
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu supported on γ-AlOOH and γ-Al2O3 for low temperature oxidation of CO and aromatic alcohols†
Zongbo Shi *a,
Qing Zhanga,
Guangpeng Yanga,
Liang Liua,
Gang Wangb,
Rizki Ekanandac,
Ismal Gamarc,
Ruilin Wangb and
Runsheng Zhuo*a
*a,
Qing Zhanga,
Guangpeng Yanga,
Liang Liua,
Gang Wangb,
Rizki Ekanandac,
Ismal Gamarc,
Ruilin Wangb and
Runsheng Zhuo*a
aREZEL Catalysts Corp., Shanghai, 201313, China. E-mail: zongbo.shi@rezel.com.cn; Fax: +86-21-38280081
bCollege of Materials Science and Engineering, Sichuan University, Chengdu 610065, China
cPT Pertamina (Persero), East Jakarta, 13920, Indonesia
First published on 10th March 2025
Abstract
0.5%Au supported on γ-AlOOH and γ-Al2O3 was used for low temperature oxidation of CO and aromatic alcohols. Various characterization techniques, including X-ray diffraction, N2 adsorption, FT-IR spectroscopy, XPS, TEM, CO2-TPD and solid-state MAS NMR, were employed to characterize these catalysts. The Au/γ-AlOOH sample has abundant hydroxyl groups and basic sites on its surface, exhibiting strong adsorption for aromatic alcohols, and outstanding activity for low-temperature oxidation of aromatic alcohols. The Au/γ-Al2O3 sample shows sufficient Au+ sites leading to high performance for low-temperature CO oxidation.
1 Introduction
Small Au nanoparticles have attracted considerable attention in the last few decades due to their high activity for catalytic oxidation of various reactions.1,2 Low temperature oxidation of carbon monoxide is the classical example of gold catalysis, as first reported by Haruta and co-workers in 1987.3,4 Recently, low temperature oxidation of alcohol on Au nanoparticles using molecular oxygen as an oxidant to the corresponding aldehydes, ketones and acids has met the demand of green synthesis of organic products.5–9The support can influence catalyst activity by participating in the reaction. For example, the synergetic catalysts of Au nanoparticles and alkaline metal oxide support promote the oxidation activity of both alcohol and CO. Costa et al. found that using MgO as a support for preparing gold catalysts enables the effective oxidation of a wide range of alcohols with molecular oxygen as the sole oxidant.10 Schüth and his coworkers synthesized gold supported on MgO, which exhibited unprecedented oxidation activity for CO even at −89 °C.11 The support can also influence the catalytic activity as it affects parameters, such as the Au particle size, Au oxidation state, and metal–support interaction.12 Reducible oxides such as TiO2, ZrO2, Fe2O3, CeO2 and Co3O4 are considered most active because of their excellent ability to provide reactive oxygen to the active gold sites.1,13 TiO2 has been reported to reduce the size of Au nanoparticles, which results in high oxidation activity of CO.14–16 The metal–support interaction between Au and the CeO2 support has also been reported, which indicated high oxidation activity of alcohol and CO.17–21 The ionic states of supported gold were stabilized by the cerium oxide and Au+ cations were claimed as the active sites in the partial oxidation of alcohols.17,18 Corma and coworkers prepared a nanocrystalline CeO2 support, which enhances the activity of Au for CO oxidation by two orders of magnitude compared to the catalysts prepared using conventional CeO2 support, by influencing the surface electronic properties and, consequently, the gold–support interaction.21
Hydroxy groups and alkalinity of support are also known to influence the stability and activity of catalytic Au nanoparticles.22–24 γ-Al2O3 is the most commonly used noble metal catalyst carrier due to its large specific surface area, which is usually obtained by interlayer dehydroxylation of γ-AlOOH at temperatures of 450–750 °C.25 γ-Al2O3 and γ-AlOOH carriers have significant differences in hydroxyl groups, surface Al3+, acidity as well as alkalinity. Huang et al. claimed that the strong alkaline sites on the surface of γ-AlOOH enable Au/γ-AlOOH to exhibit high activity in the oxidation of α,ω-diols, despite its gold particle size being much larger than that of Au/γ-Al2O3.24 The morphology of Al2O3 support also affects the activity of catalytic Au nanoparticles. Recently, the thin porous Al2O3 sheets were reported as exceptional catalyst supports for Au nanoparticles, exhibiting high activity for low-temperature CO oxidation and stabilizing Au nanoparticles at annealing temperatures up to 900 °C.26 The Au particle size and water vapor in the atmosphere is crucial for the low temperature CO oxidation performance of Au/Al2O3 catalysts,27–29 Moroz et al. discovered that the Au/Al2O3 catalysts containing Au particles with a diameter of ≤5 nm demonstrate remarkable catalytic activity for the oxidation of CO when water vapor is present under near-ambient conditions.29
Here we present an unexpected result for CO and aromatic alcohols oxidation with Au/γ-AlOOH and Au/γ-Al2O3 as catalysts. The Au/γ-AlOOH exhibits higher aromatic alcohols oxidation activity than that of Au/γ-Al2O3 catalyst, while Au/γ-Al2O3 exhibits higher CO oxidation activity. We also investigated the effects of support and Au oxidation state on the oxidation of CO and aromatic alcohols.
2 Experimental
2.1 Chemicals
NaAlO2 (Al2O3, 54.09 wt%; Na2O, 40.44 wt%) were purchased from J&K Chemical Ltd. Sodium bicarbonate, ammonium sulphate, ethanol (EtOH), HAuCl4 and KBH4 all in AR grade, were supplied by Sinopharm Chemical Reagent Co. Ltd. The chemicals were used as received, without further purification.2.2 Preparation of boehmite (γ-AlOOH) and γ-alumina (γ-Al2O3)
The hierarchically structured boehmite and γ-alumina supports with a flower-like morphology were synthesized taking bayerite as the starting raw materials following the reference.30Typical synthesis of bayerite was as follows: 20.1 g NaAlO2 was dissolved in 134.0 g of hot water, stirred and then cooled to room temperature. After that, 190 mL of 1 mol L−1 NaHCO3 solution was added drop-wise to the above solution. A white suspension was formed. The suspension was agitated for about 6 h, then filtered and washed thoroughly with hot water. The resulting filter cake was then dried at 80 °C overnight.
Typical syntheses of hierarchically structured boehmite and alumina were as follows: A mixture of 0.8 g bayerite, 0.6 g (NH4)2SO4, 2.7 g H2O and 2.3 g EtOH was placed in a 25 mL Teflon-lined stainless-steel autoclave, heated to 175 °C, maintained at the temperature for 12 h, and then cooled down naturally. The obtained white solid was recovered by filtration, washed with hot water, and dried at 80 °C overnight. The product was denoted as γ-AlOOH. The γ-Al2O3 sample was obtained by calcination of the γ-AlOOH at 550 °C for 6 h with a heating rate of 2 °C min−1.
2.3 Supporting 0.5%Au on the γ-AlOOH and γ-Al2O3
The Au/γ-AlOOH or Au/γ-Al2O3 was prepared as follows:10 g γ-AlOOH or γ-Al2O3 was added into a vessel (50 mL) containing 25 mL of 0.01 mol L−1 HAuCl4 and stirred for 2 min. Subsequently, a 5% NH3·H2O solution was added dropwise to adjust the pH of the suspension to 8–9. The suspension was further stirred for 2 h. Then, 0.27 g of KBH4 was added to reduce the Au3+ species. The obtained solid was filtered, washed with deionized water, and dried under vacuum overnight.
2.4 CO oxidation testing
The CO oxidation reaction was performed in a continuous flow fixed-bed quartz reactor under atmospheric pressure. A 0.2 g (40–60 mesh) sample was loaded and pre-treated with a 20 vol% O2/He mixture (50 mL min−1) at 100 °C for 30 min. After cooling down to 20 °C, a gas mixture with 1.0 vol% CO/2.5 vol% O2/He (50 mL min−1) was introduced. Moisture was controlled by passing the feed gas through a soda lime tube. The concentrations of CO, CO2 and O2 in the outlet streams were measured using an on-line gas chromatograph.2.5 1-Phenylethanol, benzyl alcohol and 4-methoxybenzyl alcohol oxidation testing
The oxidations of aromatic alcohols (1-phenylethanol, benzyl alcohol, or 4-methoxybenzyl alcohol) were carried out in a 25 mL glass reactor equipped with a reflux condenser and a magnetic stirrer. In a typical reaction, 100 mg of catalyst, 1 mmol of aromatic alcohols and 5 mL of tetradecane were introduced into the reactor. The air flow rate was 60 mL min−1, the string rate was 350 rpm, and the mixture was heated to 40∼70 °C and maintained at this temperature for 6∼8 h. 0.5 mmol 1,3,5-triisopropylbenzene was used as an internal standard. The liquid samples were analyzed using a Shimadzu GC-2014 gas chromatograph equipped with a 30 m DB-Wax capillary column and an FID detector.2.6 Characterization methods
Powder X-ray diffraction (XRD) patterns were collected on a Rigaku-Ultima diffractometer using a Cu Kα radiation source (λ = 0.15432 nm) in the 2θ range from 5° to 80°. Transmission electron microscopic images were conducted on TECNAI G2 F30 operating at 300 kV after the specimens were dispersed in ethanol and deposited on holey copper grids. N2 adsorption–desorption isotherms were measured at −196 °C on a Quanta chrome Autosorb-3B volumetric adsorption analyzer. Before the measurements, the samples were outgassed in the degas port of the adsorption apparatus at 150 °C for 6 h. BET specific surface area was calculated using adsorption data acquired at a relative pressure (P/P0) range of 0.05–0.30 and the total pore volume determined from the amount adsorbed at P/P0 of about 0.99. Pore size distribution (PSD) curves were calculated from the adsorption isotherm branches using the Barrett–Joyner–Halenda (BJH) algorithm. Au contents were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) on a Thermo IRIS Intrepid II XSP atomic emission spectrometer. Surface electronic state of Au was also evaluated using X-ray photoelectron spectroscopy (XPS) measurements with a Thermo Fisher Scientific ESCALAB 250Xi spectrometer with Al Kα radiation (1486.6 eV) as incident beam with a monochromator. All the spectra were obtained at room temperature, and the binding energies of elements were referenced to the adventitious C1s peak at 284.8 eV. 27Al MAS NMR spectra were measured on a VARIAN VNMRS 400WB NMR spectrometer.Temperature-programmed desorption of CO2 (CO2-TPD) testing was performed using a TP-5080 chemisorption instrument (Xianquan Co., Ltd, Tianjin, China) with a thermal conductivity detector (TCD). After pretreatment of each sample (100 mg) at 200 °C under flowing helium (25 mL min−1) for 2 h, the sample was cooled to 50 °C, and then adsorbed to saturation by introducing 10 vol% CO2/He (25 mL min−1) mixture for 10 min. CO2 physically adsorbed on the catalyst was removed by flushing the sample with helium (25 mL min−1) for 20 min. Thermal desorption of CO2 was carried out in the temperature range of 50∼200 °C increasing at a rate of 10 °C min−1.
Infrared (IR) spectra were recorded on a Nicolet Fourier transform infrared spectrometer (NEXUS 670). The self-supported wafers (about 10 mg, Φ 1 cm) were pretreated in the IR cell under vacuum at the temperature from 20 °C to 550 °C for 10 min and then the IR spectra of OH group were recorded. For IR spectra of absorbed 1-phenylethanol, the self-supported wafers were pretreated in the IR cell under vacuum at 200 °C for 10 min, after the samples were cooled down to room temperature, 1-phenylethanol vapor dozed into the IR cell. IR spectra of absorbed 1-phenylethanol were recorded under vacuum at the temperature from 20 °C to 200 °C for 10 min.
3 Results and discussions
3.1 Characterization of Au/γ-AlOOH and Au/γ-Al2O3 samples
The wide-angle XRD patterns of the Au/γ-AlOOH and Au/γ-Al2O3 are shown in Fig. 1. The diffraction peaks at 14.48°, 28.18°, 38.34° and 48.93° are related to boehmite phase (JCPDS no. 21-1307).31 The γ-Al2O3 structure (JCPDS no. 10-0425) is formed by calcining γ-AlOOH at 550 °C.32 No peaks related to the Au phase were observed, suggesting low Au concentration and small particle size, with the Au diffraction peaks overlapping with the strong peaks of γ-AlOOH and γ-Al2O3. | ||
| Fig. 1 Wide-angle XRD patterns of the as-synthesized γ-AlOOH (A, dotted line), Au/γ-AlOOH (a, solid line), γ-Al2O3 (B, dotted line) and Au/γ-Al2O3 (b, solid line). The arrows mark reflexes from Au particles. | ||
Fig. 2 shows the TEM images of Au/γ-AlOOH (left) and Au/γ-Al2O3 (right) samples. The shape of primary nanosheet (1–5 nm thick) particles of γ-AlOOH is preserved after conversion to γ-Al2O3 by calcination. Au nanoparticle can be observed in both samples, the average particle sizes of Au/γ-AlOOH sample and Au/γ-Al2O3 sample are 4.6 nm and 4.4 nm, respectively.
 | ||
| Fig. 2 TEM images (left) and Au particle size distribution (right) of the as-synthesized Au/γ-AlOOH (a) and Au/γ-Al2O3 (b). | ||
Fig. 3 presents the N2 adsorption/desorption isotherms and pore size distribution curves of the Au/γ-AlOOH and Au/γ-Al2O3 samples. Both Au/γ-AlOOH and Au/γ-Al2O3 samples show typical characteristics of type IV isotherms with the hysteresis loop, where the adsorbed amount increases continuously with rising P/P0.33
 | ||
| Fig. 3 N2 adsorption/desorption isotherms and pore size distribution curves (insert) of the as-synthesized Au/γ-AlOOH (a) and Au/γ-Al2O3 (b) samples. The curves (b) are offset a little for clarity. | ||
Table 1 summarizes the surface areas, total pore volumes and the pore diameters of the Au/γ-AlOOH and Au/γ-Al2O3 samples. The surface area of the Au/γ-AlOOH sample is 217 m2 g−1 and the total pore volume is 0.48 cm3 g−1. The surface area of the Au/γ-Al2O3 is 279 m2 g−1, and the total pore volume is 0.58 cm3 g−1. Both the surface area and pore volume of Au/γ-Al2O3 are higher than those of the Au/γ-AlOOH. Both the samples exhibit bimodal pore size distribution.34 The Au/γ-AlOOH sample has the peaks centered at 3.4 nm and 15.9 nm, respectively and the Au/γ-Al2O3 sample has those at 6.3 nm and 18.9 nm, respectively.
Fig. 4 gives 27Al NMR spectra of as-synthesized γ-AlOOH, Au/γ-AlOOH, γ-Al2O3 and Au/γ-Al2O3 samples. The resonance at ∼10 ppm is associated with octahedrally coordinated aluminum. Additional peaks at ∼68 ppm and ∼38 ppm are observed in the spectra of γ-Al2O3 and Au/γ-Al2O3 samples, indicating generation of tetrahedrally and pentahedrally coordinated aluminum after calcination of boehmite.35 Similar spectra were observed between the support and Au-loaded samples, suggesting that the addition of Au does not alter the support's structure.
 | ||
| Fig. 4 27Al NMR spectra of as-synthesized γ-AlOOH (A, dotted line), Au/γ-AlOOH (a, solid line), γ-Al2O3 (B, dotted line) and Au/γ-Al2O3 (b, solid line). | ||
The valences of Au species in Au/γ-AlOOH and Au/γ-Al2O3 samples are investigated by using XPS. As shown in Fig. 5, Au 4f peaks are chosen to compare the valence of Au in Au/γ-AlOOH and Al–Al2O3 samples. Both the Au/γ-AlOOH and Au/γ-Al2O3 samples exhibit the presence of Au0 species on the catalyst surface.36 The Au/γ-Al2O3 sample shows particular Au+ species, compared with the Au/γ-AlOOH sample. Casaletto et al. claimed that presence of Au+ species is the main requisite for achievement of the highest CO conversion at the low temperatures.17
 | ||
| Fig. 5 Au 4f XPS spectra of Au/γ-AlOOH (a) and Au/γ-Al2O3 (b). | ||
The alkalinity properties and total amounts of basic sites are investigated by using CO2 temperature-programmed desorption (CO2-TPD) technique. As shown in Fig. 6, Au/γ-AlOOH and Au/γ-Al2O3 samples display similar CO2 desorption at 110 °C, which is due to the desorption of CO2 on the weak basic sites.24 According to the peak areas of CO2 desorption in the two samples, the total basic sites of Au/γ-AlOOH are 1.6 times higher than that of Au/γ-Al2O3. Ide et al. proposed that adsorbed hydroxyl groups can activate O–H and C–H bonds, thereby increasing the oxidation rate of glycerol, even bulk gold powder becomes an active oxidation catalyst in alkaline water.37 Tang et al. found that alkali metal dopants were found to significantly delay total oxidation of CO over Co3O4 nano-catalyst,38 Gluhoi et al. found that alkali (earth) metal oxide additives act as structural promoters, which may contribute to the activation of O2 during the low-temperature oxidation of CO.39
 | ||
| Fig. 6 CO2-TPD profiles of Au/γ-AlOOH (a) and Au/γ-Al2O3 (b). | ||
Fig. 7 displays the OH stretching vibrations between 2800 cm−1 and 4000 cm−1 for the Au/γ-AlOOH and Au/γ-Al2O3 samples. Two characteristic absorption bands of OH groups of Au/γ-AlOOH were observed at 3673 cm−1 and 3668 cm−1. A broad flat peak attributed to the skeletal Al–OH of γ-AlOOH were observed at 2960∼3480 cm−1. The Au/γ-Al2O3 showed three bands at 3721 cm−1, 3671 cm−1, 3578 cm−1, which attributes to the absorption bands of OH groups of γ-Al2O3. Under vacuum condition of 20 °C and 50 °C, the absorption bands of OH groups in Au/γ-AlOOH are much higher than that in Au/γ-Al2O3. As the temperature increases, γ-AlOOH gradually transforms into γ-Al2O3, and the surface Al–OH in γ-AlOOH gradually disappears. After vacuum treatment of 550 °C, the IR spectrum (OH region) of the Au/γ-AlOOH and Au/γ-Al2O3 is basically the same, which is due to the conversion of γ-AlOOH to γ-Al2O3.40
 | ||
| Fig. 7 Change of IR spectra (OH region) of the Au/γ-AlOOH (a) and Au/γ-Al2O3 (b) as a function of temperature. | ||
The surface properties of the Au/γ-AlOOH and Au/γ-Al2O3 samples are further examined by the 1-phenylethanol adsorption. Fig. 8 displays IR spectra of 1-phenylethanol adsorbed on the Au/γ-AlOOH and Au/γ-Al2O3 samples. Both Au/γ-Al2O3 and Au/γ-AlOOH samples exhibit the peaks at 1400–1650 cm−1, which is attributed to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations
C stretching vibrations![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) from the benzene ring and α-deformation (bending vibrations) of the CH3 group.41–43 In addition, the Au/γ-AlOOH sample shows more 1-phenylethanol absorption peak than the Au/γ-Al2O3 sample, as shown in Table 2, which is consistent with the IR spectra (OH region) results. At 50 °C, the area of 1-phenylethanol absorption peak at 1443 cm−1 of Au/γ-AlOOH is 3.32 times that of Au/γ-Al2O3. Higher alcohol adsorption capacity helps to enhance alcohol oxidation activity.
from the benzene ring and α-deformation (bending vibrations) of the CH3 group.41–43 In addition, the Au/γ-AlOOH sample shows more 1-phenylethanol absorption peak than the Au/γ-Al2O3 sample, as shown in Table 2, which is consistent with the IR spectra (OH region) results. At 50 °C, the area of 1-phenylethanol absorption peak at 1443 cm−1 of Au/γ-AlOOH is 3.32 times that of Au/γ-Al2O3. Higher alcohol adsorption capacity helps to enhance alcohol oxidation activity.
 | ||
| Fig. 8 IR spectra of 1-phenylethanol absorbed on Au/γ-AlOOH (a) and Au/γ-Al2O3 (b), all samples were normalized by subtracting the peak of the vacuum adsorption sample at 200 °C. | ||
| Desorption temp. (°C) | Peak area (1443 cm−1) | |
|---|---|---|
| Au/γ-AlOOH | Au/γ-Al2O3 | |
| 20 | 8.7 | 5.0 |
| 50 | 6.3 | 1.9 |
| 100 | 5.2 | 1.7 |
| 150 | 4.3 | 1.4 |
| 200 | 2.8 | 1.0 |
3.2 Catalytic properties of Au/γ-AlOOH and Au/γ-Al2O3 samples in CO oxidation
The Au/γ-AlOOH and Au/γ-Al2O3 samples are tested for CO oxidation at 20 °C under a reaction stream with a gas composition of 1.0 vol% CO/2.5 vol% O2/He mixture. The conversion vs. reaction time is shown in Fig. 9. The initial CO conversions of the Au/γ-Al2O3 and Au/γ-AlOOH samples are 82% and 26%, respectively. The CO oxidation performance of the Au/γ-Al2O3 sample is much superior than that of the Au/γ-AlOOH sample. XPS spectra suggest that the Au/γ-Al2O3 sample contains more Au+ species than the Au/γ-AlOOH sample, which is favorable for CO2 forming. XPS spectra suggest that the Au/γ-Al2O3 sample has more Au+ species than those the Au/γ-AlOOH sample has, which is favorable for CO2 forming. These results lead to the outstanding CO oxidation performance of Au/γ-Al2O3, as shown in Scheme 1. Then, the catalytic performance of these samples tends to reduce, CO conversions of the Au/γ-Al2O3 and Au/γ-AlOOH samples decreased to about 50% and 8% after 450 hours, respectively. This result is in good agreement with the literature data,29 they hypothesized that the CO + O2 reaction occurs by inserting adsorbed CO molecules into the Aux+–OH complex to form carboxylate species, and the presence of water vapor is crucial for maintaining sufficient concentrations of Aux+–OH species.29,44 Therefore, the absence of water vapor will lead to a decrease in CO activity over time. | ||
| Fig. 9 The CO conversion of Au/γ-AlOOH (a) and Au/γ-Al2O3 (b) samples. | ||
 | ||
| Scheme 1 Proposed reaction mechanism for the oxidation of CO and alcohol over Au/γ-AlOOH and Au/γ-Al2O3 samples. | ||
3.3 Catalytic properties of Au/γ-AlOOH and Au/γ-Al2O3 samples in oxidation of alcohols
The selective oxidation of alcohols is one of the most important and fundamental transformations in organic synthesis, acted as the versatile intermediates of valuable compounds such as pharmaceuticals, agricultural chemicals, and fine chemicals. We use 1-phenylethanol, benzyl alcohol and 4-methoxybenzyl alcohol oxidation as the probe reaction of aromatic alcohols oxidation. Table 3 gives the conversion of 1-phenylethanol, benzyl alcohol and 4-methoxybenzyl alcohol oxidation on the Au/γ-AlOOH and Au/γ-Al2O3 samples, all reactions display about 100% selectivity, no other by-products can be detected. Au/γ-AlOOH sample display a conversion of 73.0% for 1-phenylethanol oxidation at 40 °C for 6 h. Chen et al. claimed a conversion of 82% and selectivity of 90% for 1-phenylethanol oxidation over Pd/SiO2–Al2O3–H2 catalyst at 150 °C for 24 h,45 and Yamaguchi et al. reported a conversion of >99% and selectivity of >90% over Ru/Al2O3 at 83 °C for 1 h.46 Herein, 1-phenylethanol can be almost completely oxidized over Au/γ-AlOOH at 40 °C for 6 h, which must be energy saving and eco-friendly. This result shows some advantages of flower-like AlOOH as support catalysis in the alcohols oxidation. Au/γ-AlOOH sample is also suitable for the oxidation of benzyl alcohol and 4-methoxybenzyl alcohol.| Entry | Substrate | Product | Temp. (°C) | Time (h) | Conv. (%) | |
|---|---|---|---|---|---|---|
| Au/γ-AlOOH | Au/γ-Al2O3 | |||||
| 1 |  |
 |
40 | 6 | 73.0 | 32.1 |
| 2 | 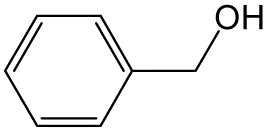 |
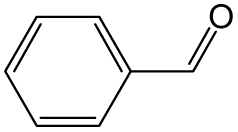 |
70 | 8 | 32.6 | 16.9 |
| 3 |  |
 |
70 | 6 | 25.4 | 14.8 |
The carrier morphology of Au/γ-AlOOH and Au/γ-Al2O3 samples is similar, and both samples exhibit a low Au concentration and small Au particle size. The Au/γ-AlOOH sample demonstrates much higher 1-phenylethanol, benzyl alcohol and 4-methoxybenzyl alcohol oxidation conversion than the Au/γ-Al2O3 sample does. This result shows some advantages of the Au/γ-AlOOH sample in the alcohols oxidation. IR spectra of OH regions and 1-phenylethanol adsorption indicate the abundant hydroxyl groups and strong adsorption capacity for alcohols of the Au/γ-AlOOH sample. Jiang et al. speculated through theoretical calculations that the –OH groups promote chemisorption of alcohol molecules on a gold cluster surface as the initial step of their oxidation by a gold cluster,47 which is confirmed by our experimental results.
3.4 Influence of support and Au oxidation state on catalytic properties
Scheme 1 exhibits the proposed reaction mechanism for the oxidation of CO and alcohol over Au/γ-AlOOH and Au/γ-Al2O3 samples. Au/γ-AlOOH and Au/γ-Al2O3 samples exhibit a low Au concentration and small Au particle size, and both samples can be used for low-temperature oxidation of CO and aromatic alcohols. During alcohol oxidation, both the carrier and gold significantly influence the reaction. The hydroxyl groups and basic sites in the carrier sometimes have a greater impact on alcohol oxidation than Au. Ide et al. proposed that bulk gold powder acts as an active catalyst for alcohol oxidation in alkaline medium.37 The Au/γ-AlOOH sample contains a large amount of OH and alkalinity, and has strong adsorption capacity for aromatic alcohols, thus exhibiting excellent low temperature oxidation activity for aromatic alcohols. Au/γ-Al2O3 possesses a large amount of Au+ which is effective in promoting the low temperature CO oxidation.174 Conclusions
Au/γ-AlOOH and Au/γ-Al2O3 samples with low Au concentrations and small Au particle sizes were synthesized for low-temperature oxidation of CO and aromatic alcohols. Under low temperature conditions, the CO oxidation conversion by the Au/γ-Al2O3 sample is about three times that of the Au/γ-AlOOH sample, while the oxidation conversions of 1-phenylethanol, benzyl alcohol, and 4-methoxybenzyl alcohol by the Au/γ-Al2O3 sample are only about half of those by the Au/γ-AlOOH sample.The high oxidation activity of Au/γ-AlOOH sample towards 1-phenylethanol, benzyl alcohol, and 4-methoxybenzyl alcohol is mainly due to its rich content of hydroxyl groups and basic sites, which facilitate the adsorption of aromatic alcohols. The high CO oxidation activity of the Au/γ-Al2O3 sample is primarily attributed to the efficient Au+ sites.
Data availability
All relevant data are within the main article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Panxi Experimental Zone Key Scientific and Technological Project, and National Key Research and Development Program Nanotechnology Specific Project (No. 2020YFA0210900). We are very grateful to Dr Xinsheng Liu and Dr Yimeng Wang for their professional knowledge and insightful comments, which have made significant contributions to the success of this study.References
- M. Alhumaimess, Z. Lin, Q. He, L. Lu, N. Dimitratos, N. F. Dummer, M. Conte, S. H. Taylor, J. K. Bartley, C. J. Kiely and G. J. Hutchings, Chem.–Eur. J., 2014, 20, 1701–1710 CrossRef CAS PubMed.
- A. Villa, A. Gaiassi, I. Rossetti, C. L. Bianchi, K. van Benthem, G. M. Veith and L. Prati, J. Catal., 2010, 275, 108–116 CrossRef CAS.
- M. Haruta, Nature, 2005, 437, 1098–1099 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405–408 CrossRef.
- P. Rodriguez, Y. Kwon and M. T. M. Koper, Nat. Chem., 2012, 4, 177–182 CrossRef CAS PubMed.
- A. Ali, D. J. Jasim, N. Rakhimov, M. J. Mohammed, M. M. Karim, A. H. Athab, A. Kumar, N. Ahmad and L. Guo, J. Mol. Struct., 2025, 1319, 139373 CrossRef CAS.
- A. Ali, M. K. Abdulameer, M. H. Mahdi, K. H. Rasool, M. S. Jabir, F. H. Zankanah, H. Majdi, A. S. Mansoor, U. K. Radi and R. Wahab, Plasmonics, 2024 DOI:10.1007/s11468-024-02481-4.
- S. Abdullaev, D. Singh, M. N. Al-Delfi, A. Kumar, Q. H. Aziz, A. Elawady, M. A. Al-Anber, A. H. Al-Rubaye, A. Ali and N. Ahmad, Appl. Organomet. Chem., 2024, 38, 7245 Search PubMed.
- A. Ali, J. M. Moradian, A. Naveed, S. N. Uz-Zaman Haider, J. Lu, S. S. A. Shah, H. Ahmed Keerio, W. Ahmad Qureshi, H. Naz, R. Wahab, F. Zhiqiang and L. Guo, J. Catal., 2024, 438, 115729 CrossRef CAS.
- V. V. Costa, M. Estrada, Y. Demidova, I. Prosvirin, V. Kriventsov, R. F. Cotta, S. Fuentes, A. Simakov and E. V. Gusevskaya, J. Catal., 2012, 292, 148–156 CrossRef CAS.
- C. J. Jia, Y. Liu, H. Bongard and F. Schüth, J. Am. Chem. Soc., 2010, 132, 1520–1522 CrossRef CAS PubMed.
- B. R. Cuenya, Thin Solid Films, 2010, 518, 3127–3150 CrossRef CAS.
- S. Li, H. Zhu, Z. Qin, G. Wang, Y. Zhang, Z. Wu, Z. Li, G. Chen, W. Dong, Z. Wu, L. Zheng, J. Zhang, T. Hu and J. Wang, Appl. Catal., B, 2014, 144, 498–506 CrossRef CAS.
- W. Yan, B. Chen, S. M. Mahurin, V. Schwartz, D. R. Mullins, A. R. Lupini, S. J. Pennycook, S. Dai and S. H. Overbury, J. Phys. Chem. B, 2005, 109, 10676–10685 CrossRef CAS PubMed.
- W. Yan, B. Chen, S. M. Mahurin, S. Dai and S. H. Overbury, Chem. Commun., 2004, 77, 1918–1919 RSC.
- K. Y. Ho and K. L. Yeung, Gold Bull., 2007, 40, 15–30 CrossRef CAS.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- A. N. Pestryakov and V. V. Lunin, Mol. Catal., 2000, 158, 325–329 CrossRef CAS.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., 2005, 117, 4134–4137 CrossRef.
- X. Y. Wang, S. P. Wang, S. R. Wang, Y. Q. Zhao, J. Huang, S. M. Zhang, W. P. Huang and S. H. Wu, Catal. Lett., 2006, 112, 115–119 CrossRef CAS.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- M. Hisamoto, R. C. Nelson, M.-Y. Lee, J. Eckert and S. L. Scott, J. Phys. Chem. C, 2009, 113, 8794–8805 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Catal. Rev., 2006, 48, 363–444 CrossRef CAS.
- J. Huang, Y. Wang, J. Zheng, W.-L. Dai and K. Fan, Appl. Catal., B, 2011, 103, 343–350 CrossRef CAS.
- K. Wefers, Alcoa Technical Paper, 1987, p. 19 Search PubMed.
- J. Wang, A. H. Lu, M. Li, W. Zhang, Y. S. Chen, D. X. Tian and W. C. Li, ACS Nano, 2013, 7, 4902–4910 CrossRef CAS PubMed.
- V. F. Anufrienko, B. L. Moroz, T. V. Larina, S. P. Ruzankin, V. I. Bukhtiyarov and V. N. Parmon, Dokl. Phys. Chem., 2007, 413, 75–80 CrossRef CAS.
- S. B. Erenburg, B. L. Moroz, N. V. Bausk, V. I. Bukhtiyarov and S. Nikitenko, Nucl. Instrum. Methods Phys. Res., Sect. A, 2007, 575, 105–108 CrossRef CAS.
- B. L. Moroz, P. A. Pyrjaev, V. I. Zaikovskii and V. I. Bukhtiyarov, Catal. Today, 2009, 114, 292–305 CrossRef.
- Z. Shi, W. Jiao, L. Chen, P. Wu, Y. Wang and M. He, Microporous Mesoporous Mater., 2016, 224, 253–261 CrossRef CAS.
- W. Q. Jiao, X. M. Liang, Y. M. Wang and M. Y. He, CrystEngComm, 2014, 16, 3348–3358 RSC.
- J. Zhang, S. Liu, J. Lin, H. Song, J. Luo, E. M. Elssfah, E. Ammar, Y. Huang, X. Ding, J. Gao, S. Qi and C. Tang, J. Phys. Chem. B, 2006, 110, 14249–14252 CrossRef CAS PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
- J. Zhang, S. Wei, J. Lin, J. Luo, S. Liu, H. Song, E. Elawad, X. Ding, J. Gao, S. Qi and C. Tang, J. Phys. Chem. B, 2006, 110, 21680–21683 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- J. García-Serrano, A. G. Galindo and U. Pal, Sol. Energy Mater. Sol. Cells, 2004, 82, 291–298 CrossRef.
- M. S. Ide and R. J. Davis, Acc. Chem. Res., 2014, 47, 825–833 CrossRef CAS PubMed.
- W. Tang, J. Weng, X. Lu, L. Wen, A. Suburamanian, C. Y. Nam and P. X. Gao, Appl. Catal., B, 2019, 256, 117859 CrossRef CAS.
- A. C. Gluhoi and B. E. Nieuwenhuys, Catal. Today, 2007, 122, 226–232 CrossRef CAS.
- G. Li, Y. Liu, D. Liu, L. Liu and C. Liu, Mater. Res. Bull., 2010, 45, 1487–1491 CrossRef CAS.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, J. Chem. Educ., 2014, 92, 826–827 Search PubMed.
- K. Shin-Ya, H. Sugeta, S. Shin, Y. Hamada, Y. Katsumoto and K. Ohno, J. Phys. Chem. A, 2007, 111, 8598–8605 CrossRef CAS PubMed.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. A. Vyvyan, Introduction to Spectroscopy, 4th edn, 2009 Search PubMed.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767–11775 CrossRef CAS.
- J. Chen, Q. Zhang, Y. Wang and H. Wan, Adv. Synth. Catal., 2008, 350, 453–464 CrossRef CAS.
- K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538–4542 CrossRef CAS PubMed.
- D. E. Jiang, S. H. Overbury and S. Dai, J. Phys. Chem. Lett., 2011, 2, 1211–1215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00527b |
| This journal is © The Royal Society of Chemistry 2025 |