
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilver nanoparticles from metallic silver via electrochemical synthesis-polyol reduction†
Chenyi Zheng abc,
Lianghong Duanabc,
Songsong Wangabc,
Qiang Wangbcd,
Qinmeng Wang*abc and
Xueyi Guo*abc
abc,
Lianghong Duanabc,
Songsong Wangabc,
Qiang Wangbcd,
Qinmeng Wang*abc and
Xueyi Guo*abc
aSchool of Metallurgy and Environment, Central South University, Changsha 410083, China. E-mail: qmwang@csu.edu.cn; xyguo@csu.edu.cn
bNational & Regional Joint Engineering Research Centre of Nonferrous Metal Resources Recycling, Changsha 410083, China
cHunan Key Laboratory of Nonferrous Metal Resources Recycling, Changsha 410083, China
dSchool of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
First published on 17th April 2025
Abstract
Silver nanoparticles (Ag NPs) possess unique physicochemical properties, making them valuable in various applications. The polyol reduction (PR) method is a prominent approach for synthesizing Ag NPs. However, traditional PR methods rely on Ag compounds like AgNO3 as feedstock to prepare precursor solution, which increases production time and costs. This study introduces a streamlined, eco-friendly technique to Ag NP synthesis via PR. Low-cost metallic Ag serves as a feedstock, and electrochemical synthesis (ES) is employed to dissolve the metallic Ag in ethylene glycol (EG), generating a precursor solution for PR. Additives are added into the precursor solution, which is then heated to synthesize Ag NPs. By utilizing the additives and the temperature-dependent reducibility of EG, Ag nanowires and purified Ag NPs are synthesized from pure and crude-Ag precursors, respectively. The ES-PR method retains the advantages of the PR method while eliminating the need for Ag compounds in precursor preparation. Additionally, H2 gas is produced as a byproduct, offering further benefits. The ES-PR method has the potential to significantly simplify the synthesis of Ag NPs via PR, facilitating the broader application of Ag NPs.
1. Introduction
Silver nanoparticles (Ag NPs) garner significant attention due to their unique physicochemical properties, such as conductivity and antimicrobial activity. Over the last decade, their global consumption has seen a notable increase,1 particularly in the electronics,2 healthcare,3 photovoltaic,4 and textile5 industries. The robust demand for Ag NPs has necessitated advancements in Ag NP synthesis techniques. Currently, the primary technologies for Ag NP synthesis include physical,6–9 electrochemical10–14 and chemical reduction15–19 processes, their characteristics are summarized in Table S1.†The chemical reduction process, renowned for its advantages in simple experimental setup and high productivity, stands as the most popular process for Ag NP synthesis. Notably, the polyol reduction (PR), a type of solvothermal method, has been recognized for its superior morphology control over Ag NPs for decades. Polyols like ethylene glycol (EG), undergo thermal decomposition, generating aldehydes and other reducing agents, these products reduce Ag+ slowly, enabling precise morphological control when appropriate additives and stabilizers are employed.20 Xia et al. prepared cubic, spherical, rod, and wire-shaped Ag NPs by adjusting polyvinyl pyrrolidone (PVP) amounts and reaction temperatures.15,19 Moreover, PR is considered to be the most effective method for large-scale synthesis of silver nanowires (Ag NWs).21
However, unlike physical and electrochemical processes that can use inexpensive metallic Ag to synthesize Ag NPs (see Table S1†), the PR method relies on Ag compounds as a feedstock for preparing precursor solution. AgNO3 is the most commonly used compound due to its high solubility in polyol. The production of AgNO3 is time-consuming and requires a large amount of H2O2 to suppress the NOx generation, as noted in ESI Note 1.†22–24 On the other hand, from the perspective of industry chain, metallic Ag is the raw-material of AgNO3, making it cheaper and greener compared to AgNO3. If metallic Ag can be directly used to produce the precursor solution for the PR method, not only can it reduce production costs and simplify processes, but it may also maintain the advantages of the PR method. To our knowledge, this possibility has received insufficient attention in the existing literature, and no studies have been reported using a feedstock other than Ag compounds to prepare the precursor for PR method.
Electrochemical synthesis (ES) is a green and sustainable method for the production of inorganic and organometallic compounds or their precursors.25,26 ES possesses the following advantages: (1) the application of electrons as an “inexhaustible” reactant. (2) Anodic dissolution (AD) of metals can be utilized to produce metal ions. (3) The electrolysis cell can be separated using an ion-exchange membrane, thereby preventing unwanted reactions involving intermediates and enriching the intermediates. For instance, Gafurov et al. prepared organonickel sigma-complexes using nickel as a sacrificial anode in an electrolyte containing 2,2′-bipyridine. Throughout the ES process, only nickel, 2,2′-bipyridine, and aromatic bromide were consumed, thereby avoiding the use of Grignard reagents and environmental hazards found in traditional methods.27 The chlor-alkali industry typically electrolyzes brine in cells separated by a cation-exchange membrane (CEM), where Cl2 gas is generated at the anode and H2 gas along with sodium hydroxide solution is produced at the cathode. The presence of the CEM prevents the O2 evolution at the anode, thereby enabling the enrichment of sodium hydroxide in the catholyte.25 Xie et al. conducted acid enrichment in a cell separated by an anion-exchange membrane (AEM), where H2SO4 was enriched to 63% in anolyte. The presence of AEM prevents the H2 evolution at the cathode.28 Notably, the AD of metallic Ag in EG solution with electrolyte has been confirmed to be viable.29 Furthermore, the stability of AEMs in EG solution has been demonstrated.30 Therefore, it is possible to prepare the precursor for the PR method directly using Ag metal as the feedstock through the ES process. This approach avoids the production of AgNO3 and thereby establishes an ES-PR method.
On the other hand, pure Ag metal is produced by electrorefining, using crude Ag as the raw-material. In 2023, Pb/Zn/Cu smelting contributed 57.5% to global Ag metal production,31 indicating a substantial crude Ag output. Electrorefining utilizes the differences in redox potentials among various metal ions to selectively reduce Ag+ at the cathode, thereby achieving purification. Intriguingly, EG exhibits temperature-dependent reducibility,32 which may allow the selective reduction of Ag+, thereby achieving purification similar to electrorefining. Although quantifying the thermodynamics of EG's reducibility presents challenges due to the complexity of the oxidized products of EG,33 Larcher et al. proposed a thermodynamic assumption to qualify the reducibility of EG. Based on their calculations, the Gibbs free energy of reduction of Ag2O is more negative than most of the metallic oxides, indicating that Ag2O more readily reduced in EG.34 Therefore, crude Ag can be used as an alternative feedstock to prepare the precursor, which can further reduce the raw material cost. Additionally, impurities in the crude-Ag precursor can be removed during the PR process.
It is noteworthy that the ES strategy can also be extended to the aqueous system for synthesizing NPs. This study focuses on the PR system, mainly due to the morphology control advantage of the PR method and the temperature-dependent reducibility of EG. Moreover, compared with the electrochemical process described in Table S1,† the ES-PR method employs direct current to prepare the precursor solution for PR, ensuring higher current efficiency (Ag+ enrichment rate discussed in Section 3.2). Additionally, the morphology control advantage of PR method is preserved.
In this study, Ag NWs and purified Ag NPs were synthesized via the ES-PR method, utilizing pure and crude Ag as feedstocks, respectively. Fig. 1 illustrates a comparative workflow between conventional AgNO3-based PR method (Fig. 1A) and the ES-PR methods (Fig. 1B and C) all starting from metallic Ag. The proposed ES-PR method employs inexpensive Ag metals as feedstocks to directly prepare precursor for PR, streamlining the processes while retaining the advantages of the PR method. Furthermore, H2 is generated as a byproduct, offering additional benefits. The ES-PR holds promise for markedly reducing the production cost and time of Ag NPs.
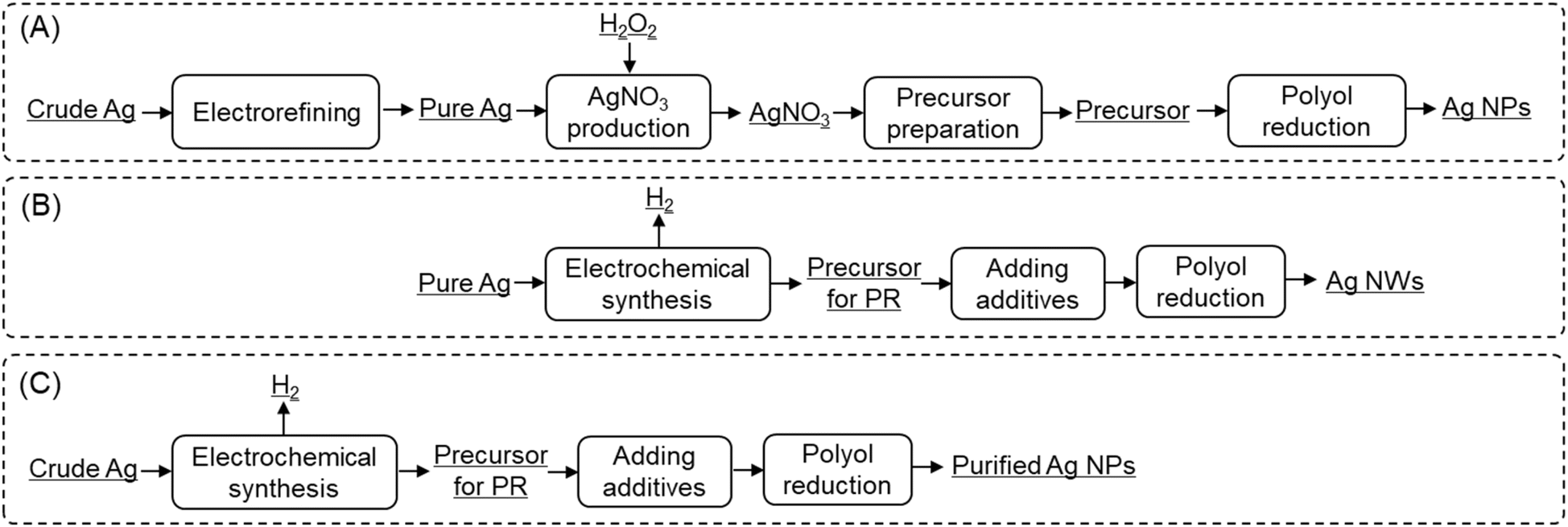 | ||
| Fig. 1 Comparative workflows for Ag NP synthesis utilizing metallic Ag as the starting material. (A) Conventional AgNO3-based polyol reduction method: AgNO3 serve as an intermediate compound for preparing precursor of PR, resulting in lengthy process and high energy consumption. (B) Ag NWs and (C) purified Ag NP synthesis employing the electrochemical synthesis-polyol reduction method in this study: the precursor for PR method can be directly produced from metallic Ag, H2 gas is generated as a byproduct. | ||
2. Material and methods
2.1 Preparation of metallic Ag samples
The pure Ag and 925-Ag sheets were purchased from Shenzhen Lifan New Material Co., Ltd. 925-Ag is an Ag–Cu–Zn alloy commonly used for silverware. It is used to simulate the application of the ES-PR method for silverware recycling. The Smelt.-Ag was prepared by melting Ag–Bi–Cu–Pb–Te mixed powders at 980 °C under argon atmosphere, the chemical composition was provided by Chenzhou Fengyue Environmental Protection Technology Co., Ltd. The Smelt.-Ag was used to simulate the application of the ES-PR method for crude Ag produced in Pb/Zn/Cu smelting. The chemical compositions of the metallic Ag feedstocks used in this study are shown in Table S2.†2.2 Electrochemical synthesis for Ag precursor solution
The ES experimental setup is shown in Fig. 2A. ES was conducted in a custom-made electrolysis cell (100 + 100 mL H-shape). The Ag sheet anodes used were 3.0 × 3.0 × 0.2 (cm) in size. Due to the small surface area and fixed spacing between the cathode and anode, the current applied in the ES process was at the mA level. The applied current could be increased when a larger electrolysis cell was employed. The electrolyte concentrations were equal in both anolyte and catholyte. A commercially available AEM, comprising a polyethylene matrix with quaternary ammonium cationic groups, was purchased from Hangzhou Huamo Technology Co., Ltd. Prior to use, the AEM was immersed in a 0.1 M aqueous solution with specific electrolyte for at least 24 hours. All electrodes underwent sandpaper polishing and ultrasonic cleaning. The pure and crude Ag anodes were encased in filter paper to prevent anodic slime from contaminating the anolyte. Heating and agitation were applied to the anolyte during ES process. Pt served as the cathode, where the gases generated were collected in a Teflon bag for further analysis. Fig. S2† shows the photographs of the ES experimental setups. | ||
| Fig. 2 Procedures and experimental setup of the electrochemical synthesis-polyol reduction method for Ag NP synthesis. (A) Metallic Ag undergoes anodic dissolution in ethylene glycol, while H2 evolution occurs at the Pt cathode, Ag+ is enriched in anolyte due to the barrier effect of anion-exchange membrane, thereby producing the precursor solution for polyol reduction. (B) The anolyte is added with additives and stabilizer to produce precursor for polyol reduction. (C) The precursor solution undergoes heating to synthesize Ag NPs via polyol reduction. | ||
2.3 Polyol reduction for Ag NP synthesis
After ES, the anolyte was analysed and then transferred to a beaker, where it was mixed with EG, additives, and stabilizers to produce precursor for PR, as shown in Fig. 2B. The PR experimental setups and reduction rate of Ag+ (R) are explained in ESI Note 2.† 20 mL precursor was introduced into a Teflon-lined stainless-steel autoclave and subjected to heating. To terminate the reaction, the autoclave was rapidly quenched in water. After cooling, the post-PR solution was diluted with deionized water and then centrifuged for subsequent analysis. The as-synthesized Ag NPs were collected by centrifugation and cleaned with ethanol and deionized water, and finally dispersed in ethanol.2.4 Instrumentation and analytical conditions
Electrochemical analyses were performed using a three-electrode electrochemical workstation (METROHM, AUTOLAB PGSTAT302N) in a 20 + 20 mL H-shape cell. Ag and Pt served as the working (WE) and counter electrode (CE), while a saturated calomel electrode (SCE) served as the reference electrode (RE). Both WE and RE were immersed in the anolyte, and the SCE was connected to the anolyte via a salt bridge filled with electrolyte-saturated EG. The electrolyte concentrations were equal in both anolyte and catholyte before ES, while the CE was placed in the catholyte. The morphologies of the Ag NPs were characterized by scanning electron microscope (SEM, JEOL, JSM-IT700HR). Chemical compositions were analyzed by inductively coupled plasma-optical emission spectrometer (ICP-OES, THERMO FISHER, iCAP PRO). Phase compositions were evaluated by X-ray diffraction (XRD, PANALYTICAL, Empyren, Cu-Kα radiation). The pH of catholyte was measured using a pH meter (LEICI, PHB-25). The cathodic gas composition was identified by gas chromatography (GC, FULI INSTRUMENTS, GC9790II). 99.99% H2 gas was prepared by electrolysis of water using a H2 generator (SHANDONG SAIKESAISI, QL-200). The NO3− concentration in post-PR solution was measured using ultraviolet-visible spectroscopy (UV-vis, SHIMADZU, UV-1800).3. Results and discussion
3.1 Anodic dissolution of Ag in EG
The AD of Ag in EG anolyte can be expressed by eqn (1).| Ag (s) → Ag+ (in anolyte) + e− | (1) |
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) were employed to investigate the Ag AD process in EG. Fig. 3A–D illustrate the CV results using NaNO3, NaCl, CH3CO2Na and Na2SO4 as the electrolytes of EG, respectively. The anodic current begins to increase at about 0.45 V versus SCE, indicating the onset potential for eqn (1). The CV curves for NaNO3 and Na2SO4 remain stable after five cycles, whereas NaCl and CH3CO2Na exhibit decrease in anodic current densities, suggesting anodic passivation. During the cathodic sweep, the NaNO3 cathodic current decrease at 0.2 V versus SCE. This decrease suggests the high solubility of anodic polarization products (AgNO3) in EG, leading to the diffusion of AD-generated Ag+ in EG. In contrast, the cathodic current of NaCl, CH3CO2Na, and Na2SO4 increase during the cathodic sweep. These increases suggest the poor solubilities of anodic polarization products (AgCl, CH3CO2Ag, and Ag2SO4) in EG,35 leading to their crystallization on the Ag electrode surface. Their reduction result in an increased cathodic current density. Consequently, NaNO3 proves to be a suitable electrolyte for the AD of Ag in EG. In practice, due to the high solubility of AgNO3 in EG, the nitrate system is almost irreplaceable for the Ag NP synthesis via PR method.
 | ||
| Fig. 3 CV and LSV curves of Ag in ethylene glycol. Pure/crude Ag and SCE were inserted into the EG anolyte, with Ag serving as WE and SCE as RE. Pt was inserted into the aqueous catholyte and served as CE. Both the catholyte and anolyte contain 0.1 M electrolyte (except in (E)) with separation by AEM, and the scan rate was 10 mV s−1. CV curves of pure Ag with (A) NaNO3, (B) NaCl, (C) CH3COONa and (D) Na2SO4 as the EG electrolyte, respectively. LSV curves showing the effects of parameters: (E) NaNO3 concentration, (F) agitation, (G) temperature, and (H) types of Ag anode on anodic current densities, respectively. | ||
On the other hand, the conductivity of the anolyte and the diffusion of Ag+ influence the anodic current densities. Fig. 3E–G elucidate the effects of various anolyte conditions on anodic current densities: NaNO3 concentration (Fig. 3E), agitation (Fig. 3F), and temperature (Fig. 3G). The addition of NaNO3 enhances the conductivity of the EG solution, while elevated temperatures and intense agitation result in higher diffusion coefficients of Ag+,36 and thereby leads to increased anodic current densities. Notably, the PVP stabilizer, used in subsequent PR process, should not be added into anolyte during ES, due to the decreased diffusion coefficient of PVP-Ag+ complexes.37 Fig. 3H presents the LSV curves for pure and crude Ag samples. Their anodic current densities are distinct, which may be attributed to the different preparation methods used for the Ag samples (rolling for sheets, casting for ingot). The grain sizes, crystal orientation and chemical composition of metal greatly influence the electrochemical corrosion behavior.38 Nevertheless, their onset potentials for AD are nearly identical, and on passivation is observed during the anodic sweep, demonstrating the feasibilities of AD process across all Ag samples.
In this section, the AD of Ag into EG is investigated. NaNO3 has been confirmed as a suitable electrolyte. In fact, the nitrate system predominates in reported PR studies due to the high solubility of AgNO3 in polyols. By employing nitrate system in ES-PR method, we can draw on the insights from the reported PR studies to guide the Ag NP synthesis, thereby promoting the practical application of ES-PR method. Nevertheless, the utilization of NaNO3 electrolyte diminishes the economic benefits of the ES-PR method, we discuss the possible process for electrolyte recovery in the following chapter.
3.2 Enrichment of Ag+ in anolyte
Based on the CV result in Fig. 3A, there is no apparent side reaction occurs during the AD in EG with NaNO3, which indicates that the Ag+ enrichment rate in the anolyte (ηA) can be calculated according to Faraday's laws. The estimation of ηA is explained in ESI Note 3.† To understand what factors that influence ηA, ES experiments were conducted, the experimental parameters and results shown in Tables S3 and S4.†The formation of byproducts besides Ag+ leads to the first type of ηA loss. The pH of catholyte significantly influences the ηA and byproduct formation, as shown in Table S3.† In #pH-1 to 5, galvanostatic electrolysis were conducted with different starting and ending catholyte pH. As shown in Fig. 4A, the ηA decrease and sediment forms in anolyte (inset photographs) under alkaline catholyte. The ending pH of the catholyte is higher than the starting pH, which is due to the H2 evolution (discussed in following section) on the Pt cathode. Fig. 4B shows the XRD patterns of the anolyte sediment, indicating that it is a mixture of Ag2O (ICSD #281041) and Ag (ICSD #64706). The Ag2O is formed by the reaction of Ag+ with OH−,39 and the Ag is shed from the surface of the AEM. Fig. 4C shows the XRD patterns and photographs of AEMs before and after electrolysis at different catholyte pH. The broad peak at 2θ = 15–25° corresponding to the polyethylene matrix of AEM.40 Under acidic catholyte (#pH-1), the XRD pattern of AEM remains almost unchanged. However, alkaline catholyte (#pH-5) lead to the degradation of AEM.41 Intriguingly, the alkaline catholyte lead to an Ag deposition (ICSD #64706) on the AEM. We suppose that hydrated electrons react with O2 to form O2− under alkaline conditions,42 which subsequently reduce Ag+ on AEM surface. Fig. 4D presents the schematic diagram of ηA loss caused by byproducts formation, Ag+ are consumed by deposition on AEM or formation of AgOH and Ag2O. These byproducts-formation-induced ηA loss can be avoided by adjusting the catholyte pH to an acidic range (pH 0.9–1.1).
 | ||
| Fig. 4 Factors influencing the Ag+ enrichment rate (ηA). Loss of ηA due to byproduct formation: (A) effect of catholyte pH on ηA, sediment is generated in anolyte when catholyte is alkaline. (B) XRD patterns of the anolyte sediment generated under alkaline catholyte. (C) XRD patterns of AEM before and after electrochemical synthesis at different catholyte pH. (D) Schematic diagram, Ag+ is consumed through deposition on AEM and AgOH and Ag2O formations under alkaline catholyte conditions. Loss of ηA due to Ag+ permeation: effect of (E) Electrolysis time, (F) electrolysis temperature and (G) applied voltage on ηA and catholyte Ag+ concentration. (H) Schematic diagram, Ag+ permeates through AEM into catholyte, resulting in Ag deposition on cathode. | ||
The permeation of Ag+ through AEM into catholyte is another factor that leads to ηA loss. The polymeric matrix of the AEM possesses elongated channels, which are lined with positively charged groups, thereby repelling the passage of Ag+. According to the Donnan membrane equilibria theory, the permselectivity of the membrane is influenced by diffusion,43 thus ES time and temperature impact the permeation of Ag+. Table S4† shows the experimental parameters and analytical results for determining the Ag+ permeations on ηA. Potentiostatic electrolysis was conducted to investigate the effect of electrolysis time on ηA, as shown in Fig. 4E (#t-1 to 5). The ηA remains stable as the electrolysis time increases, while the catholyte Ag+ concentration (wC) increases, indicating that a concentration gradient between the anolyte and catholyte promotes the permeation of Ag+. Long-time electrolysis leads to the cathodic Ag deposition. The Ag deposited on the Pt cathode could be periodically cleaned and reused in the casting process of the Ag anode. The effect of temperature on ηA is shown in Fig. 4F (#T-1 to 4), increased temperature promotes the diffusion of Ag+, leading to decreased ηA and increased catholyte Ag+ concentration. On the other hand, the external electric field (the applied voltage) also promotes the permeation of Ag+. As shown in Fig. 4G (#E-1 to 4), increased applied voltage promotes Ag+ to overcome the repelling effect of AEM, resulting in a decreased ηA. Fig. 4H presents the schematic diagram of ηA loss caused by Ag+ permeation. Due to the diffusion of Ag+, the loss of ηA induced by permeation cannot be entirely prevented, but it can be mitigated by applying appropriate ES parameter to achieve ηA > 90%. It is anticipated that advancements in membrane technology will increase the permselectivity of AEM, thereby achieving a higher ηA.
In this section, the enrichment of Ag+ in EG anolyte during ES was investigated. Catholyte pH, temperature and applied voltage are the main factors that influences ηA. The ES process effectively prepares an Ag+-containing EG solution, which indicates that the ES process is feasible to prepare the precursor for the PR process.
3.3 Ag NWs from pure Ag precursor
Ag NWs networks exhibit superior electrical conductivity and high transmittance, along with the advantage of low cost. These properties make them potential replacements for indium tin oxide in the next generation of flexible electronic devices.21 Multitude of factors have been identified to influence the Ag NWs morphology synthesized by PR, such as oxygen scavengers,44 the presence of Cl−,45 PVP concentration,45 reaction temperature,46,47 and Ag+ concentration,48 among others. Building upon this body of research, we conducted straightforward comparative experiments to validate the efficacy of Ag NWs synthesis using the pure-Ag precursor prepared by ES process through PR. The experimental parameters and analytical results for Ag NWs synthesis are presented in Table S5.†Fig. 5A shows the SEM images of Ag NWs synthesized by PR method from the pure Ag precursor prepared via ES. The optimal Ag NWs morphology is s obtained in #NW-2 with synthesis parameters: 20 mM Ag+, 5 μM FeCl3, 200 mM PVP and heated for 150 °C/8 h, while others in Fig. 5 reveal the effects of parameter change on morphology. #NW-1, 2 and 3 reveal the morphology changes of Ag NWs with varying FeCl3 concentrations. Multiply twinned particles have been proven to serve as seeds for NWs. They are susceptible to oxidative etching, leading to their dissolution and a consequent decrease in the number of Ag NW seeds.49 The oxidative etching can be mitigated by introducing Fe3+ as oxygen scavengers. During the PR process, Fe3+ were reduced to Fe2+, the Fe2+ can remove atomic oxygen from the surface of Ag, thereby preventing the oxidative etching.44 On the other hand, Cl− reacts with Ag+ to form poorly soluble AgCl, thereby slowing down the reduction rate and making anisotropic growth of Ag NWs favorable. However, high concentration of Cl− leads to the formation of AgCl and micro-sized Ag particles.45 #NW-4, 2 and 5 reveal the morphology changes of Ag NWs with varying PVP concentration. PVP strongly binds to the {100} facets of Ag, thereby hindering their growth. This selective binding allows for the preferential growth along the 〈111〉 directions, resulting in the formation of Ag NWs.50 However, at high concentrations, PVP covers all available surfaces, thereby rendering the formation of nanowires infeasible.19 #NW-6, 2 and 7 reveal the morphology changes of Ag NWs with varying reaction temperature. The reaction temperature for Ag NWs synthesis via PR is typically controlled at 140–160 °C.46,47 A lower reaction temperature increases the diameter and decreases the length of Ag NWs,46 and also leads to a low productivity (R of #NW-6 = 9.60%). Conversely, a higher reaction temperature results in the formation of nanorods.47 #NW-8, 2 and 9 reveal the morphology changes of Ag NWs with varying Ag+ concentration. Low Ag+ concentration enhances nanowires formation, though this comes at a cost to productivity. Conversely, high Ag+ concentration results in wider and shorter nanowires.48 The SEM images for determining the length of Ag NWs samples are shown in Fig. 5B.
 | ||
| Fig. 5 (A) Factors influence Ag NWs morphology. The optimal Ag NWs morphology is obtained in #NW-2 with synthesis parameters: 20 mM Ag+, 5 μM FeCl3, 200 mM PVP and heated for 150 °C/8 h, while other experiments reveal the effects of parameter change on morphology. (B) SEM images for determining the length of Ag NWs samples, 50 nanowires were measured for every sample. | ||
In this section, we successfully synthesize Ag NWs using the pure Ag precursors prepared by ES process based on the reported experimental parameters. The factors influencing the morphology of Ag NWs are well consistent with the literature, demonstrating that the morphology control advantage of PR has been preserved. Currently, we think the synthesis of Ag NWs via ES-PR method can only use the pure Ag precursor. The synthesis of Ag NWs from the crude-Ag precursor may be extremely challenging, as described in following section.
3.4 Purified Ag NPs from crude Ag precursor
Electrorefining is an important process for purifying crude Ag into pure Ag. We suppose that the impurities in the crude-Ag precursor can be removed by utilizing the temperature-dependent reducibility of EG, achieving a purification effect similar to electrorefining. This could further reduce the raw-material costs of the ES-PR method (see Fig. 1c).Tables S6 and S7† shows the experimental parameters and analytical results, and the chemical composition for Ag NPs synthesized by ES-PR method using crude-Ag precursor. To directly confirm the purification capability of EG for the crude-Ag precursor, we first heated the 925-Ag precursor without the addition of a stabilizer, and the results are shown in Fig. S4† (#925-6 to 10). Due to the high Cu concentration in the 925-Ag precursors, the purification effect of EG can be clearly observed through the color changes of the reaction products. Micro-sized Ag powders were generated after heating. Increasing reaction temperature decreases the Ag powders purity and increases the R, indicating a selective reduction of Ag+ occurs at relative low temperature (140–150 °C). However, the Ag purity and R vary dramatically with temperature, making it difficult to achieve both high purity and a high R simultaneously. Notably, commercially available PVP commonly terminates with a hydroxyl group, which allows it to act as a mild reductant as well.51 Therefore, by adding PVP as a dual-function reagent (stabilizer and mild reductant), the reducibility of the solution can be controlled precisely, and the formation of Ag NPs can be promoted.
Fig. 6 presents the effect of temperature on purity, R and morphology on the Ag NPs from crude Ag with PVP addition (Fig. 6A for 925-Ag and Fig. 6B for Smelt.-Ag). Compared to Fig. S4,† the temperature range of Ag+ reduction decreases from 140–180 °C to 110–150 °C, indicating the additional reducibility provided by PVP. Heating at 120 °C for 6 hours with 500 mM PVP addition (#Smelt.-2 and #925-2) produces relatively pure (>99.9%) Ag NPs with R > 95%, indicating the feasibility of synthesizing purified Ag NPs through the ES-PR method using crude Ag as feedstock. It is noteworthy that Cu and Bi are difficult to remove from Ag Table S7,† which is consistent with the thermodynamic calculation of Larcher.34
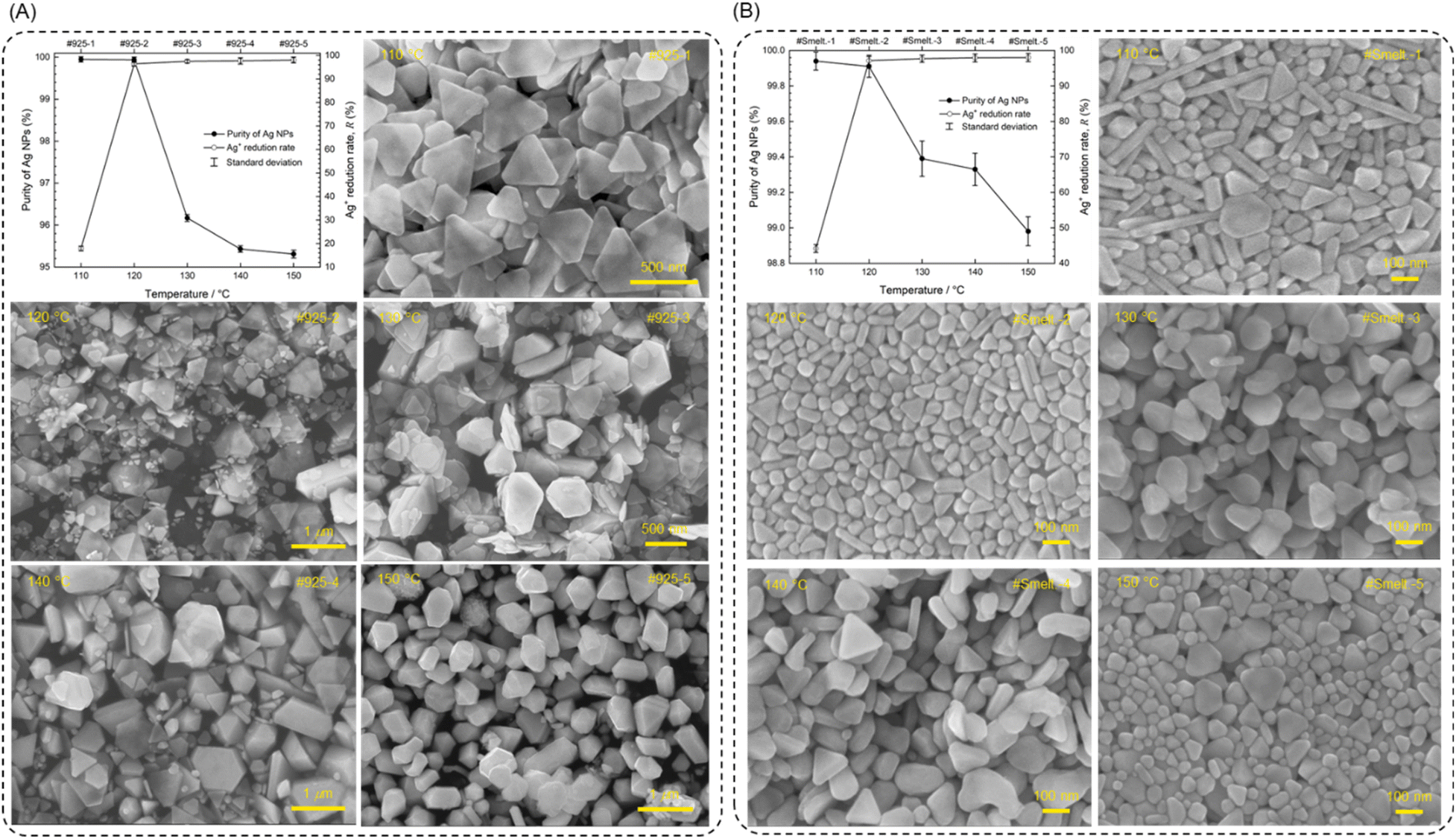 | ||
| Fig. 6 Effects of temperature on the purity, Ag+ reduction rate (R), and morphology of Ag NPs from crude Ag prepared by ES-PR method with PVP addition. The elevated reaction temperature increased the reducibility of ethylene glycol solution, resulting in an increased R and decreased Ag NPs purity. The morphologies of Ag NPs differ depending on the chemical compositions of crude Ag, indicating that impurity ions play a critical role in morphology control. (A) 925-Ag. (B) Smelt.-Ag. | ||
On the other hand, the morphologies of Ag NPs from crude Ag influence by reaction temperature and impurity ions significantly. As shown in the SEM images in Fig. 6, at 110 °C, #925-1 forms Ag nanosheets, while #Smelt.-1 is a mixture of nanorods, sheets, and particles. This may be attributed to the difference in Cu concentration between 925-Ag and smelt.-Ag. Xiong et al. have reported that Ag nanosheets can be prepared using PVP as a reducing agent, employing a mild reduction process in their study (heating in an aqueous solution at 60 °C).51 However, the #925-1 still formed uniform Ag nanosheets under stronger reducibility conditions (heating in an EG solution at 110 °C) compared to Xiong's study. We suppose that the high concentration of Cu2+ (>50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm) in #925-1 is reduced to Cu+ at 110 °C (purity of #925-1 = 99.95%), subsequently, Cu+ is re-oxidized to Cu2+ by oxygen atoms on the Ag surface, preventing the oxidative etching.52 This cyclic redox process (Cu2+ → Cu+ → Cu2+) consumes the reducing agents in solution, which leads to insufficient reducing agents to reduced Ag+. Consequently, the R of #925-1 is only 17.90%, the mild reducibility and mitigated oxidative etching results in the formation of nanosheets. In contrast, the R of #Smelt.-1 (with a lower Cu concentration) at 110 °C is 44.15%. Additionally, the Ag NPs in #Smelt.-5 at 150 °C exhibit a morphology that resembles #Smelt.-1, with a decrease in the number of nanorods in #Smelt.-5. However, #925-5 produced sub-micron polyhedral particles at 150 °C. We suppose that the larger particle size in #925-5 is due to the reduction of Cu and Zn at 150 °C (purity of #925-5 = 95.31%). The reduced Cu and Zn may deposit on some Ag NPs, facilitating their growth through the replacement of Cu and Zn with Ag+, leading to the formation of sub-micron powders. This effect will not occur in #Smelt.-5, which contains a lower Cu and Zn concentration. At 120 °C, the reducibility of the solutions is moderate, providing sufficient reducing agents to reduce Ag+, even under the cyclic redox process of Cu ions. The moderate reducibility at 120 °C does not lead to the substantial reduction of Cu2+ to Cu. Therefore, #925-2 and #Smelt.-2 produce relatively pure (>99.9%) Ag NPs with R > 95%. The above results demonstrate that impurity ions and their concentrations are crucial to Ag NPs morphology.
000 ppm) in #925-1 is reduced to Cu+ at 110 °C (purity of #925-1 = 99.95%), subsequently, Cu+ is re-oxidized to Cu2+ by oxygen atoms on the Ag surface, preventing the oxidative etching.52 This cyclic redox process (Cu2+ → Cu+ → Cu2+) consumes the reducing agents in solution, which leads to insufficient reducing agents to reduced Ag+. Consequently, the R of #925-1 is only 17.90%, the mild reducibility and mitigated oxidative etching results in the formation of nanosheets. In contrast, the R of #Smelt.-1 (with a lower Cu concentration) at 110 °C is 44.15%. Additionally, the Ag NPs in #Smelt.-5 at 150 °C exhibit a morphology that resembles #Smelt.-1, with a decrease in the number of nanorods in #Smelt.-5. However, #925-5 produced sub-micron polyhedral particles at 150 °C. We suppose that the larger particle size in #925-5 is due to the reduction of Cu and Zn at 150 °C (purity of #925-5 = 95.31%). The reduced Cu and Zn may deposit on some Ag NPs, facilitating their growth through the replacement of Cu and Zn with Ag+, leading to the formation of sub-micron powders. This effect will not occur in #Smelt.-5, which contains a lower Cu and Zn concentration. At 120 °C, the reducibility of the solutions is moderate, providing sufficient reducing agents to reduce Ag+, even under the cyclic redox process of Cu ions. The moderate reducibility at 120 °C does not lead to the substantial reduction of Cu2+ to Cu. Therefore, #925-2 and #Smelt.-2 produce relatively pure (>99.9%) Ag NPs with R > 95%. The above results demonstrate that impurity ions and their concentrations are crucial to Ag NPs morphology.
In this section, purified Ag NPs are synthesized using the crude-Ag precursor. However, controlling the morphology of these Ag NPs is challenging since impurity ions affect their shapes. Additionally, due to the variability in the chemical composition of crude Ag produced from smelting, we consider it difficult to use the crude-Ag precursor solution for preparing Ag NPs with precise morphologies. The Ag NPs generated from crude-Ag precursor may be suitable for the application in Ag conductive paste. The Ag NPs used in conductive paste (∼30–100 nm) serve as fillers within the gaps between micro-sized Ag flakes (∼4–12 μm).53,54 The size distribution of these Ag NPs is of more concern, while their morphologies are commonly not discussed, since the sintering process of the conductive paste changes their morphologies.53–55 Although there are limitations in the application of Ag NPs synthesized from crude Ag, the ultra-short synthesis route (as shown in Fig. 1c) still remains highly appealing due to its potential to remarkably reduce the production costs and time. To our knowledge, there has never been a paper that uses crude Ag as a feedstock to prepare purified Ag NPs, even when employing physical and electrochemical process.
3.5 Gaseous product and electrolyte recycling of ES-PR method
H2 is identified as the gaseous product generated during ES process. Fig. 7A shows the LSV curve revealing the H2 evolution on Pt. The onset potential at about −0.3 V versus SCE indicates the initiation of H2 evolution. Fig. 7B presents GC results of the cathodic gases generated during ME. The GC curves of cathodic gases match that of a 99.99% H2, especially when 4 V was applied, indicating H2 is the primary gaseous product. However, when the applied voltage of ES increased to 16 V, a small quantity of reddish-brown gas was observed in the gas collection bag, implying NOx formation. Consequently, to ensure that H2 remains the primary gaseous product and to prevent Ag+ permeation through AEM, high applied voltage should be avoided during the ES process. Significantly, unlike the production of AgNO3 that requires a large amount of H2O2 to suppress NOx generation, the ES process generates H2 as a byproduct, offering environmental and economic advantages. | ||
| Fig. 7 (A) LSV curve and (B) GC curves revealing the H2 evolution on Pt cathode during electrochemical synthesis. Low applied voltage ensures that H2 is the primary gaseous product. (C) UV-vis spectrum revealing the NO3− concentration change in different stage of ES-PR method, indicating the recyclability of NaNO3. (D) Proposed NaNO3 recycling process: the NaNO3 in post-PR solution is enriched into NaNO3 aqueous solution by electrodialysis. The impurities, like Cu2+, Pb2+ in NaNO3 aqueous solution are removed by ion exchange, when their concentrations are high. NaNO3 crystal is produced by heating process. | ||
On the other hand, the NaNO3 electrolyte is considered to be recyclable in the ES-PR method. The NO3− concentrations in the solutions at different stage of ES-PR method were analyzed using UV-vis, as shown in Fig. 7C. The peak at wavelength of 300 nm corresponding to the n → π* transition of NO3−.56 In the ES process, NaNO3 functions as a supporting electrolyte, facilitating the AD of Ag by enhancing the conductivity of EG without causing passivation. The NO3− concentration in anolyte increases after ES process. This increase is due to the migration of NO3− from the catholyte to the anolyte, which maintains the electrical neutrality of the system. During the precursor preparation, the anolyte is diluted by pure EG, leading to a decrease intensity of the NO3− peak. After the PR process, a peak appears at about 270 nm in the post-PR solution. This peak is likely due to aldehyde groups formed by the thermal decomposition of EG,25 which undergo an n → π* transition.57 Through peak fitting, the NO3− peak in the post-PR solution slightly decrease after PR process, indicating a small consumption of NO3−. This consumption could be due to the NOx generation caused by the dissolution of Ag0 NP in HNO3 during PR process.58 It is worth mentioned that the generation of NOx is solely related to the concentration of Ag+ in the precursor. During the synthesis of Ag NPs, the concentration of Ag+ is typically low, which means that only a small amount of NOx is emitted.58 Therefore, the NaNO3 is nearly non-consumed throughout the ES-PR method, making it recyclable.
To effectively and economically recycle NaNO3 from the post-PR solution, electrodialysis is considered an optimal choice. The feasibility of desalinating the NaNO3 solution via electrodialysis has been confirmed.59 Moreover, electrodialysis systems can be easily scaled down to small sizes, making them suitable for meeting various production capacity demands.60 In fact, small-scale electrodialysis systems are readily available on online marketplaces, with some models priced similarly to common household appliances. Fig. 7D proposes a facile electrodialysis-heating process for recycling NaNO3 from the post-PR solution. Through a combination of CEMs and AEMs in electrodialysis, the Na+ and NO3− are removed from the post-PR solution and concentrated in aqueous solution. Notably, when crude Ag is used as the feedstock in ES process, the impurity concentrations (Cu2+, Pb2+, etc.) in post-PR solution increase, these impurities can be removed through ion exchange.61 Finally, the electrolyte can be recovered through a heating process.
3.6 Anticipated developments
The PR method has demonstrated efficacy in synthesizing nanoparticles of transition and post-transition metals, such as Cu, Ni, Co, and Fe.62 We suppose that the ES-PR method can be extended to the synthesis of metallic nanoparticles beyond Ag, with the exception of certain noble metals like Au and platinum group metals, which are challenging to dissolve electrochemically. Fig. S5† shows the CV curves revealing AD of Cu, Ni, Co and Fe in EG, implying the applicability of ES-PR method among Cu, Ni, Co and Fe. Furthermore, the ES-PR strategy can be adaptable to aqueous system, establishing ES-hydrothermal and ES-aqueous reduction methods. These expansions have the potential to simplify processes for metallic nanoparticles synthesis via chemical reduction, leading to economic and environmental benefits.Based on the results above, a conceptual industrial workflow for the synthesis of metallic NPs via the ES-PR method is proposed, as shown in Fig. 8. Details of the ES and PR processes have been described previously. The addition of additives and PR process can be conducted in the same reaction container, the H2 generated by ES process is collected in a storage tank. Electrolytes are recycled via a facile electrodialysis-heating process. The ES-PR method exhibits a streamlined and eco-friendly process, with great potential to innovate the metallic NPs production via PR method.
 | ||
| Fig. 8 Conceptual industrial workflow for the synthesis of metallic nanoparticles via the ES-PR method. Crude or pure metal serves as feedstock to directly prepare precursor solutions, without the need for intermediate compounds. H2 gas is collected as a valuable byproduct. Electrolyte is recycled through an electrodialysis-heating process. | ||
4. Conclusions
This study has presented an electrochemical synthesis-polyol reduction (ES-PR) method for synthesizing Ag NPs from metallic Ag. Specifically, the method uses pure or crude Ag metal as the anode and dissolves it in ethylene glycol within a cell separated by an anion-exchange membrane. The enrichment rate of Ag+ in the anolyte is above 90% under appropriate ES parameters. Ag NWs are synthesized from the pure-Ag precursor, with reference to experimental parameters in the reported literature. Purified Ag nanoparticles (>99.9%) are synthesized from the crude-Ag precursor by utilizing the temperature-dependent reducibility of ethylene glycol. This method has the potential to establish a streamlined process for synthesizing Ag NPs via PR, eliminating the need of Ag compound for producing precursor solution. Furthermore, this method generates H2 as a byproduct during the ES process, making it an eco-friendly process. In the future, the ES strategy holds the potential to be extended to other chemical reduction techniques, including hydrothermal and aqueous systems. Furthermore, metals other than Ag, such as Cu, Ni, and Co, could also be synthesized using this technique, which could lead to economic and environmental benefits.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
C. Z. conceived the basic invention, completed the experimental concept, while X. G. and Q. W. gathered funds and materials. Q. W. and S. W. conducted the ES experiments and the analysis, and L. D. conducted the PR experiments and the analysis. C. Z., X. G., and Q. M. W. prepared the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52422410, U20A20273, W2411046), Chenzhou National Sustainable Development Agenda Innovation Demonstration Zone Construction Provincial Special Project: “Leading the Charge with Open Competition” (2023sfq59), and the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001). Additionally, we thank the Chenzhou Fengyue Environmental Protection Technology Co., Ltd for providing the chemical composition of crude Ag.Notes and references
- A. Syafiuddin, Salmiati, M. R. Salim, A. B. H. Kueh, T. Hadibarata and H. Nur, J. Chin. Chem. Soc., 2017, 64, 732–756 CrossRef CAS.
- W. Liu, R. An, C. Wang, Z. Zheng, Y. Tian, R. Xu and Z. Wang, Micromachines, 2018, 9, 346 CrossRef PubMed.
- Vidyasagar, R. R. Patel, S. K. Singh and M. Singh, Mater. Adv., 2023, 4, 1831–1849 RSC.
- S. Lee, J. Jang, T. Park, Y. M. Park, J. S. Park, Y. Kim, H. Lee, E. Jeon, D. Lee, B. Ahn and C. Chung, ACS Appl. Mater. Interfaces, 2020, 12, 6169–6175 CrossRef CAS PubMed.
- D. F. Tassw, B. Birlie and T. Mamaye, J. Text. Inst., 2024, 1–21 Search PubMed.
- M. Snellman, N. Eom, M. Ek, M. E. Messing and K. Deppert, Nanoscale Adv., 2021, 3, 3041–3052 RSC.
- M. Raffi, A. K. Rumaiz, M. U. Hassan and S. I. Shah, J. Mater. Res., 2007, 22, 3378–3384 CrossRef CAS.
- S. Shih and I. Chien, Powder Technol., 2013, 237, 436–441 CrossRef CAS.
- S. Lee, Y. Jeong, H. Oh, J. Lee, O. Y. Lee and C. Chi, Met. Mater. Int., 2009, 15, 631–636 CrossRef CAS.
- L. Scotti, G. Angelini, C. Gasbarri and T. Bucciarelli, Mater. Res. Express, 2017, 4, 105001 CrossRef.
- H. Wang, H. Wu, L. Zhong, J. Zhao and G. Li, J. Electrochem. Soc., 2017, 164, D225–D229 CrossRef CAS.
- J. Zhang, J. E. Kielbasa and D. L. Carroll, J. Mater. Res., 2009, 24, 1735–1740 CrossRef CAS.
- Y. Ding, P. Zhang and Z. Long, J. Alloys Compd., 2009, 474, 223–225 CrossRef CAS.
- J. Wang and S. Pan, Nanotechnology, 2017, 28, 425601 CrossRef PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- B. Bari, J. Lee, T. Jang, P. Won, S. H. Ko, K. Alamgir, M. Arshad and L. J. Guo, J. Mater. Chem. A, 2016, 4, 11365–11371 RSC.
- H. Wang, W. Yang, K. Li and G. Li, RSC Adv., 2018, 8, 8937–8943 RSC.
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed.
- B. Wiley, Y. Sun, B. Mayers and Y. Xia, Chem.–Eur. J., 2005, 11, 454–463 CrossRef CAS PubMed.
- F. Fiévet and R. Brayner, Nanomaterials: a danger or a promise? A chemical and biological perspective, Chapter 1: the Polyol Process, Springer, 2013, pp. 1–26 Search PubMed.
- W. Li, H. Zhang, S. Shi, J. Xu, X. Qin, Q. He, K. Yang, W. Dai, G. Liu, Q. Zhou, H. Yu, S. R. P. Silva and M. Fahlman, J. Mater. Chem. C, 2020, 8, 4636–4674 RSC.
- K. Zimmermann, Silver, Silver Compounds, and Silver Alloys, Chapter 7: Silver Compounds, Wiley-VCH, 2005, pp. 29–35 Search PubMed.
- R. Lucy, M. Serge and B. Mireille, European Patent, EP0568259A1, 1993 Search PubMed.
- Z. Liu, Y. Zhou, S. Zhang, S. Lin and D. Peng, Chinese Pat., CN111732120A, 2020 Search PubMed.
- C. Sequeira and D. Santos, J. Braz. Chem. Soc., 2009, 20, 387–406 CrossRef CAS.
- Z. N. Gafurov, A. O. Kantyukov, A. A. Kagilev, O. G. Sinyashin and D. G. Yakhvarov, Coord. Chem. Rev., 2021, 442, 213986 CrossRef CAS.
- Z. N. Gafurov, O. G. Sinyashin and D. G. Yakhvarov, Pure Appl. Chem., 2017, 89, 1089–1103 CrossRef CAS.
- R. Xie, P. Ning, G. Qu, J. Li, M. Ren, C. Du, H. Gao and Z. Li, Chem. Eng. J., 2018, 341, 298–307 CrossRef CAS.
- A. P. Abbott, G. Frisch, J. Hartley, W. O. Karim and K. S. Ryder, Prog. Nat. Sci.:Mater. Int., 2015, 25, 595–602 CrossRef CAS.
- K. Matsuoka, Y. Iriyama, T. Abe, M. Matsuoka and Z. Ogumi, J. Power Sources, 2005, 150, 27–31 CrossRef CAS.
- The Silver Institute, World Silver Survey 2024, 2024 Search PubMed.
- C. D. Sanguesa, R. H. Urbina and M. Figlarz, J. Solid State Chem., 1992, 100, 272–280 CrossRef.
- V. Livshits, M. Philosoph and E. Peled, J. Power Sources, 2008, 178, 687–691 CrossRef CAS.
- D. Larcher and R. Patrice, J. Solid State Chem., 2000, 154, 405–411 CrossRef CAS.
- G. Dzido, P. Markowski, A. Malachowska-Jutsz, K. Prusik and A. B. Jarzebski, J. Nanopart. Res., 2015, 17, 1–15 CrossRef CAS PubMed.
- S. Wang, C. Xu, Y. Hua, X. Chen, Q. Xiang, J. Li and Y. Li, J. Mol. Liq., 2023, 373, 121255 CrossRef CAS.
- G. Yang, Q. Zou, P. Wang, H. Lai, T. Lai, X. Zeng, Z. Li, J. Luo, Y. Zhang and C. Cui, J. Alloys Compd., 2021, 874, 159900 CrossRef CAS.
- R. Mishra and R. Balasubramaniam, Corros. Sci., 2004, 46, 3019–3029 CrossRef CAS.
- M. M. Murtazin, M. Y. Nesterova, S. N. Grushevskaya and A. V. Vvedenskii, Russ. J. Electrochem., 2019, 55, 680–689 CrossRef CAS.
- A. M. Musuc, M. B. Doni, L. Jecu, A. Rusu and V. T. Popa, J. Therm. Anal. Calorim., 2013, 114, 169–177 CrossRef CAS.
- Z. Sun, J. Pan, J. Guo and F. Yan, Adv. Sci., 2018, 5, 1800065 CrossRef PubMed.
- A. S. G. Mazumdar and S. N. Guha, On the electrolytic generation of hydrated electron, Bhabha Atomic Research Centre, 1975 Search PubMed.
- F. Donnan, Chem. Rev., 1924, 1, 73–90 CrossRef CAS.
- B. Wiley, Y. Sun and Y. Xia, Langmuir, 2005, 21, 8077–8080 CrossRef CAS PubMed.
- S. Coskun, B. Aksoy and H. E. Unalan, Cryst. Growth Des., 2011, 11, 4963–4969 CrossRef CAS.
- S. M. Bergin, Y. H. Chen, A. R. Rathmell, P. Charbonneau, Z. Y. Li and B. J. Wiley, Nanoscale, 2012, 4, 1996–2004 RSC.
- Y. Sun, Y. Yin, B. T. Mayers, T. Herricks and Y. Xia, Chem. Mater., 2002, 14, 4736–4745 CrossRef CAS.
- H. Ding, Y. Zhang, G. Yang, S. Zhang, L. Yu and P. Zhang, RSC Adv., 2016, 6, 8012–8096 Search PubMed.
- P. Zhang, I. Wyman, J. Hu, S. Lin, Z. Zhong, Y. Tu, Z. Huang and Y. Wei, Mater. Sci. Eng., B, 2017, 223, 1–23 CrossRef CAS.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- Y. Xiong, I. Washio, J. Chen, H. Cai, Z. Li and Y. Xia, Langmuir, 2006, 22, 8563–8570 CrossRef CAS PubMed.
- K. E. Korte, S. E. Skrabalak and Y. Xia, J. Mater. Chem., 2008, 18, 437–441 RSC.
- X. Liu, S. Wu, B. Chen, Y. Ma, Y. Huang, S. Tang and W. Liu, J. Mater. Sci.: Mater. Electron., 2021, 32, 13777–13786 CrossRef CAS.
- K. Kiełbasiński, J. Szałapak, M. Jakubowska, A. Młożniak, E. Zwierkowska, J. Krzemiński and M. Teodorczyk, Adv. Powder Technol., 2015, 26, 907–913 CrossRef.
- K. Park, D. Seo and J. Lee, Colloids Surf., A, 2008, 313, 351–354 CrossRef.
- V. Butorac, V. Simeon and V. Tomišić, Croat. Chem. Acta, 2007, 80, 533–539 CAS.
- P. S. Kalsi, Spectroscopy Of Organic Compounds, Chapter 2: Ultraviolet (UV) and Visible Spectroscopy, New age international, 2007, pp. 9–63 Search PubMed.
- D. R. Whitcomb, A. R. Clapp, P. Buhlmann, J. C. Blinn and J. Zhang, Cryst. Growth Des., 2016, 16, 1861–1868 CrossRef CAS.
- K. Weinertova, R. S. Honorato, E. Stranska and D. Nedela, Chem. Pap., 2017, 72, 469–478 CrossRef.
- H. Kariman, A. Shafieian and M. Khiadani, Desalination, 2023, 567, 116985 CrossRef CAS.
- E. Pehlivan and T. Altun, J. Hazard. Mater., 2007, 140, 299–307 CrossRef CAS PubMed.
- F. Fievet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00967g |
| This journal is © The Royal Society of Chemistry 2025 |