
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNano-foam gold-modified diamond electrochemical aptasensing platform for ultrasensitive monitoring of 17β-estradiol†
Yu Wanga,
Yibo Ma *a,
Xiaowei Dai*b,
Jia Lia,
Jian Zhanga,
Hongyu Jianga,
Maofeng Wanga and
Shenglong Yua
*a,
Xiaowei Dai*b,
Jia Lia,
Jian Zhanga,
Hongyu Jianga,
Maofeng Wanga and
Shenglong Yua
aCollege of Science, Beihua University, Jilin 132013, China. E-mail: yiboma@beihua.edu.cn
bDepartment of Reproductive Medicine Center, The Second Norman Bethune Hospital of Jilin University, Changchun 130041, China. E-mail: davidjdey@163.com
First published on 17th April 2025
Abstract
This study developed a nano-foam gold modified boron-doped diamond (NFG/BDD-Apt) electrochemical aptasensor through a synergistic electrodeposition-dealloying strategy combined with aptamer functionalization for detecting 17β-estradiol (E2) in aquatic environments. The NFG/BDD-Apt sensor was systematically characterized using SEM, Raman, and EIS to elucidate its surface morphology, molecular structure, and electrochemical properties. SEM analysis revealed the successful formation of a homogeneous three-dimensional porous NFG structure on the BDD surface, which significantly enhanced the specific surface area (1.9-fold increase vs. bare BDD) and electron transfer efficiency. Electrochemical performance evaluation through CV and DPV demonstrated superior E2 detection capabilities. Under optimized conditions, the sensor exhibited a wide linear response range from 1.0 × 10−14 to 1.0 × 10−8 mol L−1 (R2 = 0.997) with an ultralow detection limit of 1.8 × 10−15 mol L−1 (S/N = 3). NFG/BDD-Apt demonstrated exceptional selectivity (>92% specificity against common interferents) and long-term stability. This work provides a novel sensing platform combining diamond electrode advantages with nanostructured amplification effects, offering significant potential for rapid and reliable monitoring of endocrine disruptors in environmental water systems.
1. Introduction
17β-Estradiol (E2), a natural estrogen, plays a pivotal role in regulating critical physiological processes, including the menstrual cycle, follicular development, and endometrial proliferation in female reproductive systems. Beyond reproductive health, E2 significantly influences bone metabolism, cardiovascular homeostasis, and neurological functions.1–3 However, the environmental persistence of E2 residues poses escalating ecological and public health risks. Studies have demonstrated that trace concentrations of E2 (as low as ng per L levels) disrupt endocrine systems in aquatic organisms, leading to skewed sex ratios, impaired reproductive capacity in fish populations, and long-term ecosystem imbalances.4 In humans, chronic exposure to elevated estrogen levels, particularly E2, is associated with increased risks of hormone-dependent malignancies such as breast and endometrial cancers.5,6 Therefore, it is essential to establish swift, precise, and sensitive techniques for identifying E2 in water bodies to protect ecological balance and public health.Conventional techniques for identifying E2, including high-performance liquid chromatography (HPLC), gas chromatography-mass spectrometry (GC-MS), and enzyme-linked immunosorbent assays (ELISA), face inherent limitations.7–9 While HPLC and GC-MS offer high sensitivity and precision, their reliance on expensive instrumentation, complicated sample pretreatment, and prolonged analysis times renders them impractical for on-site monitoring. ELISA, despite its operational simplicity and moderate sensitivity, suffers from antibody instability and cross-reactivity issues, constraining its reliability in complex environmental matrices.
Electrochemical aptasensors have emerged as transformative alternatives, synergizing the molecular recognition prowess of aptamers with the rapid response and cost-effectiveness of electrochemical platforms.10,11 Aptamers, which are single – stranded oligonucleotides, are selected using the systematic evolution of ligands by exponential enrichment (SELEX) approach. These aptamers exhibit remarkable specificity towards target molecules. They form stable complexes with the targets through a process of structural recognition. Compared to antibodies, aptamers demonstrate enhanced thermal stability, ease of chemical modification, and negligible immunogenicity, making them ideal biorecognition elements.12–14
Boron-doped diamond (BDD) electrodes have garnered attention as advanced electrochemical substrates due to their exceptional properties: an ultra-wide potential window (∼3.7 V), near-zero background current, and remarkable chemical inertness.15,16 These attributes allow BDD electrodes to detect redox-active species across a broad potential range while minimizing interference from parasitic reactions—a critical advantage for analyzing complex environmental samples. However, the limited density of surface-active sites on pristine BDD electrodes restricts their utility in high-sensitivity biosensing applications.
The integration of nanostructured materials presents a strategic solution to amplify sensor performance. Nano-foam gold (NFG), a three-dimensional porous architecture comprising interconnected gold nanoparticles, offers exceptional surface-to-volume ratios, enhanced conductivity, and biocompatibility.17 The hierarchical porosity of NFG facilitates dense aptamer immobilization via Au–S covalent bonding while minimizing steric hindrance, thereby maximizing target accessibility. Furthermore, NFG's conductive network accelerates electron transfer kinetics, synergizing with BDD's intrinsic electrochemical advantages to create a high-performance sensing interface.18
In this study, we engineered a nano-foam gold-modified boron-doped diamond electrochemical aptasensor (NFG/BDD-Apt) through a synergistic electrodeposition-dealloying strategy. The NFG/BDD-Apt platform was systematically characterized using SEM, EIS, and Raman to validate its structural and functional properties. DPV was employed to evaluate the sensor's analytical performance for E2 detection in aqueous environments, demonstrating femtomolar sensitivity, exceptional selectivity against structural analogs, and long-term stability. This work establishes a novel paradigm for environmental estrogen monitoring by integrating diamond electrochemistry with nanomaterial engineering and molecular recognition technologies.
2. Experimental
2.1 Chemical
E2, estriol (E3), bisphenol A (BPA), HAuCl4·4H2O, and CuSO4·5H2O are all analytically pure and purchased from Shanghai Acmec Biochemical Technology Co., Ltd. Tri-(2-formylethyl) phosphine hydrochloride (TCEP), TE Buffer, and Aptamer are synthesized by Shenggong Bioengineering (Shanghai) Co., Ltd, with their 5′ ends modified with thiol groups. The aptamer is stored in dry powder form at −20 °C. Before use, it is centrifuged, shaken, and dissolved in TE buffer to reach the desired concentration. The specific base sequence is as follows:5′-SH-GCTTCCAGCTTATTGAATTACACGCAGAGGGTAGCGGCTCTGCGCATTCAATTGCTGCGCGCTGAAGCGCGGAAGC-3′
2.2 Characterization
SEM (Magellan-400) was used to observe the surface morphology of the electrodes. XRD (Bruker-D8 Advance) and laser confocal Raman spectroscopy (HR Evolution, 514 nm) were utilized to analyze the crystal structure. UV spectroscopy was performed on a UV-2550 (Shimadzu, Japan). All electrochemical experiments, including CV, DPV, and EIS, were carried out on an electrochemical workstation (Ivium-n-Stat, Netherlands).2.3 Experimental section
The schematic illustration in Fig. 1 depicts the designation and fabrication process of NFG/BDD-Apt electrodes, which are based on the advanced technology of BDD electrodes, known for their exceptional electrochemical properties and applications in water treatment. | ||
| Fig. 1 Schematic of the E2 aptasensor synthesis and preparation process. | ||
2.4 Electrochemical test
The electrochemical performance was evaluated using a conventional three-electrode system, comprising the NFG/BDD-Apt sensor as the working electrode, an Ag/AgCl reference electrode, and a Pt counter electrode. Electrochemical measurements were conducted in a solution containing 5 mM Fe(CN)63− and 0.1 M KCl as the supporting electrolyte. CV was conducted over a potential range from 0.0 to 1.2 V at a scan speed of 50 mV s−1. DPV was executed within the potential limits of 0.0 to 0.6 V, utilizing a pulse amplitude of 10 mV and a pulse duration of 10 ms. EIS was conducted over a frequency range from 10−1 to 106 Hz with an AC perturbation voltage of 5 mV.For E2 detection experiments, the NFG/BDD-Apt sensor was immersed in 0.1 M TE buffer (pH 8.0) containing varying concentrations of E2 and incubated at room temperature for 30 minutes to facilitate specific binding. The electrode was then washed extensively with ultrapure water to eliminate any E2 molecules that were not bound. The electrochemical detection was used the DPV technique. The changes in peak current were recorded and analyzed to determine the E2 concentration. This experimental protocol was designed to ensure optimal binding efficiency and measurement reproducibility, while minimizing non-specific interactions that could potentially interfere with the detection accuracy.
For sensor regeneration, the NFG/BDD-Apt electrode was rinsed with 50 mM EDTA solution to disrupt E2-aptamer complexes, followed by washing with Tris–HCl buffer and ultrapure water. This procedure effectively removed bound E2 molecules while preserving the aptamer layer.
3. Results
3.1 Deposition conditions
To investigate the electrochemical formation of the Au–Cu alloy on BDD electrodes, CV was performed under controlled electrolyte conditions. Fig. 2a demonstrates that the BDD substrate exhibits an ultra-wide potential window and minimal background current, providing an excellent foundation for subsequent modifications. As shown in Fig. 2b, underpotential deposition (UPD) of Cu occurs within 0.05 to 0.45 V, while overpotential deposition dominates below 0.05 V. These observations align with the calculated Nernst potential (0.041 V vs. Ag/AgCl, 25 °C) for Cu2+ deposition from a 20 mM solution.20 UPD is a surface-limited process where metal atoms adsorb onto a foreign substrate at potentials more positive than their thermodynamic reduction potential. This phenomenon arises from the Gibbs free energy reduction associated with surface alloy formation, as confirmed by theoretical studies.20 For instance, the lower work function of Cu compared to Au enables the formation of a Cu monolayer on the Au electrode via UPD.21,22 In the UPD region, a broad cathodic peak is observed during the negative scan, accompanied by a corresponding anodic stripping peak in the positive scan (Fig. 2b). Recent studies on Au–Cu alloy electrodeposition have further validated this behaviour.21 Although in situ DFT calculations were not performed in this study, DFT has shown that Au–Cu interfaces exhibit altered electronic structures, aligning with our experimental observations.23,24 | ||
| Fig. 2 (a) Baseline CV of a pristine BDD electrode in 0.5 M H2SO4. (b) Cu deposition behavior in 20 mM CuSO4 + 0.5 M H2SO4. (c) Au deposition profile in 20 mM KAuCl4 + 0.5 M H2SO4. (d) Co-deposition characteristics of Au–Cu alloy in 10 mM CuSO4 + 10 mM KAuCl4 + 0.5 M H2SO4. | ||
Fig. 2c reveals that Au deposition initiates at potentials more negative than 0.94 V. Notably, the absence of Cl− in the solution shifts the onset of Au deposition to more positive potentials compared to its standard reduction potential.20 A limiting diffusion current is observed below 0.62 V, followed by an oxidative current above 0.89 V during the positive scan, attributed to Au dissolution via ion dissociation.
To achieve Au–Cu co-deposition, a mixed electrolyte containing both Cu2+ and Au3+ ions was employed. The CV of Au–Cu (Fig. 2d) exhibits distinct reduction currents for both metals. Specifically, the cathodic current increases sharply below 0.26 V, indicative of UPD co-deposition of Au and Cu. Based on these findings, optimal co-deposition and dissolution potentials were selected as 0.1 V and 0.95 V, respectively.
To investigate residual Cu content, time-dependent dealloying experiments were performed (Fig. S1†). As dealloying time increased from 0 to 5000 s, the Cu oxidation peak current density decreased from 0.2636 mA cm−2 to 0.0292 mA cm−2, indicating progressive Cu removal. After 2000 s, the current stabilized at 0.0347 mA cm−2, reflecting a balance between Cu dissolution and retention in the NFG.
3.2 Structural characterization
The surface morphology of BDD and NFG/BDD electrodes was analyzed by SEM. As illustrated in Fig. 3a, the BDD substrate displays a smooth and compact surface with an average grain size of 5 μm. Following electrodeposition and dealloying, a uniformly distributed three-dimensional (3D) porous architecture was successfully constructed on the NFG/BDD electrode (Fig. 3b–d). The NFG deposition and dealloying potentials were optimized to 0.1 V and 0.95 V, respectively, based on CV results (Fig. 2). | ||
| Fig. 3 SEM images of BDD (a). NFG/BDD formed by deposition (0.1 V) and dealloying (0.95 V) at a different time: (b) 500 s/2000 s; (c) 650 s/2000 s; and (d) 1000 s/2000 s. | ||
At a deposition time of 500 s (Fig. 3b), the NFG layer exhibits a loosely packed structure with incomplete coverage of the BDD surface, leading to poor interfacial contact and compromised sensing performance. In contrast, a deposition time of 650 s yields a densely packed NFG layer with interconnected particles and uniform pore sizes averaging 50 nm (Fig. 3c). The effective surface area is greatly improved by this three-dimensional porous structure, which offers numerous active sites for electron transfer and promotes strong immobilization of biomolecules.25–27 Importantly, the hierarchical structure mitigates aptamer stacking, thereby improving loading efficiency. However, extending the deposition time to 1000 s induces particle agglomeration and interfacial stress, resulting in partial delamination of the NFG layer (Fig. 3d). As a result, 650 s was chosen as the optimum deposition time.
The crystallinity and doping degree of the electrodes were analyzed by means of XRD and Raman spectroscopy. The XRD pattern of NFG/BDD (Fig. 4a) exhibits distinct diffraction peaks for diamond (43.96°, 75.25°, and 91.39°, corresponding to (111), (220), and (311) planes) and Au (38.40°, assigned to the (111) plane).28 The Au (111) facet, known for its superior catalytic activity and smooth surface, minimizes non-specific adsorption and reduces background noise, thereby enhancing sensing performance.29 The Au (111) facet's reduced nonspecific adsorption can be attributed to its atomically smooth surface and low surface energy, which minimize the thermodynamic driving force for adsorbate binding. Theoretical studies have shown that Au (111) surfaces exhibit weaker interactions with polar molecules compared to other crystal orientations.25,30,31 This property is critical for maintaining low background noise in electrochemical measurements, as demonstrated by the sensor's ultralow detection limit (1.8 × 10−15 mol L−1) and high signal-to-noise ratio. No Cu diffraction peaks were observed in the XRD, which may be due to the residual Cu existing in the form of atomic-level surface alloy or surface clusters below the XRD detection limit after dealloying.
 | ||
| Fig. 4 (a) XRD and (b) Raman spectroscopy of BDD and NFG/BDD. | ||
Raman spectra (Fig. 4b) confirm the BDD structure via the characteristic diamond peak at 1317 cm−1, which exhibits a redshift and asymmetry due to boron doping-induced Fano effects. Broad bands at 486 cm−1 and 1204 cm−1 arise from increased local density of states caused by boron incorporation.15 After NFG deposition, additional peaks at 200–400 cm−1 and 500–800 cm−1 correspond to Au–S and Au–Au vibrational modes, respectively, confirming successful NFG modification.32–35
3.3 Electrochemical characterization
Electrochemical performance was further evaluated using CV and EIS. As shown in Fig. 5a, NFG modification reduces the peak-to-peak separation (ΔEp) from 334 mV (BDD) to 190 mV (NFG/BDD) and increases the anodic current by 1.36-fold (from 1.23 mA to 1.67 mA). The smaller the ΔEp, the greater the number of electrons transferred, indicating that the reaction proceeds more easily.36–39 The reduction in ΔEp directly reflects enhanced charge transfer kinetics due to (i) NFG's 3D porous architecture increasing electroactive surface area, (ii) metallic conductivity lowering activation barriers, and (iii) optimized mass transport through nanoscale pores. These improvements align with the Nernstian criteria for reversible systems. | ||
| Fig. 5 (a) CV curves and (b) EIS spectra of BDD and NFG/BDD electrodes. | ||
EIS results (Fig. 5b) corroborate these findings: the charge transfer resistance (Rct) of NFG/BDD decreases significantly compared to bare BDD, attributable to the high conductivity and large surface area of NFG. Subsequent aptamer functionalization slightly increases Rct due to the insulating nature of aptamers and electrostatic repulsion with Fe(CN)63−, confirming successful aptamer immobilization.
To clarify the critical role of aptamers in achieving selective E2 detection, a comparison was made between the DPV results of unfunctionalized NFG/BDD and aptamer-functionalized NFG/BDD-Apt electrodes under conditions with and without E2. The results show (Fig. S2†) that the NFG/BDD-Apt structure exhibits the best sensitivity for E2 detection.
3.4 Sensing performance of the NFG/BDD-Apt sensor
The NFG/BDD-Apt sensor was evaluated for E2 detection using DPV. As E2 concentration increases (Fig. 6a), the DPV peak current decreases progressively, attributed to E2-aptamer complex formation hindering electron transfer. A linear relationship between peak current and the logarithm of E2 concentration is observed over a wide range (1.0 × 10−14 to 1.0 × 10−8 mol L−1; Fig. 6b), with a regression equation of I(μA) = −0.0025![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lgC + 0.0920 (mol L−1), a correlation coefficient (R2) of 0.997, and a detection limit (LOD) of 1.8 × 10−15 mol L−1 (S/N = 3).
lgC + 0.0920 (mol L−1), a correlation coefficient (R2) of 0.997, and a detection limit (LOD) of 1.8 × 10−15 mol L−1 (S/N = 3).
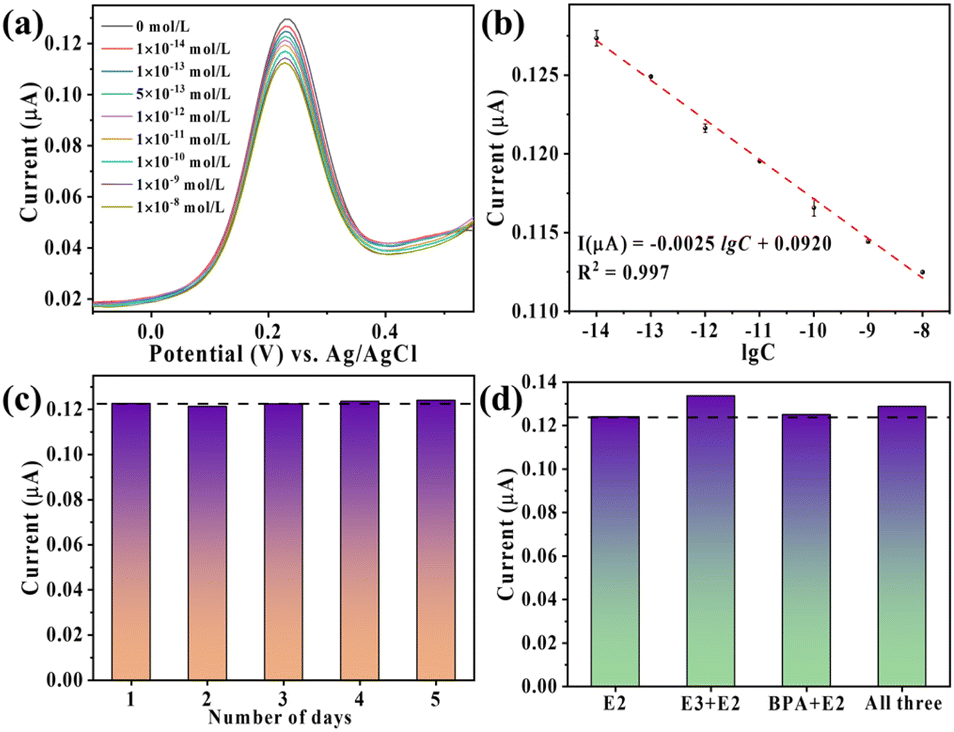 | ||
| Fig. 6 (a) DPV responses of the aptasensor during the detection of E2 with diverse concentrations; (b) calibration curve in semi-log scale; (c) stability and (d) target selectivity of NFG/BDD-Apt. | ||
Stability and selectivity tests further validate the sensor's reliability. After 5 days of storage at 4 °C, the sensor retains 95% of its initial response to 5.0 × 10−13 mol per L E2 (Fig. 6c). The selectivity was evaluated in the presence of structurally similar interfering substances, such as E3 and BPA. In mixed solutions containing 5.0 × 10−13 mol per L E2 and 5.0 × 10−11 mol per L interferents, the DPV signal deviation remains below 8% (Fig. 6d), demonstrating exceptional specificity derived from the aptamer's high affinity for E2.
The sensor retains 83.5% signal integrity after 20 regeneration cycles (EDTA/Tris–HCl protocol), with full recovery achievable via aqua regia reprocessing (Fig. S3†). Cost analysis ($0.70 per disposable unit) confirms feasibility for both reusable and single-use applications in field monitoring.
4. Mechanism
The NFG/BDD-Apt sensor's ultrahigh sensitivity arises from synergistic interactions between its components. Mixed solutions with varying concentration ratios were obtained by adding E2 to a 2 μmol per L aptamer solution. By subtracting the UV spectrum of the blank solution without E2 from that of the mixed solution, we obtained the result shown in Fig. 7a. E2 exhibits a shrinkage effect at 257 nm, which is reflected in the detection process through eqn (1) and (2).| nE2 + aptamer → E2/aptamer | (1) |
 | (2) |
 | ||
| Fig. 7 (a) UV spectra displaying the interaction of the aptamer with different concentrations of E2; (b) calibration curve derived from UV measurements. (c) CV of the BDD and NFG/BDD electrodes. | ||
The CV in 0.5 M sulfuric acid (Fig. 7c) emphasizes the exceptionally broad potential range of BDD and its minimal background current, which is essential for detecting with high sensitivity. The electrochemically active surface area of NFG/BDD (0.17 cm2), calculated from gold oxide coulometry, is 1.9-fold larger than the geometric area (0.09 cm2). This 3D porous architecture not only enhances aptamer loading but also optimizes charge transfer pathways, collectively contributing to the sensor's superior performance.
While the current study validates sensor performance under controlled laboratory conditions, future work will systematically assess temperature and humidity effects. Preliminary analysis suggests robust operation within 4–40 °C and moderate humidity (30–90% RH), supported by the thermal stability of BDD and aptamer-DNA interactions. Protective encapsulation strategies will be explored to enhance environmental resilience for field deployment.
5. Conclusions
This study developed a nano-foam gold-modified boron-doped diamond electrochemical aptasensor (NFG/BDD-Apt) for ultrasensitive 17β-estradiol (E2) detection in aquatic environments. The NFG/BDD-Apt was fabricated via electrodeposition-dealloying, integrating BDD's ultra-wide potential window and low background current with NFG's 3D porous architecture to amplify electroactive surface area (1.9× vs. bare BDD) and electron transfer efficiency. Structural characterization confirmed NFG/aptamer integration, while DPV analysis demonstrated femtomolar sensitivity (LOD: 1.8 × 10−15 mol L−1) across a broad linear range (1.0 × 10−14–1.0 × 10−8 mol L−1, R2 = 0.997). The sensor exhibited exceptional selectivity (>90% specificity against estriol and bisphenol A) and stability (95% signal retention over 5 days).By synergizing diamond electrochemistry, nanostructural engineering, and aptamer specificity, this work addresses critical limitations in conventional estrogen detection methods. The NFG/BDD-Apt platform offers a field-deployable solution for trace-level environmental monitoring, combining rapid response, robustness, and compliance with regulatory standards. Future work will explore multiplexed detection and integration with portable systems to enhance practical utility.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
YW: data curation, conceptualization, investigation, validation, writing – original draft. YM: conceptualization, methodology, writing – review & editing, formal analysis, funding acquisition. XD: methodology, investigation, writing – original draft. JL: methodology, validation. JZ: methodology, investigation, formal analysis. HJ: methodology, investigation, formal analysis. MW: conceptualization. SY: conceptualization, methodology, formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Scientific and Technological Development Program of Jilin Province, China (No. YDZJ202301ZYTS267); the National College Students' Innovation and Entrepreneurship Training Program, China (No. 202210201041). Eceshi (http://www.eceshi.com) was acknowledged for the XRD and Raman analysis.References
- M. van den Buuse, J. Sun and A. Gogos, Horm. Behav., 2025, 167, 105673 CrossRef CAS PubMed.
- E. C. Aham, A. Ravikumar, C. O. Okoye, T. P. C. Ezeorba, A. Arunjegan, G. Tamilselvan, I. U. Okagu, H. Zhang and Z. Zhang, Microchem. J., 2024, 206, 111486 CrossRef CAS.
- M. Liu, Y. Zhang, L. Feng, Q. Guo, T. Chen, C. Mu, J. Lu, Y. Cheng and X. Wu, Comp. Biochem. Physiol., Part A:Mol. Integr. Physiol., 2025, 301, 111798 CrossRef CAS PubMed.
- L. Hejji, A. Azzouz, D. Kukkar and K.-H. Kim, TrAC, Trends Anal. Chem., 2023, 166, 117194 CrossRef CAS.
- S. E. Zahra Jawad, D. Hussain, M. Najam-ul-Haq and B. Fatima, TrAC, Trends Anal. Chem., 2024, 181, 117993 CrossRef CAS.
- M. L. Báez, A. García, I. Martínez, C. González, M. Gómez and B. Rodríguez, Int. J. Electrochem. Sci., 2024, 19(4), 100538 CrossRef.
- Y. Shi, D.-D. Peng, C.-H. Shi, X. Zhang, Y.-T. Xie and B. Lu, Food Chem., 2011, 126(4), 1916–1925 CrossRef CAS PubMed.
- A. K. Tsakalof, D. C. Gkagtzis, G. N. Koukoulis and C. S. Hadjichristodoulou, Anal. Chim. Acta, 2012, 709, 73–80 CrossRef CAS PubMed.
- C. P. Silva, D. L. D. Lima, R. J. Schneider, M. Otero and V. I. Esteves, J. Environ. Manage., 2013, 124, 121–127 CrossRef CAS PubMed.
- T. Zhang, Y. Wang, R. Yuan and Y. Qi, Sens. Actuators, B, 2025, 428, 137224 CrossRef CAS.
- G. Chen, L. Wang, H. Liu, L. Wang, C. Yu and H. Li, Sens. Actuators, B, 2025, 430, 137365 CrossRef CAS.
- T. R. Tsekeli, T. I. Sebokolodi, D. S. Sipuka, F. O. G. Olorundare, S. P. Akanji, D. Nkosi and O. A. Arotiba, Electroanal. Chem., 2021, 901, 115783 CrossRef CAS.
- G. Karuppaiah, J. Velayutham, N. K. Sethy and P. Manickam, Mater. Lett., 2023, 342, 134310 CrossRef CAS.
- Z. Chang, B. Zhu, J. Liu, X. Zhu, M. Xu and J. Travas-Sejdic, Biosens. Bioelectron., 2021, 185, 113247 CrossRef CAS PubMed.
- L. Wan, Y. Liang, M. Yang, Q. Zhang, S. Xu, S. Cheng, N. Gao, D. Zhang and Y. Ma, Carbon, 2025, 233, 119835 CrossRef CAS.
- X. Wang, J. Dou, Z. Deng, M. Gong, K. Zhou, L. Ma and Q. Wei, Chem. Eng. J., 2024, 499, 156454 CrossRef CAS.
- J. Yang, Y. Hu and Y. Li, Biosens. Bioelectron., 2019, 135, 224–230 CrossRef CAS PubMed.
- C. Li, B. Jiang, H. Chen, M. Imura, L. Sang, V. Malgras, Y. Bando, T. Ahamad, S. M. Alshehri, S. Tominaka and Y. Yamauchi, Nano Res., 2016, 9(6), 1752–1762 CrossRef CAS.
- Y. Ma, J. Liu and H. Li, Biosens. Bioelectron., 2017, 92, 21–25 CrossRef CAS PubMed.
- S. Tominaka, J. Mater. Chem., 2011, 21(26), 9725 RSC.
- S. Fackler, M. Wittmann, M. K. Heubach, J. M. Hermann, L. A. Kibler and T. Jacob, Electrochim. Acta, 2024, 490, 144225 CrossRef CAS.
- T. Q. Casuse-Driovínto, A. Benavidez, N. Jemison, J. M. Cerrato, J. Feliu and F. H. Garzón, Electrochim. Acta, 2024, 489, 144220 CrossRef.
- L. Rout, A. Kumar, R. S. Dhaka, G. N. Reddy, S. Giri and P. Dash, Appl. Catal., A, 2017, 538, 107–122 CrossRef CAS.
- Q. Chang, X. Zhang and Z. Yang, Results Phys., 2023, 51, 106715 CrossRef.
- M. Ganesan, R. Keerthika Devi, A.-H. Liao, K.-Y. Lee, G. Gopalakrishnan and H.-C. Chuang, Food Chem., 2022, 396, 133722 CrossRef CAS PubMed.
- L. Li, X. Yan, Y. Liu, Y. Xing, P. Zhao, Y. Zhu, N. Liu, K. Sun, Z. Zhang and S. Zhai, Bioelectrochemistry, 2025, 161, 108833 CrossRef CAS PubMed.
- Q. Shen, J. Ding, Z. Guo, C. Wang, Y. Zhang, C. Lin, Y. Sun and L. Hang, Colloids Surf., B, 2025, 248, 114471 CrossRef CAS PubMed.
- Y. Yang, X. Chen, Y. Ma, Z. Sun, Y. Wang, Y. Zhang, P. Li and Q. Lin, Diamond Relat. Mater., 2023, 137, 110088 CrossRef CAS.
- T.-T. Zhang, Q.-L. Tang, X. Zhang, S.-P. Tang and Q. Wang, Appl. Surf. Sci., 2025, 682, 161732 CrossRef CAS.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322(5903), 932–934 CrossRef CAS PubMed.
- M. Aslam, I. S. Mulla and K. Vijayamohanan, Langmuir, 2001, 17(24), 7487–7493 CrossRef CAS.
- Y. Lu, X. Mo, G. Zhu, Y. Huang, Y. Wang, Z. Yang, L. Gao, G. Shen, Y. Wang and X. Zhao, J. Hazard. Mater., 2024, 467, 133763 CrossRef CAS PubMed.
- L. Han, X. Wang, B. Yu, X. Qin, B. Liu, X. Han, H. Yuan, B. Yu and Z. Zhao, Spectrochim. Acta, Part A, 2025, 326, 125192 CrossRef CAS PubMed.
- J. Xu, J. Wei, Y. Cui, S. Yu, S. Guo, Q. An and P. Han, Colloids Surf., A, 2024, 694, 134099 CrossRef CAS.
- G.-F. Wu, J. Zhu, G.-J. Weng, J.-J. Li and J.-W. Zhao, Talanta, 2025, 127742 CrossRef CAS PubMed.
- H. Gerischer, Angew Chem. Int. Ed. Engl., 2003, 18(4), 334–335 CrossRef.
- J. C. Myland and K. B. Oldham, Anal. Chem., 1994, 66, 1866–1872 CrossRef CAS.
- Z. Mo, Y. Pang, L. Yu and X. Shen, Food Chem., 2021, 359, 129816 CrossRef CAS PubMed.
- H. Ke, M. Liu, L. Zhuang, Z. Li, L. Fan and G. Zhao, Electrochim. Acta, 2014, 137, 146–153 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01318f |
| This journal is © The Royal Society of Chemistry 2025 |