
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual phenomena of ethylene homopolymerization catalyzed by α-diimine nickel emerging in a microflow platform†
Zeen Zhang,
Tianjiao Li,
Yong Dong and
Yangcheng Lu *
*
State Key Laboratory of Chemical Engineering, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: luyc@mail.tsinghua.edu.cn
First published on 20th February 2025
Abstract
This study focuses on ethylene homopolymerization catalyzed by α-diimine catalysts within a designed microfluidic system. Distinct from conventional batch reactors, this microfluidic configuration effectively eradicates the reaction environment heterogeneity, thereby guaranteeing homogeneous reaction conditions and decoupling the mass transfer from the reaction process. This unique setup enables precise modulation of ethylene concentration, facilitating the isolation of variables to scrutinize the effects of temperature and pressure on the polymerization. By varying residence times, it's revealed that the reaction rate is ethylene concentration-independent. Variable separation studies between concentration and temperature show an inverse relationship between temperature and branching degree (BD), contrasting traditional batch reactions due to ethylene concentration changes. Pressure studies indicate that higher pressure leads to lower BD. Different product structures' steric hindrances influence the reaction rate; greater hindrance slows it down. These novel findings suggest microreactors offer new perspectives on late transition metal-catalyzed ethylene homopolymerization, potentially enabling breakthroughs in mass production.
1. Introduction
Polyolefin materials are crucial in modern life and industry, widely used in packaging, automotive, etc. Polyethylene, the most-produced polyolefin, has chemical stability, good mechanics, and low cost. Its synthesis via ethylene homopolymerization is a key area in polymer chemistry.The development of ethylene polymerization catalysts has been remarkable. Early Ziegler–Natta1 catalysts initiated industrial polyethylene production but had limited structure regulation. Then early-transition metal metallocene complexes in combination with MAO and non-metallocene early-transition metal complexes were proved useful in the synthesis of polyolefin.2–4 Subsequently, late-transition metal catalysts exhibited a range of unique advantages.5–7 Ever since Brookhart's pioneering research on α-diimine catalyst,6 extensive studies have been carried out on this catalyst system over the past several decades.8–10 Owing to its distinctive chain walking mechanism, this particular kind of catalysts is capable of synthesizing polyolefins possessing branched chain architectures as well as diverse polymer topologies.11,12
However, traditional ethylene homopolymerization often employs batch reactors, which exhibit significant deficiencies in mass and heat transfer efficiency. During the exothermic reaction, slow heat transfer can lead to uneven local temperatures, creating temperature gradients that degrade reaction uniformity and affect the consistency of polymer properties. Simultaneously, limited mass transfer hinders adequate contact between reactants and catalysts, not only reducing reaction rates but also increasing the likelihood of side reactions due to excessively high local concentrations, making it challenging to meet the stringent requirements for high quality and performance in modern polyolefin production.
The advent of microreactor technology offers innovative ideas and effective solutions to these challenges. Microreactors are characterized by their tiny channel dimensions (typically in the micrometer range), which bestow them with a significantly high specific surface area, resulting in an order-of-magnitude improvement in mass and heat transfer efficiency compared to traditional batch reactors. In microreactors, reactants can achieve rapid mixing and uniform distribution, effectively avoiding local concentration and temperature differences, thus significantly enhancing reaction selectivity and yield.13,14 Moreover, microreactors allow for precise control of key parameters such as reaction temperature, pressure, and residence time, providing unprecedented accuracy and flexibility for optimizing reaction conditions.15 In radical polymerization reactions, they can precisely control radical concentrations and optimize the molecular weight distribution of the polymers.
In the study of ethylene homopolymerization catalyzed by α-diimine nickel catalysts, although this catalyst has shown unique potential, the interplay between mass transfer and reaction processes makes it difficult to gain clear insights into the reaction laws, hindering a deeper understanding of this catalytic system and its further application development. To address this issue, we innovatively placed the ethylene homopolymerization catalyzed by α-diimine nickel catalysts in a microreactor platform. Leveraging the characteristics of microreactors, the polymerization process proceeded effectively and smoothly, mostly conforming to common reaction characters. Surprisingly, we noticed some unusual but confirmative phenomena in a certain conditional window, such as, elevated pressure resulting in decreased reaction rate, reflecting the complexity of reaction system. We tried to interpret these phenomena at microscale, and indicated its significance for polymerization process control and design toward product functionalization or high efficiency.
2. Results and discussion
2.1 Reaction performances in a microflow platform
In the microflow system, the amount of ethylene added can be accurately controlled by regulating the flow rate of ethylene gas. Then, the mass of the product is obtained through the weighing method, and further the monomer conversion of ethylene can be calculated, which is difficult to obtain in traditional batch reactions. In this work, toluene was selected as the solvent. As a preliminary experiment, the dissolution of ethylene in toluene was observed in microflow system at first. It was found that ethylene could be sheared into small bubbles in the T-shaped micromixer and then dissolved within a few seconds. This implies that, compared with the traditional batch reactors, the dissolution of ethylene in the microfluidic system is significantly intensified, and it can be ensured that there is no presence of ethylene gas in the reaction tubes, allowing the reaction to proceed in a homogeneous state. Therefore, the influence of the mass transfer process during which ethylene gas dissolves in toluene can be eliminated, and thus the intrinsic kinetics of the reaction may be obtained.Firstly, the reliability of the polymerization platform was verified through repeated experiments. The experimental conditions are shown in Table 1. The ethylene conversion rates of the three sets of experiments were 46.59%, 45.24% and 43.99%, respectively, with the relative error being less than 3%. Meanwhile, the branching degrees (BD) of polyethylene obtained from the three sets of experiments were 128, 126 and 125, respectively, with the relative error being less than 2%. Judging from the results of both the raw material conversion and the product structure, the platform has good experimental repeatability and the experimental data are reliable. GPC were also employed to test the molecular weight and its distribution of the polymer. Under the condition (Table 1), the weight-average molecular weight (MW) of the polymer was measured to be 7.3 × 104, the number-average molecular weight (Mn) was 4.3 × 104, and the molecular weight distribution (PD) was 1.7. Compared with the data in the literature, the molecular weight and its distribution are reasonable. Since the focus of this paper is not on the discussion of molecular weight, the exploration of the laws regarding molecular weight will not be carried out in the context.
| P/MPa | T/°C | cethylene,0/% wt | ccat/‰ wt | cMAO/% wt | t/min |
|---|---|---|---|---|---|
| 0.8 | 10 | 0.75 | 0.14 | 0.13 | 10 |
By changing the reaction time at the same combination of temperature, pressure, ethylene concentration, catalyst concentration and cocatalyst concentration, the time profile of polyethylene yield and ethylene conversion were obtained, as shown in Fig. 1. Fig. 1 also shows the results of linear fitting of X–t. The fitting parameter of correlation coefficient R2 = 0.99 and the fitted straight line passes through the origin. The fitting results are in line with the characteristics of zero-order kinetics. That is, the reaction rate is independent of the ethylene concentration.
 | ||
| Fig. 1 Conversion of C2H4 versus of time refer to entries 1 to 5 in Table 2. | ||
Since in traditional batch reactions, the ethylene concentration varies with changes in pressure and temperature, the conclusions regarding the variation of the branching degree (BD) with pressure and temperature discussed in most of the literature are often the result of the coupling of multiple factors, without controlling variables. Moreover, polymerization is a highly exothermic reaction. In batch polymerization, problems such as hot spots often occur, making it rather difficult to control the reaction conditions. By taking advantage of the merits of microreactors, such as the ability to adjust the initial concentration of the system and their good heat transfer performance, the above problems can be solved.
Herein, a series of polymerization experiments in which the ethylene temperature was varied, but the pressure held at 0.8 MPa, is summarized in Table 2 and Fig. 2. The temperature range for the experiment was selected from 0 °C to 20 °C. Within this temperature range, during the reaction time of 10 minutes, the activity of the catalyst can be maintained relatively well, which means there will be no significant decrease in the reaction rate or abnormal variations in BD due to catalyst deactivation.
| Entry | P/MPa | T/°C | cethylene,0/% wt | ccat/‰ wt | cMAO/% wt | t/min | Conversion/% | BD |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.8 | 10 | 0.75 | 0.14 | 0.13 | 5 | 19.89 | 125 |
| 2 | 0.8 | 10 | 0.75 | 0.14 | 0.13 | 7 | 23.34 | 130 |
| 3 | 0.8 | 10 | 0.75 | 0.14 | 0.13 | 10 | 45.24 | 126 |
| 4 | 0.8 | 10 | 0.75 | 0.14 | 0.13 | 15 | 74.06 | 124 |
| 5 | 0.8 | 10 | 0.75 | 0.14 | 0.13 | 20 | 100 | 115 |
| 6 | 0.8 | 0 | 0.75 | 0.14 | 0.13 | 10 | 31.91 | 150 |
| 7 | 0.8 | 5 | 0.75 | 0.14 | 0.13 | 10 | 42.24 | 132 |
| 8 | 0.8 | 15 | 0.75 | 0.14 | 0.13 | 10 | 52.27 | 113 |
| 9 | 0.8 | 20 | 0.75 | 0.14 | 0.13 | 10 | 67.16 | 100 |
| 10 | 0.5 | 10 | 0.75 | 0.14 | 0.13 | 10 | 85.39 | 138 |
| 11 | 1 | 10 | 0.75 | 0.14 | 0.13 | 10 | 9.42 | 121 |
| 12 | 0.5 | 10 | 0.5 | 0.14 | 0.13 | 10 | 60.94 | 247 |
| 13 | 0.8 | 10 | 0.5 | 0.14 | 0.13 | 10 | 76.85 | 189 |
| 14 | 1 | 10 | 0.5 | 0.14 | 0.13 | 10 | 83.67 | 157 |
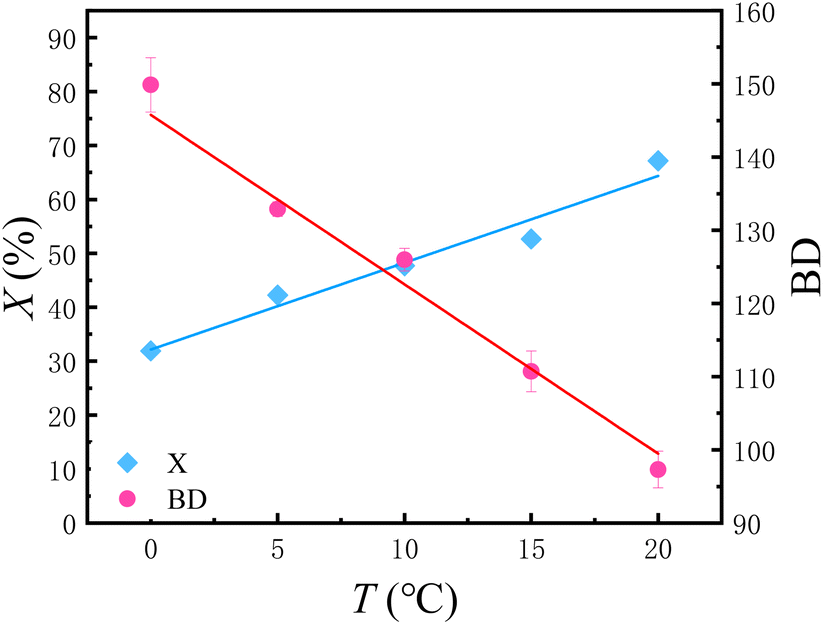 | ||
| Fig. 2 Conversion of C2H4 and BD versus temperature refer to entries 3, 6–9 in Table 2. | ||
It can be seen from the Fig. 2 that under the same reaction time, the ethylene conversion increases linearly with the increase of reaction temperature. In other words, in the case of ensuring that the catalyst does not lose activity, increasing the temperature is conducive to accelerating the reaction rate.
As for the BD, it can be seen from the Fig. 2 that BD shows a linear decreasing trend with the increase in temperature. This is contrary to the conclusions obtained in many traditional batch reactors. Derek P. Gates et al.16 once made speculations on the mechanism of ethylene polymerization catalyzed by α-diimine nickel catalysts: basically, the chain walking takes place in the cationic alkyl complex (B). As the ethylene concentration rises, the lifetime of this complex is reduced, and consequently the degree of chain walking is also decreased. Our experiment provided further evidence for this mechanism conjecture. As the temperature increases, the solubility of ethylene in toluene decreases. For traditional batch polymerization, the concentration of ethylene in the solution is exactly the solubility of ethylene in toluene under the corresponding temperature and pressure. That is to say, with the rising temperature, the concentration of ethylene drops, which is the main reason why the BD increases with the increase in temperature during batch polymerization. However, in the flow platform, the concentration of ethylene is controlled by a gas mass flowmeter, which can ensure that the ethylene concentration remains equal under different temperature conditions. Therefore, the experimental data can reflect the real impact of temperature. As the temperature rises, the movement rate of molecules accelerates and the rate of capturing ethylene increases, resulting in the decrease of BD (Fig. 3).
 | ||
| Fig. 3 Proposed mechanism of propagation for the preparation of branched polyethylene. | ||
2.2 Effect of system pressure
A series of polymerization experiments in which the system pressure was varied from 0.5 MPa to 1 MPa, but the temperature held at 10 °C, is summarized in Table 2 and Fig. 4. | ||
| Fig. 4 (a) Conversion of C2H4 and BD versus pressure with the initial concentration of C2H4 0.75% wt refer to entries 3, 10, 11 in Table 2. (b) Conversion of C2H4 and BD versus pressure with the initial concentration of C2H4 0.5% wt refer to entries 12–14 in Table 2. | ||
As shown in Fig. 4, two sets of experiments with different ethylene concentrations simultaneously demonstrated that as the pressure of the reaction system increases, the BD decreases. Regarding the conversion, at lower ethylene concentrations, the conversion increases with an increase in pressure. However, at higher ethylene concentrations, the conversion shows an opposite trend.
As for the changes in BD, with the elevation of pressure, the interaction among molecules in the liquid is enhanced and the distance between molecules becomes smaller, which makes it easier for the catalyst to capture ethylene. As a result, the chain growth rate is accelerated relative to the chain walking rate, thus leading to the reduction of BD.
As for the changes in the conversion, that is, the reaction rate, under different pressures, the two sets of experiments showed completely opposite trends. Under a higher ethylene concentration, the reaction rate decreases as the pressure increases; while under a lower ethylene concentration, the reaction rate increases with the increase in pressure. We believe that just like the steric hindrance effect of the catalyst ligand, the steric hindrance caused by the change in product structure can also significantly affect the reaction rate. When the steric hindrance is relatively large, it becomes difficult to capture ethylene and the reaction rate slows down.
Under the conditions of lower pressure and lower ethylene concentration, the degree of branching of polyethylene increases, and when the pressure is relatively low, the BD of the polymer increases sharply. According to the free volume theory, the volume of a polymer material consists of two parts: one part is the volume occupied by the molecules themselves, which is called the occupied volume; the other part is the voids between molecules, which is called the free volume. Branched polymers have a larger occupied volume and a smaller free volume compared with linear polymers, resulting in an increase in the steric hindrance of polyethylene and an enhanced shielding effect on the metal active center. When the pressure is relatively high and the concentration is also high, the polymer tends to form a straight chain. Under the action of the polymer shape and pressure, physical cross-linking occurs in the polymer, which further leads to an increase in steric hindrance and a decrease in the ethylene capture rate.
The cross-linking of polyethylene can significantly increase its thermogravimetric char yield. After the cross-linking treatment of polyethylene materials, their molecular structures become more complex and stable, which enables more carbon atoms to be retained to form char during high-temperature decomposition. Therefore, cross-linked polyethylene is superior to non-cross-linked polyethylene in terms of thermal stability, and its thermogravimetric char yield is significantly increased. By comparing the TGA results of ethylene concentrations of 0.50% wt and 0.75% wt respectively at 1 MPa under the same reaction temperature, catalyst and cocatalyst concentrations, it was found that the char yield was only 3% when the concentration was 0.50% wt, while it was as high as 14% when the concentration was 0.75% wt. This indicates that the polyethylene produced by polymerization at an ethylene concentration of 0.75% wt has more cross-linking, which is consistent with the reasons we speculated previously (Fig. 5).
 | ||
| Fig. 5 The TGA results refer to entries 11 and 14 in Table 2. | ||
3. Conclusions
In summary, a microfluidic system was designed to accomplish the ethylene homopolymerization catalyzed by α-diimine catalysts. It enabled to eliminate the impacts brought about by the heterogeneity of the reaction environment in conventional batch reactors, and achieve uniform reaction conditions. It is also capable of separating the mass transfer process from the reaction process, which makes it possible to study the intrinsic kinetics. Moreover, the concentration of the fed ethylene can be precisely controlled to separate variables and explore the influences of factors such as temperature and pressure.Through the study of polymerization reactions with different residence times, it was found that the reaction rate is independent of the ethylene concentration. Through the study of the separation of variables between concentration and temperature, it was discovered that an increase in temperature would lead to a decrease in the branching degree (BD), which is contrary to the conclusion of traditional batch reactions. The difference come from that the traditional batch reactions were deviated from the change in ethylene concentration meanwhile. Through the study of the separation of variables between concentration and pressure, it was found that the higher the pressure, the smaller the branching degree. Different product structures will bring about different steric hindrances, which in turn affect the reaction rate. The greater the steric hindrance, the stronger the shielding of the active catalytic sites and the slower the reaction rate. These unusual phenomena gave us a reminder that new platform like microreactor might provide new insights into ethylene homopolymerization catalyzed by late transition metal and potential breakthroughs in mass production.
4. Experimental sections
4.1 Materials
Ethylene (C2H4, ≥99.9%, Air Liquide (Tianjin) Co., Ltd), methylaluminoxane (MAO) (10% wt solution in toluene, Rhawn), acenaphthenequinone (C12H6O2, 98%, Macklin), 2,6-diisopropylaniline (C12H19N, 95%, Macklin), acetonitrile (C2H3N, ≥99.9%, extra dry, water ≤30 ppm, J&K), acetic acid (C2H4O2, 99.7%, Aladdin), hexane (C6H14, 97.5%, extra dry, water ≤50 ppm, J&K), acetone (C3H6O, 99.5%, Tongguang) and nickel(II) bromide ethylene glycol dimethyl ether complex ((DME)NiBr2, 97%, Adamas) were used directly without further purification. Dichloromethane (CH2Cl2, 99.9%, extra dry with molecular sieves water ≤50 ppm, J&K) and toluene (C6H5CH3, ≥99.9%, Tongguang) were purged with nitrogen and then dehydrated by adding molecular sieves with a volume fraction of 10%. Deuterated chloroform (CDCl3, (D, 99.8%)) were purchased from Adamas and was used directly as received.4.2 Equipment and procedures
 | ||
| Fig. 6 Micro-flow system. | ||
To compare batch polymerization and flow polymerization, we conducted comparative experiments under the same conditions. The experimental conditions and results are shown in Table 3. For batch polymerization, the experimental method we adopted was as follows: ethylene was continuously introduced into an autoclave filled with a toluene solution containing the catalyst and cocatalyst (which had been pre-pressurized with nitrogen to the specified pressure), and the mixture was continuously stirred. The time when ethylene started to be introduced was regarded as the starting time of the reaction. When the reaction ended, the ethylene feed was shut off and the vent valve was opened to terminate the reaction. The amount of ethylene introduced was the sum of the ethylene dissolved in toluene (estimated by the solubility) and the ethylene in the headspace (estimated by the ideal gas equation of state). Judging from the data, the mass transfer process indeed has a significant impact on the rate of the entire polymerization reaction. Microreactors, however, can separate mass transfer from the reaction, enabling the exploration of the intrinsic reaction laws. Meanwhile, by taking advantage of their enhanced mass transfer, microreactors can improve the apparent reaction rate.
| Mode | P/MPa | t/min | T/°C | ccat/‰ wt | cMAO/% wt | methylene/g | mpoly/g | Conversion/% |
|---|---|---|---|---|---|---|---|---|
| Batch | 0.1 | 1.2 | 20 | 0.14 | 0.13 | 0.00514 | 0.00235 | 45.70 |
| Flow | 0.3396 | 0.00204 | 0.59 |
4.3 Characterization
Branching degree (BD) is represented by the number of methyl groups per thousand carbon atoms (Me/1000 C). The Me/1000 C values for different polyethylene were calculated based on the ratio of the integral area of the peak attributed to the hydrogen atoms in the methyl groups to the integral area of the peaks attributed to all hydrogen atoms in the 1H-NMR spectra. The calculation formula18 shows as follows.

Among them, A1 represents the integral area of the peak assigned to the hydrogen in the methyl group in the 1H-NMR, and A2 represents the integral area of the peak assigned to the hydrogen in the methylene and methine groups.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Zeen Zhang: writing original draft, conceptualization, methodology, investigation, data curation; Tianjiao Li: investigation, validation; Yong Dong: investigation, data curation; Yangcheng Lu: supervision, conceptualization, methodology, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We appreciate financial support from the National Natural Science Foundation of China (21422603, U1662120, 21978152).References
- K. Soga and T. Shiono, Prog. Polym. Sci., 1997, 22, 1503–1546 CrossRef CAS.
- A. Andresen, H.-G. Cordes, J. Herwig, W. Kaminsky, A. Merck, R. Mottweiler, J. Pein, H. Sinn and H.-J. Vollmer, Angew Chem. Int. Ed. Engl., 1976, 15, 630–632 CrossRef.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479–1494 CrossRef CAS.
- M. Mitani, T. Nakano and T. Fujita, Chem.–Eur. J., 2003, 9, 2396–2403 CrossRef CAS PubMed.
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 744–745, 10.1039/B111252J.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- T. R. Younkin, E. F. Connor, J. I. Henderson, S. K. Friedrich, R. H. Grubbs and D. A. Bansleben, Science, 2000, 287, 460–462 CrossRef CAS PubMed.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- J. M. Kaiser and B. K. Long, Coord. Chem. Rev., 2018, 372, 141–152 CrossRef CAS.
- F. Wang and C. Chen, Polym. Chem., 2019, 10, 2354–2369 RSC.
- L. H. Shultz, D. J. Tempel and M. Brookhart, J. Am. Chem. Soc., 2001, 123, 11539–11555 CrossRef CAS PubMed.
- L. Guo, S. Dai, X. Sui and C. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- L. Falk and J. M. Commenge, Chem. Eng. Sci., 2010, 65, 405–411 CrossRef CAS.
- M. N. Kashid and L. Kiwi-Minsker, Ind. Eng. Chem. Res., 2009, 48, 6465–6485 CrossRef CAS.
- S. A. Kazemi Oskooei and D. Sinton, Lab Chip, 2010, 10, 1732–1734 RSC.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- R. van Asselt, C. J. Elsevier, W. J. J. Smeets, A. L. Spek and R. Benedix, Recl. Trav. Chim. Pays-Bas, 1994, 113, 88–98 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085–3100 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra of ligand and polymer and additional experimental details and methods. See DOI: https://doi.org/10.1039/d5ra00557d |
| This journal is © The Royal Society of Chemistry 2025 |