
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective synthesis of benzodiazepine sulfonamide-based MGAT2 inhibitors and evaluation of their antitumor activity†
Yuying Zhang‡
a,
Yalei Li‡b,
Xiaoyue Liua,
Yi Wanga,
Huachuan Zheng*c and
Dejun Zhou *a
*a
aThe Key Laboratory of Chinese Medicine Research, Development of HeBei province, Traditional Chinese Medicine Institute of Chengde Medical University, Chengde, 067000, China. E-mail: zhoudj20220307@163.com; zhoudj202207@cdmc.edu.cn
bDepartment of Surgery, The Affiliated Hospital of Chengde Medical University, Chengde 067000, China
cCancer Center, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou 121001, China. E-mail: zheng_huachuan@hotmail.com
First published on 4th July 2025
Abstract
This study synthesized benzodiazepine sulfonamide-based MGAT2 inhibitors to combat cancer recurrence and resistance. Despite modest effects, compounds 13 and 16 showed enhanced antitumor activity. The six-step synthesis method using amino acids is industrially viable, offering a basis for future drug development.
Benzenesulfonamides are a class of organic compounds with a benzene ring linked to a sulfonamide backbone, and they play a critical role in drug design because of their structural diversity and wide range of biological activities sulfonamides show various biological activities such as antibacterial, anti-inflammatory, and antitumor effects; thus, they are considered to have high potential for developing anticancer drugs.1 Benzodiazepines are formed by a parallel configuration of a benzene ring and a seven-membered iminolactam ring; they are the preferred clinical drugs for achieving sedative-hypnotic effects2 and show anxiolytic,3 anticancer,4 anticonvulsant,5 and antimicrobial6 activities.
In a previous study, benzodiazepines and sulfonamides were mixed and hybridized to design benzodiazepine sulfonamides, which can act as inhibitors of monoacylglycerol acyltransferase 2 (MGAT2),7 an enzyme with a key role in fatty acid metabolism and synthesis of triacylglycerol, a major intracellular energy storage compound.8 Abnormal lipid metabolism in tumor cells is an important feature of their rapid growth and proliferation.9 Therefore, the disruption of lipid metabolism in tumor cells by inhibiting MGAT2 could exert an inhibitory effect on tumor growth (Table 1).
| Entry | Catalyst | Dose (eq.) | Time (h) | Temperature (°C) | Yield (%) |
|---|---|---|---|---|---|
| 1 | H2SO4 | 1 | 21 | 25 | 27 |
| 2 | H2SO4 | 1.5 | 15 | 65 | 51 |
| 3 | H2SO4 | 2.0 | 15 | 65 | 55 |
| 4 | SOCl2 | 1 | 9 | 25 | 76 |
| 5 | SOCl2 | 1.2 | 4 | 25 | 96 |
| 6 | SOCl2 | 1.2 | 4 | 65 | 73 |
| 7 | SOCl2 | 2 | 4 | 25 | 90 |
Current research on MGAT2 inhibitors has focused on their application for treating metabolic diseases such as nonalcoholic steatohepatitis, obesity, and diabetes.10 Therefore, the potential and application of MGAT2 inhibitors as anticancer drugs need to be evaluated through clinical trials (Table 2).
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | 0.1 eq. Pd/C, H2, 20 °C | — |
| 2 | 0.1 eq. Pd/C, H2, 40 °C | 27 |
| 3 | 0.1 eq. Pd/C, HCO2NH4 (8 eq.), H2, 40 °C | 50 |
| 4 | 0.1 eq. Pd/C, HCO2NH4 (8 eq.), H2, 70 °C | 98 |
| 5 | 0.1 eq. Pd/C, HCO2NH4 (6 eq.), H2, 70 °C | 95 |
| 6 | HCO2NH4 (8 eq.), 180 °C | 75 |
| 7 | 0.1 eq. Pd/C, HCO2NH4 (8 eq.), 40 °C | 50 |
| 8 | 0.1 eq. Pd/C, HCO2NH4 (8 eq.), 70 °C | 96 |
| 9 | 0.2 eq. Pd/C, HCO2NH4 (8 eq.), 70 °C | 96 |
As reported earlier, benzodiazepines are usually synthesized through three routes (Fig. 1).7 Route 1: first, the amino acid is protected by the Cbz (benzyloxycarbonyl) group and then reacted with cyanuric trifluoride to produce acyl fluoride;11 this product is then condensed with 2-aminobenzoic acid to produce an amide, which is finally treated with microwaves in an acetic acid solution at 200 °C for ring closure to obtain the key benzodiazepine intermediate (A). Route 2: amino acids are condensed with indirubic anhydride to benzamide at 150 °C in a microwave, and high-temperature ring closure is performed at 200 °C with the same microwave conditions to obtain the key intermediate (A). Route 3: a condensation reaction is performed between 2-nitrobenzoyl chloride and amino acid methyl ester to obtain an amide; following hydrolysis and reduction, the ring closure reaction is performed at 200 °C in a microwave to obtain the key intermediate (A). However, all three routes have some disadvantages. For example, cyanuric trifluoride used in route 1 is costly; the condensation reaction temperature in route 2 is high, with a yield of only 42%; and in route 3, following condensation, the nitro group is reduced to the amino group by palladium on carbon (Pd/C)-catalyzed hydrogen reduction, and another step of hydrolysis is required, which affects the yield. Furthermore, a major concern with all three routes is the need for a high temperature of 200 °C for the ring closure reaction; consequently, the reaction conditions are harsh and require improvements (Table 3).
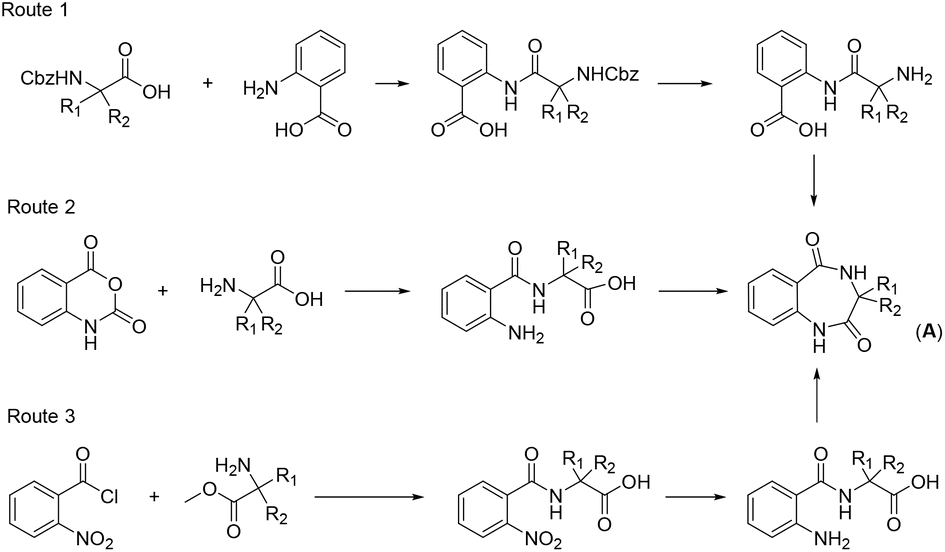 | ||
| Fig. 1 Synthesis routes for benzodiazepines reported in previous studies. | ||
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | Aniline (3 eq.), THF, 66 °C, 10 h | 45 |
| 2 | Aniline (3 eq.), CH3CN, 82 °C, 12 h | 55 |
| 3 | Aniline (3 eq.), DMSO, 100 °C, 12 h | 36 |
| 4 | Aniline (3 eq.), DMF, 100 °C, 12 h | 40 |
| 5 | Aniline (3 eq.), CH2Cl2, 40 °C, 10 h | 68 |
| 6 | Aniline (3 eq.), CHCl3, 70 °C, 9 h | 78 |
| 7 | Aniline (3 eq.), CHCl3, 80 °C, 9 h | 78 |
| 8 | Aniline (3 eq.), CHCl3, 40 °C, 9 h | 35 |
| 9 | Aniline (3 eq.), CHCl3, 60 °C, 5 h | 55 |
| 10 | Aniline (3 eq.), CHCl3, 60 °C, 11 h | 67 |
| 11 | Aniline (1 eq.), CHCl3, 60 °C, 15 h | 53 |
| 12 | Aniline (5 eq.), CHCl3, 60 °C, 8 h | 78 |
The present study aimed to determine an appropriate route to synthesize novel MGAT2 inhibitor derivatives with different substituents at positions 3 and 7 under mild reaction conditions. The key intermediates of benzodiazepines were constructed by esterification, amidation, and Pd/C-catalyzed reduction of ammonium formate by using commercially available amino acids as the starting materials. The sulfonation reaction then yielded sulfonyl chloride as the active intermediate, which was then reacted with different amines to obtain the target products (Table 4).
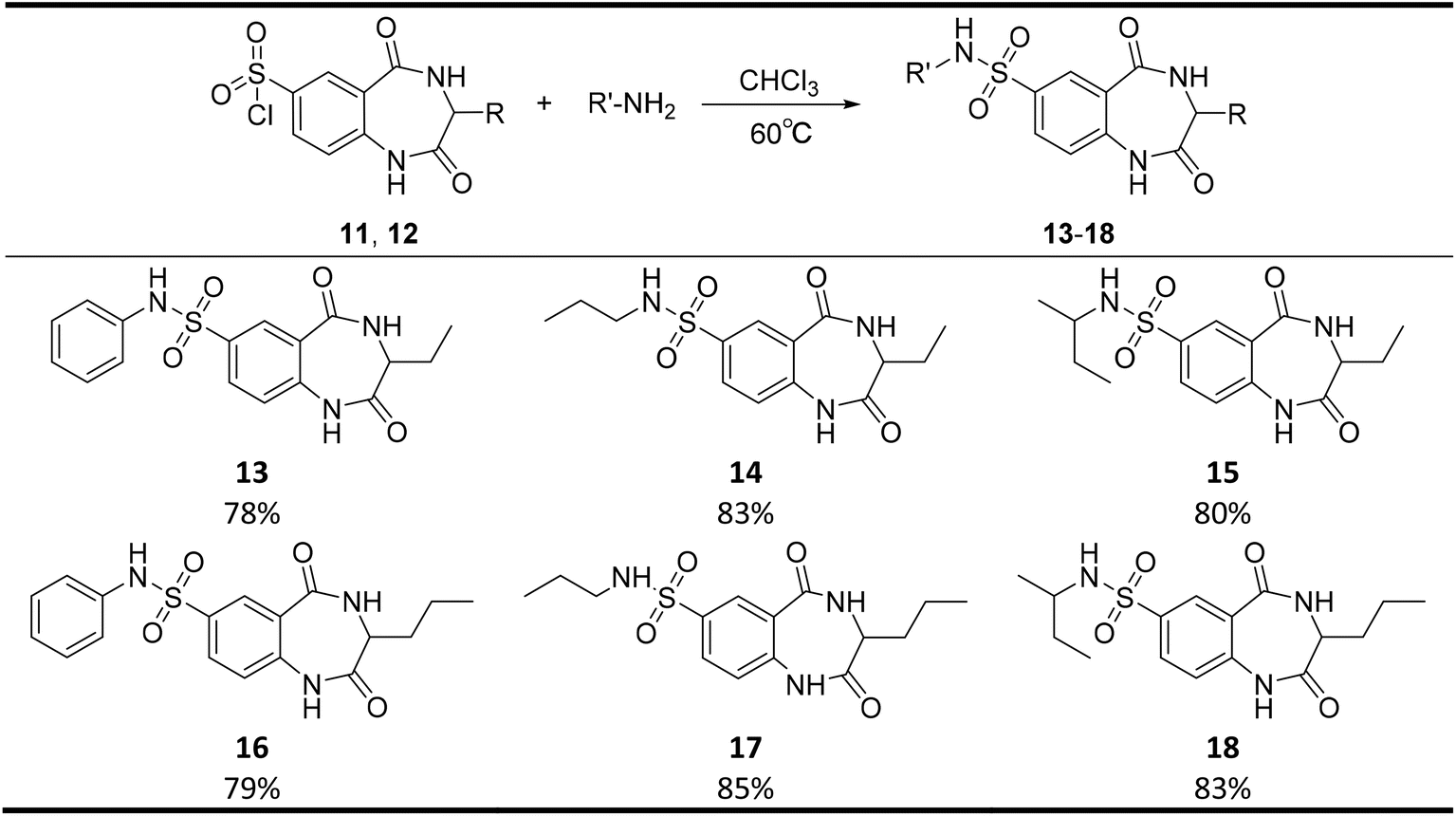 |
As shown in Scheme 1, commercially available amino acids (1, 2) were used as raw materials for the esterification reaction with methanol as the solvent in the presence of sulfoxide chloride; the obtained product was essentially completely converted to produce compounds (3, 4). Compounds (3, 4) were condensed with o-nitrobenzoyl chloride in the presence of triethylamine at room temperature to yield amides (5, 6). Compounds (5, 6) were reduced by ammonium formate; this reaction was catalyzed by Pd/C, wherein the nitro group was converted to the amino group to yield compounds (7, 8). The subsequent ring closure reaction in acetic acid yielded benzodiazepine key intermediates (9, 10). Compounds (9, 10) were then sulfonated by chlorosulfonic acid treatment to obtain sulfonyl chloride (11, 12), which was finally condensed with different amines to produce the target compounds, i.e., benzodiazepine sulfonamides (13–18).
 | ||
| Scheme 1 Reagents and conditions: (a) SOCl2 (1.2 equivalent), MeOH, reflux 4 h; (b) triethylamine (3 equivalent), CH2Cl2, 2-nitrobenzoyl chloride (1.2 equivalent), reflux, 7 h, (5) 78%, (6) 65%; (c) Pd/C (catalytic amount), ammonium formate (8 equivalent), MeOH, 70 °C, 6 h, (7) 98%, (8) 95%; (d) AcOH, 90 °C, (9) 92%, (10) 78%; (e) sulfurochloridic acid (8.5 equivalent), CHCl3, 60 °C, 15 h, (11) 96%, (12) 96%; (f) aniline or propylamine or butan-2-amine (1.1 equivalent), triethylamine (2 equivalent), CHCl3, 60 °C, 9 h. | ||
In the present study, we successfully devised a new route to efficiently synthesize benzodiazepine intermediates followed by sulfonation and sulfonylation reactions to form benzodiazepine sulfonamides with different substituents at positions 3 and 7, which are a class of compounds functioning as MGAT2 inhibitors.7 The synthesis was performed in six reaction steps. The total yields of the product with ethyl and propyl substitutions at position 3 were approximately 52–56% and 45–50%, respectively. The raw materials used for the reaction were the commonly available reagents in the market. The entire reaction was conducted at temperatures not higher than 100 °C, and ammonium formate was used as the hydrogen donor in the nitro group reduction reaction, which successfully avoided the harmful process of hydrogenation reaction. In conclusion, the developed synthesis route is economical, mild, efficient, and user-friendly and is suitable for studying pharmaceutical properties (Table 5).
|
||||||
|---|---|---|---|---|---|---|
| Compounds | R | R′ | IC50 (μg mL−1) | |||
| DLD1 | A549 | MKN45 | HepG2 | |||
| 13 | CH2CH3 | Ph | 537.1 | 521.7 | 408.7 | 250.5 |
| 14 | CH2CH3 | n-Propyl | 1276 | 1622 | 1592 | 920.8 |
| 15 | CH2CH3 | s-Butyl | 1039 | 1574 | 1862 | 896.2 |
| 16 | CH2CH2CH3 | Ph | 430.1 | 358 | 400.2 | 392.7 |
| 17 | CH2CH2CH3 | n-Propyl | 1071 | 1464 | 1546 | 910.6 |
| 18 | CH2CH2CH3 | s-Butyl | 1014 | 1511 | 1415 | 1011 |

MGAT enzyme in the re-synthesis of triacylglycerol and protects from metabolism disorders. Lang et al. demonstrated that knockout of MGAT2 in Apcmin/+ mice expedited intestinal tumor growth and progression by a significant alter the gut microbiota or inhibiting the NF-κB signaling pathway. MGAT2 silencing promoted proliferation and migration, induced G2 cell cycle arrest, and decreased apoptosis of lung cancer cells. Bioinformatics analysis showed that MGAT2 expression was upregulated in glioblastoma, colorectal and lung adenocarcinoma, closely correlated with tumor tissue, and hisological subtyping of glioblastoma. Additionally, MGAT2 expression was remarkably linked to the prognosis of hepatocellular carcinoma and colorectal cancer.12
The synthesized benzodiazepine sulfonamide compounds 13–18 were characterized by broad-spectrum analytical techniques such as NMR, mass spectrometry, and IR spectroscopy. The in vitro antitumor activities of these compounds were evaluated against the cancer cell lines DLD1, A-549, MKN45, and HepG2. These compounds showed promising activity against all the tested cell lines. The findings suggest that benzodiazepine sulfonamides are a promising new class of antitumor agents. Specifically, compound 13 effectively inhibited the growth of HepG2 tumor cells, while compound 16 effectively inhibited the growth of DLD1, A-549, and MKN45 tumor cells. The 7-substituted group R′ of compound 13 and compound 16 both contain a planar benzene ring, suggesting that the tumor cell receptors contain a planar structure that can match the plane of the benzene ring. The ethyl group at position 3 of the compound 13 is advantageous for binding to HepG tumor cell receptor, while the propyl group at position 3 of the compound 16 is advantageous for binding to DLD1, A-549 and MKN45 tumor cell receptors. Thus, both these compounds can be used as drug candidates or lead compounds for further research and development of antitumor drugs.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yuying Zhang, has done 35% of the work, including chemical experiment operation and writing article; Yalei Li has done 15% of the work, experimental operation and data organization of anti-tumor activity; Xiaoyue Liu has done 5% of the work, sample inspection; Yi Wang has done 5% of this work, a part of chemical experiment operation; Huachuan Zheng hase done 10% of the work, Guidance for biological activity experiments; Dejun Zhou has done 30% of this work, experiment content design, fund application, etc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Doctoral Initiation Fund of Chengde Medical University, grant number 202301; Hebei Natural Science Foundation, grant numbers H2023406003. We would like to thank TopEdit (https://topeditsci.com/) for its linguistic assistance during the preparation of this manuscript.Notes and references
- (a) A. Casini, A. Scozzafava and A. Mastrolorenzo, Curr. Cancer Drug Targets, 2002, 2, 55–75 CrossRef CAS; (b) F. Abbate, A. Casini and T. Owa, Bioorg. Med. Chem. Lett., 2004, 14, 217–223 CrossRef CAS; (c) M. M. Ghorab, F. A. Ragab and H. I. Heiba, Eur. J. Med. Chem., 2011, 46, 5120–5126 CrossRef CAS; (d) M. M. Ghorab, F. A. Ragab and H. I. Heiba, Arch. Pharmacal Res., 2012, 35, 59–68 CrossRef CAS.
- (a) D. De Bels, I. Bousbiat and E. Perriens, Saudi J. Anaesth., 2023, 17, 223–235 CrossRef; (b) A. N. Edinoff, C. A. Nix and J. Hollier, Neurol. Int., 2021, 13, 594–607 CrossRef CAS.
- M. P. Goldschen-Ohm, Biomolecules, 2022, 12, 1784 CrossRef CAS.
- (a) D. B. Greenberg and N. Y. Williston Park, Oncology, 1991, 5, 83–88 CAS; (b) S. J. Gawandi, V. G. Desai and S. Joshi, Bioorg. Chem., 2021, 117, 105331 CrossRef CAS; (c) W. Yu, S. Fang and X. Xie, J. Med. Chem., 2024, 67, 12835–12854 CrossRef CAS.
- A. Shafie, M. Mohammadi-Khanaposhtani and M. Asadi, Mol. Diversity, 2020, 24, 179–189 CrossRef CAS.
- (a) L. Z. Wang, X. Q. Li and Y. S. An, Org. Biomol. Chem., 2015, 13, 5497–5509 RSC; (b) Y. Liu, Z. Tong and J. Shi, Commun. Biol., 2021, 4, 1328 CrossRef CAS; (c) S. K. Mondal, S. A. Alam and G. Roymahapatra, J. Antibiot., 2024, 77, 589–599 CrossRef CAS.
- J. G. Barlind, L. K. Buckett and S. G. Crosby, Bioorg. Med. Chem. Lett., 2013, 23, 2721–2726 CrossRef CAS.
- C. E. Yen and R. V. Farese, J. Biol. Chem., 2003, 278, 18532–18537 CrossRef CAS.
- (a) P. J. McFie, A. Patel and S. J. Stone, Sci. Rep., 2022, 12, 4943 CrossRef CAS; (b) Y. Zhao, M. J. Liu and L. Zhang, Nat. Commun., 2024, 15, 9909 Search PubMed; (c) C. Calabrese, G. Miserocchi and A. De Vita, Obes. Rev., 2024, 25, e13833 Search PubMed.
- (a) P. Devasthale and D. Cheng, J. Med. Chem., 2018, 61, 9879–9888 CrossRef CAS; (b) D. Cheng, B. A. Zinker and Y. Luo, Cell Metab., 2022, 34, 1732–1748 CrossRef CAS; (c) F. Moore, W. Wang and G. Zhao, Bioorg. Med. Chem. Lett., 2023, 91, 129362 Search PubMed.
- Z. Z. Brown and C. E. Schafmeister, J. Am. Chem. Soc., 2008, 130, 14382–14383 CrossRef CAS.
- (a) Z. Qiu, C. Guo, X. Liu, S. Gao, W. Xiao, H. Cheng and L. Yin, Brain Res., 2025, 1850, 149449 CrossRef CAS; (b) D. Fu, B. Zhang, W. Fan, F. Zeng, J. Feng and X. Wang, Front. Immunol., 2024, 15, 1456719 CrossRef CAS; (c) Y. Lang, C. Zhong, L. Guo, Z. Liu, D. Zuo, X. Chen, L. Ding, B. Huang, B. Li, Y. Yuan, Y. Niu, J. Qiu and C. Qian, iScience, 2024, 27(7), 110205 CrossRef CAS; (d) Z. Wang, K. S. Embaye, Q. Yang, L. Qin, C. Zhang, L. Liu, X. Zhan, F. Zhang, X. Wang and S. Qin, J. Hepatocell. Carcinoma, 2021, 8, 119–132 CrossRef; (e) W. Lian, H. Jin, J. Cao, X. Zhang, T. Zhu, S. Zhao, S. Wu, K. Zou, X. Zhang, M. Zhang, X. Zheng and M. Peng, Cancer Cell Int., 2020, 20, 105 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01108f |
| ‡ Co-first author. |
| This journal is © The Royal Society of Chemistry 2025 |