DOI:
10.1039/D5RA01015B
(Paper)
RSC Adv., 2025,
15, 10663-10670
Predicting pKa of flexible polybasic tetra-aza macrocycles†
Received
12th February 2025
, Accepted 30th March 2025
First published on 7th April 2025
Abstract
We present physics-based pKa predictions for a library of tetra-aza macrocycles. These flexible, polybasic molecules exhibit highly charged states and substantial prototropic tautomerism, presenting a challenge for pKa prediction. Our computational protocol combines CREST/xTB conformational sampling, density functional theory (DFT) refinement in continuum solvent, and a linear empirical correction (LEC). This approach predicts known tetra-aza macrocycle pKa to within a root-mean-square deviation 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log units. This approach also provides reasonable predictions for the most stable protomers at different pH. We use this protocol to predict pKa values for four novel, synthetically achievable, previously un-synthesized tetra-aza macrocycles, providing new leads for future experiments.
log units. This approach also provides reasonable predictions for the most stable protomers at different pH. We use this protocol to predict pKa values for four novel, synthetically achievable, previously un-synthesized tetra-aza macrocycles, providing new leads for future experiments.
1. Introduction
Predicting pKa and pH-dependent speciation (prototropic tautomerization) was1 and is2 a critical component of computational medicinal chemistry.3,4 The pH-dependent speciation of drug molecules governs their solubility,5 docking poses,6 and membrane permeability.7 Modern density functional theory (DFT) approximations can routinely predict gas-phase pKa of rigid small molecules possessing a single acid/base site.8 Predicting aqueous-phase pKa requires a model for solvent.9–12 Predicting aqueous-phase pKa of large, flexible, polybasic molecules remains a challenge. Polybasic molecules are abundant in pharmaceutical design, possess prototropic tautomerism,13,14 and can be highly charged in solution.15 Flexible macrocycles can access multiple conformations within each tautomer (chameleonicity).16 Molecules that are both flexible and polybasic are particularly challenging for pKa prediction.
1.1. Tetra-aza macrocycles
Our goal in this work is to validate and use a computational protocol capable of predicting the pKa and pH-dependent speciation of flexible, polybasic, tetra-aza macrocycles (Fig. 1). Tetra-aza macrocycles combine high water solubility, tunable metal binding,17,18 and antioxidant activitiy.19,20 Tetra-aza macrocycles have been employed as catalysts, luminescent bioprobes,21 MRI contrast agents,22 and drug candidates for treating oxidative stress.23,24 Some tetra-aza macrocycles dis-aggregate amyloids.25 Other tetra-aza macrocycles have demonstrated activity as Nrf2 activators.26 The Green group has devised synthetic methods capable of accessing a broad range of substituted tetra-aza macrocycles, and has advanced these molecules' application as potential therapeutics. Initial experimental studies include the measured pKa of ten substituted macrocycles (molecules 1–10) and the protonation sites of molecules 1 and 2 (Table 1).23,27 These molecules possess between four and six acid/base sites and are positively charged at physiological pH.23,27 Proton NMR methods can be used to accurately measure the pKa and the dominant tautomers at varying pH. These studies demonstrate that substitution significantly changes the pKa.23,27,28 Synthesis and testing of the thousands of synthetically accessible tetra-aza macrocycle derivatives represents a major technical hurdle. Reliable predictions of the structures, docking poses, solubility, membrane permeability, and other properties of un-synthesized tetra-aza macrocycles could significantly accelerate development of lead compounds for the applications discussed above. Reliable prediction of pKa and charge state at physiological pH is a prerequisite for such predictions. The broad range of possible synthetically accessible macrocycles, and the demonstrated impact of chemical substitution on pKa and protonation site, motivate the use of physics-based pKa prediction protocols for tetra-aza macrocycles.
 |
| | Fig. 1 Structures of existing molecules 1–10 and previously un-synthesized molecules 11–14. Atom numbering is indicated in blue. | |
Table 1 Comparison of computed and measured pKa values for macrocycles 1–10
| Molecule |
pKa |
Experiment |
QM |
QM + LEC |
| Ref. 31 and 32. Ref. 27. Ref. 32. Ref. 23. |
| 1 |
1 |
11.37a |
10.73 |
9.63 |
| 2 |
8.22a |
3.83 |
6.15 |
| 3 |
1.61a |
−1.22 |
3.60 |
| 2 |
1 |
11.56b |
14.10 |
11.34 |
| 2 |
9.05b |
9.90 |
9.22 |
| 3 |
5.45b |
3.41 |
5.94 |
| 4 |
1.68b |
−1.90 |
3.26 |
| 3 |
1 |
10.50a |
12.25 |
10.40 |
| 2 |
7.27a |
1.73 |
5.09 |
| 3 |
1.37a |
−6.88 |
0.74 |
| 4 |
1 |
10.54a |
13.35 |
10.96 |
| 2 |
8.27a |
8.04 |
8.28 |
| 3 |
1.73a |
−4.04 |
2.18 |
| 5 |
1 |
11.14a |
10.06 |
9.30 |
| 2 |
7.47a |
3.02 |
5.74 |
| 6 |
1 |
10.6a |
12.28 |
10.42 |
| 2 |
7.00a |
3.90 |
6.18 |
| 3 |
0.85a |
−3.71 |
2.34 |
| 7 |
1 |
10.32c |
10.52 |
9.53 |
| 2 |
8.00 c |
5.87 |
7.18 |
| 3 |
1.75c |
−2.15 |
3.13 |
| 8 |
1 |
11.16b |
13.92 |
11.25 |
| 2 |
9.46b |
11.13 |
9.84 |
| 3 |
6.91b |
3.61 |
6.04 |
| 4 |
2.17b |
−6.47 |
0.95 |
| 9 |
1 |
8.27d |
8.50 |
8.50 |
| 2 |
7.36d |
5.62 |
7.05 |
| 10 |
1 |
11.31d |
16.82 |
12.71 |
| 2 |
9.35d |
15.58 |
12.08 |
| 3 |
5.25d |
3.75 |
6.11 |
| 4 |
4.21d |
2.40 |
5.43 |
| 5 |
0.98d |
−5.16 |
1.61 |
| RMSD |
|
|
3.88 |
1.21 |
1.2. Physics-based pKa prediction
Physics-based computational protocols for pKa prediction explicitly treat conformational change, making them suitable for capturing the interplay of conformation, pKa, and prototropic tautomerism in tetra-aza macrocycles. The Statistical Assessment of Modeling of Proteins and Ligands (SAMPL) physical property challenges provide a snapshot of the state-of-the-art in physics-based pKa prediction.29 In the SAMPL6 challenge, participants predicted the Gibbs free energies and pKa values of 22 N-acetylsulfonamides possessing up to three acid/base sites. The most accurate physics-based quantum mechanical (QM) methods combined conformational sampling, DFT, model solvent, and a linear empirical correction (QM + LEC). The best QM + LEC methods gave root-mean-square errors (RMSE) below 0.7log units.2 In the SAMPL7 challenge, participants predicted pKa values for 22 N-acylsulfonamides and related bioisosteres. Participants determined experimental values (“macroscopic pKa”) from the computed free energies of individual protonation tautomers (“microscopic pKa”). Only one physics-based method gave RMSE below 1log unit.29 Moreover, there was significant disagreement as to which microscopic transitions produced the measured pKa, i.e., which prototropic tautomers were most stable at each pH. In the SAMPL8 challenge, participants considered more diverse compounds including several polybasic species. A QM protocol combining conformational sampling, DFT, and the COSMO-RS solvation model yielded RMSE 1.65log units when using assignment based on the experimental transition curves.30
1.3. Overview
We use state-of-the-art physics-based computational protocols to predict the pKa of previously un-synthesized polybasic tetra-aza macrocycles. We employ protocols similar to those applied in the SAMPL6 and SAMPL7 challenges. We use these protocols to predict the pKa and pH-dependent speciation of molecules 11–14, four previously un-synthesized tetra-aza macrocycles. Table 2 reports the final predictions. We validate these protocols against the experimental pKa and pH-dependent speciation behavior of previously synthesized macrocycles. Table 1 shows the experimental pKa values and predictions of our preferred protocol. This protocol provides useful accuracy consistent with results from the SAMPL challenges. In an attempt to further refine our predictions, we systematically test the effects of the different approximations employed. We find that solvation of highly charged species is a significant source of remaining errors. Future studies will experimentally test the predictions for molecules 11–14 and will use this protocol to predict pKa values of other tetra-aza macrocycles.
Table 2 Predicted pKa values for macrocycles 11–14
| Molecule |
pKa |
Prediction |
| 11 |
1 |
7.73 |
| 2 |
5.83 |
| 12 |
1 |
10.06 |
| 2 |
9.11 |
| 13 |
1 |
8.52 |
| 2 |
7.14 |
| 14 |
1 |
6.67 |
| 2 |
6.39 |
2. Methods
Most calculations in this work use a common computational workflow. For each molecule of interest, the user provides a three-dimensional structure in which all N protonation sites are occupied, along with a list of the N exchangeable protons (ESI Fig. SI1,† blue nitrogen atoms). To illustrate, molecule 1 starts with a structure of the charge +4 species with N = 4 protonated nitrogen atoms. The approach is “black-box” in that any molecule of interest may be treated, as long as the protonated 3D structure and possible protonation sites are known. A Python script using the itertools library automatically generates all possible protonation isomers by removing all possible combinations of exchangeable protons. To illustrate, molecule 1 gives 14 protonation isomers spanning five charge states: one neutral isomer, four protonation isomers with charge +1, six isomers with charge +2, four with charge +3, and one (the original input structure) with charge +4. Spatial symmetry is not used: for example, two of the charge +1 structures for molecule 1 are redundant, with protonation on either of the two symmetry-equivalent secondary amines adjacent to the pyridine.
The computed three-dimensional structure of each protonation isomer is used as input for a metadynamics and molecular dynamics simulation using the Conformer-Rotamer Ensemble Sampling Tool (CREST).33 CREST calculations use the GFN2-xTB tight binding Hamiltonian,34 the generalized Born with surface area contributions (GBSA) continuum model for water solvent,35 and the iMTD-GC metadynamics-based exploration of conformational space employing a biasing potential expressed with the root-mean-square deviation in Cartesian space as a metric for the collective variables.36 The five lowest-energy conformations generated by CREST are refined using a Gaussian 16 DFT geometry optimization and free energy calculation.37 These calculations use density functional theory in an atomic orbital (AO) basis set to treat the molecule, and a continuum solvent model to treat the water solvent. Solvent is modeled using either the SMD or IEFPCM continuum models for water solvent, employing the default parameters for, e.g., solvent static dielectric constant.38,39 The calculations also use 6-31G(d) or def2-TZVP atomic orbital basis sets,40,41 and the B3LYP42,43 or M06-2X44 exchange-correlation functionals. All geometry optimizations and vibrational frequency calculations are performed in continuum solvent.45,46 Gibbs free energies are taken to be the free-particle-rigid-rotor-harmonic-oscillator free energy of the lowest energy conformation of each protonation state. All calculations treat temperature T = 298.15 K. The workflow is implemented as a set of Python and Perl scripts which write and process CREST and Gaussian input and output files. This implementation is freely available at the Janesko group GitHub site.
QM calculations compute the pKa as ΔG*/RT(ln10). For any acid HAn with charge n, we compute ΔG* as the Gibbs free energy of the dissociation reaction
| | |
HAn (aq) ⇄ H+ (aq) + An−1 (aq)
| (1) |
| | |
G* = Gcomput (An−1, aq) + Gexpt (H+, aq) − Gcomput (HAn, aq)
| (2) |
The computed Gibbs free energies of HAn and An−1 are taken directly from the Gaussian output files. Gexpt (H+, aq) denotes the experimental Gibbs free energy of the hydrated proton at 298.15 K and standard state concentration 1 mol L−1. This is computed as described in ref. 47.
| | |
Gexpt(H+, aq) = G0g (H+) + ΔG1atm → 1 M + ΔGaq,solv (H+)
| (3) |
The standard Gibbs energy of the gas phase proton, treated as an ideal gas at gas-phase concentration 1 bar, is taken as G0g (H+) = −6.28 kcal mol−1. This is calculated as G0g= H0g − TS0g where H0g = (5/2) RT and S0g = 26.05 cal (mol−1·K−1).48 The factor ΔG1atm → 1 M = 1.89 kcal mol−1 accounts for change of the state from 1 bar to 1 mol L−1. The aqueous solvation free energy of the proton at concentration 1 mol L−1 ΔGaq,solv (H+) = −265.9 kcal mol−1 is taken from the work of Tissandier et al.,49 corrected to treat an ideal gas at a gas-phase concentration of 1 mol L−1 dissolving as an ideal solution at a liquid-phase concentration of 1 mol L−1 as discussed by Kelley et al.50 QM + LEC calculations employ a linear empirical correction (LEC)
| | |
pKa (corrected) = a × pKa (computed) + b
| (4) |
Parameters a and b are obtained as a best-fit to the experimental data in Table 1. In addition to the tests of basis set, continuum solvent, and exchange-correlation functional discussed above, several test calculations treat other aspects of the computational workflow. Test calculations using only computed solvation free energies compute ΔG* as
| | |
HAn (aq) + H2O (aq) ⇄ H3O+ (aq) + An−1 (aq)
| (5) |
| | |
ΔG* = Gcomput (An−1, aq) + ΔGexpt (H3O+, aq) − ΔGcomput (HAn, aq) − ΔGcomput (H2O, aq)
| (6) |
Other test calculations combine eqn (5) and (6) with the pK-Yay correction.51 Test calculations employing explicit + implicit water solvent use the QCG quantum cluster growth approach to determine the conformations of the added water molecules.52 This approach uses combined metadynamics and molecular dynamics with the intermolecular force field xTB-IFF53 to grow molecule-solvent clusters, one solvent molecule at a time. The DFT approximations are tested by comparison to the CBS-QB3 ab initio composite approach as implemented in Gaussian 16.54 We compare DFT and CBS-QB3 calculations on the aqueous-phase pKa of six small molecules structurally similar to the tetra-aza macrocycles: dimethyl amine DMA, trimethyl amine TMA, pyridine, 3-hydroxy pyridine 3HP, 4-hydroxy pyridine 4HP, and phenol. Experimental pKa of these molecules are taken from the Bordwell tables.34,55–57
3. Results
3.1 Validation
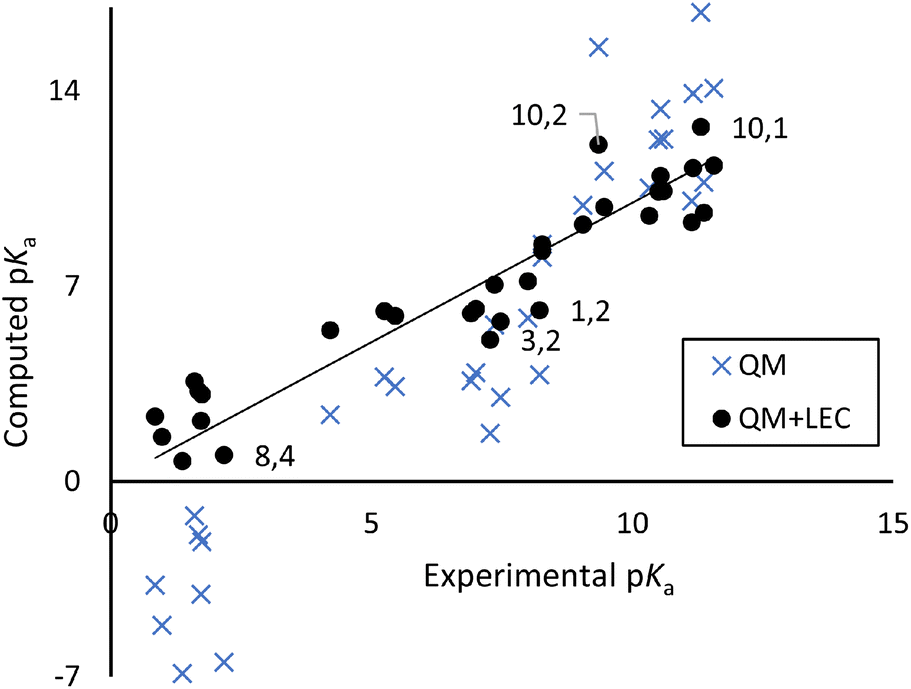
Table 1 compares measured pKa values to those computed with our preferred QM and QM+LEC method: CREST structure optimization, M06-2X/def2-TZVP structure refinement with the SMD continuum solvent model, and pKa computed from the experimental proton solvation free energy. Fig. 2 shows a scatter plot of experimental vs. QM+LEC predicted pKa. Uncorrected QM results give root-mean-square-deviation (RMSD) 3.88log units. LEC significantly improves the results, with RMSD 1.21log units comparable to those seen in previous SAMPL challenges. We regard this as significant in that the present work includes flexible species with up to six exchangeable protons and measured pKa spanning a range of ten log units.
 |
| | Fig. 2 Scatter plot of experimental vs. computed pKa from Table 1. Selected points are labelled as “molecule, pKa (n)”. | |
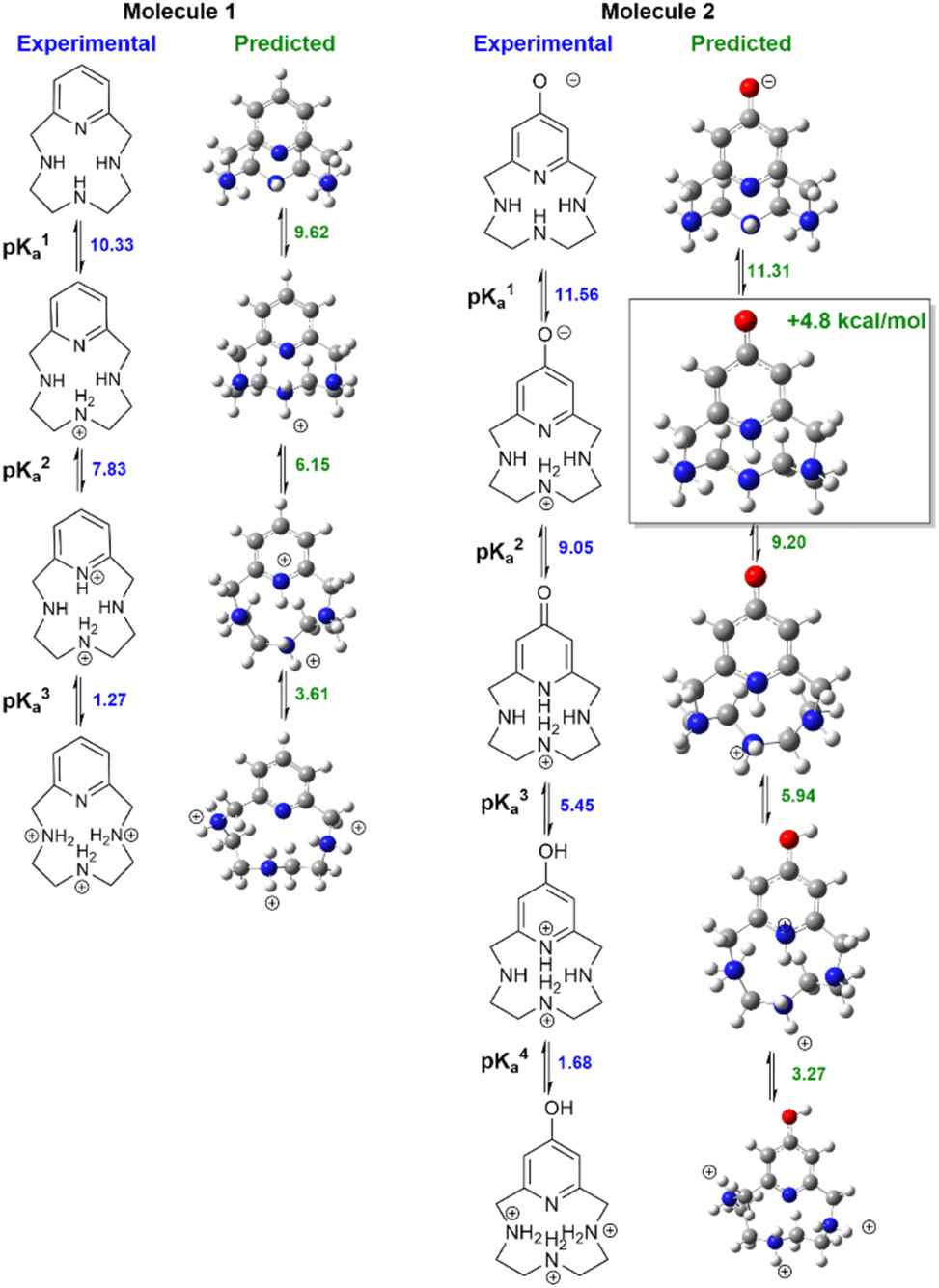
Fig. 3 compares measured lowest-energy protomers to those computed with our preferred QM+LEC method. The predicted most stable protomers show excellent agreement with experiment. The charge +1 state of molecule 1 is correctly predicted to be protonated at nitrogen N2 (see atom labeling in Fig. 1). The charge +2 state is correctly predicted to be protonated at N2 and pyridine nitrogen N4. The charge +3 state is correctly predicted to show tautomerism, protonated at nitrogen N1, N2, N3 and not protonated at pyridine nitrogen N4.
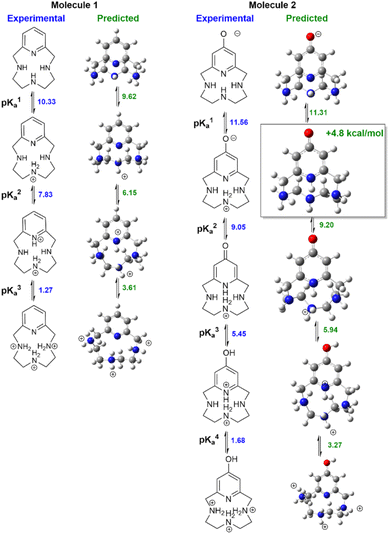 |
| | Fig. 3 Speciation of molecules 1–2. Left columns show experimentally determined stable protomers, right columns show the predicted stable protomers. | |
Our protocol also provides reasonable predictions for molecule 2. The charge +1 state is correctly predicted to have the 4-pyridone structure with protonation at N2. The charge +3 state is correctly predicted to be protonated at oxygen, yielding protonated 4-hydroxy-pyridine and protonation at N2. The charge +2 state is correctly predicted to show a significant rearrangement with protonation at N1–N3. The only discrepancy is that the most stable charge-neutral state is predicted to be the 4-pyridone tautomer. This is predicted to be 4.8 kcal mol−1 more stable than the zwitterion assigned in ref. 27. Calculations of NMR chemical shifts upon protonation confirm that the zwitterionic structure best reproduces experimental NMR titration data (ESI Table SI1†). Calculations suggest that the zwitterionic structure has a degree of proton sharing between N2 and N4 (ESI Fig. SI2†). The suggestion of proton sharing is consistent with the basicity of the moieties involved: the measured pKa of 4-pyridone in water is 11.09 at 20° (ref. 58 ), significantly higher than the measured pKa of aliphatic secondary amines (dimethylamine 10.922 at 20° ref. 59) and quite close to the measured first pKa of molecule 2. We regard this agreement as particularly significant given the discrepancies in predicted protonation tautomers seen in previous SAMPL challenges.29
3.2. Other computational protocols
Table 3 reports RMSD in pKa computed with different choices of approximate density functional, basis set, solvent model, and choice of ΔG. Here “TZP” denotes the def2-TZVP basis set and "DZ" denotes the 6-31+G basis set. For each choice, we report RMSD from QM and QM + LEC calculations and include the LEC parameters a, b (eqn (4)). The choice of density functional has a relatively small effect, with B3LYP and M06-2X giving comparable QM + LEC RMSD. The basis set has a modest effect, with TZP giving an ∼0.1log unit improvement over DZ. The solvent model significantly affects the uncorrected results, with IEFPCM giving RMSD values 6–8log units larger than those with SMD. This is true for both B3LYP and M06-2X DFT calculations. This appears to be a systematic error corrected by the LEC. QM+LEC RMSD are comparable between SMD and IEFPCM models. The source of this systematic error appears to involve an insufficient charge screening leading to progressive destabilization of highly charged species. To illustrate this in detail, we consider the difference between the first and fourth pKa for molecule 2. Experiments give a difference of 9.9log units. M06-2X/def2-TZVP calculations give a difference of 33log units for the IEFPCM model, but a difference of only 16log units for the SMD model. This difference is reflected in the slope a of the LEC, which is around 0.5 for SMD solvent and around 0.3 for IEFPCM solvent. QM calculations using only computed solvation free energies (eqn (3) and (4)) give a much larger RMSD, a result which is improved by the pK-Yay correction. Eqn (3) and (4) and the pK-Yay correction do not change the results obtained after LEC.
Table 3 RMSD in pKa (log units) computed with different model chemistries (density functional, basis set, continuum solvent), different choices of ΔG, and linear empirical corrections LEC. Column “Eqn (4) LEC a, b” denotes the optimum values of the fitted parameters in the LEC eqn (4)
| |
ΔG |
RMSD |
Eqn (4) LEC |
| Model chemistry |
QM |
QM + LEC |
a, b |
| M06-2X/TZP/SMD |
Eqn (2) |
3.88 |
1.20 |
0.50, 4.22 |
| M06-2X/DZ/SMD |
Eqn (2) |
3.75 |
1.33 |
0.52, 4.19 |
| M06-2X/TZ/PCM |
Eqn (2) |
11.54 |
1.20 |
0.28, 7.08 |
| B3LYP/TZP/SMD |
Eqn (2) |
3.84 |
1.30 |
0.49, 3.18 |
| B3LYP/TZP/PCM |
Eqn (2) |
10.36 |
1.18 |
0.28, 6.45 |
| M06-2X/TZP/SMD |
Eqn (5) |
13.7 |
1.21 |
0.28, 5.17 |
| M06-2X/TZP/SMD |
pKYaY |
9.45 |
1.21 |
0.50, −3.20 |
To confirm that the density functional and basis set have a limited effect on the accuracy, ESI Table SI2† reports a small benchmark study of rigid molecules structurally similar to tetra-aza macrocycles. For these molecules, the effect of conformational flexibility and prototropic tautomerism are minimized. RMSD obtained with the accurate ab initio composite approach CBS-QB3 in model solvent are comparable to that obtained with DFT. This strongly suggests that errors in the QM+LEC results in Table 1 arise mostly from the model solvent.
3.3 Explicit solvent
Additional insight comes from considering the role of explicit solvent. Hybrid explicit+continuum solvation models can significantly improve pKa prediction, especially for sets of related molecules where the position of explicit solvent is well-defined (e.g., two explicit water molecules hydrogen-bonded to a monocarboxylic acid).60,61 However, for flexible polybasic macrocycles, the optimum position and orientation of explicit solvent is difficult to determine a priori.
Fig. 4 presents an initial study for molecule 1, showing pKa computed (QM) with increasing numbers of explicit solvent molecules. Geometries are obtained using the quantum cluster growth (QCG) algorithm.52 ESI Fig. SI3† shows results for the hydroxypyridines used in the ab initio benchmark. Explicit solvent improves the computed pKa values, consistent with previous studies.62 The predicted low-energy solvent configurations appear chemically reasonable. However, the improvement is not monotonic with increasing number of solvent molecules. This is not a special limitation of the QCG conformation search, it is an intrinsic limitation of any hybrid explicit+continuum solvent model.
 |
| | Fig. 4 Explicit solvent calculations for molecule 1. (left) First (blue), second (red), and third (black) pKa. Horizontal dashed lines are experimental values, other lines are calculations with 0–5 explicit water molecules. (right) Computed structures for charge +2 state with 0–5 explicit water molecules. | |
3.4. Predicted pKa
We conclude by reported the predicted pKa values for novel tetra-aza macrocycles 11–14. Table 2 reports the QM+LEC results. These are computed with our preferred method: CREST/xTB structure optimization, M06-2X/def2-TZVP structure refinement with the SMD continuum solvent model, pKa computed from the experimental proton solvation free energy, and LEC a = 0.5 and b = 4.22. The computed values are chemically reasonable. Electron-withdrawing groups R = Cl and R = CF3 reduce the pKa values relative to molecule 9 (R = H). Electron-donating groups R = NMe2 and R = OMe increase the pKa values relative to molecule 9. Molecule 12 (R = NMe2) is predicted to have both pKa well above 7, suggesting that it will be doubly protonated at physiological pH. Molecules 11 and 14 (R = Cl and R = CF3) are predicted to have both pKa around or less than 7, suggesting that both molecules may have an appreciable concentration of charge-neutral tautomer at physiological pH.
4. Conclusions
Accurate prediction of the pKa values, protonation sites, and pH-dependent speciation of polybasic drug candidates remains a significant challenge in computational medicinal chemistry. Here we used black-box methods to predict the pKa and speciation of four previously un-tested tetra-aza macrocycle small molecules. These flexible molecules possess four to six acid/base sites and pose a significant challenge for pKa prediction. This work included 32 pKa values measured for 10 different aza-macrocyclic molecules as a baseline. The computational workflow employed combines exhaustive search over protonation tautomers, continuum models for water, CREST metadynamics and molecular dynamics for conformational sampling, modern density functional theory (DFT), SMD continuum solvent, and a linear empirical correction. Baseline studies give an RMSD within 1.2log units of experimentally measured values and accurate predictions of the most stable protomer at each charge state. The predicted pKa values for the previously un-tested macrocycles are chemcially reasonable. Our results highlight a significant step toward predicting the pKa of large flexible molecules.
Data availability
The data supporting this article have been included as part of the ESI.† Scripts for executing the computational workflow are available at https://github.com/bjanesko/DFTPropertyPredictionWorkflows.
Author contributions
Investigation (T. K. H, K. P., M. M., D. M. F.), formal analysis (T. K. H, D. M. F., B. G. J, K. N. G.), writing (B. G. J., K. N. G.).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge support from NIH NIGMS 2R15GM123463, Robert A. Welch Foundation grant P-2195-20240404, and Flatley Foundation through TCU. The authors are grateful for the support provided by the TCU High Performance Computing Center.
Notes and references
- J. D. Watson and F. H. C. Crick, Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid, Nature, 1953, 171(4356), 737–738 CrossRef CAS PubMed.
- M. Işık, A. S. Rustenburg, A. Rizzi, M. R. Gunner, D. L. Mobley and J. D. Chodera, Overview of the SAMPL6 pKa challenge: evaluating small molecule microscopic and macroscopic pKa predictions, J. Comput. Aided Mol. Des., 2021, 35(2), 131–166 CrossRef PubMed.
- D. Garcia Jimenez, M. Vallaro, M. Rossi Sebastiano, G. Apprato, G. D'Agostini, P. Rossetti, G. Ermondi, G. Caron and A. Chamelogk, Chromatographic Chameleonicity Quantifier to Design Orally Bioavailable Beyond-Rule-of-5 Drugs, J. Med. Chem., 2023, 66(15), 10681–10693 CrossRef CAS PubMed.
- P. D. Leeson, A. P. Bento, A. Gaulton, A. Hersey, E. J. Manners, C. J. Radoux and A. R. Leach, Target-Based Evaluation of “Drug-Like” Properties and Ligand Efficiencies, J. Med. Chem., 2021, 64(11), 7210–7230 CAS.
- B. Faller and P. Ertl, Computational approaches to determine drug solubility, Adv. Drug Deliv. Rev., 2007, 59(7), 533–545 CrossRef CAS PubMed.
- J. L. Domínguez, M. C. Villaverde and F. Sussman, Effect of pH and ligand charge state on BACE-1 fragment docking performance, J. Comput. Aided Mol. Des., 2013, 27(5), 403–417 CrossRef PubMed.
- S. Ono, M. R. Naylor, C. E. Townsend, C. Okumura, O. Okada, H.-W. Lee, R. S. Lokey and A. Cyclosporin, Conformational Complexity and Chameleonicity, J. Chem. Inf. Model., 2021, 61(11), 5601–5613 CrossRef CAS PubMed.
- M. Marín-Luna, I. Alkorta and J. Elguero, A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles, New J. Chem., 2015, 39(4), 2861–2871 RSC.
- E. L. Jones, A. J. Mlotkowski, S. P. Hebert, H. B. Schlegel and C. S. Chow, Calculations of pKa Values for a Series of Naturally Occurring Modified Nucleobases, J. Phys. Chem. A, 2022, 126(9), 1518–1529 CrossRef CAS PubMed.
- B. Thapa and H. B. Schlegel, Calculations of pKa's and Redox Potentials of Nucleobases with Explicit Waters and Polarizable Continuum Solvation, J. Phys. Chem. A, 2015, 119(21), 5134–5144 CrossRef CAS PubMed.
- J. Ho, Predicting pKa in Implicit Solvents: Current Status and Future Directions<a class, Aust. J. Chem., 2014, 67(10), 1441–1460 CrossRef CAS.
- J. Ho and M. L. Coote, A universal approach for continuum solvent pKa calculations: are we there yet?, Theor. Chem. Acc., 2010, 125(1), 3–21 Search PubMed.
- R. Casasnovas, J. Frau, J. Ortega-Castro, A. Salvà, J. Donoso and F. Muñoz, Absolute and relative pKa calculations of mono and diprotic pyridines by quantum methods, J. Mol. Struct.:THEOCHEM, 2009, 912(1), 5–12 CAS.
- A. H. Göller, Reliable gas-phase tautomer equilibria of drug-like molecule scaffolds and the issue of continuum solvation, J. Comput.-Aided Mol. Des., 2022, 36(11), 805–824 Search PubMed.
- M. Schilling and S. Luber, Determination of pKa Values via ab initio Molecular Dynamics and its Application to Transition Metal-Based Water Oxidation Catalysts, Inorganics, 2019, 7(6), 73 CrossRef CAS.
- E. Danelius, V. Poongavanam, S. Peintner, L. H. E. Wieske, M. Erdélyi and J. Kihlberg, Solution Conformations Explain the Chameleonic Behaviour of Macrocyclic Drugs, Chem.–Eur. J., 2020, 26(23), 5231–5244 CrossRef CAS PubMed.
- K. M. Lincoln, M. E. Offutt, T. D. Hayden, R. E. Saunders and K. N. Green, Structural, Spectral, and Electrochemical Properties of Nickel(II), Copper(II), and Zinc(II) Complexes Containing 12-Membered Pyridine- and Pyridol-Based Tetra-aza Macrocycles, Inorg. Chem., 2014, 53(3), 1406–1416 CAS.
- M. A. Mekhail, K. Pota, T. M. Schwartz and K. N. Green, Functionalized pyridine in pyclen-based iron (iii) complexes: evaluation of fundamental properties, RSC Adv., 2020, 10(52), 31165–31170 CAS.
- K. M. Lincoln, P. Gonzalez, T. E. Richardson, L. Rutter, D. A. Julovich, J. W. Simpkins and K. N. Green, A potent antioxidant small molecule aimed at targeting metal-based oxidative stress in neurodegenerative disorders, Chem. Commun., 2013, 49, 2712–2714 CAS.
- M. A. Mekhail, K. J. Smith, D. M. Freire, K. Pota, N. Nguyen, M. E. Burnett and K. N. Green, Increased Efficiency of a Functional SOD Mimic Achieved with Pyridine Modification on a Pyclen-Based Copper(II) Complex, Inorg. Chem., 2023, 62(14), 5415–5425 CAS.
- N. Hamon, A. Roux, M. Beyler, J. C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, Pyclen-Based Ln(III) Complexes as Highly Luminescent Bioprobes for In Vitro and In Vivo One- and Two-Photon Bioimaging Applications, J. Am. Chem. Soc., 2020, 142(22), 10184–10197 CAS.
- C. Robic, M. Port, O. Rousseaux, S. Louguet, N. Fretellier, S. Catoen, C. Factor, S. Le Greneur, C. Medina, P. Bourrinet, I. Raynal, J. M. Idée and C. Corot, Physicochemical and Pharmacokinetic Profiles of Gadopiclenol A New Macrocyclic Gadolinium Chelate With High T1 Relaxivity, Invest. Radiol., 2019, 54(8), 475–484 CrossRef CAS PubMed.
- H. M. Johnston, K. Pota, M. M. Barnett, O. Kinsinger, P. Braden, T. M. Schwartz, E. Hoffer, N. Sadagopan, N. Nguyen, Y. Yu, P. Gonzalez, G. Tircsó, H. Wu, G. Akkaraju, M. J. Chumley and K. N. Green, Enhancement of the Antioxidant Activity and Neurotherapeutic Features through Pyridol Addition to Tetraazamacrocyclic Molecules, Inorg. Chem., 2019, 58(24), 16771–16784 CrossRef CAS PubMed.
- D. M. Freire, D. Beeri, K. Pota, H. M. Johnston, P. Palacios, B. S. Pierce, B. D. Sherman and K. N. Green, Hydrogen peroxide disproportionation with manganese macrocyclic complexes of cyclen and pyclen, Inorg. Chem. Front., 2020, 7, 1573–1582 RSC.
- K. M. Lincoln, T. E. Richardson, L. Rutter, P. Gonzalez, J. W. Simpkins and K. N. Green, An N-heterocyclic amine chelate capable of antioxidant capacity and amyloid disaggregation, ACS Chem. Neurosci., 2012, 3(11), 919–927 CrossRef CAS PubMed.
- J. Zhang, Y. Yu, M. A. Mekhail, H. Wu and K. N. Green, A macrocyclic molecule with multiple antioxidative activities protects the lens from oxidative damage, Front. Chem., 2022, 10 DOI:10.3389/fchem.2022.996604.
- K. N. Green, K. Pota, G. Tircsó, R. A. Gogolák, O. Kinsinger, C. Davda, K. Blain, S. M. Brewer, P. Gonzalez, H. M. Johnston and G. Akkaraju, Dialing in on pharmacological features for a therapeutic antioxidant small molecule, Dalton Trans., 2019, 48, 12430–12439 RSC.
- A. Yepremyan, A. Mehmood, S. M. Brewer, M. M. Barnett, B. G. Janesko, G. Akkaraju, E. E. Simanek and K. N. Green, A new triazine bearing a pyrazolone group capable of copper, nickel, and zinc chelation, RSC Adv., 2018, 8(6), 3024–3035 RSC.
- T. D. Bergazin, N. Tielker, Y. Zhang, J. Mao, M. R. Gunner, K. Francisco, C. Ballatore, S. M. Kast and D. L. Mobley, Evaluation of log P, pKa, and log D predictions from the SAMPL7 blind challenge, J. Comput. Aided Mol. Des., 2021, 35(7), 771–802 CrossRef CAS PubMed.
- M. Diedenhofen, F. Eckert and S. Terzi, COSMO-RS blind prediction of distribution coefficients and aqueous pKa values from the SAMPL8 challenge, J. Comput. Aided Mol. Des., 2023, 37(8), 395–405 CrossRef CAS PubMed.
- M. A. Mekhail, K. Pota, T. M. Schwartz and K. N. Green, Functionalized pyridine in pyclen-based iron (iii) complexes: evaluation of fundamental properties, RSC Adv., 2020, 10(52), 31165–31170 RSC.
- A. Yepremyan, M. A. Mekhail, B. P. Niebuhr, K. Pota, N. Sadagopan, T. M. Schwartz and K. N. Green, Synthesis of 12-Membered Tetra-aza Macrocyclic Pyridinophanes Bearing Electron-Withdrawing Groups, J. Org. Chem., 2020, 85(7), 4988–4998 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Automated exploration of the low-energy chemical space with fast quantum chemical methods, Phys. Chem. Chem. Phys., 2020, 22(14), 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions, J. Chem. Theory Comput., 2019, 15(3), 1652–1671 CrossRef CAS PubMed.
- A. V. Onufriev and D. A. Case, Generalized Born Implicit Solvent Models for Biomolecules, Annu. Rev. Biophys., 2019, 48(1), 275–296 CrossRef CAS PubMed.
- S. Grimme, Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations, J. Chem. Theory Comput., 2019, 15(5), 2847–2862 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel; G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci; H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Williams; Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105(8), 2999–3094 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72(1), 650–654 CrossRef CAS.
- A. D. Becke, A new mixing of Hartree–Fock and local density-functional theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, A comparison of models for calculating nuclear magnetic resonance shielding tensors, J. Chem. Phys., 1996, 104(14), 5497–5509 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- J. Ho, A. Klamt and M. L. Coote, Comment on the Correct Use of Continuum Solvent Models, J. Phys. Chem. A, 2010, 114(51), 13442–13444 CAS.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation, J. Phys. Chem. B, 2011, 115(49), 14556–14562 CAS.
- B. Thapa and H. B. Schlegel, Calculations of pKa's and Redox Potentials of Nucleobases with Explicit Waters and Polarizable Continuum Solvation, J. Phys. Chem., 2015, 119(21), 5134–5144 CAS.
- G. J. Tawa, I. A. Topol, S. K. Burt, R. A. Caldwell and A. A. Rashin, Calculation of the aqueous solvation free energy of the proton, J. Chem. Phys., 1998, 109(12), 4852–4863 CAS.
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, The Proton's Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data, J. Phys. Chem., 1998, 102(40), 7787–7794 CAS.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, Aqueous Solvation Free Energies of Ions and Ion−Water Clusters Based on an Accurate Value for the Absolute Aqueous Solvation Free Energy of the Proton, J. Phys. Chem. B, 2006, 110(32), 16066–16081 CAS.
- R. W. Pereira and R. O. Ramabhadran, pK-Yay: A Black-Box Method Using Density Functional Theory and Implicit Solvation Models to Compute Aqueous pKa Values of Weak and Strong Acids, J. Phys. Chem., 2020, 124(43), 9061–9074 CAS.
- J. Gorges, S. Grimme, A. Hansen and P. Pracht, Towards understanding solvation effects on the conformational entropy of non-rigid molecules, Phys. Chem. Chem. Phys., 2022, 24(20), 12249–12259 CAS.
- S. Grimme, C. Bannwarth, E. Caldeweyher, J. Pisarek and A. Hansen, A general intermolecular force field based on tight-binding quantum chemical calculations, J. Chem. Phys., 2017, 147(16), 161708 Search PubMed.
- J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, A complete basis set model chemistry. VI. Use of density functional geometries and frequencies, J. Chem. Phys., 1999, 110(6), 2822–2827 Search PubMed.
- F. G. Bordwell, Equilibrium acidities in dimethyl sulfoxide solution, Acc. Chem. Res., 1988, 21(12), 456–463 CAS.
- F. G. Bordwell, X. Zhang and M. S. Alnajjar, Effects of adjacent acceptors and donors on the stabilities of carbon-centered radicals, J. Am. Chem. Soc., 1992, 114(20), 7623–7629 CAS.
- S. Yadav, S. Rana, M. Manish, S. Singh, A. Lynn and P. Mathur, In silico design of dehydrophenylalanine containing peptide activators of glucokinase using pharmacophore modelling, molecular dynamics and machine
learning: implications in type 2 diabetes, J. Comput. Aided Mol. Des., 2024, 39(1), 5 CrossRef PubMed.
- A. Albert and A. Hampton, Analogues of 8-hydroxyquinoline having additional cyclic nitrogen atoms. Part II. Further preparations, and some physico-chemical properties, J. Chem. Soc., 1954, 505–513 RSC.
- D. H. Everett, W. F. K. Wynne-Jones and H. B. Hartley, The dissociation constants of the methylammonium ions and the basic strengths of the methylamines in water, Proc. Roy. Soc. Lond. Math. Phys. Sci., 1941, 177(971), 499–516 CAS.
- S. Pezzola, M. Venanzi, P. Galloni, V. Conte and F. Sabuzi, Easy to Use DFT Approach for Computational pKa Determination of Carboxylic Acids, Chem.–Eur. J., 2024, 30(1), e202303167 CrossRef CAS.
- S. Pezzola, S. Tarallo, A. Iannini, M. Venanzi, P. Galloni, V. Conte and F. Sabuzi, An Accurate Approach for Computational pKa Determination of Phenolic Compounds, Molecules, 2022, 27(23), 8590 CrossRef CAS PubMed.
- S. A. Rodriguez, J. V. Tran, S. J. Sabatino and A. S. Paluch, Predicting octanol/water partition coefficients and pKa for the SAMPL7 challenge using the SM12, SM8 and SMD solvation models, J. Comput. Aided Mol. Des., 2022, 36(9), 687–705 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: M06-2X/def2TZVP/SMD geometries and free energies of all species. See DOI: https://doi.org/10.1039/d5ra01015b |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kristof Pota
a,
Kristof Pota