
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple method for Mn residue utilization for efficient peroxymonosulfate activation: enrichment of metal sites and oxygen vacancies via alkali-reduction treatment†
Jujiao Zhao a,
Xianjuan Xua,
Qiao Zhanga,
Feilan Qia,
Haoxuan Weib,
Quanfeng Wang*b,
Youzhou Hea,
Xiaoxia Baia,
Shun Guana,
Ming Zhuc and
Xingmin Wang*a
a,
Xianjuan Xua,
Qiao Zhanga,
Feilan Qia,
Haoxuan Weib,
Quanfeng Wang*b,
Youzhou Hea,
Xiaoxia Baia,
Shun Guana,
Ming Zhuc and
Xingmin Wang*a
aCollege of Environment and Resources, Chongqing Technology and Business University, Chongqing, 400067, PR China. E-mail: wxm0826@ctbu.edu.cn; Tel: +86-18046504243
bSchool of Civil and Hydraulic Engineering, Chongqing University of Science and Technology, Chongqing, 401331, PR China. E-mail: 2021020@cqust.edu.cn; Tel: +86-13527536325
cInstitute for Smart City of Chongqing University in Liyang, Chongqing University, Jiangsu 213300, PR China
First published on 2nd July 2025
Abstract
Transforming Mn residues into catalysts for activating peroxymonosulfate (PMS) for the degradation of organic pollutants is a promising method that could not only mitigate environmental pollution caused by Mn residues, but could also provide inexpensive catalysts for PMS-based wastewater treatment. Herein, a two-step method involving alkali-reduction treatment is proposed for improving the catalytic activity of Mn residues. The proposed alkali treatment successfully reduced the content of Si from 261.0 mg g−1 to 80.5 mg g−1, leading to relatively increased contents of Fe and Mn. Moreover, alkaline NaBH4 reduction treatment introduced more oxygen vacancies, further enhancing the performance. The sample treated with the two-step method exhibited rhodamine B (RhB) degradation kinetics of 0.034 min−1, which was 2.43 times higher than that of the control sample. Quenching experiments and electron paramagnetic resonance demonstrated the vital role of 1O2 in the degradation of RhB, and the mechanism was investigated. Furthermore, 18 types of degradation intermediates were detected, and the possible degradation pathway for RhB was proposed. Through employing a toxicity estimate software tool, it is indicated that RhB toxicity continuously decreases during RhB degradation. The findings of this work could shed light on the reutilization of Mn residues and the development of inexpensive catalysts for PMS-based wastewater treatment.
1. Introduction
Manganese (Mn) residues, as typical by-products of various industrial activities, such as mining and metallurgical processes, present substantial environmental challenges.1,2 The accumulation of these residues can lead to the release of heavy metals and other pollutants into the environment, contaminating soil, surface water, and groundwater.3,4 Without appropriate methods for disposing of Mn residues, serious risks could be posed to ecosystems and human health.5 Particularly, in China, the stock of Mn residues now exceeds 150 million tons and is escalating at an approximate rate of 10 million tons annually, thereby necessitating the urgent need for the effective management of Mn residues.6“Treating waste with waste” is a desired approach to solve environmental problems.7,8 Mn residues still contain a certain amount of Fe and Mn, both of which are regarded as chemical active species in advanced oxidation processes (AOPs), which are widely used for removing organic pollutants from wastewater.9–12
As one of the AOPs, peroxymonosulfate (PMS) activation has attracted much attention. With suitable catalysts, PMS can produce multiple types of reactive oxygen species (ROS), including sulfate radicals (SO4˙−), hydroxyl radicals (˙OH), and singlet oxygen (1O2), indicating its good capability in degrading a wide range of organic pollutants, from pharmaceuticals to dyes and pesticides.13–19 Many efforts have been dedicated to the development of innovative catalysts with outstanding performance for activating PMS.20 However, the widespread application of PMS activation is still limited by the high cost of catalysts, and most previous studies have either struggled to scale up their proposed processes or utilized intricate precursors for synthesis.21,22
The utilization of Mn residues as catalysts for PMS activation is highly desired since this approach converts waste materials into valuable resources for environmental remediation, thus aligning with sustainability principles.23 Several studies have shown that Mn residues can effectively activate PMS for pollutant degradation, leveraging the catalytic properties from the manganese and iron present in the residues.24 In this field, He et al. and many other researchers have conducted some remarkable studies.3,25–29 For instance, in one study a modified manganese slag/PMS system could remove 100% of diethyl dithiocarbamate in 10 min.30 Also, washing manganese slag by both Na2CO3 and HNO3 under optimized conditions led to a levofloxacin removal efficiency of 82.6% within 60 min by activating PMS.31 In another study, after calcinating Mn residues in air and subsequently treating the sample with H2SO4, the obtained catalysts could degrade 85% of tetrachlorophenol within 50 min.32 Nevertheless, most of these studies employed an acid-washing process to clean the surface and expose more active sites by removing impurities.7 However, acid washing will result in the leaching of Mn and Fe, which are crucial active sites in the catalyst.33,34 Additionally, the acid-washing process can be environmentally detrimental due to the generation of acidic waste streams that require further treatment.
To overcome the limitations associated with acid washing, alkali washing has emerged as a promising alternative process to enhance the activity of Mn residues.35 Alkali washing can remove inert composites such as SiO2, which has been revealed to be the main component in many Mn residues, while simultaneously preventing the leaching of Fe and Mn.4,35
Additionally, several studies have reported that 1O2 could be generated by PMS activated with Mn residues-derived catalysts.28,29 1O2 is considered to be a highly active agent for attacking organic pollutants with an electrophilic nature, contributing to the degradation of various organic pollutants.16,36,37 The generation of 1O2 was speculated to be associated with the existence of oxygen vacancies on the catalysts,16,38,39 thereby making the enhancement of oxygen-vacancy formation a promising method to further optimize the performance of Mn residues-derived catalysts.28,40
Herein, Mn residues were treated by alkali treatment and subsequently treated by alkaline NaBH4 solution to enhance the formation of oxygen vacancies,40,41 and the obtained catalysts were then used to active PMS for removing rhodamine B (RhB), a typical organic pollutant that is widely used in the world but is harmful to human health.42,43 Both alkali treatment and alkaline NaBH4 reduction treatment enhanced the performance of the samples and 1O2 was identified to be the main ROS involved. The catalytic mechanism for the degradation of rhodamine B was investigated and the intermediates during the degradation of RhB were revealed. Besides, toxicity assessment was also performed using the toxicity estimate software tool (T.E.S.T.).
2. Experimental
2.1 Chemicals and materials
Mn residues were obtained from companies in Xiushan, Chongqing. 2,2,6,6-Tetramethyl-4-piperidone (TEMP) and rhodamine B (RhB) were obtained from Aladdin Ltd. (Shanghai, China). Absolute ethanol (C2H5OH, 99.7%), sodium hydroxide (NaOH, >98%), sulfuric acid (H2SO4, 98%), methanol (CH3OH, 99.7%), L-histidine, sodium chloride (NaCl, 98%), sodium borohydride (NaBH4, 98%) and sodium dihydrogen phosphate (NaH2PO4, 98%) were obtained from Chongqing Chuandong Co. These chemicals were used as received without any further purification. The river water was taken from the Chongqing section of the Yangtze River. The lake water was obtained from Cuihu Lake in the campus of Chongqing Technology and Business University.2.2 Preparation of the samples
The collected Mn residues without any treatment were named as MS.In a typical process, MS was first calcined at 350 °C in air for 3 h and then washed by water. The obtained sample was then milled into a powder. Then, 10 g calcined MS powder was dispersed in 100 mL of 4 mol per L NaOH solution for 1 h at a temperature of 60 °C. After that, the sample was washed by water and further calcined at 500 °C for 3 h with increasing the temperature at a rate of 5 °C min−1. The alkali-treated and calcined sample was denoted as ACMS.
Another sample was prepared and treated by the similar calcination process but without the alkali treatment for contrast group and this was denoted as CMS.
The ACMS was further treated by alkaline NaBH4 solution to improve its performance. In a typical process, 1 g ACMS sample was immersed in 500 mL 200 mmol per L alkaline NaBH4 solution (pH 11, room temperature) for 150 s and filtrated immediately. The obtained sample was then dried at 60 °C and denoted as ACMS-R.
CMS was also treated by the same process to obtain another sample, which was denoted as CMS-R.
2.3 Characterization methods
The morphology of the samples was observed using a ZEISS Sigma 300 scanning electron microscopy (SEM) system. Transmission electron microscopy (TEM) images were collected on a Talos F200S instrument. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping was performed using the same TEM equipment. X-ray diffraction (XRD) patterns were obtained on a Rigaku Ultima IV (Japan) at a scan rate of 2° min−1. X-ray photoelectron spectroscopy (XPS) analysis was conducted on an ESCALAB 250Xi spectrometer (USA), using an Al Kα X-ray source (1486.6 eV). N2 adsorption–desorption isotherms were recorded on a Micromeritics ASAP2460 system (USA) and degassing was performed at 200 °C for 8 h. Electron paramagnetic resonance (EPR) measurements were performed using a Bruker EMXmicro-6/1/P/L instrument with TEMP as the spin-trapping agent. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) was performed using a PerkinElmer Optima 5300 DV instrument.2.4 Procedures and analysis
A 250 mL beaker was used as the reactor and a 100 mL solution of 50 mg L−1 RhB was added to the beaker. A magnetic stirrer was employed at a stirring speed of 400 rpm to evenly disperse the catalysts during the process. The temperature was controlled by the heating function of the magnetic stirrer. The pH was adjusted by adding NaOH or H2SO4. The reaction time was recorded and the count started after the addition of PMS. The solution samples for detection were collected and filtered through a 0.22 μm filter. Next, 0.5 mL quenching solution mixed with methanol and L-histidine was added into the sample to terminate the oxidation reaction. The concentration of RhB was detected by UV-vis spectroscopy (UV2365, UNICO, Shanghai, China) at 554 nm. The degradation intermediates of RhB were identified by liquid chromatography-mass spectrometry (LC-MS) (Agilent LC1290-6550-QTOF with a C-18 column, mobile phase: acetonitrile/water (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, v/v), flow rate: 0.3 mL min−1). The total organic carbon (TOC) removal efficiency was revealed using a Shimadzu TOC-L-CPH system. The toxicity estimate software tool (T.E.S.T.) was employed to evaluate the change of toxicity during the degradation of RhB.
:40, v/v), flow rate: 0.3 mL min−1). The total organic carbon (TOC) removal efficiency was revealed using a Shimadzu TOC-L-CPH system. The toxicity estimate software tool (T.E.S.T.) was employed to evaluate the change of toxicity during the degradation of RhB.
3. Results and discussion
3.1 Characterization of the catalysts
The SEM images with different magnifications of CMS, ACMS, and ACMS-R can be seen in Fig. 1. The SEM images of CMS (Fig. 1a and b) show the presence of many cracks with a rugged structure. The larger magnification image shows the morphology consisted of nanoparticles. ACMS, the sample prepared with the alkali-washing step, presented a much similar structure. After the alkali-reduction treatment by NaBH4 solution, the structure of ACMS-R also did not change much. TEM images of ACMS-R were also recorded (Fig. S1†), which further demonstrated that ACMS-R exhibited a nanoparticles structure. | ||
| Fig. 1 (a) and (b) SEM images of CMS; (c) and (d) SEM images of ACMS; (e) and (f) SEM images of ACMS-R. | ||
The EDS elemental mapping provided further insight into the compositional homogeneity of the ACMS-R. A uniform distribution of Mn and Fe could be observed in the ACMS-R sample, as depicted in Fig. 2. Since Mn and Fe were regarded as the active elements for activating PMS, this uniformity could be favorable for the activity of the catalysts.
 | ||
| Fig. 2 (a)–(c) EDS elemental mapping images of ACMS-R. | ||
The phase composition of the samples was revealed by XRD measurements, as shown in Fig. 3a. The preponderant crystalline structures of CMS were SiO2 (JCPDS. 70-3755), Ca3Si2O7 (JCPDS. 22-0539) and CaSO4 (JCPDS. 72-0916). Generally, these substances are regarded as having poor catalytic activity toward PMS activation.32 After alkali treatment, the signals for CaSO4 disappeared in the XRD pattern of ACMS; meanwhile, peaks corresponding to CaFe5O7 (JCPDS. 30-0257) and CaMn(Si2O6) (JCPDS. 74-0336) could be observed. It is worth noting that CaSO4 could react with NaOH, leading to the recrystallization of Ca ions. On the other hand, the intensity of diffraction peaks of the quartz phase materials might then be reduced, resulting in the observation of the peaks corresponding to Fe, Mn-containing substances. However, the XRD pattern of ACMS-R was exactly similar to that of ACMS, indicating the alkali-reduction treatment by NaBH4 solution did not change the crystalline structure of the samples. The N2 adsorption–desorption isotherms of CMS, ACMS, and ACMS-R are depicted in Fig. 3b and the Brunauer–Emmett–Teller (BET) surface area was calculated. CMS, ACMS, and ACMS-R exhibited BET surface areas of 13.5, 15.1, and 18.5 m2 g−1, respectively. These results revealed the increase in the BET surface area after alkali treatment and alkaline NaBH4 solution treatment. A larger BET surface area will increase the contact area for capturing PMS molecules, which could facilitate the degradation efficiency of organic pollutants with PMS activation.
 | ||
| Fig. 3 (a) XRD patterns of CMS, ACMS, and ACMS-R; (b) N2 adsorption–desorption isotherms of CMS, ACMS, and ACMS-R. | ||
Alkali treatment was expected to reduce the content of inactive materials and avoid the leaching of Mn and Fe. To further reveal is this was true, ICP-AES was employed and the contents of Mn, Fe, and Si in MS, CMS and ACMS were investigated, and the results can be seen in Table S1.† MS contained a high concentration of S (1527.9 mg g−1), which was in the form of soluble sulfates, leading to relatively lower contents of the other elements. Significantly, the content of Si decreased from 261.0 mg g−1 to 80.5 mg g−1, while the contents of Fe and Mn in ACMS were obviously higher than those in CMS. These results demonstrate that the alkali treatment could reduce the content of Si element, leading to the increased relative contents of Fe and Mn, both of which are regarded as active in PMS activation.
The detailed element information of the samples was investigated by XPS. As shown in Fig. 4a, the Fe 2p3/2 peak could be deconvoluted into two peaks at 710.6 and 712.6 eV corresponding to Fe2+ and Fe3+, respectively.29 The Fe2+/Fe3+ ratios of CMS, ACMS, and ACMS-R were 62.3:37.7, 46.2:53.8, and 56.6:43.4, respectively. After the alkali treatment, the ACMS sample presented a lower Fe2+ content compared to CMS. The alkali treatment reduced the content of Si element and resulted in the reconstruction of the metal species, which might facilitate the oxidation of Fe species. With further treating ACMS with alkaline NaBH4 solution, the Fe2+ content in the ACMS-R sample increased, which could be due to the reductive ability of NaBH4. A similar trend could also be observed in the valence conversion of Mn in the samples. As depicted in Fig. 4b, the Mn 2p3/2 spectra of the three samples could be deconvoluted into three peaks corresponding to Mn2+ (641.0 eV), Mn3+ (642.2 eV), and Mn4+ (644.3 eV), respectively.28,29 The Mn2+ content of ACMS-R was 50.7% of the total Mn, which was much higher than that of ACMS (28.6%). The O 1s spectra of ACMS and ACMS-R can be seen in Fig. S2.† The observed peak at 529.7 eV could be due to the oxygen in the lattice (OL).28,29 The peak at 531.6 eV could be attributed to the oxygen atoms with a low coordination toward metal ions, which originated from the existence of oxygen vacancies.12,44 Thus, these kinds of oxygen atoms are regarded as OV in this manuscript. The peak at 532.7 eV represented the oxygen in the adsorbed molecules (Oads) on the surface (such as H2O).14,44 The OV/OL ratio of ACMS was 4.14, while that value for ACMS-R was 4.75. The increased OV/OL ratio indicated the successful formation of more oxygen vacancies during the alkali-reduction treatment by NaBH4 solution, which was in accordance with previous reports.41
 | ||
| Fig. 4 (a) XPS Fe 2p spectra of CMS, ACMS, and ACMS-R. (b) XPS Mn 2p spectra of CMS, ACMS, and ACMS-R. | ||
3.2 Catalytic performances of the samples
The catalytic performances of the prepared samples were investigated by degrading RhB under similar conditions, as shown in Fig. 5 and S3.† The oxidation of RhB by PMS alone was first evaluated and 27.7% of RhB was removed within 30 min. The adsorption of ACMS-R was next investigated and ACMS-R could only adsorb a small amount of RhB (1.6%), demonstrating the removal of RhB mainly depended on the oxidation process. As displayed in Fig. 5a, by adding CMS as the catalyst, merely 32.2% of RhB was removed within 30 min, revealing the poor activity of CMS for PMS activation. ACMS, the sample prepared with alkali treatment, presented a RhB removal efficiency of 56.5% within 30 min, which was significantly higher than that of CMS, indicating the enhanced performance of ACMS. Treating the sample with alkaline NaBH4 solution further increased the performance since the ACMS-R could remove 65.9% of RhB within 30 min while an even higher removal efficiency of 98.3% could be achieved within 120 min. To clearly evaluate the enhancement of the alkali-reduction treatment by NaBH4 solution, the CMS was also treated by alkaline NaBH4 solution and the obtained CMS-R exhibited poor activity compared to ACMS-R. As shown in Fig. 5b, the calculated RhB degradation kinetics rates for CMS, CMS-R, ACMS, and ACMS-R were 0.014, 0.015, 0.024, and 0.034 min−1, respectively. The RhB degradation kinetics rate of ACMS-R was 2.43 times higher than that of CMS, further indicating the good performance of ACMS-R. | ||
| Fig. 5 (a) RhB degradation in PMS activated with the CMS, CMS-R, ACMS, and ACMS-R as catalysts, respectively; (b) corresponding fitted kinetics curves. Experimental conditions: [RhB]0 = 50 mg L−1, [catalyst] = 800 mg L−1, [PMS] = 400 mg L−1, pH0 = 7, T = 25 °C. | ||
The effects of several parameters on the degradation of RhB with ACMS-R as the catalyst were investigated. RhB removal by PMS activated with ACMS-R as a catalyst under different catalyst dosages was conducted, as depicted in Fig. 6a. When increasing the catalyst dosage from 400 to 800 mg L−1, the RhB removal efficiency increased from 84.3% to 95.2% within 120 min. This could be due to the larger amount of active sites under the higher catalyst dosages. However, further increasing the catalyst dosages to 1200 and 1600 mg L−1 resulted in a decrease in the RhB removal efficiency. It has been reported that the degradation of organic pollutants in these system is driven by the active radicals, thus, too high a catalyst dosage might lead to the self-capture of radicals on the surface of the catalyst, resulting in a poorer performance of the catalyst for removing organic pollutants.9
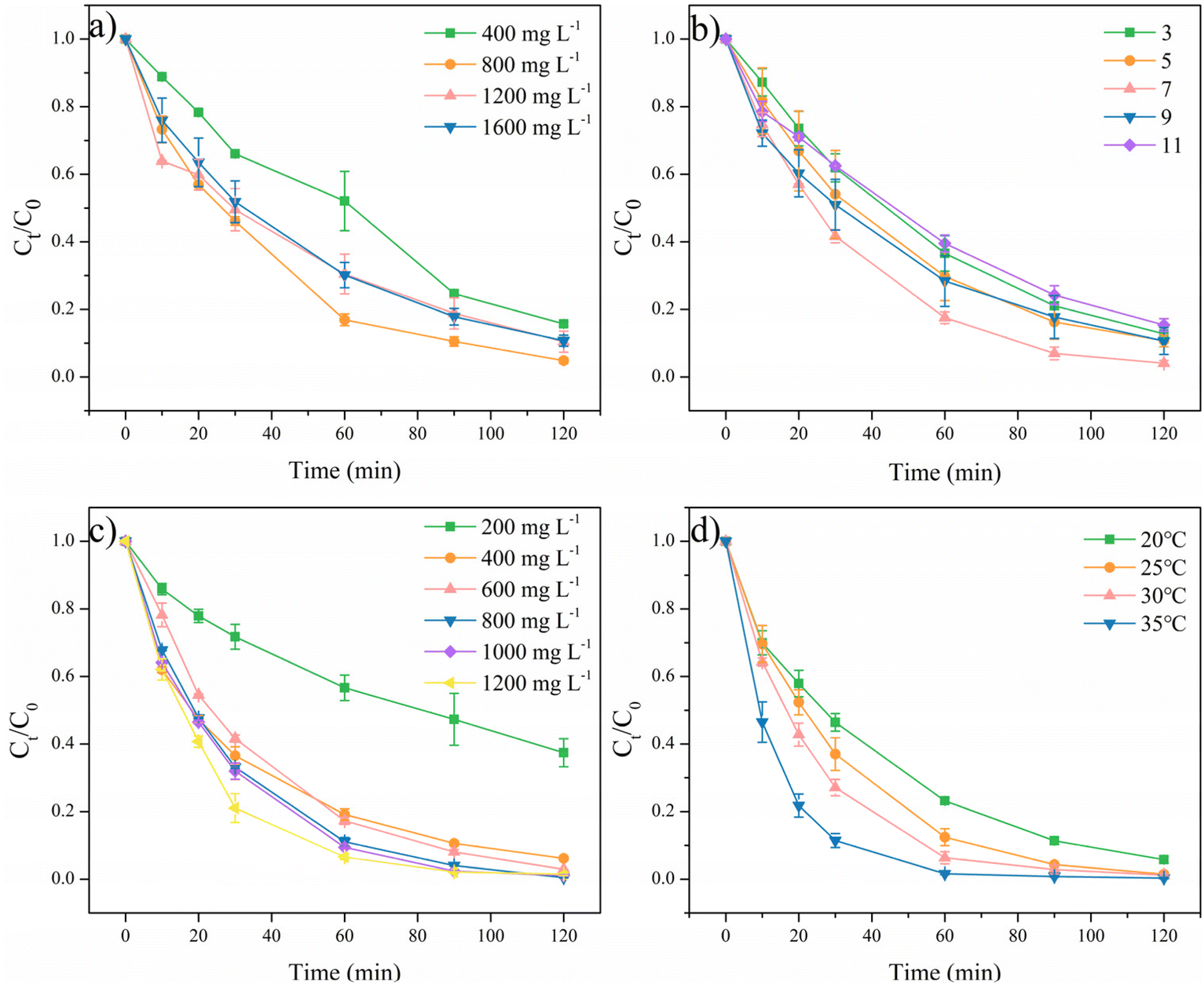 | ||
| Fig. 6 Effects of the (a) catalyst dosage, (b) pH, (c) PMS concentration, and (d) temperature. Experimental conditions: [RhB]0 = 50 mg L−1, [catalyst] = 800 mg L−1 (for b, c, and d), [PMS] = 400 mg L−1 (for a and c) and 800 mg L−1 for d, pH0 = 7 (for a, b, and d), T = 25 °C (for a, b, and c). | ||
It is widely known that the pH value has a significantly influence on the performance of PMS activation. As shown in Fig. 6b, within 30 min, the ACMS-R/PMS system presented RhB removal efficiencies of 38.1%, 46.0%, 58.4%, 49.0%, and 37.5% at pH 3, 5, 7, 9, and 11, respectively. High RhB removal efficiency above 84.3% with pH values ranging from 3 to 11 could be achieved by the ACMS-R/PMS system within 120 min, indicating the effectiveness of ACMS-R. It could be noticed that the highest RhB removal efficiency of 96.0% was achieved at the pH value of 7, while acidic or alkaline conditions slightly hindered the performance of the ACMS-R/PMS system. It is known that the pKa2 of PMS is 9.4, which means that the dominant PMS species would be SO25− when the pH value is higher than 9.4 (ref. 14). Since SO25− is harder to be activated than HSO5−, the lower activity under alkaline conditions might be due to this phenomenon.45 Meanwhile, it has also been reported that H2SO5 would be more significant at low pH values and H2SO5 is more difficult to activate compared to HSO5−, leading to a decrease in performance under acidic conditions.46
Fig. 6c displays the performances at various PMS concentrations. With 200 mg L−1 PMS, only 62.6% RhB could be removed within 120 min. The RhB removal efficiencies within 60 min were 43.4%, 80.9%, 82.7%, 89.0%, 90.6%, and 93.4% when the PMS concentrations were 200, 400, 600, 800, 1000, and 1200 mg L−1, respectively, indicating that the RhB removal efficiency increased with increasing the PMS concentration. Significantly, a high RhB removal efficiency of 99.5% could be obtained with 800 mg L−1 PMS within 120 min. Nevertheless, further increasing the PMS concentration did not lead to an obvious enhancement of performance, which could be attributed to the self-quenching of PMS.
Based on the above results, the optimized conditions for the ACMS-R/PMS system were revealed to be 800 mg L−1 catalyst, 800 mg L−1 PMS, and pH 7. The performance of the ACMS-R/PMS system was compared to other systems for RhB degradation, as shown in Table S2†.47–50 ACMS-R exhibited better activity than other catalysts derived from waste, such as zinc oxide dust and sludge.
Under this condition, the RhB removal was performed at different temperatures (Fig. 6d) and the apparent activation energy (Ea) was calculated according to the Arrhenius equation (eqn (1)):
|
lnk = lnA − Ea/RT
| (1) |
3.3 Catalytic mechanism of ACMS-R
The catalytic mechanism of ACMS-R for activating PMS was investigated. Quenching experiments were conducted with adding methanol and L-histidine to identify the reactive oxidative species in the ACMS-R/PMS system. It is known that methanol has a rapid reactive kinetics with both SO4˙− and ˙OH (k˙OH = 9.7 × 108 mol L−1 s−1, kSO4˙– = 1.1 × 107 mol L−1 s−1).34 As shown in Fig. 7a, by adding 0.5 M methanol, 83.0% RhB removal was achieved within 60 min, while that in the control group was 89.0%. The presence of methanol just slightly decreased the RhB removal efficiency, implying that both SO4˙− and ˙OH did not play vital roles in the degradation of RhB. However, 1O2, which could be generated when the catalysts have oxygen vacancies, has been reported to be effective for removing RhB.16 Thus, L-histidine was added to capture 1O2 and it was noticed that the RhB removal efficiency significantly decreased to 0% in the presence of 0.05 M L-histidine and 0.5 M methanol. These results indicated that 1O2 played an important role in removing RhB in the ACMS-R/PMS system.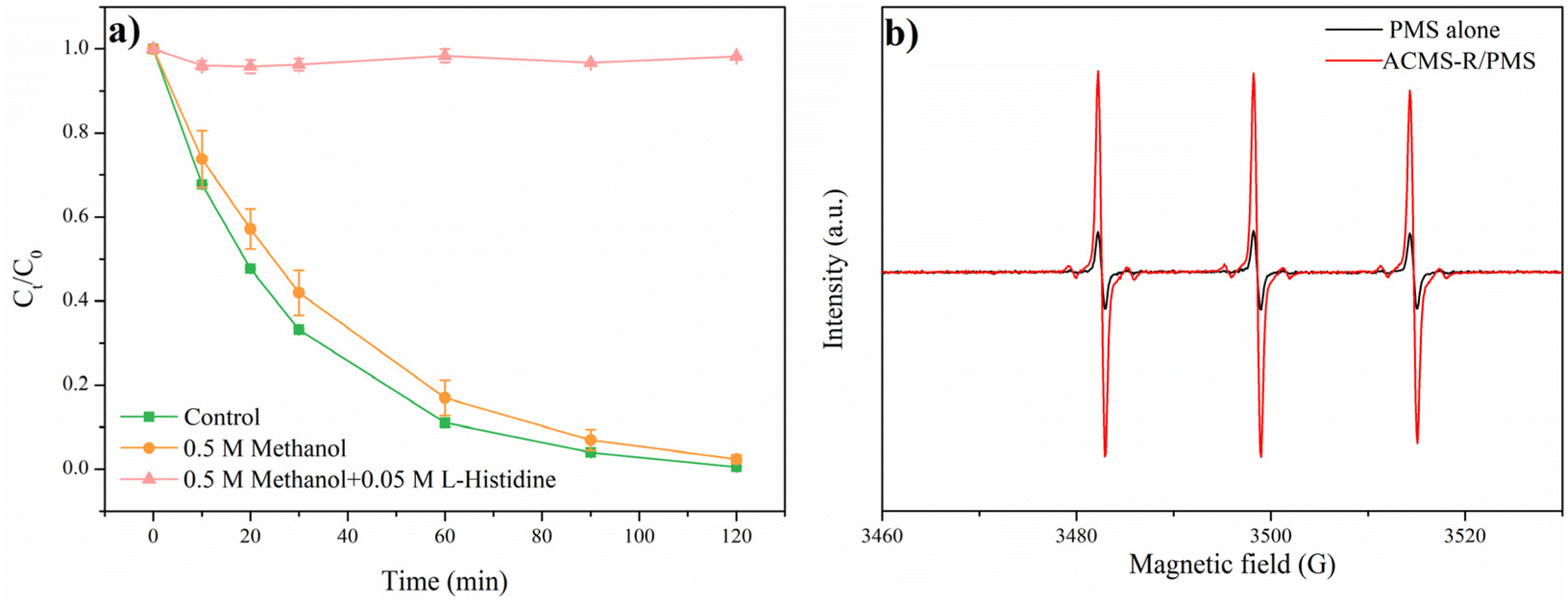 | ||
| Fig. 7 (a) Quenching experiments with ACMS-R as the catalyst. Experimental conditions: [catalyst] = 800 mg L−1, [PMS] = 800 mg L−1, [RhB]0 = 50 mg L−1, pH = 7, T = 25 °C, [methanol] = 0.5 M, [L-histidine] = 0.05 M. (b) EPR measurements of the ACMS-R/PMS system with TEMP as the spin-trapping agent. | ||
To further demonstrate the generation of 1O2, EPR was employed and the results could be seen in Fig. 7b. Without ACMS-R, the solution with PMS alone exhibited the typical triplet peak corresponding to the TEMP-1O2 adduct.15 This could be attributed to the reaction between SO25− and HSO5−, which has been widely reported.17 Remarkably, after adding the ACMS-R into the PMS solution, the signal increased significantly. These results further indicated the generation 1O2 of in the ACMS-R/PMS system.
The better performance of ACMS-R is further discussed herein. Taking the chemical compositions of ACMS-R into consideration, it is reasonable to speculate that Fe and Mn are the active species. First, the alkali treatment conducted in this study could reduce the content of other inert compositions in the Mn residues, such as Si-based materials, leading to the relatively increased contents of Fe and Mn, as demonstrated by the ICP-AES results. Treating the sample with alkaline NaBH4 solution could introduce more OV into the sample, and it is reported that the OV content is correlated to the generation of 1O2. As revealed above, the dominant oxidative species in the ACMS-R/PMS system was 1O2 and the OV/OL ratio in ACMS-R was 4.75, which was much higher than that of ACMS (4.14). Thus, the better performance of ACMS-R compared to CMS and ACMS could be attributed to the higher contents of Fe and Mn and the improved content of OV. To further reveal the chemical conversion during the reaction, the XPS spectrum of ACMS-R after the degradation of RhB was measured and is shown in Fig. 8. The OV/OL ratio decreased from 4.75 to 3.12 after the reaction, implying the vital role of oxygen vacancies in the generation of 1O2. As reported in the literature, the weakly-bound oxygen, which is represented by OV in this manuscript, could be transformed into active oxygen (O*), and the O* could react with HSO5− to form 1O2 (eqn (2) and (3)).12,28 The valence conversion of Fe and Mn during the reaction was also investigated. The relative intensity ratio of Fe2+ and Fe3+ changed from 56.6:43.4 for fresh ACMS-R to 46.1:53.9 after the reaction, while those of Mn2+, Mn3+, and Mn4+ changed from 50.7:15.9:33.4 for the initial ACMS-R to 38.5:34.6:26.9 for the used ACMS-R. These results indicated that both Fe and Mn were involved in the reaction. Since the dominant ROS in the degradation of RhB in the ACMS-R/PMS system was 1O2, it is speculated that the PMS activation mechanism involved the transformation of Mn species from high valence to low valence, which could result in the formation of SO5˙− (eqn (4)–(6)).44 Consequently, the accumulated SO5˙− could be favorable for the generation 1O2 of through the reaction shown in eqn (7).37,44 On the other hand, it could be noticed that the low-valence species (Fe2+ and Mn2+) increased. This could be attributed to the decreased content of OV, which need more low-valence metals for coordination.16,37,51
| OV → O* | (2) |
| O* + HSO5− → HSO4− + 1O2 | (3) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mn(IV) + HSO5− → Mn(III) + SO5˙− + H+ Mn(IV) + HSO5− → Mn(III) + SO5˙− + H+
| (4) |
|
Mn(III) + HSO5− → Mn(II) + SO5˙− + H+
| (5) |
|
Fe(III) + HSO5− → Fe(II) + SO5˙− + H+
| (6) |
| 2SO5˙− + 2H2O → 4H+ + 2SO24− + 21O2 | (7) |
 | ||
| Fig. 8 (a) XPS Fe 2p, (b) Mn 2p, and (c) O 1s spectra of ACMS-R after the reaction. | ||
3.4 Applicability and reusability of ACMS-R
As the catalyst was prepared using solid waste as precursors, the applicability and reusability of ACMS-R are very important. Anion substances widely exist in actual water bodies and could influence the PMS activation performance. As shown in Fig. 9a, with the presence of 25 mmol per L Cl− or 25 mmol per L H2PO4−, the RhB removal efficiencies in the ACMS-R/PMS system within 60 min were 98.2% and 93.0%, respectively. Both of those were slightly higher than that of the control group (89.0%). These results demonstrated the good performance of ACMS-R in complex water bodies with different anions. | ||
| Fig. 9 (a) Effects of Cl− and H2PO4− on RhB degradation by the ACMS-R/PMS system. Experimental conditions: [RhB]0 = 50 mg L−1, [catalyst] = 800 mg L−1, [PMS] = 800 mg L−1 [anions] = 25 mmol L−1, pH = 7, T = 25 °C. (b) Stability experiments of ACMS-R. Experimental conditions: [RhB]0 = 50 mg L−1, [catalyst] = 800 mg L−1, [PMS] = 800 mg L−1, pH = 7, T = 25 °C. | ||
Next, the stability of ACMS-R was investigated and the results are presented in Fig. 9b. After five cycles under the same conditions, a high RhB removal efficiency of 94.4% could still be achieved within 120 min, indicating the good reusability of ACMS-R.
To further reveal the performance of ACMS-R in real water in actual water bodies, two typical actual water body samples were gathered for conducting the RhB degradation experiments. River water was collected from the Yangtze River and lake water was obtained from Cuihu Lake in the campus of Chongqing Technology and Business University. First, 50 mg L−1 RhB was added in to these water body samples and the pH was adjusted to 7 before the reaction. As depicted in Fig. S5,† within 60 min, the ACMS-R/PMS system could remove 98.7% of the RhB in the lake water and 97.5% of the RhB in the river water. These results demonstrated that ACMS-R has excellent activity for RhB degradation in actual water bodies.
3.5 Major degradation intermediates
To comprehensively elucidate the potential degradation mechanisms of RhB, LC-MS was employed for analysis of the degradation intermediates. The corresponding mass data are presented in Table S3,† with a comprehensive list of the intermediates encountered along the RhB degradation pathway. Based on the detected intermediates, the RhB degradation pathway was constructed (Fig. 10). The experimental results demonstrated that the degradation of RhB involved a complex interplay of various chemical reactions, encompassing deethylation, deamination, dealkylation, decarboxylation, chromophore cleavage, ring-opening, and ultimate mineralization.52 At the initial stage of degradation, oxidative species primarily targeted the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, initiating a deethylation reaction, which transformed RhB (m/z = 443) into P1, P2, and P3 (m/z = 387).53 Subsequently, the continued action of the oxidative species progressively removed the ethyl groups from these intermediates, sequentially yielding P4, P5 (m/z = 359), and ultimately P6 (m/z = 331) upon detachment of the final ethyl group. Following the rupture of the –NH3 bond, P6 was transformed into P7 (m/z = 301).52 Furthermore, the central carbon atom of P7 was attacked, leading to the formation of P8 (m/z = 269), P9 (m/z = 279), P10 (m/z = 283), P11 (m/z = 214), and P12 (m/z = 182).48 As degradation progressed, the chromophore structure underwent disruption, resulting in the formation of a series of monocyclic aromatic compounds, including P13 (m/z = 154), P14 (m/z = 122), and P15 (m/z = 94).54 Subsequently, oxidative species persisted in the oxidation process, catalyzing the ring–opening reactions of the macromolecular organic intermediates, giving rise to alcohols and acids, such as P16 (m/z = 90), P17 (m/z = 90), and P18 (m/z = 116).54 The final stage of degradation involved mineralization, where the alcohols and acids were thoroughly decomposed into small inorganic molecules, including NO3−, NH4+, CO2, and H2O.52,55 To demonstrate the mineralization of RhB during the reaction, TOC values were measured, as shown in Fig. S6.† Within 120 min, the TOC removal efficiency reached 24.2% in the ACMS-R/PMS system, indicating the mineralization of RhB by PMS activated with ACMS-R as the catalyst.
N bond, initiating a deethylation reaction, which transformed RhB (m/z = 443) into P1, P2, and P3 (m/z = 387).53 Subsequently, the continued action of the oxidative species progressively removed the ethyl groups from these intermediates, sequentially yielding P4, P5 (m/z = 359), and ultimately P6 (m/z = 331) upon detachment of the final ethyl group. Following the rupture of the –NH3 bond, P6 was transformed into P7 (m/z = 301).52 Furthermore, the central carbon atom of P7 was attacked, leading to the formation of P8 (m/z = 269), P9 (m/z = 279), P10 (m/z = 283), P11 (m/z = 214), and P12 (m/z = 182).48 As degradation progressed, the chromophore structure underwent disruption, resulting in the formation of a series of monocyclic aromatic compounds, including P13 (m/z = 154), P14 (m/z = 122), and P15 (m/z = 94).54 Subsequently, oxidative species persisted in the oxidation process, catalyzing the ring–opening reactions of the macromolecular organic intermediates, giving rise to alcohols and acids, such as P16 (m/z = 90), P17 (m/z = 90), and P18 (m/z = 116).54 The final stage of degradation involved mineralization, where the alcohols and acids were thoroughly decomposed into small inorganic molecules, including NO3−, NH4+, CO2, and H2O.52,55 To demonstrate the mineralization of RhB during the reaction, TOC values were measured, as shown in Fig. S6.† Within 120 min, the TOC removal efficiency reached 24.2% in the ACMS-R/PMS system, indicating the mineralization of RhB by PMS activated with ACMS-R as the catalyst.
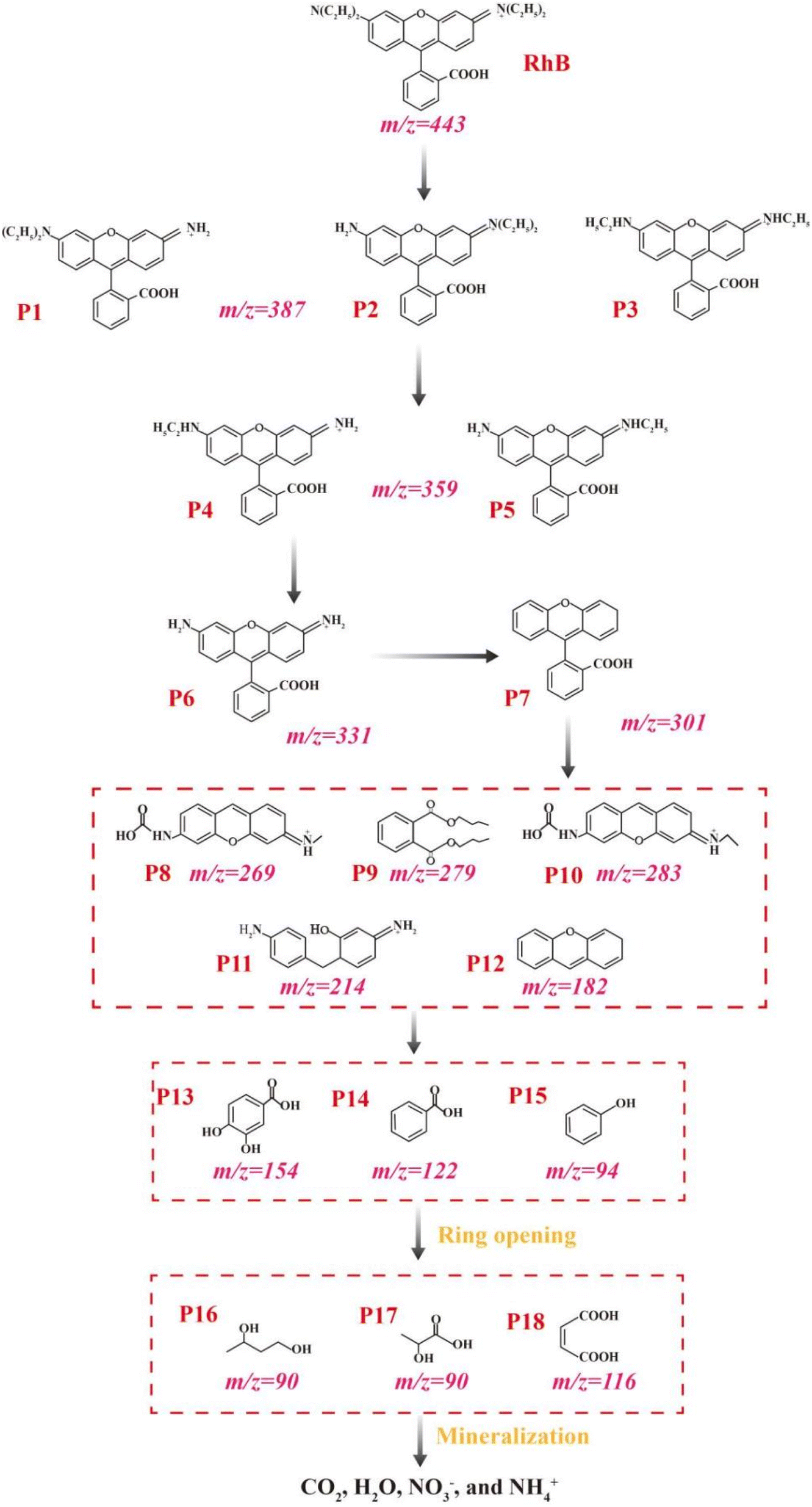 | ||
| Fig. 10 Possible RhB degradation pathways in the ACMS-R/PMS system. | ||
3.6 Toxicity assessment
To evaluate the environmental implications, a quantitative structure–activity relationship (QSAR) approach was employed to utilize the toxicity estimate software tool (T.E.S.T.) to conduct toxicity analysis and predict the toxicity changes during the RhB degradation process. Specifically, the fathead minnow LC50 (96 h), daphnia magna LC50 (48 h), oral rat LD50, bioconcentration factor, developmental toxicity, and mutagenicity were investigated.56 The results are presented in Fig. S7 and Table S4.† The LC50 (96 h), LC50 (48 h), developmental toxicity, and mutagenicity of RhB were determined to be 0.04 mg L−1, 4.63 mg L−1, 0.95, and 0.26, respectively, predicting it as “very toxic,” “toxic,” and “developmentally toxic”.56 For fathead minnows (Fig. S7a†), all the intermediates exhibited lower toxicity than RhB. For the daphnia magna (Fig. S7b†), only intermediates P11 (1.87 mg L−1) and P12 (1.92 mg L−1) surpassed the toxicity of RhB. For oral rat (Fig. S7c†), most intermediates displayed lower toxicity than RhB (924.86 mg kg−1). Regarding the bioaccumulation factors (Fig. S7d†), only the intermediate P7 (27.57) had a higher bioconcentration factor than RhB (15.48). In terms of the developmental toxicity (Fig. S7e†), most intermediates were less toxic than RhB (0.95). For mutagenicity (Fig. S7f†), the intermediates of the terminal degradation pathways exhibited low mutagenicity, classified as “mutagenicity negative”.56 In summary, despite a few intermediates, like, P2 exhibiting higher toxicity than RhB, the majority of the intermediates presented lower toxicity, indicating that RhB could be oxidized into less toxic products in the ACMS-R/PMS system.4. Conclusions
A two-step method involving alkali treatment and NaBH4 alkali-reduction treatment was developed to transform Mn residues into catalysts for activating PMS toward the degradation of RhB with good performance. The alkali treatment removed the Si and prevented the leaching of Fe and Mn, which are regarded as the active species. The alkaline NaBH4 solution treatment successfully increased the content of oxygen vacancies. The optimized catalyst presented good performance with 1O2 as the dominant ROS. Overall, 18 kinds of degradation intermediates were revealed and the possible degradation pathways were proposed. Toxicity assessment by T.E.S.T. demonstrated the significantly decreased toxicity during the degradation of RhB. Furthermore, the ACMS-R/PMS system was effective when tested with water samples from actual water bodies, such as lake water and river water, and the system also exhibited high reusability during five recycles.Data availability
The authors do not have permission to share data.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 52300032], Natural Science Foundation of Chongqing [grant numbers CSTB2023NSCQ-MSX0700, CSTB2023NSCQ-MSX0789, and CSTB2024NSCQ-MSX1255], Chongqing Municipal Education Commission [grant numbers KJQN202300838, KJZD-M202200802, KJQN202201548, KJQN202401554, and KJQN202401548] and Start-up Foundation of High-level Talents in Chongqing Technology and Business University [grant number 2356017].References
- F. Wang, G. Long, K. Ma, X. Zeng, Z. Tang, R. Dong, J. He, M. Shangguan, Q. Hu, R. K. Liew, Y. Li and J. Zhou, Environ. Chem. Lett., 2023, 21, 2251–2284 CrossRef CAS.
- H. Su, W. Zhou, X. Lyu, X. Liu, W. Gao, C. Li and S. Li, Miner. Eng., 2023, 202, 108264 CrossRef CAS.
- R. Ji, T.-J. Liu, L.-L. Kang, Y.-T. Wang, J.-G. Li, F.-P. Wang, Q. Yu, X.-M. Wang, H. Liu, H.-W. Guo, W.-L. Xu, Y.-N. Zeng and Z. Fang, J. Clean. Product., 2022, 380, 135076 CrossRef CAS.
- F. Wang, G. Long and J. L. Zhou, J. Hazard. Mater., 2024, 465, 133419 CrossRef CAS PubMed.
- F. Wang, G. Long and J. L. Zhou, Sci. Total Environ., 2023, 894, 165049 CrossRef CAS PubMed.
- J. Shu, Y. Deng, X. Wei, M. Chen, Y. Yang and Z. Deng, ACS ES&T Water, 2023, 3, 2229–2237 Search PubMed.
- X. Li, H. Liu, Y. Zhang, J. Mahlknecht and C. Wang, J. Environ. Manage., 2024, 352, 120051 CrossRef CAS PubMed.
- C. Chen, L. Liu, W. Li, Y. Lan and Y. Li, J. Hazard. Mater., 2022, 439, 129572 CrossRef CAS PubMed.
- S. Sun, S. Song, S. Yang, Y.-L. He, Y. Shi, P. Zhou, Z.-k. Xiong, Y. Liu, H. Zhang, Y. Du, C.-S. He and B. Lai, Chin. Chem. Lett., 2024, 35, 109242 CrossRef CAS.
- Z. Ha, M. Ma, X. Tan, Y. Lan, Y. Lin, T. C. Zhang and D. Du, Environ. Res., 2023, 234, 116607 CrossRef CAS PubMed.
- A. Xu, S. Fan, T. Meng, R. Zhang, Y. Zhang, S. Pan and Y. Zhang, Appl. Catal., B, 2022, 318, 121833 CrossRef CAS.
- Y. Chen, X. Bai, Y. Ji and D. Chen, J. Hazard. Mater., 2023, 441, 129912 CrossRef CAS PubMed.
- Q. He, X. Liao, G. Li, Y. He and J. Shen, Chem. Phys., 2023, 571, 111912 CrossRef CAS.
- S. Zhu, H. Li, L. Wang, Z. Cai, Q. Wang, S. Shen, X. Li and J. Deng, Chem. Eng. J., 2023, 458, 141415 CrossRef CAS.
- J. Yan, L. Gong, S. Chai, C. Guo, W. Zhang and H. Wan, Chem. Eng. J., 2023, 458, 141456 CrossRef CAS.
- J. Zhao, F. Li, H. Wei, H. Ai, L. Gu, J. Chen, L. Zhang, M. Chi and J. Zhai, Chem. Eng. J., 2021, 409, 128150 CrossRef CAS.
- Z. Yang, X. Tan and C. Zhang, Sep. Purif. Technol., 2024, 336, 126320 CrossRef CAS.
- L. Zhao, J. Zhang, Z. Zhang, T. Wei, J. Wang, J. Ma, Y. Ren and H. Zhang, J. Colloid Interface Sci., 2022, 623, 520–531 CrossRef CAS PubMed.
- W. Chen, X. Guo, M. Wu, Z. Liu, C. Yang, H. Xie and J. Chen, Sep. Purif. Technol., 2024, 335, 126263 CrossRef CAS.
- W. Zhou, M. Li and Y. Liu, J. Environ. Manage., 2024, 351, 119876 CrossRef CAS PubMed.
- M. Li, Y. Zhang, Y. Liu, Y. Chen, Z. You, L. Zhang, Y. Liang and Z. He, J. Clean. Product., 2023, 428, 139408 CrossRef CAS.
- X. Zhou, Y. Wang, X. Wang and Y. Zhang, J. Environ. Chem. Eng., 2023, 11, 110120 CrossRef CAS.
- M. Wu, W. Sun, X. Meng, J. Kang and Y. Yang, ACS ES&T Water, 2023, 3, 3265–3273 Search PubMed.
- M. Li, Z. He, H. Zhong, W. Sun, M. Ye and Y. Tang, J. Environ. Manage., 2023, 328, 116945 CrossRef CAS PubMed.
- M. Li, Z. He, H. Zhong, L. Hu and W. Sun, Environ. Sci.: Nano, 2022, 9, 1037–1051 RSC.
- H. Liang, W. Wang, W. Liang, X. Deng, X. Ruan, D. Zhang and Y. Yang, J. Environ. Chem. Eng., 2023, 11, 109126 CrossRef CAS.
- C. Quimbaya-Ñañez, E. A. Serna-Galvis, J. Silva-Agredo, L. Huerta, R. A. Torres-Palma and Y. Ávila-Torres, J. Environ. Chem. Eng., 2024, 12, 112015 CrossRef.
- M. Li, Z. He, H. Zhong, W. Sun, L. Hu and M. Luo, Chem. Eng. J., 2022, 441, 136024 CrossRef CAS.
- H. Shen, M. Luo, J. Wang, M. Li, Z. He, H. Zhong, W. Sun, M. Ye and Y. Tang, Chem. Eng. J., 2023, 472, 144915 CrossRef CAS.
- M. Li, H. Zhong, Z. He, L. Hu, W. Sun, P. Loganathan and D. Xiong, Chem. Eng. J., 2021, 413, 127478 CrossRef CAS.
- M. Li, F. Huang, L. Hu, W. Sun, E. Li, D. Xiong, H. Zhong and Z. He, Chem. Eng. J., 2020, 401, 126085 CrossRef CAS.
- X. Zhou, C. Luo, J. Wang, H. Wang, Z. Chen, S. Wang and Z. Chen, Sci. Total Environ., 2021, 762, 143120 CrossRef CAS PubMed.
- J. Ni, Q. Zhang, X. Fu, Q. Zhang, S. Wu, Z. Xiong, J. Wang and Q. Zeng, ACS Appl. Nano Mater., 2023, 6, 13492–13502 CrossRef.
- J. Liang, M. Guo, Y. Xue, J.-n. Gu, J. Li, F. Shi, X. Guo, X. Min, J. Jia, K. Li and T. Sun, Chem. Eng. J., 2022, 435, 135000 CrossRef CAS.
- C. Pan, Y. Sun, Y. Dong, H. Hou, M. F. Kai and J. Lan, J. Hazard. Mater., 2024, 465, 133198 CrossRef CAS PubMed.
- Z. Yang, Z. Zhou, X. Tan, G. Zeng and C. Zhang, J. Mater. Sci. Technol., 2025, 204, 224–237 CrossRef CAS.
- Y. Guan, G. Zhou, L. Li, Y. Jiang, J. Yin, C. Liu, L. Zhang, Q. Han and E. H. Ang, Sep. Purif. Technol., 2024, 335, 126072 CrossRef CAS.
- T. C. Khiem, N. N. Huy, E. Kwon, S. Wacławek, A. Ebrahimi, W.-D. Oh, S. Ghotekar, Y. F. Tsang, W.-H. Chen and K.-Y. A. Lin, Chem. Eng. J., 2023, 476, 146404 CrossRef CAS.
- A. Khan, H. Ullah, Q. Wu, W. Gong, L. Ma, S. Zhao, A. Xu and X. Li, Chem. Eng. J., 2023, 472, 145112 CrossRef CAS.
- M. Xu, Q. Zhang, Z. Zhu, Y. Xue, T. Zhang and J. Hong, J. Clean. Product., 2022, 377, 134258 CrossRef CAS.
- Y. Zhao, L. Wu, P. Hong, X. He, S. Gao, Y. Yu, B. Liao and H. Pang, Appl. Surf. Sci., 2024, 645, 158864 CrossRef CAS.
- L. Yu, G. Zhang, C. Liu, H. Lan, H. Liu and J. Qu, ACS Catal., 2018, 8, 1090–1096 CrossRef CAS.
- C. Gong, F. Chen, Q. Yang, K. Luo, F. Yao, S. Wang, X. Wang, J. Wu, X. Li, D. Wang and G. Zeng, Chem. Eng. J., 2017, 321, 222–232 CrossRef CAS.
- M. Chen, T. Yang, L. Zhao, X. Shi, R. Li, L. Ma, Y. Huang, Y. Wang and S.-c. Lee, Appl. Surf. Sci., 2024, 645, 158835 CrossRef CAS.
- X. Duan, C. Su, J. Miao, Y. Zhong, Z. Shao, S. Wang and H. Sun, Appl. Catal., B, 2018, 220, 626–634 CrossRef CAS.
- J. Huang, Y. Dai, K. Singewald, C.-C. Liu, S. Saxena and H. Zhang, Chem. Eng. J., 2019, 370, 906–915 CrossRef CAS.
- X. Shan, Z. Xie, X. Ma, Y. Xu, S. Zhang, Q. Jia, R. Wan and H. Tao, Microporous Mesoporous Mater., 2022, 337, 111948 CrossRef.
- Y. Huang, X. Chen, Y. Fan, C. Wang, Y. Cao, W. Peng, B. Fu, J. Liu and M. Hu, Miner. Eng., 2024, 214, 108781 CrossRef CAS.
- Z. Huang, T. Wang, M. Shen, Z. Huang, Y. Chong and L. Cui, Chem. Eng. J., 2019, 369, 784–792 CrossRef CAS.
- L. Zheng, S. Cao, M. Tang and M. Ge, Water, Air, Soil Pollut., 2025, 236, 65 CrossRef CAS.
- G. Fang, J. Zhou, Y. Cai, S. Liu, X. Tan, A. Pan and S. Liang, J. Mater. Chem. A, 2017, 5, 13983–13993 RSC.
- Y. Ren, J. Wang, G. Qu, N. Ren, P. Lu, X. Chen, Z. Wang, Y. Yang and Y. Hu, Sep. Purif. Technol., 2023, 314, 123616 CrossRef CAS.
- H. Cheng, X. Li, C. Huang, J. Zhu, P. Wang, H. Cao, C. Feng, D. Ling, H. Liu and M. Cheng, J. Clean. Product., 2023, 393, 136354 CrossRef CAS.
- T. Xu, L. Fu, H. Lu, M. Zhang, W. Wang, B. Hu, Y. Zhou and G. Yu, J. Clean. Product., 2023, 401, 136794 CrossRef CAS.
- J. Zhang, M. Yan, G. Sun, X. Li, B. Hao and K. Liu, Chemosphere, 2022, 304, 135318 CrossRef CAS PubMed.
- Y. Liu, X. Zhang, X. Li and Z. Zhou, J. Water Process Eng., 2024, 57, 104588 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01113b |
| This journal is © The Royal Society of Chemistry 2025 |