DOI:
10.1039/D5RA01209K
(Paper)
RSC Adv., 2025,
15, 14594-14603
Electron structure variations in hindered phenolic antioxidant induced enhancement of thermo-oxidative aging performance in polyamide 6†
Received
19th February 2025
, Accepted 29th April 2025
First published on 6th May 2025
Abstract
PA6 is widely used but vulnerable to thermo-oxidative aging, impacting its durability. Hindered phenols are key antioxidants that extend the service life of PA6 by slowing this aging process. However, the effect of molecular structural differences on their antioxidant activity is still elusive, impeding their development and application. Here, two antioxidants, 2-((3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionyl)oxy)ethyl acrylate (PEA) and 2-((3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionyl)oxy)propyl acrylate (PPA), were utilized to enhance the thermo-oxidative aging resistance of PA6. Their primary difference is that PPA possesses an electron-donating group in its molecular structure. The methyl functional group in the PPA molecule can increase the electron cloud density around the benzene ring, resulting in PPA having a higher free radical scavenging rate compared to PEA. In addition, in contrast to PEA, the low volatility and mobility of PPA ensure that its antioxidant activity can be fully utilized during aging. These factors collectively support the excellent antioxidant activity of PPA (PPA > PEA). As a result, the PA6/PPA composite maintains 88% of its original tensile strength after 12 days of continuous aging, whereas PA6/PEA and PA6/1010 (commercial antioxidant) composites retained 70% and methyl 30%, respectively. These encouraging findings provide a theoretical basis for the synthesis of antioxidants.
1 Introduction
Polyamide (PA) is a thermoplastic engineering plastic with high mechanical strength and good crystalline properties, as well as corrosion and abrasion resistance, etc. It is widely used in 5 G electronics, the automotive industry, and the textile industry.1,2 Polyamide (PA) is susceptible to thermo-oxidative aging when subjected to elevated temperatures and the presence of oxygen, resulting in a deterioration in its mechanical properties and consequently impairing the material's service longevity.3–6 The addition of antioxidants has been demonstrated to effectively decelerate the aging process.7,8 Hindered phenols, commonly utilized as additives, effectively trap peroxyl and macromolecular radicals, thereby terminating chain reactions of polymers.9–12 The inactivation of free radicals by hindered phenols predominantly involves an atom transfer mechanism.13 In the mechanism, the rate of removing hydrogen atoms is a key indicator for the antioxidant activity of hindered phenols, which is related to the O–H bond dissociation enthalpy (BDE) of the phenol hydroxyl group.14 Judging the antioxidant activity of hindered phenols by calculating the O–H BDE of phenol hydroxyl has been demonstrated, but few practical applications have been made, and in this work, we will address the above theses.
The introduction of functional groups into the structure of conventional hindered phenols can effectively improve their antioxidant activity.15,16 Zhang et al. found that the phenolic hydroxyl group and carbamate contained in the antioxidant-hindered phenol functionalized graphene oxide (HPFGO) have a synergistic effect, which makes the composites exhibit excellent antioxidant properties. The phenolic hydroxyl group in HPFGO can provide hydrogen atoms to capture ROO˙ radicals and are further converted to benzoquinone radicals. The resulting radicals can further capture the hydrogen atoms on the carbamate to continue to capture free radicals, which improves the number of free radicals captured by HPFGO and increases the antioxidant efficiency.17,18 Li et al. prepared hindered phenol PAMAM (dendritic antioxidants) containing phenolic hydroxyl and tertiary amine groups and demonstrated that the two groups also have the same synergistic effect in hindered phenol. They can not only provide hydrogen atoms to scavenge free radicals but also scavenge the free radicals by electron transfer and thus have more excellent antioxidant properties.19 In summary, hindered phenols containing functional group have a bifunctional antioxidant mechanism for the scavenging of free radicals,20 which is conducive to the enhancement of the antioxidant properties of hindered phenols. However, the effect of the bridging structure of functional groups differences on their antioxidant activity is still elusive, impeding their development and application.
In this work, an acrylate group was introduced in the structure of hindered phenol. And a methyl group was introduced near the ester group to study the effect of the electron-donating group methyl and acrylate group on the antioxidant activity of hindered phenol. The effect of hindered phenolic bonding structure on free radical scavenging activity was studied by comparison of scavenging capacity and scavenging rate of 1,1-diphenyl-2-picrylhydrazyl (DPPH) radicals. The ability of hindered phenols to provide hydrogen atoms was investigated by analyzing the electron cloud density of phenol hydroxyl and the O–H BDE. The interaction of the antioxidant with PA6 was analyzed by solubility parameters, and the antimigration resistance of the antioxidant in PA6 was evaluated. The antioxidant effect of antioxidants in PA6 was studied using thermo-oxidative aging experiment. The introduction of methyl groups into the structure of acrylate-bonded hindered phenols was found to improve the free radical scavenging ability of the hindered phenols. It has good compatibility with PA6, exhibits low mobility, and shows a good antioxidant effect.
2 Experimental sections
2.1 Materials
3,5-Di-tert-butyl-4-hydroxyphenylpropionic acid (AO), hydroxyethyl acrylate (HEA), 4-dimethylaminopyridine (DMAP), and N,N-dicyclohexylcarbodiimide (DCC) were obtained from Aladdin Biochemistry and Technology Co. (Shanghai, China). Hydroxypropyl acrylate (HPA) was purchased from Shanghai McLean Biochemical Technology Co. (Shanghai, China). Polyamide 6 (PA6, YH-800) was purchased from Yueyang Petrochemical Company (Yueyang, China). Methylene chloride anhydrous was purchased from Anhui Zesheng Technology Co. (Anhui, China). Dichloromethane and methanol were procured from Fuyu Fine Chemical Co., Ltd (Tianjin, China). All materials were used without further purification.
2.2 Synthesis of antioxidants
Two hindered phenolic antioxidants, 2-((3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionyl)oxy)ethyl acrylate (PEA) and 2-((3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionyl)oxy)propyl acrylate (PPA), were synthesized by esterification of HEA, HPA, and AO (Fig. 1). To illustrate the synthesis process, the synthesis of PEA is used as an example. Initially, 2.7819 g of AO and 1.1605 g of HEA were introduced into a three-necked flask and dissolved in 40 ml of anhydrous dichloromethane. This mixture process was carried out in a water bath maintained at 0–5 °C. After 30 minutes of reaction at this temperature and nitrogen atmosphere, 3.0949 g of DCC was dissolved in 40 ml of anhydrous dichloromethane solution and slowly added to the reaction system. After this addition, 0.1221 g of DMAP was introduced into the mixture, and the reaction was continued at room temperature for 36 h to obtain the product. The product was purified by column chromatography with dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol = 20:1 eluent and further was washed using vacuum evaporation eluent. The purified product was vacuum dried at 60 °C for 12 hours to obtain a light-yellow viscous oil with a yield of 70.80%.
:methanol = 20:1 eluent and further was washed using vacuum evaporation eluent. The purified product was vacuum dried at 60 °C for 12 hours to obtain a light-yellow viscous oil with a yield of 70.80%.
 |
| | Fig. 1 Synthesis of PEA and PPA. | |
2.3 Preparation of PA6/antioxidant composites
PA6 was dehydrated in a vacuum oven at 80 °C for 24 hours. Subsequently, 1 wt% of antioxidant (PEA or PPA) was added to the PA6 and mixed uniformly. The mixture was extruded in a miniature twin-screw extruder (LHFD1-130718) at 210–240 °C with a rotation speed of 240 rpm under melt blending extrusion. The resulting pellets were dried in a vacuum oven at 80 °C for 24 h and molded at 250 °C using an injection molding machine (CJ80M3V). The obtained samples were denoted as PA6, PA6/PEA, and PA6/PPA. Each sample group was divided into 6 portions and further dried overnight to standardize their thermal history. Subsequently, the samples were aged in an oven at 150 °C for a duration of 0, 1, 2, 4, 8, and 12 days.
2.4 Characterization
The chemical structures of the synthesized samples were characterized by Fourier Infrared Spectroscopy (FTIR), Nuclear Magnetic Resonance Hydrogen Spectroscopy (1H NMR), Carbon Spectroscopy (13C NMR), and Thermogravimetric Analyzer (TG). FTIR spectra were recorded on the ATR scanning surface using a Nicolet iS50 FTIR spectrophotometer (Thermo Fisher Scientific, USA). 1H NMR and 13C MNR were tested on a Bruker Avance NEO 400 spectrometer (Bruker, Rheinstetten, Germany) with deuterated chloroform (Chloroform-d) as solvent. Thermogravimetric tests were performed on a thermogravimetric analyzer (TGA55, TA Instruments, USA) with a nitrogen atmosphere. The thermal cracking products of PA6 and stabilized PA6 were observed by thermal cracking gas chromatography (7890B-5977A, Agilent Technologies) and a thermogravimetric-infrared (TG-IR) system. The free radical scavenging rate was characterized by a dual-beam ultraviolet spectrometer (UV6300, China). The ethanol solutions were prepared with a DPPH concentration of 0.2 mmol l−1 and different concentration ratios of hindered phenol to DPPH, and the absorbance was measured by scanning the solution at 516 nm for 90 min. The absorbance was calculated according to eqn (1).| |
 | (1) |
where D is the free radical scavenging rate, Int represents spectrum intensity, and AO represents antioxidant. The tensile tests were performed by a microcomputer-controlled electronic mechanical testing machine (SNAS, China). Notched impact strength tests were performed by a mechanical testing machine (ZBC8400-B, China). Each data point is the average of four samples. The melt crystallization behavior of PA6 and stabilized PA6 was carried out by the differential scanning calorimetry (DSC, TA instruments, America). The structure of the fracture surface was characterized by a scanning electron microscope (SEM, QUANTA250FEG, FEI Company, America) equipped with an energy spectrometer.
2.5 Calculations of density functional theory (DFT)
The DFT calculations were performed on a Gaussian 16 platform. Initial structural and frequency optimizations of the material were conducted at the B3LYP/6-31g(d) level, preceding enthalpy calculations. The bond dissociation enthalpy (BDE) of the O–H bond in the parent phenol was determined by the enthalpy difference between the phenoxy radical and hydrogen atom and the initial phenol molecule (eqn (2)).21| | |
BDE(O−H) = E(PhO˙) + E(H˙) − E(PhOH)
| (2) |
3 Results and discussion
3.1 Characterization of antioxidants
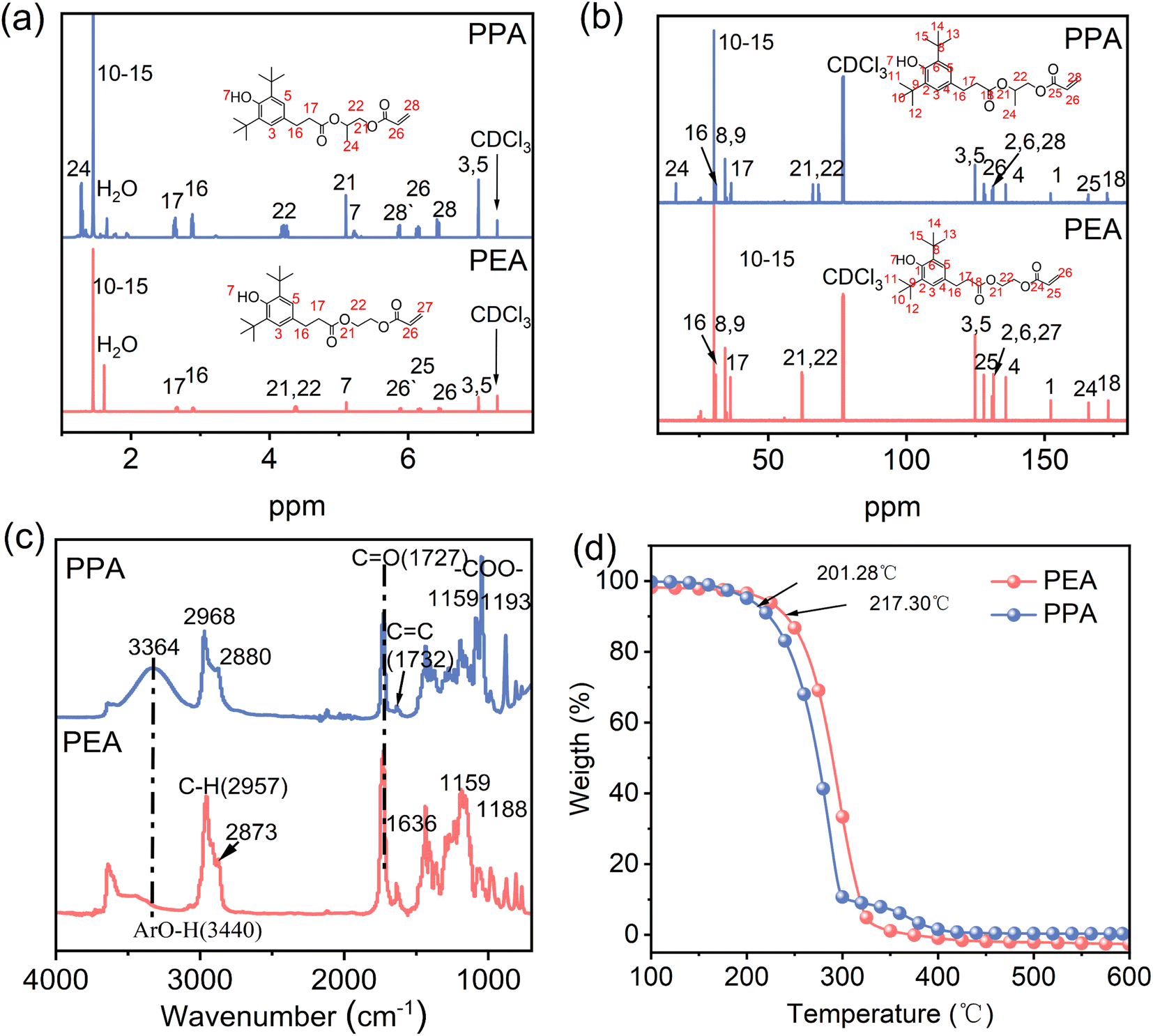
The chemical structure of the synthesized two hindered phenols (PEA, PPA) is confirmed by 1H NMR, 13C NMR, and FTIR patterns. For 1H NMR spectra of PEA, the peak at 6.43–6.48/5.13–6.20/5.87–5.90 ppm and 4.35–4.38/4.38–4.41 ppm can be attributed to the hydrogen on the carbon–carbon double bond in the olefin and the hydrogen between the ester groups, respectively (Fig. 2a). For 13C NMR spectra of PEA, the peak at 62.11/62.30 ppm and 127.98/131.48 ppm can be attributed to carbon between ester groups and carbon on double bond (Fig. 2b). For FTIR of PEA, the peaks at 1732 and 1636 cm−1 correspond to the telescopic vibration peak of the carbonyl group in the ester group and the telescopic vibration peak of the carbon–carbon double bond in olefin, respectively. The peaks at 2968 and 2880 cm−1 are the telescopic vibration peaks of the hydrocarbon bond (Fig. 2c). Similarly, the corresponding atom shifts and FTIR peak of PPA can be corroborated in the 1H NMR, 13C NMR, and FTIR spectra (Fig. 2a–c). These results confirm that the HEA and HPA have been introduced into the molecular structure of hindered phenols. Notably, PPA differs from PEA by the presence of an extra methyl group, and the atomic shifts of 1H NMR (1.23–1.28 ppm) and 13C NMR (16.52 ppm) spectra serve as evidence supporting this structure (Fig. 2a and b). The thermal stability of PEA and PPA is further estimated by TG, as shown in Fig. 2d. The initial and maximum decomposition temperatures of PEA are 217.30 °C and 296.40 °C, respectively (Fig. S1, ESI†). The initial and maximum decomposition temperatures of PPA are 288 °C and 296 °C, respectively (Fig. S1, ESI†). The results exhibit that the methyl group in the PPA leads to a slightly lower degradation temperature of PPA than that of PEA. Nevertheless, the introduction of phenolic hydroxyl para-long chains increased the degradation temperature of hindered phenols to some extent compared to monomeric AO (Fig. S2 and Table S1, ESI†). In addition, both antioxidants are classified as the one-step destructive type, undergoing a single degradation process. (Fig. S3, ESI†) presents the TG analysis of PEA and PPA within the temperature range of 25–200 °C. This plot does not exhibit a similar pattern of water peaks, suggesting that the water content in the antioxidants PEA and PPA is negligible.22
 |
| | Fig. 2 (a) 1H NMR spectra, (b) 13C NMR spectra, (c) FT-IR curves, and (d) TG curves of the PEA and PPA. | |
3.2 DPPH radical scavenging analysis of antioxidants
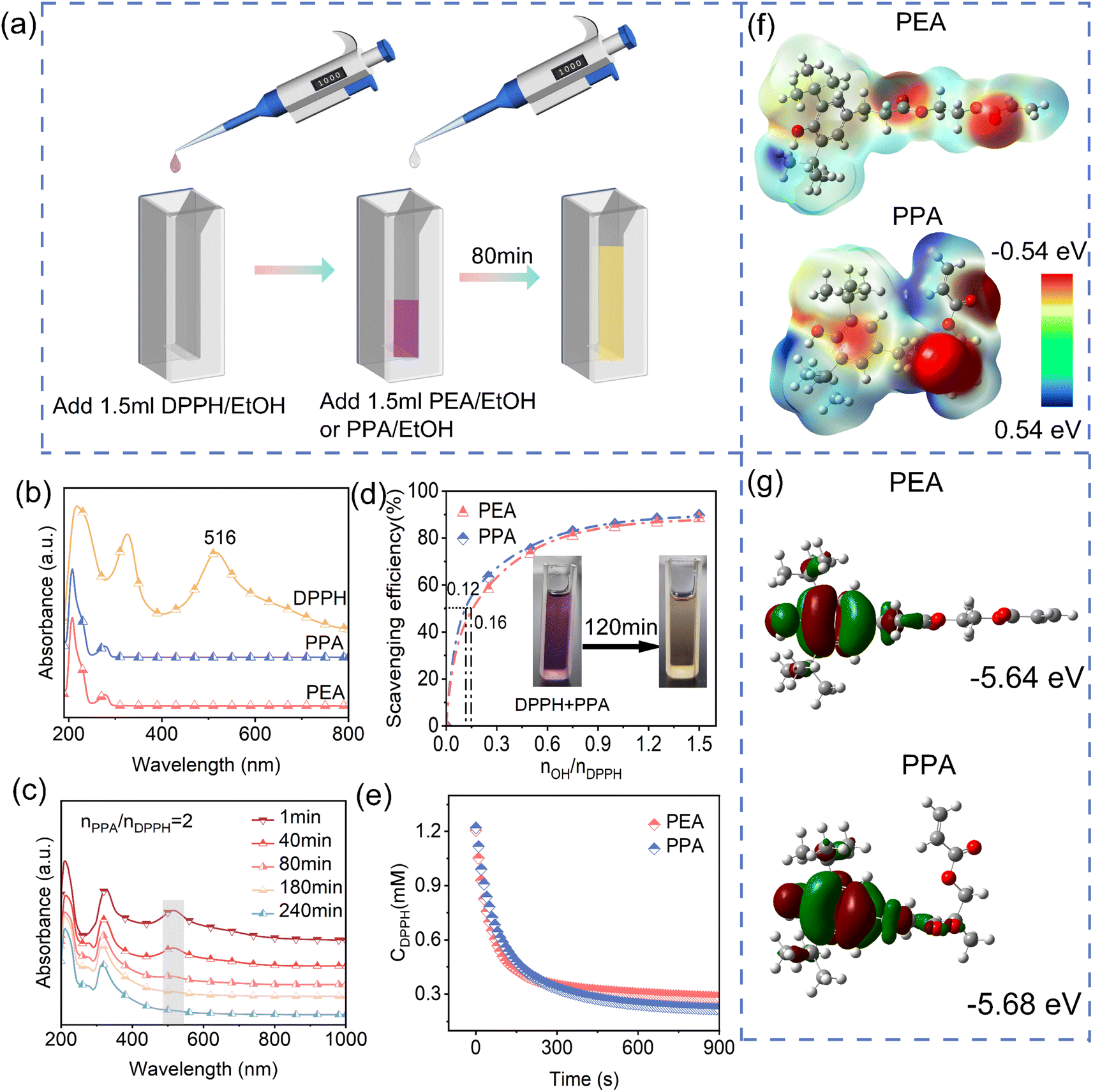
To evaluate the potential of PEA and PPA as antioxidants, free radical scavenging experiments were performed (Fig. 3a; detailed experiment process; please see characterization section). The DPPH/EtOH solution has a distinct absorbance peak at 516 nm, while the ethanol solution of the antioxidant has no distinct absorbance peak there (Fig. 3b), so the free radical scavenging rate can be calculated by testing the absorbance of the mixed solution at 516 nm.23 Taking PPA as an example, the absorbance peak at 516 nm gradually disappeared with the extension of the reaction time between the antioxidant and DPPH (Fig. 3c and Table S2, ESI†), virtually vanishing after 240 minutes, which indicates the completion of DPPH radical scavenging. The antioxidant efficiency of PEA and PPA as a function of concentration is shown in Fig. 3d and Table S3, ESI.† The free radical scavenging rate increased gradually with increasing concentrations of antioxidants. More importantly, the molar mass of the antioxidant required to scavenge 50% DPPH free radicals (EC50) is PEA > PPA, and the DPPH scavenging rate is PPA > PEA, which indicates both PPA and PEA have excellent free radical trapping capacity. Hence, both PPA and PEA can be used as antioxidants to trap free radicals generated by the degradation of polymer compounds, thereby terminating chain reactions. When the concentration of antioxidants was much greater than that of free radicals, the concentration of DPPH radicals decreased rapidly with time (Fig. 3e). The kinetic fitting parameters for the reaction of DPPH radicals with antioxidants show that the reaction of PEA or PPA with DPPH radicals is a secondary kinetic reaction.24 By comparing the reaction rates, it was found that the reaction rate constants for scavenging DPPH radicals are PPA > PEA (Table S4, ESI†), indicating that PPA with a methyl group has a faster radical scavenging rate.
 |
| | Fig. 3 (a) Experimental procedure of free radical scavenging. UV absorption spectra of (b) antioxidants and DPPH radicals and (c) PPA with DPPH radical solution at different reaction times. (d) Free radical scavenging rate of antioxidants at different concentrations, (e) DPPH radical concentration as a function of the reaction same, (f) electrostatic formulae and (g) HOMO enthalpy levels of PEA and PPA. | |
To further understand the structural origin of PPA's efficient free radical scavenging or trapping ability, the BDE of the phenolic hydroxyl group in PPA and PEA was calculated (please see experimental sections, eqn (2)).25 As shown in (ESI Table S5†), the O–H BDE of the PEA and PPA are 307.23 and 301.95 kJ mol−1, respectively. This result reveals that the introduction of a methyl group into PPA can reduce the BDE of the phenolic hydroxyl group. This phenomenon is attributable to the methyl group acting as an electron-donating substituent, which enhances the electron cloud density around the phenolic hydroxyl group. This increase in electron density facilitates a more rapid rate of hydrogen atom removal and diminishes the enthalpy barrier necessary for the reaction.14,26,27 Consequently, PPA exhibits a greater free radical scavenging capacity compared to PEA. The HOMO–LUMO energy gap (ΔE) represents the electron transfer capacity (Table S6, ESI†), a smaller enthalpy gap signifies a stronger electron transfer capacity, facilitating the transition of electrons from the HOMO to the LUMO. Given that the ΔE of PPA is smaller than that of PEA, PPA facilitates electron transport more effectively, thereby accelerating the reaction rate for capturing free radicals and enhancing antioxidant capacity.28–30
3.3 Long-term oxidative stability of PA6/antioxidant composites
To manifest the antioxidant activity of PPA and PEA in practice, the PA6/PPA and PA6/PEA composites were prepared, and the corresponding stress–strain curve were investigated at different aging times (Fig. 4a–f). The corresponding yield points of PA6, PA6/PEA, and PA6/PPA are shown in Table S7 (ESI).† The fracture behavior of PA6 transitioned from ductile to brittle after just 2 days of aging (Fig. 4c), while the PA6/PEA and PA6/PPA composites exhibited similar transitions later in the aging process, occurring on the 8th day (Figure. 4e) and 12th day (Fig. 4f), respectively. As shown in (Fig. 4g–i), the tensile strength retention rate, elongation at break and notched impact strength retention rate of PA6/PPA and PA6/PEA before aging are slightly higher than that of pure PA6. After one day of aging treatment, the PA6, PA6/PPA, and PA6/PEA composites exhibit an increase in tensile strength (Fig. 4g). This enhancement is attributed to the predominant cross-linking reactions among the PA6 molecular chains during the early stages of aging.31 As aging time extended, the tensile strength of PA6 began to deteriorate on 2nd day of the aging process and was only 74% of their original strength, while PA6/PPA and PA6/PEA composites remained relatively stable. It was not until the 12th day of aging that a sharp decline in tensile strength was observed for both composites. These outcomes suggest that both PPA and PEA are effective in enhancing the thermal oxidation resistance of PA6 during the aging process. Notably, the tensile strength of the PA6/PEA and PA6/PPA composites after 12 days of aging was 70% and 88% of their original strength (Fig. 4g), respectively, indicating that PPA has superior resistance to thermo-oxidative aging compared to PEA. In addition, the PA6/1010 composite exhibited a substantial degradation in tensile strength after only 4th day of thermo-oxidative aging and was only 40% of their original strength, markedly underperforming in comparison to the PA6/PPA and PA6/PEA composites. As shown in Fig. 4h, the elongation at break of PA6/PEA and PA6/PPA before aging is higher than that of PA6 and PA6/1010. As the aging process progressed, the samples changed from ductile to brittle fracture, resulting in a decrease in the elongation at break. PA6 exhibited a marked reduction in elongation at break on the first day following aging. In contrast, PA6/PEA and PA6/PPA composites demonstrated a significant decrease in the elongation at break on the 8th day of aging, while PA6/1010 composites showed this reduction on the 4th day. This observation indicates that the presence of PEA and PPA on the PA6 molecular chain exerts a protective effect, which is superior to that of 1010. In terms of notched impact strength retention rate (Fig. 4i), the PA6 and PA6/1010 composites experienced a significant reduction in strength on the 1st and 4th days after aging, decreasing to 35% and 30% of their original strength, respectively. In contrast, the strength of the PA6/PPA and PA6/PEA composites showed a gradual decline over the aging period. Until the 12th day of aging, the notched impact strength of PEA and PPA was about 32% and 54% of their original strength. The fracture morphology of the samples demonstrated that PA6 exhibited brittle fracture characteristics (radial shape) as early as the 2nd day of aging,32 while the composite materials did not show brittle fracture before the 1st day of aging (Fig. 4j). These results further demonstrate the excellent antioxidant properties of PPA and PEA. Similarly, the PA6/PPA composite exhibited significantly superior performance compared to the PA6/PEA composite after the 12th day of aging, which indicates the PPA has superior antioxidant properties compared to PEA. This is consistent with the previously mentioned effect of the methyl groups in the PPA molecular chain on the electron density around the phenolic hydroxyl groups, thereby enhancing the free radical scavenging ability of PPA.
 |
| | Fig. 4 Stress–strain curves of different samples in different aging days: (a) 0 day, (b) 1 day, (c) 2 days, (d) 4 days, (e) 8 days, (f) 12 days. (g) Tensile strength retention rate. (h) Elongation at break. (i) Notched impact strength retention rate. (j) Notched impact morphology at different aging days. | |
3.4 Crystallographic, thermal cracking, anti-migration, and carbonyl index analysis of PA6 and PA6/antioxidant composites
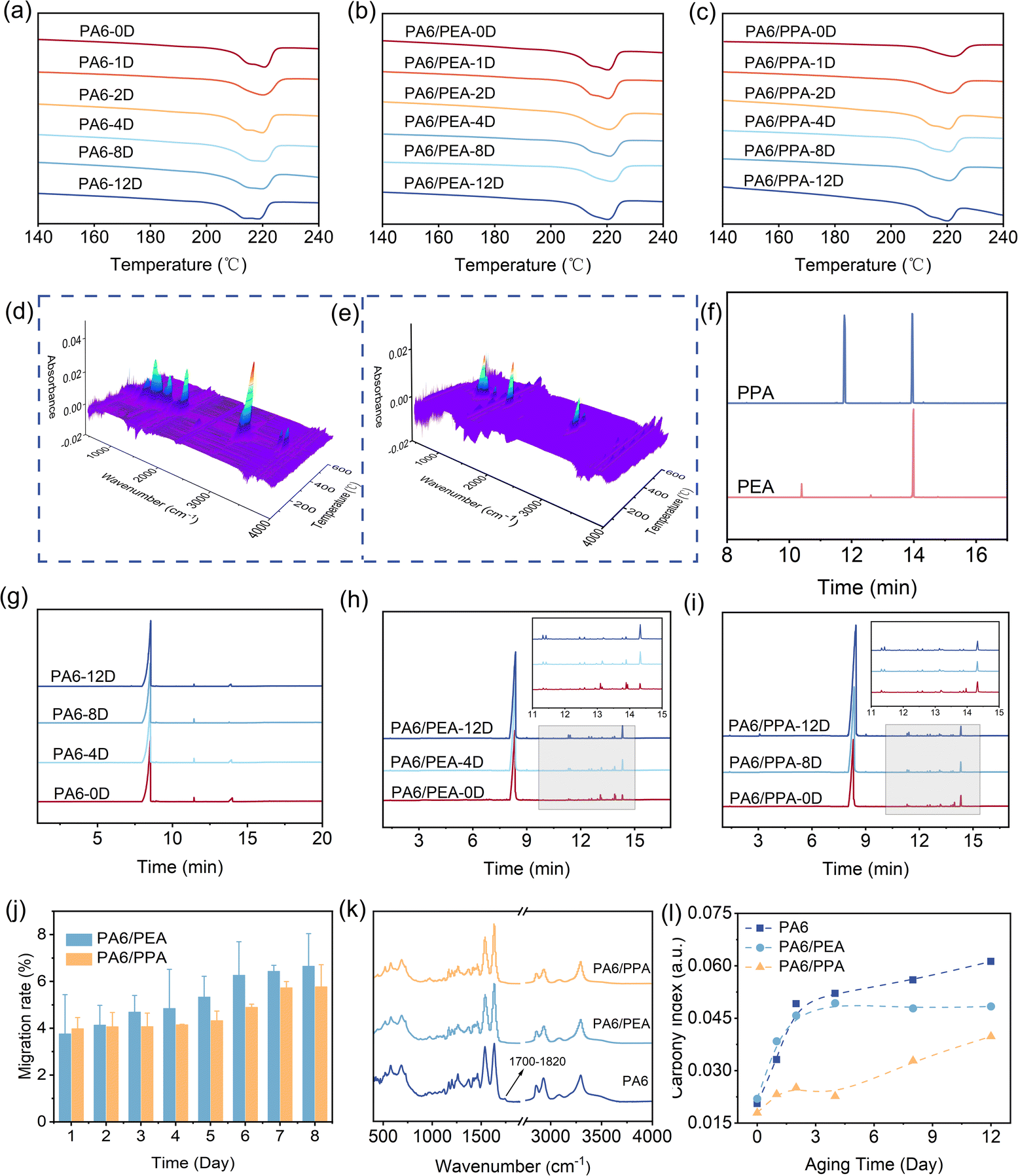
The degradation of PA6 could lead to changes in its degree of crystallinity and melting temperature.31 Therefore, the DSC is employed to further analyze the thermo-oxidative aging behavior of the samples (ESI Tables S11 and 12†). As shown in (Fig. 5a–c), as the aging process progresses, all samples exhibit two all melting peaks at 214 °C and 220 °C, corresponding to γ-crystal and α-crystal of PA6, respectively. The results reveal that the breakage of PA6 molecular chains has disrupted the hydrogen bonding structure between PA6 molecules, leading to the formation of unstable γ-crystals. PA6, PA6/PEA, PA6/PPA all with the extension of aging time, the three α crystal types gradually increased, the stabilized PA6 α crystal type of the growth rate is greater than PA6, and α is more stable than γ (ESI Table S11†).33,34 The observed phenomenon could be attributed to the efficacious radical scavenging capabilities of PEA and PPA, which suppress the fragmentation of molecular chains within PA6. This suppression allows PA6 molecular chains to have enough time and is liable to form a stable α-crystalline structure.
 |
| | Fig. 5 DSC melt curves for different aging days of (a) PA6, (b) PA6/PEA, (c) PA6/PPA. TG-IR couplings: (d) PEA, (e) PPA. Chromatograms: (f) PEA and PPA, (g) PA6, (h) PA6/PEA, (i) PA6/PPA. (j) PEA and PPA mobility with time. (k) FT-IR curves of PA6 and stabilized PA6 aged for 12 days. (l) PA6 and stabilized PA6 carbonyl index. | |
To further assess the thermal stability of PEA and PPA during the thermo-oxidative aging process, their cleavage behavior was characterized by TG-IR analysis and cleavage mass spectrometry. As shown in Fig. 5d and e, the compounds PEA and PPA exhibit a maximum decomposition temperature of 296.40 °C and 287.28 °C, respectively, indicating that their thermal stability meets the processing and service requirements for PA6. The cleavage mass spectrometry shows the emergence of fragments for PEA and PPA at 10.395 min and 11.767 min, respectively, demonstrating that PEA is more volatile than PPA (Fig. 5f and ESI Tables S13–S17†). This characteristic enables PPA to retain a greater amount of its effective components during the thermo-oxidative aging process. In addition, the presence of long-chain linkages in the hydroxyl pairs in the PPA fragments enhanced its antioxidant activity to some extent.5 By comparing the percentage of antioxidants in PA6 for different aging times (Fig. 5g–i and ESI Table S18†), it was also found that PPA had a higher percentage of antioxidants in PA6. Furthermore, the mobility experiment is carried out to investigate the stability of PEA and PPA in PA6,35 detailed experiment method, please see ESI Note S4.† As shown in Fig. 5j, the initial mobility of PPA in PA6 is slightly higher than that of PPA, and the initial mobility is calculated from the standard curve (Fig. S4 and Table S19, ESI†). However, as the aging time increases, the mobility rate of PEA surpasses that of PPA. The reason for this result is that PPA has a solubility parameter more closely aligned with that of PA6, resulting in better compatibility between PPA and PA6 (ESI Note S2 and Tables S8–10†).36 These outcomes collectively substantiate the fact that PPA possesses superior thermo-oxidative aging resistance in PA6 compared to PEA.
The carboxyl index of PA6 during the aging process can be utilized to further assess the thermo-oxidative aging performance of the sample. As shown in Fig. 5k and l, the pure PA6 shows a new characteristic absorbance peak in the range of 1650–1800 cm−1 that intensifies with aging time, corresponding to the characteristic signal peak of carbonyls.37 This observation can be attributed to the molecular restructuring within the PA6 during aging, where carbon and oxygen atoms within the molecular chain engage in single bond formation, resulting in the formation of complex functional groups, such as esters, aldehydes, and ketones.8 In contrast, the absence of a distinct carbonyl peak in the FT-IR spectrum of PA6/PEA and PA6/PPA composites indicates that the incorporation of PEA and PPA effectively mitigated the thermo-oxidative aging of PA6 (Fig. 5k). This phenomenon can be attributed to the previously demonstrated free radical scavenging capabilities of PEA and PPA, which capture reactive free radicals and generate stable free radicals, thus inhibiting the thermo-oxidative aging effect of PA6. In addition, compared with the carbonyl indices at the 12th day of PA6, PA6/PEA and PA6/PPA composites PA6/PPA exhibit the lowest carbonyl index (Fig. 5l), which further underscores that PPA has superior thermo-oxidative aging resistance for PA6 compared to PEA.
3.5 Antioxidation mechanism
The antioxidant mechanisms of PPA and PEA were further analyzed, as shown in Fig. 6. There is a certain interaction between the amide group in PA6 and the ester group of the antioxidant38 enhancing the compatibility of PA6/PEA and PA6/PPA composites. In addition, the PEA and PPA molecules contain two active groups, phenol hydroxyl and acrylate, which have a unique bifunctional stabilization mechanism. The acrylate group can capture the free radicals generated by the PA6, and the hydrogen in the phenol hydroxyl group can form an intramolecular hydrogen bond with the oxygen in the acrylate group, which effectively prevents the thermal degradation of PA6.20,39 For antioxidant PPA, the introduction of a methyl group can change the stable state structure of the molecule, resulting in an increase in the electron cloud density around the benzene ring and a decrease in the ΔE and dissociation enthalpy. This change leads to the fact that PPA can provide a faster dehydrogenation ability and a higher free radical scavenging rate compared to PEA, thereby enhancing the antioxidant activity of PPA.27 In addition, compared with PEA, the PPA in the PA6 matrix exhibits excellent stability of PPA during the aging process, as evidenced by its lower volatility and mobility. These factors collectively determine that the antioxidant activity of PPA is higher than that of PEA.
 |
| | Fig. 6 Mechanism of resistance to thermal and oxygen aging of PEA and PPA. | |
3.6 Conclusion
In summary, this work elucidates the effect of electron-donating groups (methyl groups) on the antioxidant performance of two hindered phenolic antioxidants (PEA and PPA) containing acrylate groups. The methyl group in the PPA near the ester group can increase the electron cloud density of the hindered phenol-benzene ring and decrease the ΔE and dissociation enthalpy, which leads to the fact that the antioxidant PPA has higher free radical scavenging ability and a faster scavenging rate. Additionally, in contrast to antioxidant PEA, the PPA in the PA6 matrix exhibits low volatility and mobility, allowing it to maintain a higher content of antioxidants during the aging process, thereby further enhancing the antioxidant properties of PA6. These facts lead to that PA6/PPA composite maintains 88% of its original tensile strength after 12 days of continuous aging, whereas PA6/PEA composite only retained 70%. Notably, the PA6/1010 composite (1010 is a commercial antioxidant) retained only 30% of its origin tensile strength at the 12th day of aging. This encouraging find attests to the exceptional application potential of the PPA antioxidant.
Data availability
Data will be made available on request.
Author contributions
Bingxue Guo: data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. Jinfen Lou: data curation, formal analysis, software, writing – review & editing. Tingting Kuang: data curation, methodology, supervision. Yanping Wang: data curation, formal analysis, investigation. Kai Zhang: data curation, project administration, resources. Jun Qin: data curation, resources, software. Min He: funding acquisition, project administration, supervision, writing – review & editing. Yufei Liu: data curation, project administration, writing – review & editing. Yongji Gong: data curation, methodology, project administration, writing – review & editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52163009); the Guizhou Provincial Science and Technology Program Guizhou Science and Platform Talents (QKHPTRC-CXTD [2023] 012); the Guizhou Provincial Science and Technology Program Project [2023] General 009.
References
- J. J. Tian, X. Liu, L. Ye, Z. Zhang, E. C. Quinn, C. Shi, L. J. Broadbelt, T. J. Marks and E. Y. X Chen, Angew Chem. Int. Ed. Engl., 2024, 63, e202320214 CrossRef CAS.
- G. L. Sullivan, J. Delgado-Gallardo, T. M. Watson and S. Sarp, Water Res., 2021, 196, 117033 CrossRef CAS PubMed.
- O. M. McIntee, N. Sreedhar, B. C. Welch, V. M. Bright, A. Roy, M. Paul and A. R. Greenberg, Polymer, 2024, 308, 127350 CrossRef CAS.
- W. Zhao, J. He, P. Yu, X. Jiang and L. Zhang, Polym. Degrad. Stab., 2023, 207, 110223 CrossRef CAS.
- T. Kajiyama and Y. Ohkatsu, Polym. Degrad. Stab., 2001, 3, 445–452 CrossRef.
- Y. Zhong, K. Liu, W. Chen and W. Lu, Polymer, 2022, 262, 125495 CrossRef CAS.
- T. M. Wu, C. J. Chen, Y. L. Lai and T. W. Shyr, Advanced Materials Research, 2014, 834, 156–159 Search PubMed.
- A. Rjeb, L. Tajounte, M. C. El Idrissi, S. Letarte, A. Adnot, D. Roy, Y. Claire, A. Périchaud and J. Kaloustian, J. Appl. Polym. Sci., 2000, 77, 1742–1748 CrossRef CAS.
- M. C. Foti, J. Pharm. Pharmacol., 2007, 29, 1673–1685 CrossRef PubMed.
- J. Mayer, E. Metzsch-Zilligen and R. Pfaendner, Polym. Degrad. Stab., 2022, 200, 109954 CrossRef CAS.
- X. P. Wang, F. H. Yao and J. Chen, J. Appl. Polym. Sci., 2023, 140(30), e54085 CrossRef CAS.
- O. Okamba-Diogo, F. Fernagut, J. Guilment, F. Pery, B. Fayolle and E. Richaud, Polym. Degrad. Stab., 2020, 179, 109206 CrossRef CAS.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS PubMed.
- A. P. Vafiadis and E. G. Bakalbassis, Chem. Phys., 2005, 316, 195–204 CrossRef CAS.
- J. Zhang, Q. Ke, J. Bai and M. Yang, Polym. Degrad. Stab., 2023, 218, 110550 CrossRef CAS.
- W. Wu, H. Li, S. Yang, X. Lai, H. Fan and X. Zeng, Sci. Res. Essays, 2018, 50, 372–387 CAS.
- H. Xie, H. Li, X. Lai, W. Wu and X. Zeng, Macromol. Mater. Eng., 2015, 300, 893–900 CrossRef CAS.
- L. Zhang, H. Li, X. Lai, W. Wu and X. Zeng, Mater. Lett., 2018, 210, 239–242 CrossRef CAS.
- C. Li, P. Sun, H. Yu, N. Zhang and J. Wang, RSC Adv., 2017, 7, 1869–1876 RSC.
- S. I. Achigo, M. Sasaki, Y. Takahashi, F. Kojima, T. Takada and T. Okita, Polym. Degrad. Stab., 1988, 22, 63–77 CrossRef.
- G. Brigati, M. Lucarini, V. Mugnaini and G. F. Pedulli, J. Org. Chem., 2002, 67, 4828–4832 CrossRef CAS.
- M. R. Caruso, M. M. Calvino, P. Šiler, L. Cába, S. Milioto, L. Lisuzzo, G. Lazzara and G. Cavallaro, Small, 2025, 21, 2405215 CrossRef CAS.
- A. C. Akinmoladun, E. O. Ibukun and I. A. Dan-Ologe, Sci. Res. Essays, 2007, 2, 191–194 Search PubMed.
- R. Guitard, V. Nardello-Rataj and J. M. Aubry, Int. J. Mol. Sci., 2016, 17, 1220 CrossRef PubMed.
- J. Wu, Y. Zhou and X. Xu, Int. J. Quantum Chem., 2015, 115, 1021–1031 CrossRef CAS.
- J. Chen, J. Yang, L. Ma, J. Li, N. Shahzad and C. K. Kim, Sci. Rep., 2020, 10, 2611 CrossRef CAS.
- X. C. Weng and Y. Huang, Grasas Aceites, 2014, 65, e51 Search PubMed.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS PubMed.
- Y. Xiao, F. Fu, Y. Wei, S. Shi and Y. Shan, Antioxidants, 2022, 11, 1014 CrossRef CAS PubMed.
- M. Rouhani, Comput. Theor. Chem., 2021, 1195, 113096 CrossRef CAS.
- Y. Shu, L. Ye and T. Yang, J. Appl. Polym. Sci., 2008, 110, 945–957 CrossRef CAS.
- J. Lou, M. He, Y. Liu, K. Zhang, L. He, S. Qin and J. Yu, Polymer, 2024, 290, 126553 CrossRef CAS.
- R. Li, K. Shi, L. Ye and G. Li, Composites, Part B, 2019, 162, 11–20 CrossRef CAS.
- Y. Zhang, Y. Zhang, S. Liu, A. Huang, Z. Chi, J. Xu and J. Economy, J. Appl. Polym. Sci., 2011, 120, 1885–1891 CrossRef CAS.
- K. Shi, L. Ye and G. Li, J. Therm. Anal. Calorim., 2015, 119, 1747–1757 CrossRef CAS.
- Z. Kulinski, E. Piorkowska, K. Gadzinowska and M. Stasiak, Biomacromolecules, 2006, 7, 2128–2135 CrossRef CAS.
- C. Li, J. Wang, M. Ning and H. Zhang, J. Appl. Polym. Sci., 2012, 124, 4127–4135 CrossRef CAS.
- L. Xia, B. Shentu and Z. Weng, Polym. Eng. Sci., 2014, 54, 2197–2206 CrossRef CAS.
- S. Yachigo, M. Sasaki, K. Ida, K. Inoue, S. Tanaka, H. Yoshiaki, F. Emiko and Y. Kazunori, Polym. Degrad. Stab., 1993, 39, 329–343 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Jun Qin
b,
Jun Qin