OH)3NO3 and CuO, showed a significant decrease after deactivation, whereas the Cu+ (Cu3P) peaks exhibited a notable increase. The curve fitting results demonstrated that the Cu+ content in the fresh adsorbent was zero, while it increased to 69.2% in the deactivated adsorbent (Fig. S2(a) and (b)†). This trend was likely due to chemical interactions between Cu2(OH)3NO3, CuO, O2, and PH3 on the surface of the adsorbent, leading to the transformation of Cu2(OH)3NO3 and CuO into Cu3P.
BET analysis of the fresh and deactivated adsorbents further supported the conclusions drawn from the XRD and XPS results. Fig. S2 and S3 and Table S2† present the BET characterization results of the fresh and deactivated adsorbents. The nitrogen adsorption/desorption curves and pore size distribution analyses confirmed that the nitrogen adsorption/desorption profile of the deactivated adsorbent remained unchanged, with a predominant pore size in the range of 0.5–2 nm. This indicated that the porosity of the adsorbent was not significantly compromised during the PH3 adsorption–oxidation process, retaining its predominantly microporous structure. However, compared with the fresh adsorbent, there was a noticeable decrease in the specific surface area, total pore volume, and pore diameter of the deactivated adsorbent (Table S2†). This reduction was mainly due to the accumulation of by-products on the surface and within the pore structure. The accumulation of these substances could block the pores and obscure the adsorption sites, ultimately leading to a decline in adsorbent activity.
According to the experimental and characterization results, the adsorption mechanism of PH3 on the Cu30Ce1:40/HZSM-5-[N13] adsorbent in an air environment could be reasonably determined. When PH3 was captured by the Cu30Ce1:40/HZSM-5-[N13] adsorbent, it rapidly transformed into Cu3P, P2O5, H2O, and phosphate, according to the following reaction process. (1) PH3 was first activated by Cu2(OH)3NO3/CuO, generating P3− ions (PH3 → P3−), (2) the P3− ions reacted with Cu2(OH)3NO3/CuO to form Cu3P, (3) excess P3− further reacted with O2− ions released from Cu2(OH)3NO3/CuO, producing P2O5 and phosphates (Cu (PO3)2 and Cu2P2O7), and finally (4) H+ ions dissociated from PH3 and combined with O2− to generate H2O. The chemical reactions could be summarized as follows:
| | |
2P3− + 3Cu2(OH)3NO3 → 2Cu3P + 3NO3− + 9H+ + 3O2− + 6e−,
| (R2a) |
| | |
P3− + 3CuO + 3e− → Cu3P + 3O2−,
| (R2b) |
| | |
2P3− + 5O2− → P2O5↑+ 16e−,
| (R3) |
| | |
P3− + 3O2− → PO3− + 8e−,
| (R4) |
| | |
2P3− + 7O2− → P2O74− + 16e−,
| (R5) |
3.6 Regeneration of deactivated adsorbents
According to environmental protection and economic considerations, the deactivated adsorbent had to undergo regeneration treatment. Based on the XRD and XPS analysis results, the main cause of adsorbent deactivation was the accumulation of reaction products, such as Cu3P, P2O5, Cu (PO3)2, and Cu2P2O7. These products could be easily decomposed or transformed at high temperatures. During the regeneration process, Cu3P, Cu (PO3)2, and Cu2P2O7 could react with oxygen at high temperatures (450 °C), leading to their decomposition or transformation and thereby restoring the adsorbent's activity. The decomposition and transformation reactions of the products during the regeneration process are given by:| | |
4Cu3P + 11O2 → 12CuO + 2P2O5↑,
| (R7) |
| | |
Cu3(PO4)2 → 3CuO + P2O5↑,
| (R8) |
| | |
Cu2P2O7 → 2CuO + P2O5↑.
| (R9) |
Furthermore, during the regeneration process, P2O5 sublimated into a gaseous form and condensed inside the reaction tube, forming a waxy substance. This process helped restore the active species, thereby recovering the adsorption activity of the adsorbent. As shown in Fig. 10, the adsorption capacity of the adsorbent decreased with the number of regeneration cycles. After three regeneration cycles, the phosphorus capacity of the regenerated adsorbent reached 90.2 mg g−1, which was approximately 67% of the breakthrough capacity of the fresh adsorbent. Compared with the 70.4 mg g−1 of the Cu30/HZSM-5-[B] adsorbent without modification, the regenerated adsorbent still exhibited a significant advantage. This indicated that the heating regeneration method was feasible for the reuse of deactivated adsorbents and demonstrated good stability in restoring adsorption activity.
 |
| | Fig. 10 Effect of regeneration times on PH3 removal performance: (a) PH3 breakthrough curve and (b) PH3 breakthrough adsorption capacity. | |
4 Conclusions
To enhance PH3 removal efficiency in the recycled hydrogen of polysilicon reduction furnaces, a novel adsorbent was prepared in this work through hydrogen nitrate impregnation and Ce doping. Among the adsorbent materials tested, the Cu30Ce1:40/HZSM-5-[N13] variant, containing 30% copper, 13% hydrogen nitrate, and a cerium-to-copper molar ratio of 1:40, demonstrated the best performance in eliminating PH3, with a breakthrough capacity of 135.2 mg g−1 for PH3. After three regeneration cycles, the adsorbent still maintained ideal performance and demonstrated a significant advantage compared with the unmodified adsorbent. FT-IR and XRD techniques indicated that the introduction of hydrogen nitrate led to the formation of Cu2(OH)3NO3, which significantly enhanced the oxidative properties of the adsorbent. BET, NH3-TPD, H2-TPR, and XRD analyses revealed that the infusion of Ce notably increased the specific surface area of the adsorbent, increased the number of acid sites, enhanced the quantity of surface-reactive oxygen species, and promoted the dispersion of reactive species. These factors were critical in improving the adsorption of PH3. Moreover, Ce incorporation changed the adsorbent's microstructure, exposing more active sites. XRD and XPS analyses revealed that the primary active species were Cu2(OH)3NO3 and CuO. The main cause of the adsorbent's reduced effectiveness was attributed to the depletion of these critical components (Cu2(OH)3NO3 and CuO) and the accumulation of byproducts (Cu3P, P2O5, Cu (PO3)2, and Cu2P2O7) on the surface and within the pore structure.
Data availability
The data underlying the results of this study can be obtained from the corresponding author upon reasonable request.
Author contributions
Zhiyuan Liu: writing – original draft, validation, methodology, investigation, formal analysis, data curation, conceptualization. Guoqiang Huang: writing – review & editing, supervision, conceptualization.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Our research was supported by Tianjin University.
References
- S. Gaskin, L. Heath, D. Pisaniello, R. Evans, J. W. Edwards, M. Logan and C. Baxter, Toxicol. Ind. Health, 2017, 33, 289–296 CrossRef CAS PubMed.
- K. Yan, G. Tong, W. Gao, B. Li, W. Sun and L. Zhao, AlChE J., 2024, 70, e18438 CrossRef CAS.
- D. Chen, Guangzhou Chem. Ind., 2016, 44, 181–182 Search PubMed.
- T. V. Pavlova, G. M. Zhidomirov and K. N. Eltsov, J. Phys. Chem. C, 2018, 122, 1741–1745 CrossRef CAS.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang and C. Zhao, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- X. Xu and G. Huang, Ind. Eng. Chem. Res., 2016, 55, 1380–1386 CrossRef CAS.
- C. C. Lin, K. A. Dambrowitz and S. M. Kuznicki, Can. J. Chem. Eng., 2012, 90, 207–216 CrossRef CAS.
- C. Martínez and A. Corma, Coord. Chem. Rev., 2011, 255, 1558–1580 CrossRef.
- Q. Li, H. Li, X. Zong, H. Sun, Y. Liu, Z. Zhan, S. Mei, Y. Qi, Y. Huang and Y. Ye, Sci. Total Environ., 2024, 912, 169534 CrossRef CAS PubMed.
- Q. Li, F. Pan, W. Li, D. Li, H. Xu, D. Xia and A. Li, Polymers, 2018, 10, 1136 CrossRef PubMed.
- E. G. Caswell, US Pat., US2971607, 1961 Search PubMed.
- B. E. Langley, US Pat., US2987139A, 1961 Search PubMed.
- J. Feng, F. Wang, C. Wang, K. Li, X. Sun and P. Ning, ACS Appl. Mater. Interfaces, 2021, 13, 24670–24681 CrossRef CAS PubMed.
- S. Li, J. Hao, P. Ning, C. Wang, K. Li, L. Tang, X. Sun, D. Zhang, Y. Mei and Y. Wang, Sep. Purif. Technol., 2017, 180, 23–35 CrossRef CAS.
- Y. Wang, Q. Lin, C. Wang, K. Li, X. Sun, X. Song, Y. Gao and P. Ning, J. Environ. Sci., 2021, 104, 277–287 CrossRef CAS PubMed.
- J. Feng, L. Ma, C. Wang, Y. Ma, X. Sun, L. Jia, P. Ning, R. Zhang, F. Wang and K. Li, Small, 2023, 19, 2301169 CrossRef CAS PubMed.
- Y. He, L. Ye, W. Cen, D. Sun and J. Li, Chem. Eng. J., 2024, 503, 158250 CrossRef.
- L. Jia, X. Yang, K. Hu, J. Feng, F. Wang, K. Li, X. Sun, F. Wang and P. Ning, Chem. Eng. J., 2023, 457, 141277 CrossRef CAS.
- X. Xu, G. Huang and S. Qi, Chem. Eng. J., 2017, 316, 563–572 CrossRef CAS.
- Y. Zhou, T. He, S. Liu and G. Huang, Ind. Eng. Chem. Res., 2018, 57, 15122–15131 CrossRef CAS.
- Z. Luo, Q. Liang, Y. Qi and G. Huang, Sep. Purif. Technol., 2024, 344, 127148 CrossRef CAS.
- T. Cao, Z. Zhou, Q. Chen, Z. Li, S. Xu, J. Wang, M. Xu, T. Bisson and Z. Xu, Fuel Process. Technol., 2017, 160, 158–169 CrossRef CAS.
- T. Liu, M. Li, X. Bo and M. Zhou, ACS Sustainable Chem. Eng., 2018, 6, 11457–11465 CrossRef CAS.
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS.
- J. Feng, K. Li, S. Li, C. Wang, P. Ning, X. Sun and Y. Wang, Res. Chem. Intermed., 2020, 46, 329–346 CrossRef CAS.
- S. Li, K. Li, J. Hao, P. Ning, L. Tang and X. Sun, Chem. Eng. J., 2016, 302, 69–76 CrossRef CAS.
- X. Song, S. Li, K. Li, P. Ning, C. Wang, X. Sun and Y. Wang, Microporous Mesoporous Mater., 2018, 259, 89–98 CrossRef CAS.
- J. Feng, F. Wang, C. Wang, K. Li, P. Ning, X. Sun and L. Jia, Sep. Purif. Technol., 2021, 277, 119420 CrossRef CAS.
- Y. Wang, P. Ning, R. Zhao, K. Li, C. Wang, X. Sun, X. Song and Q. Lin, Front. Environ. Sci. Eng., 2021, 15, 1–10 CrossRef.
- K. Ma, K. Li, P. Ning, J. Feng, J. Bao, L. Shi, X. Wang and X. Sun, J. Environ. Sci., 2025, 151, 161–173 CrossRef CAS PubMed.
- Y. Wang, Q. Ying, Y. Zhang, Y. Liu and Z. Wu, Appl. Catal., A, 2020, 590, 117373 CrossRef CAS.
- Y. Zeng, T. Wang, S. Zhang, Y. Wang and Q. Zhong, Appl. Surf. Sci., 2017, 411, 227–234 CrossRef CAS.
- Y. Zhang, X. Yan, B. Niu and J. Zhao, Green Chem., 2016, 18, 3139–3151 RSC.
- C. Yang, M. Florent, G. de Falco, H. Fan and T. J. Bandosz, Chem. Eng. J., 2020, 394, 124906 CrossRef CAS.
- S. Allahyari, M. Haghighi, A. Ebadi and S. Hosseinzadeh, Ultrason. Sonochem., 2014, 21, 663–673 CrossRef CAS PubMed.
- I. O. Ali, S. A. El-Molla, I. A. Ibraheem and T. M. Salama, Microporous Mesoporous Mater., 2014, 197, 48–57 CrossRef CAS.
- B. Huang, G. Liu, P. Wang, X. Zhao and H. Xu, Processes, 2019, 7, 167 CrossRef CAS.
- Y. Yan, X. Yang, P. Ning, C. Wang, X. Sun, F. Wang, P. Gao and K. Li, J. Environ. Sci., 2025, 148, 476–488 CrossRef CAS PubMed.
- P.-O. Larsson and A. Andersson, J. Catal., 1998, 179, 72–89 CrossRef CAS.
- N. Ping, Y. Honghong, Y. Qiongfen, T. Xiaolong, Y. Liping and Y. Zhiqing, J. Rare Earths, 2010, 28, 581–586 CrossRef.
- T. Meng, Y. Lin and Z. Ma, Mater. Chem. Phys., 2015, 163, 293–300 CrossRef CAS.
- H. Pan, Y. Jian, C. Chen, C. He, Z. Hao, Z. Shen and H. Liu, Environ. Sci. Technol., 2017, 51, 6288–6297 CrossRef CAS PubMed.
- X. Xing, N. Li, D. Liu, J. Cheng and Z. Hao, Front. Environ. Sci. Eng., 2022, 16, 125 CrossRef CAS.
- V. Subbaramaiah, V. C. Srivastava and I. D. Mall, Ind. Eng. Chem. Res., 2013, 52, 9021–9029 CrossRef CAS.
- F. Cavani, Catal. Today, 1998, 41, 73–86 CrossRef CAS.
- C. Dong, H. Wang, H. Du, J. Peng, Y. Cai, S. Guo, J. Zhang, C. Samart and M. Ding, Mol. Catal., 2020, 482, 110755 CrossRef CAS.
- O. Tursunov, L. Kustov and Z. Tilyabaev, J. Petrol. Sci. Eng., 2019, 180, 773–778 CrossRef CAS.
- R. Ma, S. Zhang, L. Li, P. Gu, T. Wen, A. Khan, S. Li, B. Li, S. Wang and X. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9699–9708 CrossRef CAS.
- Z. Ren, S. Quan, Y. Zhu, L. Chen, W. Deng and B. Zhang, RSC Adv., 2015, 5, 29734–29740 RSC.
- X. Tang, J. Xue and C. Xing, Appl. Surf. Sci., 2020, 530, 147264 CrossRef CAS.
- K. Zhang, Z. Xiong, S. Li, B. Yan, J. Wang and Y. Du, J. Alloys Compd., 2017, 706, 89–96 CrossRef CAS.
- Y. Tang, J. Feng, P. Ning, F. Wang, X. Sun and K. Li, Chem. Eng. J., 2023, 461, 142078 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Guoqiang Huang
* and
Guoqiang Huang