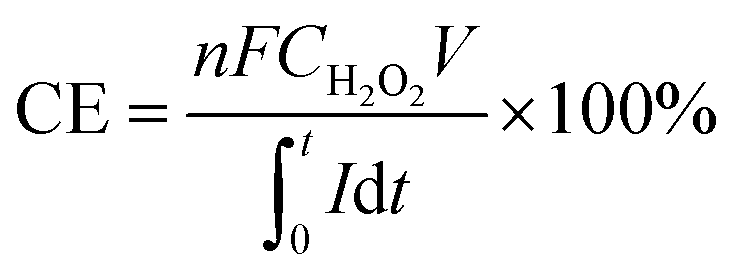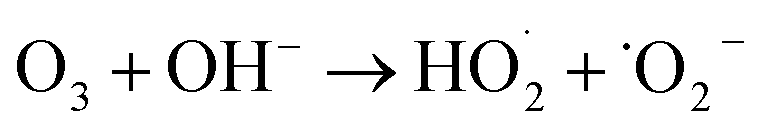DOI:
10.1039/D5RA00939A
(Paper)
RSC Adv., 2025,
15, 10273-10286
Treatment of cationic red X-GRL in high-salt printing and dyeing wastewater by an electrocatalytic ozonation system: treatment efficiency and degradation mechanism†
Received
8th February 2025
, Accepted 21st March 2025
First published on 3rd April 2025
Abstract
High salt concentrations in printing and dyeing wastewaters significantly influence pollutant removal. The function of the electrocatalytic ozonation (MgMnxOy–GAC/EP) system in removing pollutants from high-salt printing and dyeing wastewater was investigated. Under high NaCl concentration, the H2O2 yield in the electrochemical system was maintained at approximately 53 mg L−1. Under optimal treatment conditions, the degradation efficiency of cationic red X-GRL in the MgMnxOy–GAC/EP system reached 100% within 16 min, and the mineralization efficiency achieved 90.8% within 60 min. The specific energy consumption of the MgMnxOy–GAC/EP system was 0.027 kW h per gCOD. The SF of the MgMnxOy–GAC/EP system was 13.04, indicating that MgMnxOy–GAC, EO and O3 had a remarkable synergistic effect in the removal of cationic red X-GRL. The existence of ˙OH, ˙Cl, ˙O2− and 1O2 in the MgMnxOy–GAC/EP system was demonstrated by quenching and electron paramagnetic resonance experiments. Based on these outcomes, the degradation mechanism of cationic red X-GRL in the MgMnxOy–GAC/EP system under high salt conditions was proposed, which was the action mechanism of multiple free radicals mainly dominated by ˙O2− and 1O2. After repeated experiments, the MgMnxOy–GAC/EP system accomplished a COD removal efficiency of 84%, which signified its relatively high stability. The MgMnxOy–GAC/EP system achieved a COD removal efficiency of approximately 86% for diverse pollutants. Overall, this study revealed that the MgMnxOy–GAC/EP system has novel prospects for the treatment of organic pollutants in high-salt wastewater.
1 Introduction
Dyeing and finishing processes in the textile industry have played a crucial role in the global economy.1 These processes are major water consumption processes that use water as the primary medium for dye and chemical application.2,3 This has contributed to approximately 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of textile dye wastewater annually, significantly straining global water resources.4,5 Moreover, they are rich in various inorganic salt components (including K+, Ca2+, Na+, Mg2+, CO32−, NO3−, Cl− and SO42−).6 The salinity in high-salt wastewater adversely affects biological treatment systems, leading to floating sludge, microbial metabolism inhibition, and reduced organic pollutant removal efficiency.7 Meanwhile, the wastewater contained persistent pollutants such as dyes and additives that were resistant to biological degradation and photodegradation. This resistance has made the treatment of high-salt printing and dyeing wastewater challenging when conventional biological methods are used.8,9
000 tons of textile dye wastewater annually, significantly straining global water resources.4,5 Moreover, they are rich in various inorganic salt components (including K+, Ca2+, Na+, Mg2+, CO32−, NO3−, Cl− and SO42−).6 The salinity in high-salt wastewater adversely affects biological treatment systems, leading to floating sludge, microbial metabolism inhibition, and reduced organic pollutant removal efficiency.7 Meanwhile, the wastewater contained persistent pollutants such as dyes and additives that were resistant to biological degradation and photodegradation. This resistance has made the treatment of high-salt printing and dyeing wastewater challenging when conventional biological methods are used.8,9
Advanced oxidation processes (AOPs) effectively remove recalcitrant organic pollutants from wastewater.10 These include Fenton processes,11 ozone processes,12 photocatalytic processes,13 electrochemical processes14 and persulfate processes.15 These technologies typically generate highly oxidizing free radicals (such as ˙OH, 1O2, and ˙O2−), which can oxidize and degrade large molecules in water into low-toxicity or non-toxic small molecules.16 They could even directly mineralize these pollutants into CO2 and H2O.17 However, the reactions of these free radicals with pollutants are generally not selective, making them susceptible to interference from the inorganic salt components present in wastewater.18 For instance, Miao et al. employed high gravity technology to enhance the Co–Mn/γ-Al2O3 catalytic ozone oxidation for the degradation of high-salt phenol wastewater, achieving a phenol mineralization efficiency of 90.02% within 80 min.19 They investigated the influence of salinity on phenol degradation under high salt conditions. It was observed that salts tended to accumulate at the active sites of the catalysts, thereby influencing the degradation and mineralization efficiency of phenol. Wang et al. fabricated a meshed boron-doped diamond (BDD)/boron-doped graphene (BG)/Ti electrode for the electrocatalytic degradation of high-salt printing and dyeing wastewater.20 This electrode could remove 93.43% of methyl orange at an initial concentration of 15 mg L−1 within 60 s. Zhu et al. prepared a hierarchical porous PbO2 electrode on a fluorine-doped tin oxide template via direct electrodeposition.21 Electrochemical degradation experiments were conducted on various pollutants, achieving removal rates of 86.5%, 72.0%, and 68.8% for surfactants (sodium n-dodecyl sulfate, SDS), pharmaceutical drugs (diclofenac sodium salt, DSS), and dye pollutants (rhodamine B, RhB), respectively. Chen et al. developed a Co–PEG–PbO2 electrode modified with Co2+ and polyethylene glycol to enhance the electrocatalytic performance and stability of PbO2 electrodes. This innovation was achieved through polystyrene microsphere-assisted electrodeposition combined with surface structure optimization and active material doping strategies.22 This electrode was employed for the efficient degradation of p-chlorophenol in water. The modified electrode exhibited superior performance, achieving a p-chlorophenol removal efficiency of 97.34%, a TOC removal efficiency of 64.25%, a mineralization current efficiency of 23.45%, and a reduced cell voltage compared to conventional electrodes. However, electrochemical degradation exhibits limited treatment efficiency, high energy consumption, and difficulty in mineralizing refractory organic pollutants.23 Hence, there was an imperative need to explore a system capable of effectively eliminating difficult-to-degrade pollutants from high salt printing and dyeing wastewater.
Recently, electrocatalytic ozonation technology has attracted considerable attention in water treatment.24 As a new advanced oxidation technology that combined the advantages of electrocatalysis and ozone catalytic oxidation, it was extensively employed to remove organic pollutants produced in water treatment procedures.25 Due to the high salt content in printing and dyeing wastewater, it can act as a natural electrolyte for electrocatalytic ozonation technology without the addition of external electrolytes, thereby avoiding secondary pollution.26 The electrocatalytic cathode is capable of generating H2O2 through a two-electron reduction pathway, and H2O2 can react with O3 to form ˙OH.27 Meanwhile, the catalyst can catalyze both O3 and H2O2, significantly enhancing the treatment efficiency of pollutants.28 Furthermore, the intrinsically high electrical conductivity of high salt wastewater significantly reduces the internal resistance of the electrochemical system, improves the current transfer efficiency, thereby lowering energy consumption and strengthening the H2O2 production capacity at the cathode. The active chlorine species such as ClO− and ClO2− produced by the anodic oxidation of Cl− in synergy with free radicals like ˙OH and ˙O2− effectively degrade complex pollutants, thereby improving the electrochemical efficiency under high salt conditions.
Nevertheless, within an electrocatalytic ozonation system, the salt ions existing in high-salt wastewater influence the degradation of organic pollutants to a certain extent. For instance, Xie et al. developed a methodology to alleviate the high energy consumption and low mass transfer characteristics of electro-peroxidation (EP) for the in situ generation of H2O2, and it was observed that the degradation and mineralization efficiencies of 100 mg per L phenol at 15 min and 60 min were 100% and 72.1%, respectively.29 Under high salt conditions, the mineralization efficiency of phenol by Fe0@CS/EP was relatively low.
Previous studies indicated that in the electrochemical treatment of high-salt wastewater, Cl− was oxidized at the anode to form Cl2, which further reacted to form ClO3− and ClO2− with significant bleaching properties. This phenomenon effectively promotes the degradation of pollutants. In acidic conditions, ˙Cl was generated through the reaction between Cl− and ˙OH. Since the oxidation–reduction potential of ˙Cl (2.4 V) was lower than that of ˙OH (2.8 V), the presence of Cl− inhibited the degradation of organic pollutants30,31
Lu et al. also elucidated the dual effects of inorganic salts such as NaCl on the reaction process in heterogeneous catalytic ozonation processes.32 First, these salts reduced the saturation of ozone in the liquid phase, accelerated the depletion of ozone and hydroxyl radicals, and thereby inhibited the reaction. Second, the increase in salinity reduced bubble coalescence and reduced bubble size, thereby enhancing the gas–liquid mass transfer efficiency. However, the removal efficiency of pollutants and the impact of salt ions on the electrocatalytic ozonation system under high salt conditions require further exploration.
This study conducted a detailed investigation into the treatment efficiency of an electrocatalytic ozonation system for pollutants under high salt conditions. Initially, an electrochemical reactor was constructed to explore how the high salt content in printing and dyeing wastewater influenced the electrochemical synthesis of H2O2. The characteristics of graphite felt electrodes were studied using SEM, XRD and XPS. Subsequently, an inner-circulation fluidized bed reactor was constructed, merging the electrochemical generation of H2O2 with catalytic ozonation to form an electrocatalytic ozonation system. Cationic red X-GRL, a typical pollutant in high-salt printing and dyeing wastewaters, was selected for use in this study. The effects of ozone concentration, current density, catalyst dosage, and initial pH on the degradation of cationic red X-GRL were examined. The analyses of ˙OH, ˙O2−, and 1O2 was conducted through free radical quenching experiments. The changes in free and non-free radicals during different treatment processes were analyzed using EPR technology, and the influence of high salt on free radicals was investigated. Additionally, the contributions of O3 oxidation alone, catalytic ozone oxidation, and electro-peroxidation reactions to the degradation of cationic red X-GRL were quantitatively evaluated. The mechanism of the electrocatalytic ozonation system for the degradation of cationic red X-GRL was also proposed. Finally, the stability and environmental adaptability of the electrocatalytic ozonation system were assessed.
2 Materials and methods
2.1 Chemicals and materials
The following chemicals and reagents were ACS reagent grade or higher and did not require further purification. All the materials are provided in Text S1.† All solutions were prepared using ultra-pure water. The water quality parameters of the high-salt printing and dyeing wastewater are presented in Table S1.†
2.2 Preparation of modified graphite felt (GF) electrode
The pretreated GF was subjected to electrochemical treatment in a 1 mol per L C3H6O3 solution with a constant current density of 150 A m−2 for anodic oxidation for 30 min. After removal, the GF was rinsed with pure water and dried in an oven at 70 °C for 12 h. The dried GF was subsequently deposited in a g-C3N4 dispersion at a current density of 12.5 A m−2 for 30 min. Finally, the prepared electrode was dried in an oven at 70 °C for 12 h.
2.3 Preparation of the catalyst
A certain quantity of treated activated carbon particles was weighed and immersed in a mixed solution containing a certain concentration of Mg(NO3)2 and Mn(NO3)2. The mixture was left to stand at room temperature for 6 h. Subsequently, under stirring, KMnO4 solution and sodium NaOH were slowly added to form MnO2 and Mg(OH)2 precipitates. After incorporating all reagents, the mixture was thoroughly stirred for 5 min and left to stand for another 6 h. Subsequently, the supernatant was decanted, and the sample was washed with deionized water 2–3 times to eliminate substances that were not firmly loaded on the carrier. The precursor of the obtained catalyst sample was dried in an oven at 80 °C for 12 h. Subsequently, it was placed in a tube furnace and calcined at 845 °C for 3 h in a nitrogen atmosphere, ultimately yielding the MgMnxOy–GAC catalyst.
2.4 Experimental procedures
The electrochemical hydrogen peroxide production was conducted in a 120 mL electrochemical system (ES), as shown in Fig. S1.† The reactor was designed for flexible assembly and disassembly to accommodate varying numbers of electrodes. The modified graphite felt was used as the cathode (the preparation method is presented in Text S2†), and SnO4–RuO2/Ti was used as the anode. The H2O2 yield was assessed under continuous flow conditions. Pure oxygen (99.99%) was passed through an ozone generator (Anseros COM-AD-01) and then dispersed into the water solution at a flow rate of 200 mL min−1 using titanium diffusers (diameter 15 mm, length 20 mm, porosity 35%), with an initial pH of 7.7, a current density of 22.5 A m−2, and a hydraulic retention time (HRT) of 300 s. Unless otherwise specified, no adjustments were made.
An organic glass container with a volume of 6 L was used. A digital DC power supply (KXN-305D, Zhao Xin, China) was used, with SnO4–RuO2/Ti as the anode and graphite felt (6 cm × 5 cm) as the cathode spaced 3 cm apart. The laboratory-prepared MgMnxOy–GAC (preparation method is presented in Text S3†) catalyst was also used. The pollutant treatment was performed using electrochemical oxidation (EO), ozonation (O3), electro-peroxidation (EP), MgMnxOy–GAC adsorption, electrochemical catalytic oxidation (MgMnxOy–GAC/EO), catalytic ozonation (MgMnxOy–GAC/O3), and electrocatalytic ozonation (MgMnxOy–GAC/EP) processes. Among the O3, EP, MgMnxOy–GAC/O3, and MgMnxOy–GAC/EP processes, pure oxygen (99.9%) as the feed gas passed through the ozone generator and was subsequently dispersed into the pollutant solution at a rate of 800 mL min−1 via the bottom titanium aeration plate (length: 60 mm, width: 60 mm, pore size: 1 μm). The tail gas was adsorbed by a KI solution (2 wt%). A schematic of the experimental setup is shown in Fig. S2.† At regular intervals, 10 mL samples of the liquid were collected and extinguished with 300 μL sodium thiosulfate solution. Each experiment was repeated at least three times, and the results were plotted as the mean of duplicates, with error bars indicating deviations from the mean. The initial pH was 7.7, the current density was 22.5 A m−2, the flow rate was 800 mL min−1, the NaCl concentration was 12000 mg L−1, the catalyst concentration was 10 g L−1, and the O3 concentration was 24 mg L−1. Unless in the parameter optimization experiments, these conditions were adopted.
2.5 Characterization
Structural identification was achieved by X-ray diffraction (XRD) using a DX-2700 instrument (Dandong Haoyuan) with Cu Kα radiation, covering a 0° to 80° range at a step size of 5°. Elemental composition was analyzed by X-ray photoelectron spectroscopy (XPS) using a USA Thermo Scientific K-Alpha instrument. Field emission scanning electron microscopy (FE-SEM) was used to examine electrode morphology using a MERLIN Compact instrument (Zeiss, Germany). Cyclic voltammetry tests were performed using a CHI660 electrochemical workstation (Shanghai Chenhua).
2.6 Analytical methods
The concentration of H2O2 was quantified using titanium salt spectrophotometry with a UV-vis spectrophotometer.33 The concentration of cationic red X-GRL was measured using UV-vis spectrophotometry,34 and the COD value of the solution was assessed using the potassium dichromate method.35 Electron paramagnetic resonance measurements were conducted using an EMX nano instrument (Germany Bruker). The capturing agents of ˙OH, ˙O2− and ˙Cl are 5,5-dimethyl-1-pyrrolidine-N-oxide (DMPO). The 1O2 capturing agent was 4-amino-2,2,6,6-tetramethylpiperidine (TEMP). The capture experiments were conducted by adding excess tert-butanol (TBA, ˙OH scavenger), isopropanol (IPA, ˙OH scavenger), p-benzoquinone (p-BQ, ˙O2− scavenger), and furfuryl alcohol (FFA, 1O2 scavenger) as scavengers.36 The presence of ˙OH was determined by fluorescence spectrophotometry using terephthalic acid as a probe,37 while the Cl− concentration in the solution was analyzed via ion chromatography.
The current efficiency of H2O2 production can be determined using the following formula: eqn (1).
| |
 | (1) |
In this equation,
n represents the charge transfer number (dimensionless);
F is the Faraday constant (C mol
−1);
C denotes the H
2O
2 concentration (mol L
−1);
V stands for the volume of continuous flow sampling (L);
I signifies the constant current (A); and
t indicates the reaction time (s).
The dynamic equation eqn (2) determines the first-order rate constants.
where
C0 represents the initial pollutant concentration (mg L
−1),
Ct denotes the pollutant concentration at
t min (mg L
−1), and
k is the rate constant (min
−1).
Define the synergistic factor (SF) as a function of the rate constants of multiple processes involved in the coupled system, which can be determined using eqn (3).
| |
 | (3) |
The specific energy consumption (SEC, kW h per g COD) for the EP, MgMnxOy–GAC/O3, MgMnxOy–GAC/EO, and MgMnxOy–GAC/EP processes for COD removal was determined using eqn (4).
| |
 | (4) |
where
V represents the solution volume (L), COD
0 and COD
t denote the initial and current COD values of the solution (mg L
−1) at
t = 0 and
t,
I denotes the electric current (A),
t is the reaction time (h),
U represents the average battery voltage (V),
CO3 denotes the concentration of ozone in the gas phase, and
r is the energy required to produce ozone (10 kW h per kg O
3).
3 Results and discussions
3.1 Effect of NaCl concentration on H2O2 production in ES
This study investigated the effect of different NaCl concentrations on H2O2 generation in ES. As shown in Fig. 1a, when the NaCl concentration increased from 1500 to 4500 mg L−1, the H2O2 concentration increased from 44.85 to 53.46 mg L−1 in the ES. As the NaCl concentration was further increased, the H2O2 concentration remained relatively stable. This was attributed to the fact that the rate of electron transfer was accelerated with increasing salinity.38 Nevertheless, when the salinity exceeded a certain value, more Cl− was adsorbed on the electrode surface, thereby reducing the number of active sites on the electrode.39 In conclusion, salinity significantly influences the production of H2O2 in the ES system and is capable of enhancing the production of H2O2 within a certain concentration range. Beyond this range, the effect of further increasing the salinity on the improvement of H2O2 production was limited in the ES system. As shown in Fig. 1b, when the NaCl concentration increased from 1500 to 4500 mg L−1, the voltage decreased from 29.2 to 11.48 V in the ES. Concurrently, the current efficiency increased from 79.3% to 95.53%. The reduction in voltage suppressed the occurrence of side reactions on the electrode surface.40 This enhanced the production of H2O2 on the cathode surface, which was further supported by the observed improvement in current efficiency. As the NaCl concentration increased further, the voltage in the ES slightly decreased, and the current efficiency experienced a slight increase. These results are in accordance with the stable trend of H2O2 concentration under high salt conditions. The results obtained in this experiment indicate that the electrochemical synthesis of H2O2 can be effectively conducted even under high salt conditions.
 |
| | Fig. 1 Effect of NaCl concentration on ES (a) H2O2 concentration and (b) voltage and current efficiency. | |
3.2 Effect of high salt on graphite felt cathode in ES
The surface morphologies of the graphite felt electrodes before (GF) and after (GF-1) use were compared by FESEM. As shown in Fig. 2a, granular substances were observed on the relatively smooth surface of the GF. In contrast, significant grooves and pores were formed on the surface of GF-1 (Fig. 2b) due to acid corrosion. This structural change significantly increased the specific surface area of the electrode, thereby increasing the number of active sites on the graphite felt.
 |
| | Fig. 2 SEM images of GF (a) and GF-1 (b) and (c) XRD, XPS spectral peaks of (d) C 1s of original, (e) C 1s of reacted, (f) O 1s of original, (g) O 1s of reacted, (h) N 1s of original, (i) N 1s of reacted, (j) CV curves. | |
Fig. 2c shows that the XRD patterns of GF exhibit diffraction peaks at 25.7° and 43.26° of 2θ. These peaks correspond to the (002) and (100) crystal planes of graphite, respectively, indicating the microcrystalline structure of the material.41 The XRD pattern of GF-1 remained unchanged at the same position, indicating that the GF electrode crystal structure was stable under high salt conditions. This result indicated that the main structure of the GF was not affected under high salt conditions, thereby confirming the stability of the ES process in such situations.
GF and GF-1 were further characterized by XPS analysis. As shown in Fig. 2d and e, in the C 1s spectrum, the proportion of C–C bonds significantly increased from 64.06% in GF to 86.66% in GF-1. It has been reported that the C–C bond in carbon materials serves as an active site for the electrocatalytic oxygen reduction reaction.42 As shown in Fig. 2f and g, in the O 1s spectrum, the proportion of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O bonds increased from 18.8% in GF to 34.69% in GF-1. It has been shown that carbon atoms in OC–O groups have strong positive charges, which are beneficial for the adsorption of oxygen using the Pauling mode.43 Therefore, the increase in the proportion of C–C bonds and OC–O bonds enhanced the electrochemical activity of GF, thereby enhancing the ability of ES to produce H2O2. As shown in Fig. 2h and i, in the N 1s spectrum, the N–(C)3 bond was formed by polyvinylpyrrolidone (PVP) adhesion between carbon nitride and the electrode. Meanwhile, the C–O–N bond was formed through the interaction between CO on the surface of the graphite felt electrode and pyridine N in g-C3N4. The proportion of C–O–N bonds increased from 12.22% in GF to 32.56% in GF-1, and the proportion of N–(C)3 bonds decreased from 56.86% in GF to 28.58% in GF-1. The change in the proportion of chemical bonds promoted the transition of the binding force between the electrode and g-C3N4 from an adhesive force to a stronger interatomic force, indicating a more stable binding between g-C3N4 and graphite felt, which was conducive to enhancing the electrochemical activity of the electrode.
C–O bonds increased from 18.8% in GF to 34.69% in GF-1. It has been shown that carbon atoms in OC–O groups have strong positive charges, which are beneficial for the adsorption of oxygen using the Pauling mode.43 Therefore, the increase in the proportion of C–C bonds and OC–O bonds enhanced the electrochemical activity of GF, thereby enhancing the ability of ES to produce H2O2. As shown in Fig. 2h and i, in the N 1s spectrum, the N–(C)3 bond was formed by polyvinylpyrrolidone (PVP) adhesion between carbon nitride and the electrode. Meanwhile, the C–O–N bond was formed through the interaction between CO on the surface of the graphite felt electrode and pyridine N in g-C3N4. The proportion of C–O–N bonds increased from 12.22% in GF to 32.56% in GF-1, and the proportion of N–(C)3 bonds decreased from 56.86% in GF to 28.58% in GF-1. The change in the proportion of chemical bonds promoted the transition of the binding force between the electrode and g-C3N4 from an adhesive force to a stronger interatomic force, indicating a more stable binding between g-C3N4 and graphite felt, which was conducive to enhancing the electrochemical activity of the electrode.
The CV curves of the different samples are shown in Fig. 2j. The CV curve of GF exhibited weak redox peaks, indicating its limited catalytic activity for ORR. After acid etching, the CV curve of GF-1 shows a significantly enhanced reduction in peak current density and more distinct redox features, indicating a remarkable improvement in its 2e− ORR activity.44 This phenomenon may be attributed to the etching effect of acid on the surface of graphite felt, which exposes more active sites on the carbon substrate and forms abundant edge defect structures, thereby optimizing the adsorption and activation of oxygen molecules.45
3.3 Cationic red X-GRL degradation performance analysis
3.3.1 Cationic red X-GRL removal by different processes. The degradation efficiency of cationic red X-GRL under high salt conditions was investigated. As shown in Fig. 3a and b, at 12 min, the degradation efficiency of cationic red X-GRL in EO was 20.8%, with the kinetic constant k of 0.006 min−1. In the MgMnxOy–GAC/EO system, the cationic red X-GRL degradation efficiency was 74%, and the kinetic constant k was 0.09 min−1. The results demonstrated that the addition of MgMnxOy–GAC enhanced the degradation efficiency of the EO system. This was possibly attributed to the promotion of H2O2 decomposition by MgMnxOy–GAC, thereby generating more ˙OH. Additionally, the degradation efficiency of cationic red X-GRL in the ozonation process alone was 43%, and the kinetic constant k was 0.046 min−1. This result was attributed to the lower oxidation efficiency of O3 in the ozonation process alone. However, in the MgMnxOy–GAC/O3 system, the degradation efficiency of cationic red X-GRL was 97%, and the kinetic constant k was 0.317 min−1, indicating that the addition of MgMnxOy–GAC could significantly improve the oxidation efficiency of O3. The degradation efficiency of cationic red X-GRL in MgMnxOy–GAC/EP reached 100%. The results demonstrated that the MgMnxOy–GAC/EP system performed superiorly in degrading cationic red X-GRL under high salt conditions.
 |
| | Fig. 3 Different processes for cationic red X-GRL removal. (a) Cationic red X-GRL degradation, (b) COD removal, (c) reaction rate constant, (d) SF, and (e) SEC in various processes. | |
The mineralization degree of organic pollutants is a crucial factor in assessing the oxidation efficiency of AOPs. As shown in Fig. 3c, compared with the other systems, the COD removal efficiency of MgMnxOy–GAC/EP was 90.8% at 60 min. Based on the kinetic constants, the interactions between MgMnxOy–GAC, EO and O3 in the coupled system were evaluated by SF. As shown in Fig. 3d, the SF of the EP, MgMnxOy–GAC/O3 and MgMnxOy–GAC/EO systems were 0.79, 6.76 and 4.65, respectively, while the SF of the MgMnxOy–GAC/EP system was 13.04. These results suggested that MgMnxOy–GAC, EO and O3 had a remarkable synergistic effect in removing cationic red X-GRL. Meanwhile, the SEC of the MgMnxOy–GAC/EP system was 0.027 kW h per g COD (Fig. 3e). Therefore, the MgMnxOy–GAC/EP system not only improved the COD removal efficiency but also reduced the specific energy consumption.
3.3.2 Effects of key operational parameters on MgMnxOy–GAC/EP system. This study investigated the effect of different initial pH on the removal efficiency of cationic red X-GRL in the MgMnxOy–GAC/EP system under high salt conditions. As shown in Fig. 4a and b, the degradation efficiency of cationic red X-GRL significantly improved with the increase in pH from 4.7 to 7.7, and its kinetic constant k increased from 0.120 to 0.257 min−1. The COD removal efficiency also increased from 51.4% to 69.85%. The COD removal efficiency decreased as the pH increased to 8.7. It has been shown that under acidic conditions, the oxidation of pollutants is attributed to direct oxidation by O3.46 However, under alkaline conditions, the HO2− produced by the decomposition of H2O2 facilitated the decomposition of O3 into ˙OH.47 Owing to the higher oxidation potential of ˙OH compared to O3, the degradation efficiency of cationic red X-GRL was superior under alkaline conditions compared to acidic conditions. At higher pH values, the decrease in COD removal efficiency can be attributed to the dissociation of O3 and H2O2 into H2O.48 Therefore, the optimal pH of the MgMnxOy–GAC/EP system was 7.7.
 |
| | Fig. 4 Effect of (a and b) pH, (c and d) current density, (e and f) ozone concentration, and (g and h) catalyst dosage on the removal efficiency of cationic red X-GRL in the MgMnxOy–GAC/EP synergistic system. The inset patterns show the reaction rate constant k at 24 min. Except for the investigated parameter, the other parameters were set as follows: T = 293 K, initial pH = 7.7, cationic red X-GRL = 160 mg L−1, catalyst = 6 g L−1, current density = 22.5 A m−2, O3 concentration = 24 mg L−1, flow rate = 800 mL min−1, and NaCl concentration = 12000 mg L−1. | |
The effect of current density on the removal efficiency of cationic red X-GRL in the MgMnxOy–GAC/EP system under high salt conditions is shown in Fig. 4c and d. When the current density increased from 17.5 A m−2 to 22.5 A m−2, cation red X-GRL was almost completely removed at 24 min, and the kinetic constant k increased from 0.188 to 0.257 min−1. The COD removal efficiency then increased from 57.19% to 69.85% after 60 min. Higher current density could enhance the electrochemical production capacity of H2O2, thereby promoting the reaction of H2O2 with O3 to generate ˙OH.49 This approach can improve the degradation efficiency of cationic red. However, as the current density was further increased, the COD removal efficiency decreased. This phenomenon was attributed to the high current density, which may have generated a large amount of H2O2. If the supply of O3 was insufficient to react with the generated H2O2, the ˙OH would have been quenched by excess H2O2.50 Therefore, in the MgMnxOy–GAC/EP system, the optimal current density was 22.5 A m−2.
The effect of the O3 concentration on the removal efficiency of cationic red X-GRL under high salt conditions in the MgMnxOy–GAC/EP system is shown in Fig. 4e. The removal efficiency of cationic red X-GRL significantly improved as the ozone concentration increased from 8 to 20 mg L−1 and the kinetic constant k increased from 0.129 to 0.244 min−1. At 60 min, the COD removal efficiency increased from 58.75% to 69.68% (Fig. 4f). The mass transfer of O3 from the gas phase to the liquid phase was facilitated by an increase in the concentration of gas-phase O3, thereby increasing the concentration of dissolved O3.51 However, as the O3 concentration was further increased, the kinetic constant k and the COD removal efficiency slightly increased. Therefore, in the MgMnxOy–GAC/EP system, the optimal O3 concentration was 20 mg L−1.
The effect of catalyst dosage on the removal efficiency of cationic red X-GRL in MgMnxOy–GAC/EP system was studied. As shown in Fig. 4g and h, as the catalyst dosage increased, the degradation and mineralization efficiency of cationic red X-GRL significantly increased, indicating that the catalyst significantly promoted the degradation of cationic red X-GRL. As the amount of catalyst increased, more active sites were provided, thereby accelerating the decomposition of O3 and producing more reactive oxygen species (ROS).52 When the catalyst dosage was 8 g L−1, the degradation efficiency of cationic red X-GRL reached 100%, with a kinetic constant k of 0.399 min−1, and a COD removal efficiency of 89.27%. When the catalyst dosage was increased from 8 to 10 g L−1, the kinetic constant k and the COD removal efficiency slightly increased. This phenomenon was attributed to the fact that the reaction was limited by the O3 concentration.53 The number of active sites on the surface of the 8 g per L MgMnxOy–GAC catalyst was sufficient for a 20 mg per L O3 concentration. Therefore, in the MgMnxOy–GAC/EP system, the optimal catalyst dosage was determined to be 8 g L−1.
3.4 Characterization of MgMnxOy–GAC
The morphological evolution of Mn–GAC and MgMnxOy–GAC catalysts before and after the degradation of cationic red X-GRL was systematically investigated by FE-SEM. The Mn–GAC catalyst exhibited obvious surface corrosion after the reaction (Fig. 5a and b), characterized by a significant increase in porosity and partial collapse of the pore structure, indicating damage to the structural integrity under oxidative conditions. In contrast, MgMnxOy–GAC maintained a good surface morphology with negligible changes in the pore structure (Fig. 5c and d), demonstrating the stabilizing effect of Mg doping. The enhanced stability is attributed to the formation of strong Mg–O–Mn interface bonds induced by Mg incorporation. These bonds not only facilitate efficient oxygen electron transfer but also enhance the structural stability of the MgMnxOy–GAC catalyst.
 |
| | Fig. 5 SEM comparison of Mn–GAC and MgMnxOy–GAC catalysts before and after cationic red degradation. (a and b) Surface morphology before and after Mn–GAC degradation, (c and d) surface morphology of MgMnxOy–GAC before and after degradation, and (e) XRD pattern. | |
The structural evolution of MgMnxOy–GAC and MgMnxOy–GAC-1 catalysts was characterized by XRD (Fig. 5e). Both samples exhibited characteristic diffraction peaks of graphite carbon at 23° and 43° (JCPDS no. 26-1076). For the MgMnxOy–GAC catalyst, the diffraction peaks at 34.9°, 40.5°, 58.7°, 70.1° and 73.7° can be attributed to the (111), (200), (220), (311) and (222) crystal planes of MnO (JCPDS no. 07-0230). Notably, although the MgMnxOy–GAC-1 catalyst retained the same MnO phase as MgMnxOy–GAC, the intensity of its diffraction peaks was significantly reduced. This phenomenon may be due to the partial dissolution or surface etching of the active component (MnO) during the corrosion process, which results in grain refinement.54
3.5 Mechanism discussion
3.5.1 Identification of major ROS. To investigate the role of ROS in the MgMnxOy–GAC/EP system, various concentrations of scavengers, including tert-butanol (TBA), isopropyl alcohol (IPA), p-benzoquinone (p-BQ), and furfuryl alcohol (FFA), were used. As shown in Fig. 6a, the addition of 5 mM TBA led to a slight decrease in the degradation efficiency of cationic red X-GRL. However, increasing the TBA concentration to 10 mM and 20 mM did not significantly decrease the degradation efficiency of cationic red X-GRL. This result indicates that excess TBA can completely quench ˙OH in the system. As shown in Fig. 6b, the introduction of 5 mM FFA significantly affected the degradation efficiency of cationic red X-GRL. When the FFA concentration was further increased to 20 mM, the degradation efficiency of cationic red X-GRL slightly decreased. Additionally, the introduction of 5, 10, and 20 mM p-BQ caused a significant decrease in the degradation efficiency of cationic red X-GRL (Fig. 6c). These phenomena suggest the crucial roles of ˙O2− and 1O2 in the degradation of cationic red X-GRL. Furthermore, IPA was used as a quencher to further investigate the role of ˙OH in the degradation of cationic red X-GRL. Specifically, the degradation efficiency of cationic red X-GRL did not significantly decrease with the addition of an excess of IPA (Fig. 6d). Overall, the degradation of cationic red X-GRL was inhibited after the addition of p-BQ and FFA, revealing the key roles of ˙O2− and 1O2 in the degradation of cationic red X-GRL. Additionally, the addition of TBA and IPA reduced the degradation efficiency of cationic red X-GRL, indicating that ˙OH also participates in the degradation of cationic red X-GRL in the MgMnxOy–GAC/EP system. Fig. 6e shows that 1O2 and ˙O2− play a dominant role in the degradation of cationic red X-GRL. To better clarify the contribution of different active species to the degradation of cationic red X-GRL, the inhibition efficiency (η) of various scavengers was calculated (eqn (5)).55| | |
η = (1 − k0/ks) × 100%
| (5) |
where k0 represents the degradation efficiency constant in the absence of scavengers and ks is the degradation efficiency constant in the presence of scavengers. As shown in Fig. 6f, the inhibition efficiencies of TBA and IPA were 44% and 57%, respectively, indicating that ˙OH contributed little to the degradation of cationic red X-GRL in the MgMnxOy–GAC/EP system. The inhibition efficiencies of FFA and p-BQ were 84% and 87%, respectively. Therefore, ˙O2− and 1O2 play a leading role in MgMnxOy–GAC/EP system.
 |
| | Fig. 6 Effect of various scavengers on cationic red X-GRL degradation: (a) TBA, (b) FFA, (c) p-BQ, and (d) IPA. (e) Comparison effect of various scavengers at the same concentration (f) its k with the addition of different scavengers in MgMnxOy–GAC/EP. (g) EPR spectra of DMPO–˙OH, DMPO–˙Cl, DMPO–˙O2− and TEMP–1O2 under different reaction times in the MgMnxOy–GAC/EP system. (h) Removal rate of COD and the change in Cl− concentration within 60 min in the MgMnxOy–GAC/EP system. | |
To further clarify the role of the ROS in MgMnxOy–GAC/EP system, an EPR analysis of the active species within the system was performed. As shown in Fig. 6g, signals for DMPO–˙OH, DMPO–˙O2− and TEMP–1O2 were detected after 10 min, confirming the free radical quenching experiment results. Meanwhile, the presence of DMPO–˙Cl was detected, indicating that a process in which Cl− reacted with ˙OH to form ˙Cl existed in the MgMnxOy–GAC/EP system. The signal of DMPO–˙Cl was gradually strengthened during the reaction, whereas the concentration of Cl− decreased accordingly (Fig. 6h). This phenomenon indicates that ˙OH reacted with Cl−, resulting in the consumption of ˙OH. Under high salt conditions, multiple free radical and non-free radical substances were present in the MgMnxOy–GAC/EP system, which synergistically acted on the degradation of cation red X-GRL.
3.5.2 Proposed mechanism for the degradation of cationic red X-GRL in MgMnxOy–GAC/EP system. In this study, XPS analysis was performed to examine the changes in the oxidation valence states on the surface of MgMnxOy–GAC before and after the reaction, as shown in Fig. 7a. The Mn2+ and Mn4+ contents increased from 29.1% and 8.6% to 38.6% and 58%, respectively, and the Mn3+ content decreased from 62.3% to 3.4%. These results show that Mn2+, Mn3+ and Mn4+ species are actively involved in the catalytic reaction.
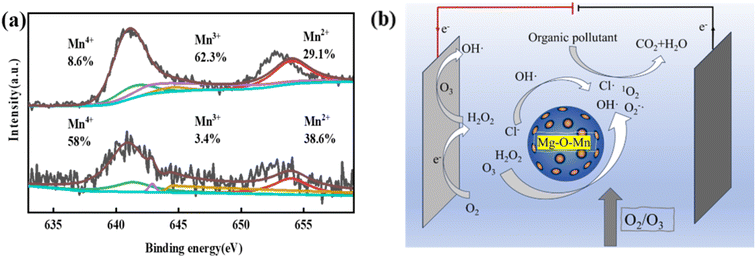 |
| | Fig. 7 (a) Changes in the valence state of Mn and (b) mechanism of pollutant degradation by the MgMnxOy–GAC/EP system. | |
The degradation mechanism of cationic red X-GRL under high salt conditions is shown in Fig. 7b. The O2/O3 mixture was introduced into high-salt printing and dyeing wastewater. At the cathode surface, this gas mixture was reduced to produce H2O2 and ˙OH (eqn (6)–(8)).29 These substances further reacted with O3 to produce ˙O2− and 1O2 (eqn (9)–(13)). In addition, O3 in the liquid phase was adsorbed by MgMnxOy–GAC, thus catalyzing the formation of ˙OH.56 Meanwhile, low-valence Mn ions on the catalyst surface react with O3 and H2O2 to produce high-valence Mn ions, ˙OH and ˙O2− (eqn (14)–(17)). The Mg–O–Mn chemical bond within MgMnxOy significantly increases the electron transfer rate.24 ˙OH in MgMnxOy–GAC/EP system reacted with Cl− to form ˙Cl (eqn (18)).57
In summary, the degradation of cationic red X-GRL by the MgMnxOy–GAC/EP system under high salt conditions was divided into three aspects: (1) active substances such as ˙OH, ˙O2− and 1O2 were formed due to the reaction of the O2/O3 mixture on the cathode surface. (2) O3 and H2O2 were efficiently catalyzed by the MgMnxOy–GAC to produce ˙OH, ˙O2− and 1O2. (3) ˙Cl was generated as a consequence of the reaction between ˙OH and Cl−. Ultimately, the cationic red X-GRL was mineralized into H2O and CO2 by these free radicals under high salt conditions.
| | |
O2 + 2H+ + 2e− → H2O2
| (6) |
| | |
˙O3− + H2O → ˙OH + O2 + OH−
| (8) |
| |
 | (10) |
| |
 | (11) |
| |
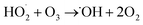 | (12) |
| | |
˙O2− + ˙OH → 1O2 + OH−
| (13) |
| |
 | (14) |
| | |
Mn3+ + H2O2 → Mn4+ + ˙OH + OH−
| (16) |
| | |
4Mn4+ + 2O2− → 4Mn3+ + O2
| (17) |
| | |
Cl− + ˙OH → ˙Cl + OH−
| (18) |
3.6 Potential application of MgMnxOy–GAC/EP
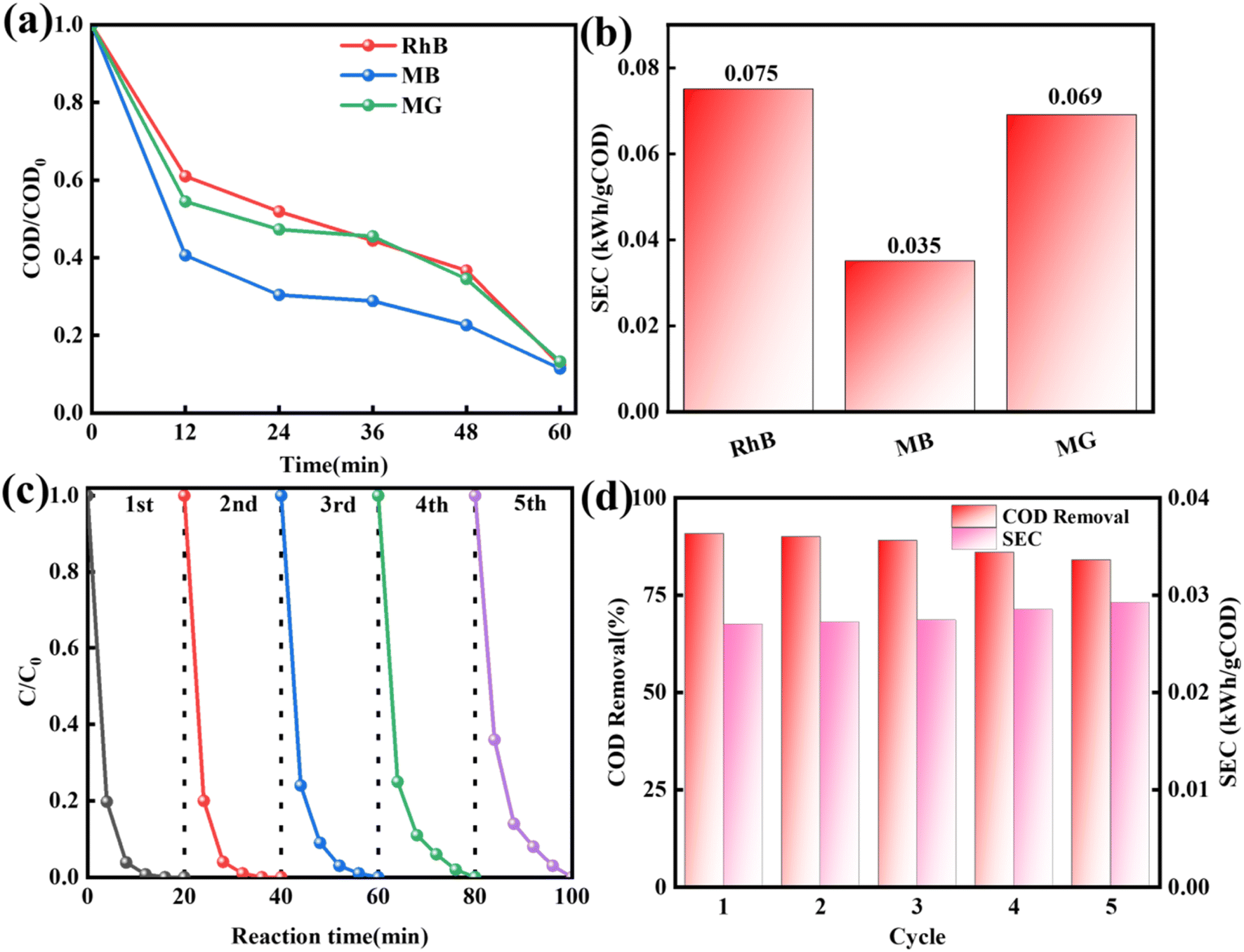
The effectiveness of the MgMnxOy–GAC/EP system for degrading different dyes under high salt conditions was assessed. As shown in Fig. 8a, in MgMnxOy–GAC/EP system, the COD removal efficiencies of rhodamine B (RhB), methylene blue (MB), and malachite green (MG) were 87.6%, 88.6%, and 86.8%, respectively. As shown in Fig. 8b, the SEC values for RhB, MB, and MG in MgMnxOy–GAC/EP system were 0.075, 0.035 and 0.069 kW h per gCOD, respectively.
 |
| | Fig. 8 (a) Removal rate of COD in different high-salt printing and dyeing wastewaters, (b) SEC of different high-salt printing and dyeing wastewaters, (c) cyclic experiment, and (d) COD removal rate and SEC in the cyclic experiment. | |
The stability of the MgMnxOy–GAC/EP system in wastewater treatment is a critical parameter. As shown in Fig. 8c, the degradation efficiency of cationic red X-GRL remained constant at 100% after five cycles. The COD removal efficiency and SEC were slightly different (Fig. 8d). Consequently, the MgMnxOy–GAC/EP system demonstrated excellent reactivity, low energy consumption, and high strength for wastewater treatment. It is well suited for removing multiple pollutants in high-salt printing and dyeing wastewater.
4 Conclusions
According to this study, the MgMnxOy–GAC/EP system was capable of effectively eliminating organic pollutants in high-salt printing and dyeing wastewater. Under high salt conditions, H2O2 generation in the electrochemical system proceeded effectively. The cationic red X-GRL was thoroughly eliminated by the MgMnxOy–GAC/EP system within 16 min, and a mineralization efficiency of over 90% was attained within 60 min. Moreover, the SF of the MgMnxOy–GAC/EP system was 13.04, highlighting the synergistic effect of H2O2, O3 and MgMnxOy–GAC in removing cationic red X-GRL under high salt conditions. The degradation of cationic red X-GRL in the MgMnxOy–GAC/EP system mainly relied on the synergistic mechanism of multiple free radicals dominated by 1O2 and ˙O2−. After replication experiments, the COD removal efficiency remained at approximately 84%, indicating the relatively high stability of the MgMnxOy–GAC/EP system. The removal effects of different pollutants under high salt printing and dyeing wastewater by the MgMnxOy–GAC/EP system were evaluated. This study provides a feasible approach for treating organic pollutants under high salt conditions.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Zhiwei Shang: validation, investigation, methodology, software, writing-original draft; Jing Zhu: methodology, supervision; Guoyu Zhang: conceptualization, supervision, writing, review and editing; Feng Liu: conceptualization, supervision, writing, review and editing; Jiapeng Zhang: supervision; Weirun Li: supervision; Wenye Li: supervision; Fanbo Zeng: supervision; Shuyu Zhao: supervision; Hong You: funding acquisition, supervision; Zhipeng Li: funding acquisition, supervision.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was financially supported by the State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology (No. 2022TS10), the Taishan Industrial Experts Program, and the Natural Science Foundation of Shandong Province of China (No. ZR2023ME212).
References
- G. B. Fernandes, R. D. Alves and L. T. Marconsini, et al., Catal. Today, 2025, 445, 115050, DOI:10.1016/j.cattod.2024.115050.
- O. Tong, S. Shao, Y. Zhang, Y. Chen and S. L. Liu, et al., Clean Technol. Environ., 2012, 14, 857–868, DOI:10.1007/s10098-012-0453-x.
- C. C. Pei, A. A. Aryee, K. K. Zhu, R. Wang and R. P. Han, Sep. Purif. Technol., 2025, 355, 129625, DOI:10.1016/j.seppur.2024.129625.
- Y. L. Pang and A. Z. Abdullah, Clean:Soil, Air, Water, 2013, 41, 751–764, DOI:10.1002/clen.201000318.
- X. Bai, K. F. Lin and Q. Wang, Chem. Eng. J., 2024, 499, 155365, DOI:10.1016/j.cej.2024.155365.
- Z. U. Khan, M. Moronshing and M. Shestakova, et al., Desalination, 2023, 548, 116254, DOI:10.1016/j.desal.2022.116254.
- Y. Y. Zhao, X. M. Zhuang and S. Ahmad, et al., World J. Microbiol. Biotechnol., 2020, 36(3), 37, DOI:10.1007/s11274-020-02815-4.
- J. Cui, Y. Feng, B. Xu, W. Zhang and L. Tan, Process Saf. Environ. Prot., 2023, 179, 835–846, DOI:10.1016/j.psep.2023.09.062.
- J. Wang, H. Liu, D. Ma, Y. Wang and G. Yao, et al., Chemosphere, 2021, 268, 128796, DOI:10.1016/j.chemosphere.2020.128796.
- L. Cheng, Y. Mao, L. Jiang, R. Ma and J. Ma, et al., Chemosphere, 2023, 334, 138964, DOI:10.1016/j.chemosphere.2023.138964.
- S. F. Kang, C. H. Liao and H. P. Hung, J. Hazard. Mater., 1999, 65, 317–333, DOI:10.1016/S0304-3894(99)00005-9.
- J. Zhe, H. He, Z. W. Yi, Z. W. Guo and H. S. Xu, et al., Sep. Purif. Technol., 2025, 356, 129812, DOI:10.1016/j.seppur.2024.129812.
- Y. Zhao, Y. H. Wang, H. B. Chi and Y. A. Zhang, et al., Appl. Catal., B, 2022, 318, 121858, DOI:10.1016/j.apcatb.2022.121858.
- D. Y. Kong, B. Liang, H. Yun, H. Y. Cheng and J. C. Ma, et al., Water Res., 2015, 72, 281–292, DOI:10.1016/j.watres.2015.01.025.
- S. Li, X. D. Jiang, W. C. Xu and M. Li, et al., Water Res., 2024, 267, 122456, DOI:10.1016/j.watres.2024.122456.
- Y. Ge, X. C. Zhong, K. Wang and L. L. Huang, et al., Sep. Purif. Technol., 2025, 357, 129876, DOI:10.1016/j.seppur.2024.129876.
- S. T. Cheng, J. X. Song and L. H. Yu, et al., Sep. Purif. Technol., 2025, 353, 128356, DOI:10.1016/j.seppur.2024.128356.
- Z. Y. Song, X. Q. Wu, Q. Q. He and P. X. Xiang, et al., J. Environ. Chem. Eng., 2024, 12, 112164, DOI:10.1016/j.jece.2024.112164.
- F. Miao, T. Cheng, L. Wang, K. Li and M. Bao, et al., Chem. Eng. Sci., 2024, 292, 120019, DOI:10.1016/j.ces.2024.120019.
- B. Wang, M. Li, C. Li, X. Xuan and H. Li, J. Environ. Chem. Eng., 2024, 12, 113254, DOI:10.1016/j.jece.2024.113254.
- Y. F. Zhu, M. T. Chen and Y. R. Feng, et al., Small Struct., 2025 DOI:10.1002/sstr.202400389.
- B. Y. Chen, Q. Yu, Z. Chen, W. Zhu and S. T. Li, et al., Sep. Purif. Technol., 2021, 279, 119792, DOI:10.1016/j.seppur.2021.119792.
- X. Zhang, Q. Yang, Z. Li and W. Tang, et al., Sep. Purif. Technol., 2025, 353, 128368, DOI:10.1016/j.seppur.2024.128368.
- Q. Shi, J. Ren, H. Wang, S. Li and Z. Bian, Chem. Eng. J., 2024, 490(151476) DOI:10.1016/j.cej.2024.151476.
- N. Abd Rahman, K. T. Wong and C. E. Choong, et al., Appl. Surf. Sci., 2025, 679, 161174, DOI:10.1016/j.apsusc.2024.161174.
- B. Y. Shi, L. Zhang, H. Y. Sun and J. A. Ren, et al., Chemosphere, 2022, 304, 135258, DOI:10.1016/j.chemosphere.2022.135258.
- Y. J. Guo, C. Cao, L. X. Zheng and Y. X. Zhong, et al., Sep. Purif. Technol., 2025, 356, 129811, DOI:10.1016/j.seppur.2024.129811.
- M. Ghahrchi and A. Rezaee, Chemosphere, 2021, 263, 127858, DOI:10.1016/j.chemosphere.2020.127858.
- J. Xie, Y. Zheng, Q. Zhang and S. Li, et al., Appl. Catal., B, 2023, 320, 121935, DOI:10.1016/j.apcatb.2022.121935.
- Y. Zhang, H. Ma, X. Chen and W. Wang, et al., J. Hazard. Mater., 2023, 443, 130250, DOI:10.1016/j.jhazmat.2022.130250.
- Y. Yang, J. J. Pignatello, J. Ma and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 2344–2351, DOI:10.1021/es404118q.
- K. Lu, T. Ren, N. Yan, X. Huang and X. Zhang, Environ. Sci. Technol., 2023, 57, 18888–18897, DOI:10.1021/acs.est.3c00595.
- Z. Chen and J. J. Pignatello, Water Res., 2024, 253, 121256, DOI:10.1016/j.watres.2024.121256.
- W. R. Zhao, Q. W. Liao, J. Zhang and Y. A. Yang, et al., Chem. Eng. J., 2011, 171, 628–639, DOI:10.1016/j.cej.2011.03.105.
- Y. Zheng, S. Qiu, F. Deng, Y. Zhu, G. Li and F. Ma, Sep. Purif. Technol., 2019, 224, 463–474, DOI:10.1016/j.seppur.2019.05.054.
- X. Chen, L. Wang, J. Jin, W. Sun and Z. Yang, et al., Sep. Purif. Technol., 2024, 347, 127600, DOI:10.1016/j.seppur.2024.127600.
- J. C. Wu, Y. Zhao, K. Li, S. Muhammad, M. Z. Ju and L. H. Liu, et al., TrAC, Trends Anal. Chem., 2022, 157, 116734, DOI:10.1016/j.trac.2022.116734.
- Y. S. Zhang, H. M. Duan, N. Wang and D. D. Wei, et al., Chem. Eng. J., 2023, 477, 146903, DOI:10.1016/j.cej.2023.146903.
- R. S. Zou, B. Rezaei and X. Y. Yang, et al., Green Chem., 2024, 26, 8367–8382, 10.1039/d4gc02220c.
- X. L. Fan and C. S. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566, 10.1039/d1cs00450f.
- D. Song, J. Li, Z. Wang and C. Zhao, Appl. Surf. Sci., 2020, 532, 147450, DOI:10.1016/j.apsusc.2020.147450.
- Z. Pan, K. Wang, Y. Wang, P. Tsiakaras and S. Song, Appl. Catal., B, 2018, 237, 392–400, DOI:10.1016/j.apcatb.2018.05.079.
- H. Xu, Z. Zhang, H. Guo, X. Lin, N. Li and W. Xu, J. Taiwan Inst. Chem. Eng., 2021, 125, 387–393, DOI:10.1016/j.jtice.2021.06.028.
- A. Abbas, X. E. Eng, N. Ee and F. Saleem, et al., J. Energy Storage, 2021, 41, 102848, DOI:10.1016/j.est.2021.102848.
- H. Ren, Y. Su, S. Zhao, N. Chen and C. Li, et al., J. Energy Storage, 2024, 75, 109592, DOI:10.1016/j.est.2023.109592.
- J. Y. Gu, J. X. Xie and S. S. Li, et al., Chem. Eng. J., 2023, 452, 139597, DOI:10.1016/j.cej.2022.139597.
- W. Q. Guo, Q. L. Wu and X. J. Zhou, et al., RSC Adv., 2015, 5, 52695–52702, 10.1039/c5ra07951a.
- H. Zhang, S. Li, C. Zhang, X. Ren and M. Zhou, Chemosphere, 2024, 365, 143330, DOI:10.1016/j.chemosphere.2024.143330.
- S. Gharibian, H. Hazrati and M. Rostamizadeh, Chem. Eng. Process., 2020, 150, 107880, DOI:10.1016/j.cep.2020.107880.
- G. Xia, Y. Wang, B. Wang and J. Huang, et al., Water Res., 2017, 118, 26–38, DOI:10.1016/j.watres.2017.04.005.
- Z. Lin, W. Yao, Y. Wang, G. Yu and S. Deng, et al., Water Res., 2016, 88, 691–702, DOI:10.1016/j.watres.2015.11.005.
- T. T. Cao, J. T. Xu, S. Z. Fu and Q. H. Zeng, et al., J. Hazard. Mater., 2024, 480, 136076, DOI:10.1016/j.jhazmat.2024.136076.
- J. Zhang, Z. Xiong, J. Wei, Y. Song, Y. Ren, D. Xu and B. Lai, Chem. Eng. J., 2019, 383, 123144, DOI:10.1016/j.cej.2019.123144.
- X. T. Li, Z. S. Wang, J. Z. Ma and H. He, Environ. Sci. Technol., 2025, 59(10), 5318–5326, DOI:10.1021/acs.est.4c11016.
- Z. Xiong, B. Lai and P. Yang, Water Res., 2018, 140, 12–23, DOI:10.1016/j.watres.2018.04.030.
- H. Zhu, W. C. Ma and H. J. Han, et al., Chem. Eng. J., 2017, 327, 91–99, DOI:10.1016/j.cej.2017.06.025.
- G. Zeng, L. An, M. Jiang and W. Li, et al., J. Hazard. Mater., 2024, 480, 136084, DOI:10.1016/j.jhazmat.2024.136084.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Feng Liu*ab,
Jiapeng Zhanga,
Weirun Lib,
Wenye Lia,
Fanbo Zenga,
Shuyu Zhaoa,
Hong Youab and
Zhipeng Liab
*ab,
Feng Liu*ab,
Jiapeng Zhanga,
Weirun Lib,
Wenye Lia,
Fanbo Zenga,
Shuyu Zhaoa,
Hong Youab and
Zhipeng Liab