
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzimidazole(s): synthons, bioactive lead structures, total synthesis, and the profiling of major bioactive categories†
Lotfi M. Aroua *a,
Fahad M. Almindereja,
Hind R. Almuhaylana,
Abdulelah H. Alosaimia,
Faten Medinib,
Hamdoon A. Mohammedc,
Suliman A. Almahmoudc,
Riaz A. Khan*c and
Nejib H. Meknide
*a,
Fahad M. Almindereja,
Hind R. Almuhaylana,
Abdulelah H. Alosaimia,
Faten Medinib,
Hamdoon A. Mohammedc,
Suliman A. Almahmoudc,
Riaz A. Khan*c and
Nejib H. Meknide
aDepartment of Chemistry, College of Science, Qassim University, Box: 6644, Qassim 51452, Kingdom of Saudi Arabia. E-mail: lm.aroua@qu.edu.sa
bLaboratory of Aromatic and Medicinal Plants, Biotechnology Center of Borj-Cedria, Carthage University, 2050, Tunis, Tunisia
cDepartment of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, Qassim University, Qassim 51452, Saudi Arabia. E-mail: ri.khan@qu.edu.sa
dLaboratory of Bio-Organic, Structural and Polymer Chemistry (LR99ES14), Department of Chemistry, Faculty of Sciences, University of Tunis El-Manar, El-Manar 2092, Tunis, Tunisia. E-mail: aroua.lotfi@yahoo.com
eDepartment of Fundamental Science, High Institute of Medical Technologies of Tunis, El Manar University, Tunis 1006, Tunisia
First published on 28th March 2025
Abstract
Benzimidazole, a fused bicyclic compound with benzene and pentacyclic 1,3-diazole moeities, has a simple aromatic heterocyclic structure. The moiety has become an indispensable anchor for the development of new pharmacologically active products, and has yielded several therapeutic agents with anticancer, antihypertensive, antimicrobial, antifungal and antiulcer effects. Benzimidazoles, as synthetically feasible and pharmacophoric synthons, have been relentlessly pursued for the preparation of new analogues and derivatives, and they have successfully developed into some of the most sought-after and vital pharmacophores for drug discovery. The use of varied substituents and differing patterns around the benzimidazole nucleus has provided a wide spectrum of biological activities. In addition, the benzimidazole moiety constitutes a building block for the production of several drugs, drug candidates, new chemical entities, and lead molecules. The importance of this nucleus for bioactivity, e.g., antibacterial, antitubercular, antidiabetic, anticancer, antifungal, anti-inflammatory, analgesic, antioxidant, antihistaminic, and antimalarial activity, has led us to take note and provide an overview of the synthetic development approaches for various benzimidazole derivatives together with their biological actions. This review is projected to further assist in the design and development of new benzimidazole-based compounds for new and optimized pharmacologically active products towards new drug-development strategies.
1. An introduction to benzimidazole: a saga of chemistry and pharmacology
Extensive research work on vitamin B12 highlighted the structure and importance of the benzimidazole nucleus. The 1,3-positions of a diazole ring were fused with the 4,5-positions of an aromatic benzene ring. This structure constitutes the basic benzimidazole ring.1 Other benzo-substituted diazoles, with 1,2- and 2,3-substitutions of the five-membered aromatic diazole ring, keeping the fused benzene as an unsubstituted heterocyclic ring, have been categorized as indazole and benzopyrazole, respectively. The IUPAC name for benzimidazole is 1H-1,3-benzimidazole. However, several other names have also been used, including azaindole; benzimidazole; benzo-glyoxaline; benzimidazole; BZI; 1,3-diazaindene; and 3-azaindole. The molecular formula is C7H6N2 with a molecular weight of 118.1359. The IUPAC standard InChI key is 1S/C7H6N2/c1-2-4-7-6(3-1)8-5-9-7/h1-5H,(H,8,9). The preliminary synthesis of benzimidazole has been reported from the condensation of phenylenediamine with formic acid, or its equivalent, trimethyl orthoformate. The benzimidazole nucleus has been found to be stable for the further build-up of extended new molecules incorporating the central benzimidazole ring; these played a pivotal role in finding new leads and developing new drugs and new drug templates. These provided diverse and impactful bioactivities; some examples are under pharmacological development and some have found clinical use as referral standards and drugs.Progressive work involving the benzimidazole nucleus led to the development of several synthetic strategies to prepare benzimidazole-based, structurally diverse compounds with multiple bioactivities. From the standpoint of synthesis, different approaches utilizing various synthons and starting materials belonging to acid-, ester-, ortho-ester-, nitrile-, acid-chloride-, and orthoformate-based molecular frameworks have been used.2 A plethora of compounds containing the benzimidazole bicyclic ring structure in their molecular framework have displayed prominent biological activity profiles with high therapeutic potentials in almost all fields of pharmacology and therapeutics.3 Benzimidazoles, as structural isosteres of nucleotides, have plentifully structures that can feasibly interact with polymers of biological origins, culminating in a broad spectrum of pharmacologically active compounds with lowered toxicity and better therapeutic outcomes.4
Over the past decades, numerous studies describing syntheses of chemical systems incorporating the benzimidazole nucleus as part of their synthetic strategies have been prepared, modified, and reported. Notwithstanding advances in synthetic strategies and protocols, direct and traditional patterns of convergent and cumulative synthesis, disconnection and retrosynthetic tactics, divergent and ligation-like approaches for synthesis/semi-synthesis, and bulk-scale preparation have also been used. A wide and diverse range of biological activity evaluations of benzimidazole-based structures, molecular templates, new chemical entities, and desired metal complexes has been reported.5 Studies reporting several classes of bioactivities, including antimicrobial,6–11 anthelminthic,12–14 antithrombotic,15,16 antiplatelet,17–19 anticoagulant,20–23 anti-inflammatory,24–28 antiulcer,29,30 antifungal,31–33 acetylcholinesterase34–37 antiprotozoal,4,38,39 antitubercular,40–43 antileishmanial,44–46 antimycobacterial,47,48 antiviral,49,50 anti-HIV,51,52 and antitumor53,54 activities, are abundantly available. Additionally, benzimidazole targets have been described as inhibitors of hepatitis C55–57 and as an indoleamine-2,3-dioxygenase-1 (IDO1) inhibitor, predicted from in silico SAR (structure–activity relationship) approaches through structure-based virtual screening. This culminated in obtaining in vivo biological activity profiles of several compounds.58 Benzimidazole-structure-templated compounds have also been known to be antihypertensive in action,59,60 in addition to acting as a Zika virus inhibitor,61 an in vitro α-glucosidase inhibitor,62,63 a NOX2 antagonist,64 and as antiglycation,65 antioxidant,66–68 antileukemic,69–72 and antitubercular73–77 agents. Recently, the benzimidazole molecular template has also been reported to be a potent anticancer entity.78–83 More recently, the benzimidazole structural motif has been observed to be antihypertensive,84,85 a non-nucleoside reverse transcriptase inhibitor,86–89 an anticonvulsant,90–93 ulcerogenic,94–97 a non-peptide angiotensin-II receptor antagonist,98–100 an AMP-activated protein kinase activator,101–103 and an antileukemic activity agent69,101–103 in terms of its biological activity profiles.
The simple and complex structural moieties derived from the benzimidazole structural template, which are present in various compounds as sub-structural entities, have exponentially increased in number, showing vastly different biological properties.104 The benzimidazole core template has proven to be an exceptional chemical structure that has manifested diverse ranges and types of biological and therapeutic activities.105 In the past, many works reporting the importance of chemical systems incorporating a benzimidazole nucleus have broadly been elaborated on. The diverse biological activities of benzimidazoles, and their derived structures have culminated in the development of several drugs which have been introduced to the market, e.g., albendazole (antimicrobial); omeprazole (antiulcer); bendamustine, nocodazole, and abemaciclib (antitumor); enviradine (antiviral); candesartan (antihypertensive); and benoxaprofen analogues (anti-inflammatory),106,107 to name a few.
2. Biological activity profiles of benzimidazole-based compounds: the current status
2.1 Antimicrobial activity
The gradual but continuous emergence of a number of drug-resistant microbial strains over the last several decades has pushed researchers towards searching for and developing new antimicrobial agents that are capable of combating this situation.108,109 The benzimidazole pharmacophore as a core structure has contributed to the molecular template of several leads which, together with benzimidazole derivatives, have served as part of several active substances showing significant antimicrobial activity.110 Recently, the elegant synthesis of 2-substituted benzimidazole, benzoxazole, and benzothiazole derivatives was achieved from the reaction of p-N,N-diethylamino-salicylaldehyde 1 with o-phenylenediamine, o-aminophenol, and o-amino-thiophenol, respectively, with PCl3 in ethanol as the reaction medium. The 95% yield of product 2 and its derivatives provided ample material for bioactivity evaluations (Scheme 1). This established the potential of the benzimidazole structural unit as an antibacterial and antifungal template for developing new chemical entities exhibiting antimicrobial activity against several bacteria. These benzoxazole and benzothiazole derivatives displayed good antimicrobial activity against E. coli and S. aureus, while the developed benzothiazole derivatives showed remarkable antifungal activity potential against Candida albicans and Aspergillus niger.111 | ||
| Scheme 1 The synthesis of 2-substituted benzimidazole templates, benzoxazole 2, and benzothiazole derivatives. | ||
The development of 2,5-disubstituted benzimidazole derivatives as products 3, 4 and 5, obtained from substituted o-phenylenediamine and appropriately substituted aldehydes, was realized under microwave-assisted synthesis conditions in the presence of Na2S2O5. Moderate-to-high yields in the range of ∼90% were enough for bioactivity testing of the synthesized benzimidazoles, which exhibited antifungal and antibacterial activity against E. coli ATCC 25922, S. aureus ATCC 25923 and ATCC 3933, and S. epidermidis ATCC 12228. The MICs of 2-(3-bromothiophen-2-yl)-5-chloro-1H-benzimidazole and 5-bromo-2-(3-bromothiophen-2-yl)-1H-benzimidazole were found to be <4 μg mL−1, while the lowest activity level was 2 μg mL−1 (Scheme 2).112
 | ||
| Scheme 2 The synthesis of the 2,5-disubstituted benzimidazoles 3, 4 and 5. | ||
The synthesis of the benzimidazole bidental ligand 6 was achieved from the condensation of 2-(4-aminophenyl)benzimidazole with a 5-bromosalicylaldehyde derivative. Metal complexation of ligand 6 yielded the dimeric meta-complex 7 (80% yields). The (E)-2-((4-(1H-benzo[d]imidazol-2-yl)phenylimino)-methyl)-4-bromophenol ligand 6 and corresponding Zn(II), Ni(II), and Cu(II) complexes, 7, exhibited antibacterial activity against Gram-positive Micrococcus luteus and Gram-negative Escherichia coli and Enterobacter aerogenes. Evaluations of the antibacterial activities of Ni(II), Zn(II), and Cu(II) complexes demonstrated their moderate-to-excellent levels of activity (Scheme 3),113 therefore providing evidence for the antimicrobial potential of benzimidazole as a core component.
 | ||
| Scheme 3 The synthesis of the benzimidazole ligand 6 and its Zn(II), Pd(II), Ni(II) and Cu(II) metallic complexes 7. | ||
In a series of mono- and di-substituted benzimidazole derivatives reported by Ajani et al., the 2-(2-aminophenyl)- and 2-(benzyl-N-phenylsulfonyl)-benzimidazole derivatives 8 and 9 were synthesized from the NH4Cl-catalyzed condensation of o-phenylene-diamine with the corresponding carboxylic acid to produce the desired product. The synthesized compounds were tested against four bacterial strains, namely, Staphylococcus aureus, Bacillus licheniformis, Proteus vulgaris, and Pseudomonas aeruginosa. 2-(1H-Benzimizadol-2-yl)-aniline 8 and 2-benzyl-1-(phenylsulfonyl)-1H-benzimidazole 9 showed high antibacterial activities with an MIC value of 15.63 mg mL−1 for both compounds (Scheme S-1, ESI file†).114
The synthesis of the potent antibacterial 2-substituted benzimidazoles, N-5-aryl(1,3,5-triazinane-4-thione), and oxadiazinane-4-thione derivatives, were achieved from the iodine-catalysed condensation reaction of thiourea with the 2-benzimidazolyl-ethenone 10, leading the formation of corresponding derivative, 2-aminothiazol-5-benzimidazolyl, product 11. This latter was reacted with aryl isothiocyanate leading to the urea-based product 12, which underwent a series of reactions of formol, and the mixture of methylamine/formol to produce the benzimidazolyl oxadiazinane, and the benzimidazolyl triazinane derivatives 13 and 14, respectively. The microbial growth inhibition efficacy of the synthesized benzimidazole derivatives 13 and 14 was evaluated after screening six different types of bacterial strains, i.e., Bacillus subtilis MTCC 441, Bacillus cereus ATCC 9372, Staphylococcus aureus ATCC 96, Escherichia coli ATCC 8739, Klebsiella pneumoniae MTCC 109, and Salmonella typhi ATCC 4420. These tested compounds possessed notable antimicrobial activities (Scheme S-2, ESI file†).115
The syntheses of 2-substituted benzimidazolyl isoxazole-5-one, compound 16, pyrazol-3-one, compound 17, and pyrimidin-4-one, compound 18, were realized through cycloaddition reactions of hydrazono-ethyl acetoacetate 15 with NH2OH·HCl, hydrazine, urea, and thiourea entities. These differently substituted benzimidazole end-product compounds exhibited interesting antimicrobial activities (Scheme S-3, ESI file†).116
The synthesis of methylene-N-aryl 19, pyrazolo-3-one 20, and the (4-fluorophenyl)-piperazin-2-substituted benzimidazole derivative 21 were achieved via the nucleophilic substitution of 2-chloromethylene benzimidazole. These products showed weak antimicrobial and cytotoxic activities (Scheme S-4, ESI file†).117 On the other hand, several azo-substituted benzimidazole derivatives, namely benzoxazole and benzothiazole, 22 and 23, were prepared from electrophilic substitution reactions of diaza-sulfonyl-benzimidazole salt with the corresponding aromatic derivatives to yield the desired products. These poly-heterocyclic compounds exhibited significant antimicrobial, antibacterial, and antitubercular activities (Scheme S-5, ESI file†).7 As another set of compounds based on a new triaryl benzimidazole scaffold, the derivatives 4-iodo- and 5-bromo-phenyl-N-aryl-azidine were prepared via electrophilic tri-aza-aromatic substitutions. The synthesized products, 23 and 24, demonstrated high levels of bioactivity against MRSA and VRE bacteria (Scheme S-6, ESI file†).118
The synthesis of a polyaromatic bis(benzimidazolyl) carbamide derivative, 26, spaced with a bi-aryl-pyridin-phenyl derivative was accomplished through a multi-step series of reactions. The prepared derivative, 26, manifested strong bioactivity towards the tested bacteria (Scheme S-7, ESI file†).119 Contextually, a series of N-sulfoxyamide benzimidazole derivatives was prepared from the reaction of benzimidazole derivatives with chlorosulfonyl aromatic derivatives at moderate temperature, which exhibited strong antibacterial activity towards the tested bacteria (Scheme S-8, ESI file†).120 Also, antibacterial agents from a new series of non-symmetrically substituted p-nitro-benzyl-containing benzimidazole N-heterocyclic carbene–silver(I) complexes, 34 and 35, were prepared through silver oxide metalation from N-(p-nitrobenzyl)-N-(alkyl)benzimidazoliminium hexafluorophosphate under mild conditions (Scheme S-9, ESI file†).121 The synthesis of the benzimidazole compound 36 was followed by reactions with hydrazine and carbon disulphide, with cyclisation as the final step (Scheme S-10, ESI file†).122 The antibacterially active benzimidazole-cored compound 4-amino-5-[2-(1H-benzimidazol-2-yl)-3-(4-chloroanilino)propyl]-2,4-dihydro-3H-1,2,4-triazole-3-thione, 37, was synthesized in four steps.
The polyaromatic heterocyclic benzimidazole derivatives 39, were obtained from the reaction of different 2-, 3- and 4-substituted benzimidazole moieties (obtained from Michael cycloaddition with 1-naphthalic carbazide as a starting material) with the substituted benzimidazole-2-enone derivatives 38. The p-nitro- and p-chloro-substituted products showed the highest antibacterial activity levels (Scheme S11, ESI file†).123 Moreover, the synthesis of 2-(3-fluorobenzyl)-1H-benzimidazole derivatives, containing various substituted functional groups and heterocyclic ring moieties, was carried out via the N-nucleophilic substitution reaction of the starting material 2-(4-fluorobenzyl) benzimidazole 40 with various electrophilic functional groups and heterocycles in several steps. The reaction scheme shows the synthetic route and the prepared products, 41–44, which were obtained in high yields (∼80%). These products have been reported to show high antibacterial efficacy (Scheme 4).124
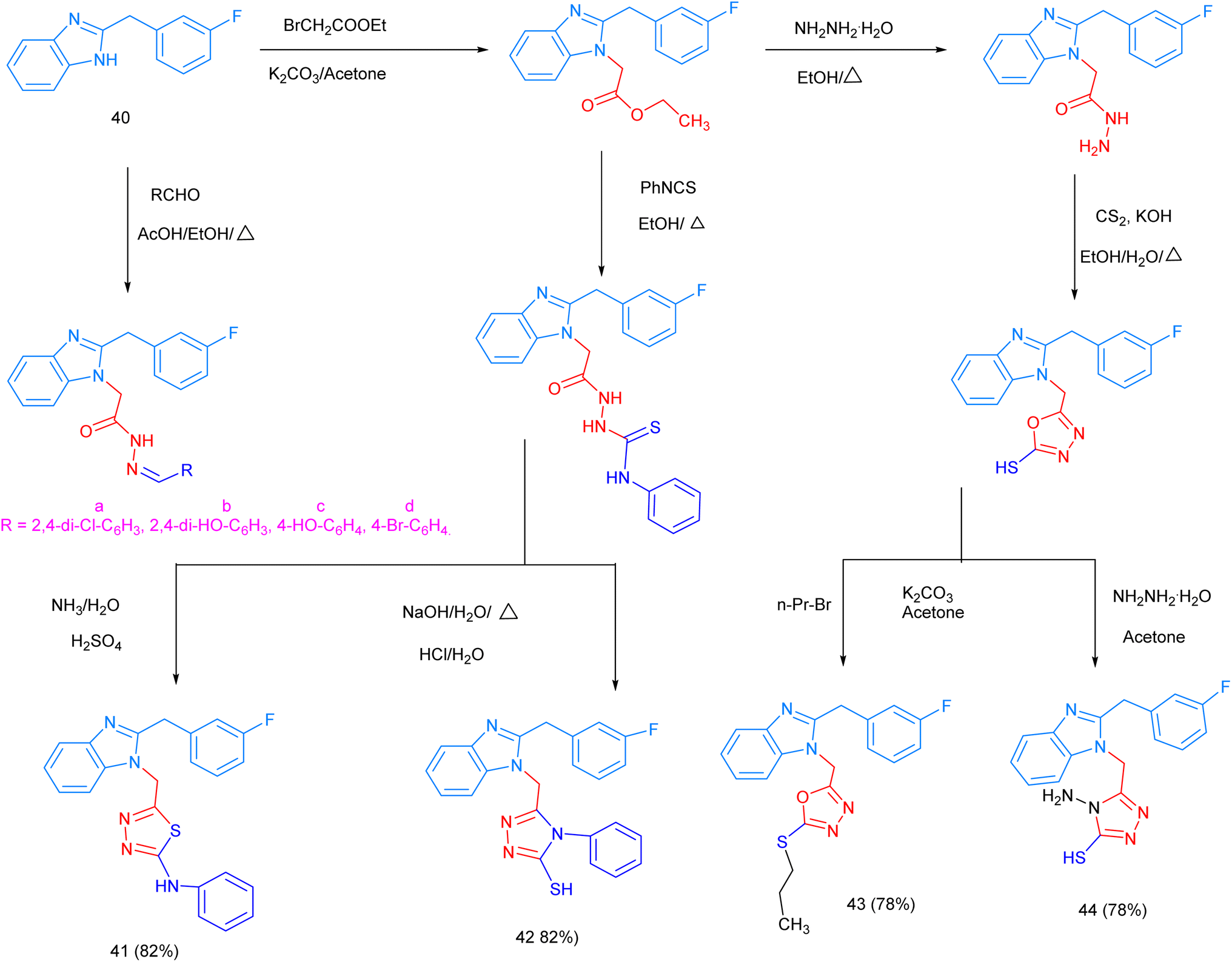 | ||
| Scheme 4 The synthesis of 2-(3-fluorobenzyl)-1H-benzimidazole derivatives with various functional groups and heterocyclic moieties. | ||
In a nutshell, the 2,5-disubstituted benzimidazole derivatives showed high levels of bioaction at doses as low as 2 μg mL−1.
2.2 Antiviral activity
Benzimidazole-based templated core compounds represent one of the major target molecules for manifesting high levels of antiviral activity.125 A series of 2-substituted benzimidazole-N-carbamates was prepared through the conversion of trichloro-carbon-functionalized 2-trichlorocarbon benzimidazole, i.e., compound 45, to obtain amide or ester groups, yielding the products 46, and 47, which was further followed by N-acylation with different electrophilic reagents to produce the benzimidazole derivatives 48–56. These products displayed antiviral activity, and compound 48 was among the most active compared with other compounds of the collection, while compound 57 presented moderate levels of antiviral activity, in contrast to compound 47, which was inactive (Scheme S-12, ESI file†).126Another series of benzimidazoles compounds with tetracyclic fused structures, 60–66, homologues to steroids in the structural patterning of the tetracyclic set-up, was successfully synthesized. The last step of the four stages resulted in the formation of 2-substituted benzimidazole templated core products through the condensation of 1,3-bis(methoxycarbonyl)-2-methyl-2-thiopseudourea with the o-phenylene diamine derivatives 58 and 59 in an acidic environment. The biological activity results indicated that these compounds possessed antiviral activity against human cytomegalovirus (CMV) and varicella-zoster virus (VZV) (Scheme S-13, ESI file†).127
The condensation of 2,3-diaminobenzoic acid with different aldehydes, followed by amidation, provided a new class of benzimidazole derivatives, which were designated as 2-pyridyl-1H-benzimidazole-4-carboxamide derivatives, 67–84. The compounds showed noticeable antiviral activity. Compounds 78 and 79 displayed strong and selective antiviral activity against coxsackievirus B3 in Vero cells under in vitro conditions (Scheme S-14, ESI file†).128 Further structural expansion of the selected heterocyclic rings, via the condensation of 2,3-diaminobenzamidines with heterocyclic 4-imidazol-, 4-pyrol- and 4-pyridine-carboxaldehydes in absolute ethanol, yielded the corresponding imidazole, pyrrole, and pyridine benzimidazole structures 85–93. Biological activity studies showed that the compounds containing pyridine rings, compounds 91–93, displayed strong antiviral activity against RNA-replicating enteroviruses, whilst compound 88 manifested activity against all four types of tested viruses (Scheme S-15, ESI file†).129 Newer derivatives, 2-chloro-, 2-bromo- and 2-iodo-5,6-dichlorobenzimidazole ribonucleosides, 101–106, obtained in moderate-to-high yields (68–90%), were prepared by substituting the nitrogen atom of 5,6-dichlorobenzimidazole-2-amines with bis(trimethylsilyl)acetamide, followed by reaction with 1,2,3,5-tetra-O-acetyl-β-D-ribofuranose (TAR). The compounds, 101–106, showed interesting antiviral activity against two types of viruses. The brominated compound exhibited four times more antiviral activity than the compound containing the chlorine atom (Scheme 5).130
 | ||
| Scheme 5 The synthesis of 2-chloro-, bromo-, and iodo-5,6-dichlorobenzimidazole ribonucleoside derivatives. | ||
The anomeric carbon atom of tetra-acetate ribose assisted the synthesis of a new set of benzimidazole N-riboses, 5-chloro-2-methoxy, 2-thioalcoxy, and 2-thiones 110–121, through the N-acetylation of the 2-chloro-5-nitrobenzimidazole compound 107. The synthesized products elicited higher levels of antiviral activity against different types of viruses, except compounds 114 and 117, which were weakly active against the HCM virus and possessed no cytotoxicity within their antiviral dose range (Scheme S-16, ESI file†).131 Another set of reactions involving ribose anomeric acetate was used to produce 5′-modified 2,5,6-trichlorobenzimidazole ribonucleoside compounds, 123–127, via the N-condensation of the 2,4,5-trichlorobenzimidazole substrate 122. The methanol group of the ribonucleoside was converted to azidomethyl and chloromethyl groups, yielding compound 128. The newly synthesized products also showed antiviral activity against certain other viruses, especially against HCM (Scheme S-17, ESI file†).132
The introduction of sulpho-coumarins to the benzimidazole nucleus led to the synthesis of two series of 2-sulfurmethylene-coumarine- and 2-sulfurmethylene-coumarine-N-(2,3,5-triacetatooxypyran-4-yl)-benzimidazole 5,6-disubstituted derivatives, 131a–k, and 134a–e, which were synthesized from the double condensation of the benzimidazole-2-thione products 129a–f with the 3,4,5,6-tetracetoxypyrane and 3-chloromethyl-chromen-2-one compounds 130a–c. The obtained products showed high activity against HCV, especially the compound 2-[(6-bromocoumarin-3-yl)methylene-thio]-5-fluorobenzimidazole, 131i, and its derivative, 1-[(2,3,4,6-tetra-o-acetyl)glucopyranos-1-yl]-2-[(6-bromocoumarin-3-yl)methylenethio]benzimidazole, 134c (Scheme S-18, ESI file†).133 Additionally, in a single step, the 1-alkoxy-2-alkylbenzimidazole compounds 146–150 were produced via reacting the synthon 2-methyl-6-nitro-phenylamine, 145, with primary iodo-alkanes in the presence of NaH as a strong base. The products 146–150 exhibited antiviral activity against certain viruses. Antiviral testing indicated that compound 148 was the most effective anti-HIV-1 product in the series (Scheme S-19, ESI file†).134 Thus, a number of benzimidazole-based products, especially ribofuranose-containing and brominated products, and imidazole, pyrrole, and pyridine benzimidazole structures showed significant antiviral activity.
2.3 Antifungal compounds
The versatility of the benzimidazole molecular framework has been confirmed based on the strong antifungal activities135,136 of the prepared derivatives. New analogues containing tertiary amine, substituted benzimidazole, and triazole moieties have been synthesized, and screened against Candida albicans spores with significant levels of bioaction. Among the N-methyl- and N-phenyl-substituted benzimidazole derivatives, 144a–i, containing an asymmetric carbon atom bound to C2, hydrogen, chlorine, or the hydroxyl group present on C5, the tertiary amine and 1,2,4-triazole substructures were synthesized in moderate yields through the reaction of difluoro-phenyl amino methyl triazole with the 2-chloromethylene benzimidazole derivatives 142 and 143. The products manifested strong antifungal activity (Scheme 6).137 However, the inherent toxicity of this series of compounds and the anti-bacterial resistance pushed the discovery of newer derivatives with improved antifungal activity. | ||
| Scheme 6 The synthesis of asymmetric-carbon-containing 144a–i, where a benzimidazole moiety is C2-bonded with an N-methylene 1,2,4-triazole-N-(2,4-difluorophenyl) derivative. | ||
Compound 147, a 4-amino-5-phenyl-2,4-dihydro-[1,2,4]triazol-3-one benzimidazole derivative, was successfully prepared through the substitution of 5-chloro-2-(1-chlorobenzyl)-1H-benzoimidazole 145 with amino-triazolone, compound 146. Compound 147 was evaluated for its antifungal properties against Candida glabrata. The compound possessed a polar side chain and OH and SH groups, which culminated in it exhibiting higher potential for antifungal action (Scheme S-20, ESI file†).138
Another interesting new series of benzimidazole-based Schiff base derivatives, 150, was synthesized via the condensation of 1,6-disubstituted benzimidazole-2-carbaldehydes, 148, and phenyl hydrazine derivative compounds, 149. The antifungal activities of the final compounds were tested. The bioassay results indicated that noticeable inhibitory activity against R. solani and M. oryzaein was shown by most of the synthesized compounds. The highest in vitro inhibition activities, which exceeded the reference drug's EC50 value at 1.20 μg mL−1 and 1.85 μg mL−1, respectively, were shown by the compounds bearing 2,4-difluoro groups in their structures (Scheme S-21, ESI file†).139
Moreover, recently, 10 new 2-aryl benzimidazole derivatives, 151a–g, were also synthesized through the cycloaddition of substituted phenyl carbaldehydes and 4-substituted 1,2-phenelenediamine, catalysed in the presence of H2SO4/SiO2 and reacted under microwave heating. The antifungal activities were evaluated against several filamentous fungi, i.e., Candida albicans, C. dubliniensis, C. parapsilosis, C. krusei, C. tropicalis, Cryptococcus neoformanse, Aspergillus flavus, Aspergillus clavatus, Alternaria alternate, Microsporum canis, and Trichophyton mentagrophytes. Among the synthesized derivatives, the compound containing fluorine at the para-position of the benzene ring exhibited the highest antifungal activity at a dose of 8.64 μg mL−1 (Scheme S-22, ESI file†).140
Thus, the pharmacophoric value of the benzimidazole molecular template was also demonstrated in terms of antifungal potency. Nonetheless, another benzimidazole derivative, a benzimidazole triazole, 156, was obtained through a sequence of five steps. The benzimidazole compound, 152, obtained through the condensation of 4-methyl-o-phenylenediamine with p-formyl methyl benzoate, after undergoing a reaction with hydrazine, produced the hydrazide 153. The hydrazide 153 further underwent a reaction with isothiocyanate to yield the corresponding thiosemicarbazide 154. Cyclization in a basic medium produced the corresponding 4-substituted-5-[4-(5-methyl-1H-benzoimidazol-2-yl)-phenyl]-4H-[1,2,4]triazole-3-thiol compound 155, which, finally, upon reacting with 2-bromo-1-phenylethanone yielded the final product 156. All the new compounds in the synthetic sequence were evaluated for their antifungal activity against Candida glabrata, Candida krusei, Candida parapsilosis, and Candida albicans. According to biological evaluation assays, the majority of the derivatives showed moderate to strong antifungal activity against all the tested fungal strains. Compounds possessing 3,4-dihydroxy phenyl groups manifested the highest inhibitory activities against the fungal strains, with MIC50 values ranging from 0.78 to 1.56 μg mL−1. The products were also non-toxic at their bio-effective concentrations (Scheme S-23, ESI file†).141
Furthermore, various benzimidazolium N-phenyl methylthioformates, 158 and 159, spirano-benzimidazolium, 160, methyl dithioformic esters, 161 and 162, and a 2-ethane nitrile 1,3-disubstituted benzimidazole compound, 163, were synthesized. Synthesis was achieved by reacting the 3,1′-disubstituted-1,3′-diphenylethyl-2,2′-[2,2′]bibenzoimidazolylidene compound 157 with isothiocyanate, isocyanate, carbon disulphide, and acetonitrile, which produced a number of products, 158–163, of which compound 160 was the most active compound as an antifungal product (Scheme S-24, ESI file†).142 Another synthetic scheme involving the use of a benzimidazole molecular template produced 1,2-bis-(2-mercapto-benzoimidazol-1-yl)-ethane-1,2-dione, compound 165, via the double condensation of the mercapto-benzimidazole synthon derivative 164 upon reacting with diethyl carbonate. Reaction with copper(II) and nickel(II) metal complexes of bis(ethane diamine) yielded the corresponding diiminic bis-(2-thiol benzimidazole) complexes 166. The Cu(II) complex was far more active against fungi than the Ni(II) analogue due to the effects of the metal ion on the cells (Scheme S-25, ESI file†).143 Another interesting benzimidazole–oxadiazole framework-based derivative, 170, was produced as an antifungal agent. The corresponding carbohydrazide compound 168 was transformed, in the first step, via condensation with p-formyl methyl benzoate and 4-substituted o-phenylene-diamine, followed by the reaction of hydrazine hydrate with the corresponding benzimidazole phenyl ester 167. Cyclization with carbon disulphide led to the mercapto-oxadiazole product 169, which, when reacted with various derivatives of phenacyl bromides, produced multiple components. The compounds 186h and 186p were found to be promising candidates for further development towards the treatment of fungal infections (Scheme S-26, ESI file†).144 Additionally, a series of fused thiazolo-benzimidazole benzoxazole products, 173, was also synthesized in three consecutive steps. The first step involved the reaction of 5-substituted 2-mercapto-benzimidazole with benzofuran-2-yl-ethanone in an acidic medium, leading to the corresponding sulfanyl ketone 171. Compound 171 was later converted through an intramolecular cycloaddition reaction, catalysed in the presence of PPA (polyphosphoric acid), to the corresponding 3-benzofuran-2-yl-benzo[4,5]imidazo[2,1-b]thiazole compound 172. Further, through a one-pot three-component reaction, the latter compound reacted with a secondary amine and formaldehyde to yield the fused benzimidazole–thiazole analogue 173. Biological activity testing results showed that the dibromo-substituted compounds were the most active products against the tested fungi, compared to the mono-bromo-substituted compounds (Scheme S-27, ESI file†).145 A synthetically interesting agricultural fungicide, a benzo-[4,5]-imidazo[1,2-d][1,2,4]triazine derivative, was also synthesized through the condensation of 2-chloroacetic acid or 2-bromopropionic acid with o-phenylene-diamine to form the corresponding benzimidazole product 174, which, when reacted with substituted phenyl-hydrazine, produced the corresponding hydrazino-benzimidazole compound 176. Compound 176, when treated with an excess of a mixture of chloroformate and triethylamine, was converted to yield a 1,2,4-triazol-3-one fused benzimidazole through an intramolecular cyclization reaction. The product exhibited fungicidal activity at 50 μg mL−1 (Scheme S-28, ESI file†).146
A series of 2-chloromethyl-1H-benzimidazole derivatives was achieved through multistep reactions. The initial step consisted of the condensation of 2-chloroacetic acid with o-phenylenediamine derivatives in acidic media, leading to 2-chloromethylbenzimidazole structures. These intermediates were later reacted with acyl-chloride and methyl sulphate to produce N-methylated benzimidazole and N-acylated benzimidazole products, 178 and 181. Benzimidazole derivatives also reacted with 2-chloro-N-methyl-N-(2-methylamino-phenyl)-acetamide, 179, and o-phenylenediamine, thereby yielding the corresponding targeted benzimidazole derivatives 180a–q and 183a–n. Several of these alkoxymethyl-1H-benzimidazole products were effective against a number of agricultural fungi (Scheme S-29, ESI file†).147 An extension of substituted o-phenylene-diamine and substituted phenyl acrylic acids also produced compounds as a series of novel 5-(nitro/bromo)-substituted 2-styryl benzimidazole derivatives. In this context, compound 184 was synthesized in ethylene glycol, which showed that bromo-containing compounds were more biologically active than nitro-containing compounds against fungi for this specific molecular framework (Scheme S-30, ESI file†).148
Furthermore, a library of 2-benzimidazolylimino-5-arylidene-4-thiazolidinones, compounds 188a–k, was also synthesized. The approach started with the condensation of chloro-acetyl chloride to 2-aminobenzimidazole, yielding the corresponding chloro-acetamide, compound 185, which underwent intramolecular cyclization to produce the fused tricyclic compound 1H-1,3a,8-triaza-cyclopenta[a]inden-2-one, 186. Compound 185 was also reacted with ammonium thiocyanate to yield 2-(1H-benzoimidazol-2-ylamino)-thiazol-4-one, compound 187, which, when treated with substituted benzaldehydes, produced the targeted pharmacophores 188a–k. These compounds were tested for their potential antifungal effects and showed positive activity against agricultural fungi, especially against the phytopathogens B. elliptica, Fusarium graminearum,P. nicotianae, and R. solani (Scheme S-31, ESI file†).140 A simple and effective approach towards the synthesis of the benzimidazole derivative 189 was established using o-phenylene-diamine and a substituted aldehyde, which were reacted in the presence of a mild catalyst, nano-SnCl4/SiO2. The compound was active in bioactivity testing against various yeasts and filamentous fungi, based on the broth microdilution method of testing (Scheme S-32, ESI file†).149
2.4 Anti-protozonal activities
Strong levels of antiprotozoal activity are reportedly shown by benzimidazole-derived compounds.150,151 A series of benzimidazole pentamidine compounds was synthesized in which the peripheral amidine groups of the pentamidine moiety were transformed into substituted benzimidazole groups, which led to better antiprotozoal activity being shown by the products, especially compound 190. Another concurrent approach produced compounds 190a–j in 68–90% yields, with diversified substitutions; these also exhibited desired levels of antiprotozoal activity (Scheme 7).152 | ||
| Scheme 7 The synthesis of the spaced bis(2-arylbenzimidazole)s 190a–j. | ||
2-(2-Amino-4(5)-nitro-1H-benzimidazol-1-yl)-N-arylacetamides were also synthesized as antiprotozoal compounds. First, bromo-cyanogen was reacted with activated 4-nitro-o-phenylenediamine to produce the corresponding product 2-amino-5-nitrobenzimidazole. The intermediate was then condensed with 2-chloro-N-aryl-acetamide to afford a mixture of regio-isomeric products, 4- and 5-nitrobenzimidazole-N-aryl-acetamides, as the compounds 191 and 192. These compounds were found to be antiprotozoal products (Scheme S-33, ESI file†).153 In another attempt, a synthetic strategy for biphenyl benzimidazole diamidines was developed, which primarily consisted of the synthesis of 5-cyano-2-(cyanoaryl)benzimidazoles, 194a–i, through the condensation of a substituted 3′-formyl-biphenyl-carbonitrile, 3a, with 3,4-phenylene-diamines, yielding the desired active products. The two cyano groups of compounds 194a–i, when treated with hydrochloric hydroxylamine, were transformed into the corresponding N-hydroxy amidines (compounds 195a–i). These intermediates were later reduced into the corresponding amidine groups (compounds 196a–i). These compounds displayed remarkable biological activities, and compounds 196c, 196d, 196f, 196h, and 196i were good antiprotozoal products. Also, there were three compounds, 196f, 196h, and 196i, which showed significant improvements in bioactivity compared to the furamidines IIa and III (Scheme S-34, ESI file†).154
Among other antiprotozoal products, the synthetic approach to 2-{[2-(1H-imidazol-1-yl)ethyl]sulfanyl}-1H-benzimidazole has been outlined in detail. The compound 2-mercapto-1,5,6-trisubstituted-1H-benzoimidazole, 197, which was obtained from the condensation of carbon disulphide with the corresponding 1,2-diaminobenzene, showed bioactivity. Compound 197 reacted with 1-(2-chloro-ethyl)-1H-imidazole to yield the corresponding thioether 198. The developed series of compounds, 198d–g, was treated with methyl iodide to produce the corresponding benzimidazolium salts. These benzimidazole derivatives manifested higher biological activities than the reference standard compound. Compounds 198p–s were the most active of all in the series and showed strong antiprotozoal activities (Scheme S-35, ESI file†).155 Contextually, sulphur heterocycles containing a series of benzimidazole derivatives of thieno[2,3-d]-pyrimidin-4-ones, prepared through the condensation of substituted benzimidazole, 2-mercaptobenzimidazole, and 2-mercaptomethylbenzimidazole with 2-(2-chloro-ethyl)-5,6-disubstuted-3H-thieno[2,3-d]pyrimidin-4-one, showed considerable levels of antiprotozoal activity (Scheme S-36, ESI file†).156
The synthetic route to N-methylated benzimidazole esters and amides, 205–208, involved the synthesis of an intermediate, 5,6-disubstituted-1-methylbenzimidazole-2-carbaldehyde, compound 204, which was prepared in two steps from o-phenylenediamine and 2-hydroxyacetic acid; it was easily converted to the corresponding ester and amide derivatives. Bioactivity testing exhibited moderate antiprotozoal activity, thereby indicating that the ester analogue compounds were more active than others in the series (Scheme S-37, ESI file†).157 Another series of new benzimidazole–benzothiazole entities connected with an amide moiety, obtained from N-methyl-2-nitro benzoate, was synthesized through saponification in the first step, followed by condensation with 2-amino-5-nitrothiazole, yielding the product. The study also mentioned that the compounds 211 and 212 were strongly active as antiprotozoal agents (Scheme S-38, ESI file†).158
Moreover, the condensation of o-phenylenediamine with both 3-trifluoromethly cinnamic acid and iso-nicotinic acid yielded the corresponding 2-substituted-benzimidazole intermediates 213 and 214, which were further reacted with aminopyridines under microwave conditions, in the presence of formaldehyde, yielding the corresponding 2,3-disubstituted benzimidazoles, 215 and 216, which possessed high antiprotozoal activity (Scheme S-39, ESI file†).40
2.5 Antimalarial compounds
Several benzimidazole derivatives were proposed, synthesized, and investigated for their bioactivity as antimalarial agents against several malaria-causing pathogenic Plasmodium species,159 e.g., P. falciparum, P. ovale, P. malariae, P. vivax, and P. knowlesi.169 For the synthesis of antimalarial agents, 6-O-, and N-substituted diamino pyridine derivatives, 217, were condensed with cyanogen bromide to yield the 2-aminobenzimidazole analogue compounds 218, which were later reacted with phenacyl bromide to produce the 6-substituted 2-amino-3-benzoylbenzimidazole compounds 219. Upon reduction, they yielded the corresponding alcohols, 220. Many 2-amino-pyridin-benzimidazole derivative products were also produced, which displayed antimalarial activity. These newly synthesized products were quick to kill pathogens, and showed an absence of cross-resistance, together with a low frequency of pathogenic resistance re-emergence. The products also showed excellent oral bioavailability (Scheme 8).160 These products had the distinction of being active and non-cross-resistant against the studied pathogens. | ||
| Scheme 8 The synthesis of 3-N-trisubstitued phenylethanol-3H-imidazopyridin-2-amines in three steps from substituted diamino-pyridines. | ||
Recently, a new antimalarial compound, identified as the fused pyrido[1,2-a] benzimidazole 223, was synthesized under microwave conditions through the nucleophilic substitution of the chlorine atom of the tetracyclic compound 222, obtained from 3,4-disubstituted aniline 221. The bioactivity was tested under in vivo and in vitro conditions (Scheme S-40, ESI file†).161
2.6 Antitubercular compounds
New benzimidazole derivatives containing nitrogen heterocycles are conspicuously present in the antitubercular drug domain.162 Heterocycles containing N-atom(s) have been pursued in search of new chemical entities and new drug lead(s) for developing newer anti-tubercular agents.163 In this context, a library of 5-methyl-carboxylate-substituted benzimidazole derivatives, 225, was prepared via the condensation of N-alkyl diaminobenzoate with sodium phenyl methane sulfonate with DMF as the solvent at 90 °C, with high yields (75–84%). The antitubercular activities of derivative compounds with the base structure 225 were evaluated against mycobacteria. The results demonstrated that the most effective compound contained a trifluoromethyl group in its structure (Scheme 9).164 | ||
| Scheme 9 The synthesis of 5-methyl-carboxylate 2,3-disubstituted benzimidazoles 225. | ||
Moreover, a novel series of compounds, 2-styryl-benzoyl-1H-benzimidazols, was synthesized from the base compound 30 by the condensation of 3,4-diaminobenzophenone with malonic acid (method 1) or acetic acid to produce the corresponding 2-methyl benzimidazole derivatives, which were later reacted with aromatic aldehydes (method 2). 5-Nitro homologous structures based on template 31 were prepared. Antitubercular activity screening of the products showed better bioactivity with electron-donating-group substitution, i.e., Cl, O, and S, in the structural frameworks (Scheme S-41, ESI file†).165
2.7 Anti-inflammatory and analgesic compounds
Benzimidazole-core-derived compounds have also been shown to possess anti-inflammatory activity in parallel to nonsteroidal anti-inflammatory drugs (NSAIDs). The compound N-(4-(2-phenyl-1H-benzo[d]imidazol-1-yl) phenyl)acetamide, 232, was synthesized according to Scheme 10.166 The 2-p-substituted-aryl benzimidazoles 228 were reacted with p-nitro-fluorobenzene to produce the corresponding 3-p-nitrophenyl-2-p-substituted-aryl benzimidazole compounds 229a–g. When treated with concentrated HCl, the cyano group of compound 229a was transformed into the carboxylic acid derivative 230. In the presence of HCl and Sn, compounds 229c–g underwent the reduction of the nitro group into an amine. This functional group was then transformed into the corresponding acetamide by simple treatment with acetyl chloride to produce the target compound 232. During bioactivity testing, 1,2-diphenyl benzimidazoles (DPBIs) with para-nitro and para-bromo substitutions at N1-Ph and C2-Ph (232, R = p-NO2, R = p-Br), obtained in relatively low yields (48–57%), exhibited high anti-inflammatory activities under in vitro and in vivo conditions.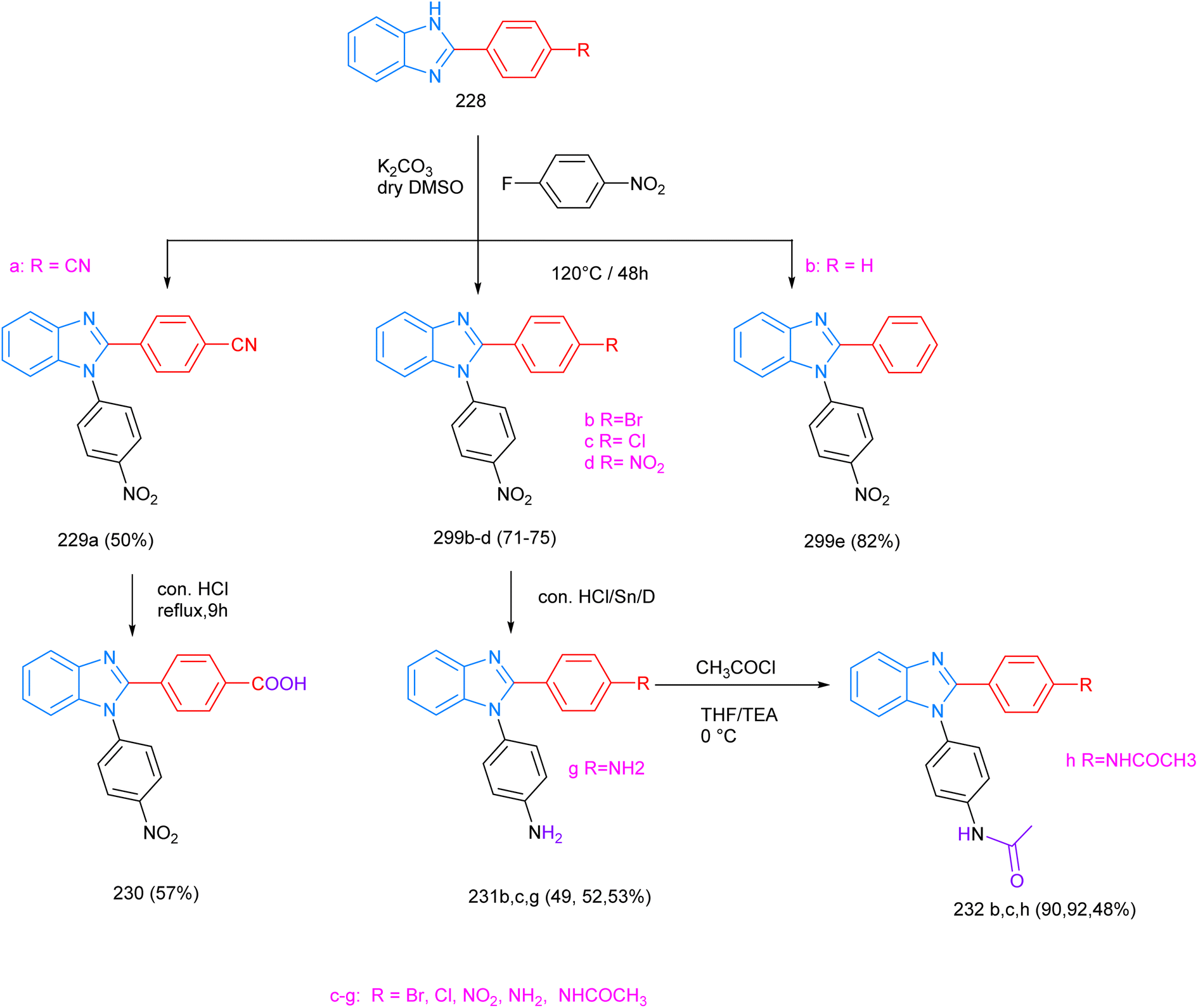 | ||
| Scheme 10 The synthetic approach to 3-p-acetamide phenyl-2-p-substituted-aryl benzimidazoles 232. | ||
Additionally, 2-methylthiobenzimidazole compounds with different combinations of aromatic and heterocyclic substituents were synthesized in multiple steps. This approach started with the synthesis of 4-(2-methylthiobenzoimidazol-1-yl)-thiazol-2-ylamine 233, which was obtained from the treatment of 2-methylthio-1H-benzoimidazole with chloro-acetyl chloride, thiourea and hydroxylamine. The heterocyclic nitrogen atom of substrate 233 acted as a nucleophile to yield the iminic salt 234. In certain other reactions, the primary amine functional group of the same substrate reacted as a nucleophilic species with halogen derivatives to generate the corresponding amino-ketones 235a–b and the amide 236. In particular, compound 234 underwent intramolecular cycloaddition upon refluxing in (2 N) HCl solution and then washing with aq. NH4OH to yield the fused heterobicyclic imidazo[2,1-b] thiazole benzimidazole product 237. In turn, compound 235b underwent a reaction with phenyl thiosemicarbazide to produce the corresponding product 238, which was reacted with p-substituted and unsubstituted phenacyl bromide and methyl bromo acetate to afford the target products 239 and 240, thereby incorporating a second thiazole ring (Scheme S-42, ESI file†).167 In vitro screening showed that benzimidazole–thiazole hybrids linked to the acetyl moiety, phenyl thiosemicarbazone, 1,3-thiazolines, 4-thiazolidinedione, and 1,3-thiazoline substituted with p-chlorophenyl moieties were the highest inhibitors of the COX-2 enzyme. The benzimidazole-thiazole derivative possessing a 4-thiazolidinedione substructure exhibited the highest inhibition under in vivo assay conditions.
Furthermore, a novel compound, the 2-methylaminobenzimidazole derivative 241, was also synthesized via the N-alkylation of 2-chloromethylbenzoimidazole and p-substituted aniline. The analgesic and in vivo anti-inflammatory biological activities were tested. Most of the new compounds showed potent analgesic and anti-inflammatory activity compared to the corresponding reference drugs (Scheme S-43, ESI file†).168 Moreover, a number of substituted 2-(2-hydroxynaphtyl)-3-alkyl benzimidazole derivatives, 241, were synthesized via the condensation of substituted o-phenylenediamines with 2-hydroxynaphthaldehyde. The appropriately obtained benzimidazole derivative 242 was later reacted with an alkyl iodide. The target compound 243 possessed analgesic activity (Scheme S-44, ESI file†).169 The products 241 and 243 and benzimidazole–thiazole extended products were active at higher levels as analgesic and anti-inflammatory compounds. The thiazole derivative was the highest level COX-2 inhibitor.
2.8 Antihistamine compounds
Another set of benzimidazole-derived multiple-ring expanded phenyl-pyrazole conjugated benzimidazole-quinoxaline derivatives was synthesized. The products showed considerable levels of antihistaminic activity. The synthetic processes utilized the condensation of benzimidazole, 2,3-diphenylquinoxaline, and formaldehyde to yield the corresponding methylene-spaced hybrid benzimidazole-quinoxaline derivative 244. The obtained product was reacted with chloroacetone to introduce a methyl ketone group, forming molecule 245, which was further condensed with p-substituted benzaldehyde(s), leading to the corresponding aromatic enone-derivative-based products 247 in 45–80% yields. The compounds were later reacted with aryl carbazide to afford the corresponding N-substituted 4,5-dihydropyrazolic benzimidazole-quinoxaline derivatives 247 (Scheme 11).170 | ||
| Scheme 11 The synthetic route to N-substituted 4,5-dihydropyrazolic benzimidazole-quinoxaline derivatives 247. | ||
Another series of extended-ring benzimidazole derivatives, 2-(3-aminopiperidin)-benzimidazoles, was proposed as a H1-antihistamine for insomnia therapy. One of these new derivatives exhibited activity equivalent to the currently used H1-antihistamine. The synthetic route was implemented through the condensation of o-phenylenediamine with piperidine-1,3-dicarboxylic acid 1-tert-butyl ester, yielding the corresponding 2-substituted benzimidazole derivative 248, which underwent two successive alkylation reactions with halogen derivatives to yield the expected target product 250 (Scheme S-45, ESI file†).171 The 2-(3-aminopiperidin)-benzimidazole was very active as a H1-antihistamine, and it holds promise for the future.
2.9 Antihypertensive activity
A set of 5-nitro benzimidazoles bearing 1,4-disubstituted and 1,5-disubstituted indole components was prepared through the N-acylation of 4-nitro-o-phenylenediamine, yielding the amidic compound 251, which underwent an intramolecular cyclization reaction to produce the corresponding benzimidazole product 252. Condensation with (4-(bromomethyl)-1H-indol-1-yl)(phenyl)methanone 253 yielded the corresponding product 1-(1H-indol-4-ylmethyl)-5-nitro-2-alkyl-1H-benzoimidazole 254. The nitrogen atom of the indole moiety was reacted with 2-fluorobenzonitrile, which led to the corresponding benzimidazole–indole–benzonitrile, product 255. Product 255 was refluxed under basic conditions to transform it into a carboxylic acid, the final target product 256 (Scheme S-46, ESI file†).172 The results of bioactivity studies showed antihypertensive activity. The compound 11,2-(4-((2-butyl-5-nitro-1H-benzo[d]imidazole-1-yl) methyl)-1H-indol-1-yl)benzoic acid, 256c, was the most active product.Additionally, another series of 6-substituted benzimidazole–indole derivatives was prepared from 1,4-disubstituted or 1,5-disubstituted indole and benzoic acid moieties. According to Scheme S-47,† the 2-(2-substituted-7-methyl-3H-benzoimidazol-5-yl)-4,6-disubstituted benzoxazole 257 reacted with the 4- or 5-bromomethyl-1-(2-methylcarboxylate) indoles 258a–b to yield the corresponding N-alkylated indole carboxylate derivatives 259 and 260, which, when converted by refluxing in basic media, produced the targeted benzimidazole–oxazole–indole carboxylic acid derivatives 261 and 262. The bioactivity results indicated that compounds 261b and 262b effectively reduced blood pressure, and they were considered effective against hypertension (Scheme S-47, ESI file†).173 Another di-substituted benzimidazole product, 2,5-disubstituted benzimidazole 263, was produced by condensing 4-substituted o-phenylenediamine with salicylaldehyde and p-anisaldehyde under microwave irradiation conditions. The expected target products manifested antihypertensive activity. The reaction was extended to differently substituted compounds. Among the prepared products, compounds 263c–f were the most active of the series (Scheme S-48, ESI file†).174 Other structurally extended benzimidazole compounds, bis-benzimidazole–indole–benzoic acid derivatives 266, were reported in terms of their synthesis and biological activity. The 2-(4-bromomethyl-indol-1-yl)-benzonitrile 264 was reacted with 2′-substituted-1,7′-dimethyl-1H,3′H-[2,5′]bis-benzoimidazolyl, which yielded the corresponding product 265; after hydrolysis in a basic medium, this yielded the expected target product, 266. Among these benzimidazole derivatives, compound 266c was an effective antihypertensive product (Scheme S-49, ESI file†).175
Another series of benzimidazole analogues, 4′-2-butyl-5-sulfamoylbenzoimidazolmethyl-biphenyl-2-carboxylic acids 269, was prepared when an amine was mixed with 5-chlorosulfonyl benzimidazole 268, which was obtained by treating 4-(2-butyl-benzoimidazol-1-ylmethyl)-biphenyl-2-carboxylic acid 267 with chlorosulfonic acid. Among the series of 5-alkylsulfamoyl benzimidazole derivatives, compounds 269g and 269h showed significant antihypertensive activity (Scheme S-50, ESI file†).176 The compounds in the series 266 and 269 exhibited promising anti-hypertensive activity.
2.10 Antidiabetic compounds
A benzimidazole core template has also been utilized as a bioactive pharmacophore for developing antidiabetic compounds. Hyperglycaemia is widespread in the global population and is almost endemic to a large extent. Several drug candidates and lead compounds have been developed to maintain glucose levels below a recommended control value and within prescribed limits. Benzimidazole derivatives demonstrated high levels of antidiabetic activity.177 Two new compounds, namely 3,6-dimethyl-5-oxo-pyrido[3,4-f][1,2,4]triazepino[2,3-a]benzimidazole 271 and 10-amino-2-methyl-4-oxo-pyrimido[1,2-a]benzimidazole 272, were synthesized. When reacted with DMF–DMA, 6-methyl-7H,9H-4b,5,9,10-tetra-aza-benzo[a]azulen-8-one, compound 270, containing a benzimidazole moiety, transformed into the fused tetra-heterocyclic product 271 in 58% yield. After treatment with an excess of methyl sulphate in an aqueous medium, the same substrate underwent an intramolecular rearrangement, yielding the fused tricyclic amino product 272 in 67% yield. Compound 271 exhibited high biological activity against both α-amylase and α-glucosidase enzymes, while the latter derivative, compound 272, showed comparatively lower levels of bioactivity against both enzymes (Scheme 12).178 | ||
| Scheme 12 The synthesis of 3,6-dimethyl-5-oxo-pyrido[3,4-f][1,2,4]triazepino[2,3-a]benzimidazole 271 and 10-amino-2-methyl-4-oxo-pyrimido[1,2-a]benzimidazole 272, which are described to be antidiabetic agents. | ||
2-Mercapto-benzimidazole, a sulfurized benzimidazole derivative, was utilized for producing another set of antidiabetic compounds; it was reacted with ethyl 2-chloroacetate through a nucleophilic reaction to yield the thio-ester 273. When treated with hydrazine hydrate, the thio-ester functional group was transformed into the corresponding thio-semicarbazide–benzimidazole, intermediate 274, which later underwent a reaction with various carboxylic acid derivatives in the presence of POCl3, forming the corresponding 1,3,4-oxadiazole-based structure 275. However, when compound 274 was reacted with an aldehyde followed by a reaction with 2-thioacetic acid, this yielded the amidic thiazolidin-4-one benzimidazole product 277. The thiazolidinone 275 and oxadiazole 277 products, possessing a 2-mercapto benzimidazole nucleus, were tested in vivo for their antidiabetic activity using the oral glucose tolerance test (OGTT), and superior antidiabetic activity was shown by the compounds with NO2, OH, and Cl substituents (Scheme S-51, ESI file†).179
Based on predictive studies, the compound (5Z)-5-[3(4)-(1H-benzimidazol-2-yl-methoxy)benzylidene]-1,3-thiazolidine-2,4-dione 279 was synthesized when 2-chloromethylbenzimidazole reacted with phenols to form the equivalent benzimidazole intermediate coupled with a phenoxy group, 278. This compound underwent an electrophilic substitution at the meta-position with a thiazolidine-2,4-dione moiety. The three newly synthesized benzimidazole-pharmacophore-possessing compounds showed antihyperglycemic activity related to insulin sensitization (Scheme S-52, ESI file†).180
Another novel antidiabetic product, N-substituted-2-[4-(2-substituted-benzoimidazol-1-ylmethyl)-[1,2,3] triazol-1-yl]-acetamide 280, was synthesized. The condensation of 2-substituted benzimidazole with propargyl bromide yielded the corresponding N-propargylated benzimidazole intermediate. The intermediates were later reacted with a mixture of sodium azide and 2-bromo-ketone, in the presence of copper(I) as a catalyst, through a click chemistry approach to yield the 1,4-disubstituted 1,2,3-triazole product 280. The new analogues of product 280 were screened for their antidiabetic activity. All the compounds manifested significant activity toward the α-amylase and α-glucosidase enzymes in antidiabetic testing (Scheme S-53, ESI file†).181 26 new derivatives resulting from the structural diversification of fused benzimidazoles and oxadiazoles were synthesized. The synthetic approach involved the condensation of 3-methy-o-phenylenediamine and 4-formyl-methylbenzoate, leading to the corresponding 4-(7-methyl-1H-benzoimidazol-2-yl)-methyl benzoate product 281, which was transformed into the hydrazide intermediate 282, which, when treated with hydrazine hydrate and different aryl carbaldehydes, produced the hydrazone derivative 283. The hydrazine compound underwent intramolecular cyclisation and, catalysed by PhI(AcO)2, was transformed into the 2-substituted-[1,3,4]oxadiazole product 284. The product and its several derivatives displayed strong enzymatic inhibition activity against α-glucosidase. The compound activities were also validated through molecular docking studies (Scheme S-54, ESI file†).182
Recently, a library of benzimidazole–triazolothiadiazole derivatives was prepared. The compound 4,5-dimethyl-o-phenylenediamine was reacted with 4-carboxybenzaldehyde to produce the corresponding compound 4,5-dimethyl-benzimidazole-benzoic acid 285. When treated with thiosemicarbazide, the obtained compound was converted into 4-amino-4H-benzimidazole-[1,2,4]triazole-3-thiol 286, which was then reacted with carboxylic acid in the presence of POCl3 to produce the fused bicyclic 6-substituted-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole benzimidazole derivative 287. The product 287 and its derivatives were evaluated as β-glucuronidase inhibiting agents, and they manifested higher antidiabetic activity compared to the reference standard agent D-saccharic acid 1,4-lactone (Scheme S-55 ESI file†).183
Another new benzimidazole-based derivative, benzoyl aryl benzimidazole 289, was synthesized using ammonium chloride or a mixture of ammonium chloride and sodium metabisulfite as a catalyst. The compound 3,4-diamino benzophenone 288 and appropriately substituted aryl aldehydes were reacted. In vitro antidiabetic assays of product 289 and its derivatives/analogues showed good-to-exceptionally-high antidiabetic activity against α-amylase and β-glucosidase. The target benzimidazole, an analogue of product 289, having a hydroxyl group at the p-position of the phenyl part of the structure of the product 289, demonstrated significant inhibitory activity against α-amylase (IC50 = 12.09 ± 0.38 M) and β-glucosidase (IC50 = 11.02 ± 0.04 M) in comparison to the reference standard acarbose (Scheme S-56, ESI file†).184
Another series of newer benzimidazole derivatives, combining sulphur and hydrazide functionalities, which had shown good antidiabetic activity, was proposed and synthesized. The compound 2-thioester hydrazide 290 was reacted with carboxylic acid in the presence of POCl3 to yield the 1,3,4-oxadiazole intermediate 291. When treated with aldehydes, the compound yielded the benzimidazole hydrazine product 292. The compound 292 and its derivatives were reacted with mercaptoacetic acid in DMF in a reducing medium to yield the corresponding benzoimidazol-2-yl-thio-N-2-methyl-4-oxothiazolidin-3-yl-acetamide product 293. The set of benzimidazole derivatives showed remarkable antidiabetic activity. The bioactivity test results favoured the four compounds 291c, 291d, 291h and 291i, which were highly active (Scheme S-57, ESI file†).185
Another set of benzimidazole analogues, derived from 7-methyl-3H-imidazo[4,5-b] pyridine 295, were prepared through the condensation of 4-methyl-pyridine-o-diamine 294 with aromatic aldehydes under microwave heating conditions. The compound 295 was oxidized using KMnO4/NaOH to yield the corresponding carboxylic acid derivative 296, which was reacted with a 4-substituted pyridinic primary amine to allow the production of the secondary amide target 297. The final products, imidazopyridine analogues of benzimidazole, were thus prepared, and their antidiabetic activity was tested. The products displayed strong effects in terms of reducing blood glucose levels in experimental models (Scheme S-58, ESI file†).186
Another series of antidiabetic products, starting from the condensation of 5-nitrophenylenediamine with [4-(4-formyl-phenyl)-cyclohexyl]-ethyl ethanoate 298, produced the desired compound 5-amino-3-substituted benzimidazole 299, which acted as a nucleophile with different electrophilic species to produce the corresponding N-alkylated-benzimidazole phenyl cyclohexyl ethyl ethanoate compound series 300a–p from the appropriately derived intermediates 298 and 299. The compound 300k exhibited distinctive antidiabetic activity (Scheme S-59, ESI file†).187
N-Benzyl, N-benzoyl, and N-diphenyl benzimidazole derivatives, compounds 302, 303 and 304, were synthesized through reactions involving the fused tri-heterocyclic benzimidazole structure 301, providing a range of functionalized bromide derivatives as the products. Among the synthesized benzimidazole derivatives, compounds 302-2b and 302-3b displayed the highest antidiabetic activity during biotesting (Scheme S-60, ESI file†).188
Another type of benzimidazole-derived products, metal-complexed compounds, was prepared with the aim of finding better antidiabetic agents. The compound 1-(2-methyl-benzoimidazol-1-ylmethyl)-1H-benzotriazole was complexed with Cu(II) chloride, Cu(II) nitrate, and Zn(II) chloride to yield the corresponding bidentate metal complexed species 305, 306 and 307, respectively. The compounds [Cu(mbmb)2Cl2] (305) [Cu(mbmb)2(NO3)2] (306), and [Zn(mbmb)2Cl2] (307), which structurally contained 1-[(2-methyl-1H-benzoimidazol-1-yl) methyl]-1H-benzotriazole as part of the final metal-complexed products, were tested against three enzymes, namely α-amylase, α-glucosidase, and β-galactosidase, for their antidiabetic efficacy. The compound 305 showed a higher level of activity as an antidiabetic product in α-amylase testing, while the compound 307 showed no significant level of antidiabetic activity (Scheme S-61, ESI file†).189
Notwithstanding that benzimidazole-templated structures offer a wide spectrum of biological activity as potent antidiabetics, the development of benzimidazole-based antidiabetic products is still ongoing. A series of newer products was obtained from the condensation of differently substituted o-phenylenediamines with aryl aldehydes in the presence of sodium metabisulfite to yield the disubstituted aryl benzimidazole based structures 308, with 88%, 91%, and 69% yields, respectively, of the products 308c, 308d, and 308i, where the chloro-derivative 308d was the highest-yield product. In vitro screening of these products manifested considerable levels of α-amylase inhibition, particularly the chloro- and fluoro-substituted products 308c, 308d and 308i, all of which showed high IC50 values, ranging from 1.86 ± 0.08 M to 3.16 ± 0.31 M, compared to the standard acarbose (IC50 = 1.46 ± 0.26 M) (Scheme 13).190
 | ||
| Scheme 13 The synthesis of disubstituted aryl-benzimidazoles 308, which can act as antidiabetic agents. | ||
The synthesis of the 2-aryl benzimidazole derivative 309 was achieved via the condensation of 4,5-disubstituted-1,2-aminobenzene under reflux in DMF using Na2S2O5. The yields of the products 309a–k varied from low to moderate to high levels. In vitro testing of the α-amylase inhibitory activity followed. All the products exhibited α-amylase inhibitory potential compared to the reference standard acarbose (IC50 = 1.46 ± 0.26 M), with IC50 values ranging from 1.48 ± 0.38 M to 2.99 ± 0.14 M for the products (Scheme 14).191 A number of products, including the 2-aryl, Cu-metal-complexed, and benzoyl aryl benzimidazoles, showed anti-diabetic activity nearly on par with the reference standard compound used for antidiabetic activity.
 | ||
| Scheme 14 The synthesis of the targeted 2-arylbenzimidazole derivatives 309. | ||
2.11 Anticancer compounds
Benzimidazole-templated compounds have also been found to be some of the most significant heterocyclic structures in terms of showing considerable levels of cytotoxic effects under in vitro and in vivo experimental conditions. The anticancer properties of benzimidazole products have been studied against several cancer cell lines.192 Among the developed compounds, a new series based on the ethyl-(1,2-disubstituted)-5-carboxylate benzimidazole derivative 310, which was obtained from the condensation of 3,4-diamino ethyl benzoates (produced after three synthetic steps) with 4-substituted benzaldehydes in an aqueous medium and in the presence of sodium bisulphite solution in 76–90% yields, showed potent sirtuin inhibition activity (SIRT1/SIRT2). Among the new derivatives, ethyl-2-(4-(dimethylamino)-phenyl)-1-phenyl-1H-benzo[d]-imidazole-5-carboxylate displayed high SIRT2 inhibition, with an IC50 value of 26.85 ± 1.92 μM, with the product showing antitumor activity against three cancer cell lines, i.e., colon (HCT-116), breast (MDA-MB-468), and leukaemia (CCRF-CEM) (Scheme 15).193 | ||
| Scheme 15 The synthesis of 5-ethyl-carboxylate-2-(4-substituted phenyl)benzimidazoles 310, which can act as anticancer agents. | ||
Another series of benzimidazole derivatives, compounds 311, 312 and 314, incorporating tetracyclic sub-structures, was synthesized through the one-step microwave-assisted condensation of substituted 1,2-diaminobenzene with cyclohexane, 1,2-cyclohex-4-ene carboxylic anhydride, and (1-carboxymethyl-cyclohexyl)-acetic acid. The anticancer activities of all the synthesized compounds were established against breast (T47D), lung (NCl H-522), colon (HCT-15), ovarian (PA-1) and liver (Hep G2) cancer cell lines. Some of the synthesized benzimidazole derivatives exhibited satisfactory anti-proliferative activity, with IC50 values ranging from 7.5 ± 0.3 μM to 14.6 ± 0.4 μM (Scheme S-62, ESI file†).194 Also, several other variants of structurally diverse heterocyclic compounds containing benzimidazole and pyrazole substructures were synthesized by condensing 1,3-disubstituted pyrazole-4-carbaldehyde 314 with (1H-benzoimidazol-2-yl)-acetonitrile. The cyano-benzimidazole–pyrazole compound 315 was successful against the pancreatic cancer cell lines SW1990 and AsPCl, with IC50 values of 30.9 ± 0.77 μM and 32.8 ± 3.44 μM, respectively, when compared with gemcitabine as a reference standard drug. The compound containing a p-fluorophenyl substituent was among the most active products (Scheme S-63, ESI file†).81
Recently, more structurally complex benzimidazoles based on a 2-((imidazole/benzimidazol-2-yl)thio)-aryl ethanone template, 316, were obtained by neutralizing mercapto-keto-imidazolium sulphate salts, which were produced by the reaction of 2-mercapto-1H-imidazole and 3-oxo-3-aryl ethyl propionoate. The anticancer activity was evaluated in cell-based assays against the human breast cancer cell lines T4-7D and MCF-7, and compared with the cell-free cyclin-dependent kinase 2 assay. The test results indicated that these products possessed good antiproliferative activity (Scheme S-64, ESI file†).195
As a type of metallo-organic product, manganese-based hexacoordinated complexes were produced using 1-benzyl-1H-benzimidazoles as ligands and 2,2′-bipyridine as co-ligands, having the general formula, Mn(CO)3(bpy)L. These complexes showed promising anticancer activity in preliminary testing (Scheme S-65, ESI file†).196
Also, 2-amino-benzoimidazole and 1-methyl-imidazole-2-carbaldehyde were reacted to lead to the corresponding Schiff base, compound 318. The obtained product was used as a ligand to prepare copper(II)- and zinc(II)-complexed structures having the framework of compound 319. The prepared complexes showed anticancer effects. The copper complex was also active as an anti-breast-cancer complex (Scheme S-66, ESI file†).197
Another newer series of benzimidazole compounds derived from {5-[4-(methyl-oxetan-3-yl-amino)-benzoyl]-1H-benzoimidazol-2-yl}-melthyle carbamate, 320, was prepared by the condensation of 4-substituted-o-phenylenediamine with 1,3-bis(methoxycarbonyl)-2-methyl-2-thiopseudourea in methanol under microwave irradiation. The benzimidazole-derived product, with water soluble characteristics, showed effects against lung and prostate cancers when administered orally (Scheme S-67, ESI file†).198
Yet another benzimidazole derivative, 326, was prepared in seven steps starting from salicylic acid. Upon treatment with acetic acid, compound 324 was converted to the corresponding benzimidazole intermediate, which was transformed into 2′-(2-hydroxyphenyl)-1H,1′H-[2,4′]bibenzoimidazolyl-4-methyle carboxylate, compound 326, under hydrogenation using H2/Pd-C. The benzimidazole-based compounds 324, 325 and 326 manifested significant biological activities against the human lung cancer cell line A-549 and epithelial and HeLa cell lines (Scheme S-68, ESI file†).199
In a similar manner, 2-hydroxynaphtalene imino-benzimodazole, compound 327, was intuitively produced by condensing methyl-2-amino-1H-benzoimidazol with 2-hydroxy naphthaldehyde in ethanol. The resulting product was employed as a ligand for coordination with zinc(II) (69%), cobalt(II) (74%), and copper(II) (82%) salts, showing effects against liver, skin, colon, breast, and cervical cancer cell lines; the compound containing Zn(II) was far more active than the compounds containing Cu(II) and Co(II) as part of the metal-complexed structure (Scheme S-69, ESI file†).200
A series of benzimidazole-biphenyl-pyrazolo-ethenones based on the structure template 332 was prepared. Benzimidazole–acetic hydrazide reacted with the chalcone 331 to produce the compounds. The target set of benzimidazole structures 332 showed interesting anticancer activities. The product 332a proved its efficacy against lung cancer cell lines (Scheme S-70, ESI file†).201
The 5-chloro- and carboxy-1H benzoimidazol-2-yl-acetonitriles 333a and b were used as starting materials to synthesize diversely functionalized, heterocyclic acetonitrile products, including thiazolidin-4-one 334b, 4-amino-3-substituted-3H-thiazole-2-thiones 335a1 and 335a2, 3-substituted-5-oxo-thiazolidin-2-ylidenes 338a1, 338a2, 339a1, and 339a2, 3,4-disubstituted-3H-thiazol-2-ylidenes 340a, b1, and b2, and 3-substituted-[1,3]thiazinan-2-ylidenes 341a and b. After being evaluated under in vitro conditions against human cell lines expressing human colon carcinoma (HCT 116), human breast adenocarcinoma (MCF7), and human hepatocellular carcinoma (HEPG2), the produced compounds showed anticancer activity, with IC50 values of less than 10 μg mL−1 (Scheme S-71, ESI file†).202
For another interesting series of anticancer compounds, the chalcone precursor 342 was obtained by reacting 2-acetyl benzimidazole with a substituted aromatic aldehyde to produce several derivative intermediates. These intermediates were then cyclo-condensed using hydrazine hydrate and phenyl hydrazine in two different processes, producing the pyrazoline derivatives 343a–g and 344a–g, respectively. It was discovered that the 2-[5-(3,4-dimethoxyphenyl)-1-phenyl-4,5-dihydro-1H-3-pyrazolyl] 344f was the most active product in the series. After closely examining the substitutions, it was inferred that the electron-donating group (–OCH3) on the phenyl ring at the fifth position of the pyrazoline moiety played a significant role in the anticancer activity (Scheme S-72, ESI file†).203
With the aim of synthesizing new thiazolidinedione compounds, both conventional and microwave-assisted approaches were used. The reaction of 3,4-diamino methyl benzoate with tetra-substituted benzaldehydes in the presence of Na2S2O5 as a reducing agent produced the 2-phenyl-substituted benzimidazolyl methyl carboxylate products 345a–d. The ester groups of these compounds were reduced to primary alcohols to yield the products 346a–d, which underwent moderate oxidation reactions to lead to the corresponding aldehydes 347a–d. The products 347a–d were used as starting materials to undergo condensation with 2-(2,4-dioxo-thiazolidin-3-yl) derivatives to yield the 5-methylenebenzimidazole-3-substituted thiazolidine-2,4-dione structures 349a–t, 350a–d, 351a–d and 352. Interestingly, a panel of human cancer cell lines, including breast (MDAMB231), prostate (PC-3), cervical (HeLa), lung (A549), bone (HT1080), and kidney (HeK-293T), were utilised to assess the in vitro cytotoxic potentials of some of these newly synthesized compounds 349n, 349p and 349q, and these products showed strong cytotoxic effects. Anticancer efficacy toward PC-3, HeLa, A549, and HT1080 cancer cells was exhibited, with IC50 values ranging from 0.096 to 0.63 μM. In contrast to the tested cancer cell lines, the majority of the products was determined to be ineffective against normal HeK-293T kidney cells. The treatment of the cell lines with compounds 349p and 349q resulted in the desired morphological characteristics of apoptosis, i.e., nuclear fragmentation and cell shrinkage. Additionally, the test products caused the assembly of F-actin proteins to be disrupted, which inhibited cell migration. As demonstrated by Hoechst and DCFH-DA staining and mitochondrial membrane and annexin binding tests, the growth of cancer cells was suppressed, inducing apoptosis in the A549 cell line (Scheme S-73, ESI file†).204
Furthermore, two novel ruthenium–DMSO-based metal complexes with 2-aminophenyl benzimidazole were also synthesized. The ruthenium–DMSO-based complexes 353 and 354 were synthesized by the complexation of 2-(2-aminophenyl)-benzimidazole with RuCl3 salt in DMSO. In compound 353, ruthenium is coordinated with the benzimidazole nitrogen atom, aniline nitrogen, two chlorides, and two DMSO molecules. In compound 354, ruthenium is connected to the benzimidazole nitrogen atom, aniline nitrogen, three chlorides, and one DMSO species. The anticarcinogenic activity of these products was tested under in vitro and in vivo conditions. The in vitro screening was performed against the human breast cancer cell line MCF7, the human colorectal cancer cells Caco-2, and the normal human liver cell line THLE-2. The metal complex 353 displayed mild in vitro anticancer activity with lower toxicity towards normal cells, while the other complex 354 manifested high inhibition potential under in vivo conditions (Scheme 16).205
 | ||
| Scheme 16 The ruthenium–DMSO-based complexes 353 and 354. | ||
Moreover, Schiff bases derived from 2-aminophenyl benzimidazoles were also synthesized as probable anticancer agents. The compounds 3-{[2-(1H-benzoimidazol-2-yl)-phenylimino]-methyl}-phenyl-2-ol 355 and naphthalen-2-ol 356 (HL1 and HL2) were complexed with CuCl2·2H2O in the presence of 2-mercaptobenzothiazole or 2-aminobenzothiazol as a sulphur source, with sulphur being a contributor to cytotoxicity. The four obtained complexes, compounds 357–360, showed tetrahedral coordination. Based on in vitro anticancer activity testing against A-549, Caco-2, HT29 and RPE-1 cell lines, the best anticancer activity results were obtained with complex 358, which presented higher activities towards both the A-549 and Caco-2 cell lines, indicating good selectivity for the human lung cancer cell line A-549 and moderate selectivity for the colorectal cell line Caco-2 at doses of 10.9 and 15.7 μM, respectively (Scheme 17).83 The importance of these metal complexes is well understood in the anticancer realm, especially as anti-lung-cancer agents. However, compound 358 needs to be evaluated under in vivo conditions. In general, the metal complex compounds discussed in this section are superior in terms of bioactivity during preliminary testing under in vitro conditions and seem promising for future development. Thus, in the anti-cancer area of pharmacological activity, benzimidazole-based metal-complexed compounds hold promise for the future.
 | ||
| Scheme 17 The synthesis of Schiff base complexes derived from 2-aminophenyl benzimidazoles. | ||
Additionally, other Schiff bases were produced from 2-aminomethylbenzimidazole and naphthaldehyde, compound 361. The intended complexes were synthesized by reacting one equivalent of an ethanolic solution of the ligand (H2L) with one equivalent of a metal chloride salt (ZrCl4, FeCl3·6H2O, and CdCl2·6H2O). Based on in vitro antitumor activity testing of the complexes, the cadmium complex 362 showed significant cytotoxicity against MCF-7, Hep G2 and HCT 116 cell lines. The iron complex product 363 also presented strong cytotoxicity towards the Hep G2 and HCT cell lines, together with moderate activity against the MCF-7 cell line (Scheme S-74, ESI file†).206
2.12 Antioxidant compounds
For the design and synthesis of several antioxidant compounds, benzimidazole-templated molecules were chosen. Detrimental reactive oxygen and nitrogen species (ROS and RNS) and other free radicals, considered as the progenitors of physiological malfunction and biological disorders in living beings, have been effectively neutralized by large numbers of chemical entities, including both naturally sourced and synthetic products.207–210 Certain amines possessing N–H bonds have also functioned as antioxidant molecules,211,212 and, against this backdrop, benzimidazole products were designed and synthesized as antioxidant agents. In one synthetic approach, sodium bisulphite was reacted with 4-hydroxybenzaldehyde to yield hydroxy methane sodium sulfonate, which reacted under in situ conditions with 4-substituted o-phenylenediamines to yield the 2,4-disubstituted benzimidazole derivatives of compound 365. Compound 365 underwent the Mannich reaction at the ortho position of the phenolic group with formaldehyde and a tertiary amine derivative to reach the target compound based on template 366, though in low yields. The benzimidazole derivatives worked well as potent antioxidants, and the Mannich-benzimidazole derivatives' phenolic function was experimentally validated, showing them to be strong antioxidant agents. The results showed that most of these derivatives (Scheme 18) can be referred to as free radical scavengers and potent antioxidants.213 | ||
| Scheme 18 The synthesis of the Mannich-benzimidazole derivatives 366 possessing phenolic functionality, which were tested as antioxidant agents. | ||
Moreover, combining N-methyl-o-phenylenediamine or 2-amino-phenol with aromatic aldehydes and aromatic acids, in the presence of polyphosphoric acid (PPA) as an effective catalyst and solvent, under an efficient synthetic protocol allowed the formation of novel 2-substituted benzimidazole and benzoxazole derivatives; these were confirmed to have the structure 367, and were potential antimicrobial and antioxidant agents. When compared to conventional medications, the newly synthesized benzoxazole and benzimidazole derivatives exhibited good to excellent antibacterial and antioxidant properties (Scheme S-75, ESI file†).214
Another set of benzimidazole-component-based products was synthesized to act as strong antioxidants. The reaction of benzimidazole-based derivatives with thiazole analogues provided products 368, based on alkyl-thiourea and 2-bromo-1-(1-methyl-1H-benzoimidazol-2-yl)-ethanone, which exhibited exceptional antioxidant activity in comparison to the tested reference standard antioxidant molecule. In vitro antioxidant testing of the final products showed significantly stronger radical scavenging capacity than the widely recognized antioxidant standard BHA (Scheme S-76, ESI file†).215
In another recently reported study, antioxidant compounds based on 5,6-dimethyl-2-phenyl-1H-benzimidazole derivatives were successfully synthesized. The compound 5,6-dimethyl-2-phenyl-1H-benzoimidazole was reacted with 2-bromoethyl ethanoate, and the ester group was transformed into the corresponding hydrazide 369, whereupon the intermediate 369 was used as the starting material for various extended reactions aimed towards the targeted preparations of antioxidant compounds. Upon reacting with benzaldehyde, isothiocyanate, and carbon disulphide, the hydrazide compound afforded the hydrazone 370, thiosemicarbazide 371, and 2-mercapto-[1,3,4]-oxadiazole 372, respectively. The compounds 371 were hydrolysed under basic conditions to produce the [1,2,4]-triazole-3-thiol benzimidazole compounds 373; then, in an acidic medium, the reaction yielded [5-(5,6-dimethyl-2-phenyl-benzoimidazol-1-ylmethyl)-[1,3,4]-thiadiazol-2-yl] alkyl amine compounds 374. Most of the new compounds exhibited moderate levels of antioxidant activity. Higher levels of antioxidant activity were reported for the compounds possessing carbothioamide- and 1,2,4-triazole-derivative-based substructures (Scheme S-77, ESI file†).216 Meanwhile, another class of antioxidant compounds that included indole-tethered benzimidazole-based 1,2,3-triazoles was also developed. Reactions of propargyl bromide with 1H-indole-3-carbaldehyde, followed by condensation with o-phenylenediamine, yielded the corresponding 2-(1-prop-2-ynyl-1H-indol-3-yl)-1H-benzoimidazole product 375, which was again reacted with aromatic azides through a click chemistry reaction. It was catalysed by Cu(I) and led to the 2-[1-(1-aryl-1H-[1,2,3]-triazol-4-ylmethyl)-1H-indol-3-yl]-1H-benzoimidazole compound 376 and its derivatives. The obtained derivatives showed antioxidant radical scavenging potential, with IC50 values ranging from 8.50 to 10.05 μg mL−1. It was observed that the presence of 4-methyl, 2-methoxy, and 4-methoxy substituents on the phenyl ring resulted in strong antioxidant activity (Scheme S-78, ESI file†).217
Another series of compounds with strong antioxidant activity was synthesized incorporating the benzimidazole core as the principal pharmacophoric and molecular template. In the presence of the reducing reagent Na2S2O5, arylaldehydes were condensed with 3,4-diamino-benzene, benzoic acid, benzonitrile, and sulfonic acid to produce the corresponding 2-aryl-5-(nitrile, carboxylic acid, and sulfonic acid) benzimidazole products 377a–d. The three categories of aryl benzimidazole derivatives were evaluated for their antioxidant properties against various radicals using DPPH, FRAP, and ORAC assays, revealing varying levels of radical scavenging activity. The results indicated that the number and positions of hydroxy groups on the 2-aryl part, as well as the presence of a diethylamino or 2-styryl group, correlated well with the high antioxidant activity (Scheme S-79, ESI file†).218
Two newer series, 2-(aryl)-6-morpholin-4-yl-benzimidazole derivatives 378 and 4-methylpiperazin-1-yl-benzimidazole derivatives 379, were synthesized from 5-morpholin-4-yl- and 5-(4-methylpiperazin-1-yl)-2-nitroaniline using aryl aldehydes. The process was realized via a one-pot synthetic protocol through the reduction of the nitro group, followed by a cyclization step with sodium hydrosulphite as the reducing agent, adopting both conventional and microwave energy techniques for all the reactions carried out in this synthesis. The benzimidazole-based morpholines and piperazines possessed the potential to be used as a pharmaceutical source for antioxidant action, which was demonstrated by the reported results. A majority of the synthesized products showed high scavenging activity based on the CUPRAC, FRAP, DPPH, and ABTS methods of antioxidant activity testing. It was specifically the compounds containing morpholine and piperazine rings at the C-6 position of the benzimidazole ring that were the most active products (Scheme S-80, ESI file†).219
A new series of benzimidazoles, obtained using the aza-Michael reaction, in the form of 1,3-disubstituted benzimidazole-2-thiones was synthesized. Carbon disulphide was condensed with 4-substituted-o-phenylenediamine to produce the 5-substituted-1,3-dihydrobenzoimidazole-2-thione. Under reflux in DMF with methyl acrylate, the precursor afforded the corresponding bis(N-methyl propanoate) products 380a–e, which were further converted to their corresponding propanoic hydrazides 381a–d. The biological test results revealed that 1,3-disubstituted benzimidazole-2-thione was an effective oxidative stress inhibitor that was found to work for liver regenerative treatment (Scheme S-81, ESI file†).220
Pyrazolyl benzimidazole derivatives were also prepared in a two-step process. The synthesis of N-substituted pyrazolyl benzimidazoles was achieved through the condensation of o-phenylenediamine with 1-phenyl-3-(4-substituted-phenyl)-1H-pyrazole-4-carbaldehydes, in the presence of zinc chloride, which afforded the 2-[1,2-disubstituted-1H-pyrazol-4-yl]-1H-benzoimidazole structures 382a–h in very high yields, ranging from 85% to 92%. Later transformations of these compounds 382a–h resulted in the corresponding N-alkylated products 383a–l. These products were obtained upon reacting 382a–h with a variety of alkyl bromide derivatives, in high yields (81% to 99%). These products also demonstrated strong antioxidant activity (Scheme 19 and Table 1).221
 | ||
| Scheme 19 The synthesis of two series of new N-substituted pyrazole-containing benzimidazoles 382 and 383. | ||
| Compound number | Substituents | Yield (%) | Compound number | Substituents | Yield (%) | ||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R1 | R2 | ||||
| 382a | H | — | 91 | 383a | H | n-C5H11 | 89 |
| 382b | NO2 | — | 92 | 383b | H | –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
94 |
| 382c | –OCH3 | — | 89 | 383c | H | Bn | 98 |
| 382d | Cl | — | 85 | 383d | NO2 | n-C5H11 | 83 |
| 382e | OH | — | 90 | 383e | NO2 | –CHCH2 |
96 |
| 382f | NH2 | — | 92 | 383f | NO2 | Bn | 99 |
| 382g | CH3 | — | 87 | 383g | –OCH3 | n-C5H11 | 81 |
| 382h | Br | — | 85 | 383h | OCH3 | –CHCH2 |
85 |
| 383i | OCH3 | Bn | 90 | ||||
| 383j | Cl | n-C5H11 | 81 | ||||
| 383k | Cl | –CHCH2 |
87 | ||||
| 383l | Cl | Bn | 92 | ||||
Another set of 2-(4-substituted-phenyl)-5-methyl-1H-benzoimidazoles derivatives 384a–e was synthesized through the condensation of 4-methyl-o-phenylenediamine and 4-substituted benzaldehydes in the presence of sodium metabisulfite, with absolute ethanol as the solvent, at room temperature. The products were tested for their antioxidant activity employing the DPPH free radical scavenging assay. These products demonstrated substantial antioxidant action, with IC50 values of 1.054–19.05 μg mL−1, when compared with the reference standard BHT (26.96 μg mL−1) (Scheme S-82, ESI file†).222 These products are prospective candidates for further development as antioxidants, which are key progenitors for managing a number of physiological disorders, including cancers, diabetes and neurological malfunctions.
The synthesis of another target compound, bis-(N-allyl-1H-benzoimidazol-2-yl-methyl)-benzylamine, compound 386, was achieved. It involved, firstly, the preparation of benzyl dicarbamic acid via the condensation of benzylamine in the presence of excess 2-chloroacetic acid. Secondly, the dicarbamic acid was reacted with o-phenylenediamine, leading to the bis(N-methyl-2-benzimidazolyl)-benzyl amine intermediate 385. This compound 385 subsequently underwent double N-allylation with allyl bromide. The obtained product 386 was used as a ligand to produce Ni(II) complexes, which showed significant antioxidant activity compared with known natural antioxidants, i.e., mannitol and vitamin C (Scheme S-83, ESI file†).223
Yet, another set of new benzimidazole carboxamide derivatives was also synthesized as antioxidant agents. In the presence of acetic acid, 4-(bromomethyl)-biphenyl-2-carboxylic acid, compound 387, was reacted with tetraethyl orthocarbonate to produce the 1-[(2′-cyanobiphenyl-4-yl)-methyl]-5-carboxylate-2-ethoxybenzimidazole-derivative intermediate 388. The hydrolysis of compound 388 in sodium hydroxide solution yielded compound 389. The target 390 was effectively produced by reacting different substituted benzylamines with derivatives of compound 389 in the presence of TBTU and DPIA. The reagent 1,1-diphenyl-2-picrylhydrazyl (DPPH) was used to investigate the free radical scavenging capacity of the synthesized compounds 390a–j in comparison with ascorbic acid as a standard antioxidant substrate. These results showed that five compounds, i.e., 390a, 390c, 390d, 390f and 390i, had higher antioxidant activity (Scheme S-84, ESI file†).224
Two new metal-complexed compounds, the 2-(4′-thiazolyl)benzimidazole-based Cu(II)-dipeptide complexes [Cu(Gly–Gly)(TBZCl)]·4H2O] 391 and [Cu(Gly-L-Leu)(TBZCl) H2O] 392, were synthesized (Scheme S-85, ESI file†). The dipeptide glycylglycine reacted, in the presence of sodium hydroxide, with 2-thiazol-2-yl-1H-benzoimidazole and copper(II) chloride, yielding the tetrahydrate and monohydrate complexes 391 and 392. In these structures, copper metal chelated with the oxygen atom of the carboxylate and the nitrogen atom of the primary amine, and it bonded with the two nitrogen-atom-based imine (NC) moieties of the 2-thiazol-2-yl-1H-benzoimidazole compounds. The study mentioned the excellent antioxidant capabilities of these final products.225
3. Structure–activity relationships in benzimidazole derivatives
A survey of benzimidazole-based compounds, lead structures, and new chemical entities reflects the versatile nature of benzimidazole and its broadly diverse derivatives as essential pharmacophores in numerous biologically active heterocyclic compounds, with a diverse range of pharmacological activities. The NH group in benzimidazole exhibits the characteristics of being both significantly acidic and mildly basic in nature.226,227 The compound possessed the capacity to allow salt preparation. The notion that the benzimidazole moiety holds significant potential in the pharmaceutical domain for the advancement of innovative medicinal compounds with a range of pharmacological activities not only illustrates its significance as a near-universal and highly versatile pharmacophore but also poses challenges in terms of choice with regards to the best structural selections to complement the benzimidazole pharmacophore to obtain the intended bioactivity.The structure–activity relationships (SARs) of some of the most potent benzimidazole compounds in terms of anticancer activity are presented. Even though there are many potential causes of cancer, the two most prevalent ones are thought to be aberrant enzyme activity and genetic abnormalities. There are approximately 277 different types of cancers that have been identified, and lung cancer is the most common among men and the leading cause of cancer-related deaths. Colorectal and prostate cancers are the next most prevalent cancers in terms of incidence, while hepatocellular and stomach cancers are leading causes of mortality. Compared to males, females are more likely to die from breast, colorectal, lung and cervical cancers.228 According to Satija et al.,229 a number of potential targets exist for anticancer therapy, primarily involving topoisomerase-II, which mediates DNA cleavage and cell death; serine/threonine kinase inhibitors, which induce cell arrest; tyrosine kinase inhibitors, which prevent angiogenesis; tubulin polymerization inhibitors, which are known to produce mitotic arrest; COX inhibitors, which prevent tumor invasion; and PDGFs, which mediate apoptosis and proliferation.
Compounds with piperazine linked to benzimidazole–pyrimidine hybrids were prepared and tested for cytotoxicity by Sana et al.,230 and at concentrations ranging from 2.21 to 7.29 μM, the amine-linked benzimidazole–pyrimidine demonstrated the highest levels of cytotoxic activity against A549 human lung cancer cell lines. Compound 393 showed promising activity as an anticancer agent against A549 cell lines. Based on the results of structure–activity relationship (SAR) tests, products with an amine linkage showed superior anticancer activity. The cytotoxicity against A549 cell lines was increased by sterically hindering the trifluoromethyl substituent at the C2-position of the benzimidazole ring (Fig. 1).
 | ||
| Fig. 1 Compound 393, which is an anticancer product with activity against A549 cell lines. | ||
The 1,2-diarylbenzimidazoles 394 showed anticancer activity, with an IC50 value of 8.47 μM and GI50 values ranging from 0.71 to 2.41 μM. Testing with HepG2 and HeLa cells confirmed that apoptosis halted tumor cells in the G2/M phase, and molecular docking studies confirmed the binding capacity to tubulin crystals. The para-substitution of the compounds improved their bioactivity compared to ortho- or meta-substitution. The presence of 3,4-(OCH3)2 resulted in the highest activity compared with other ortho-para di-substitutions, but the activity of meta–para di-substituted compounds was found to be much higher231 (Fig. 2).
 | ||
| Fig. 2 Enhanced in vitro activity against HeLa and Hepg2 cell lines was shown owing to meta–para-disubstitutions. | ||
A new set of CHK-2 enzyme inhibitors 395, obtained by substituting pyrazole for an aryl moiety in 2-aryl-1H-benzimidazoles, was synthesized. Products showed anticancer efficacy, with a range of IC50 values from 52.8 μM to 5.5 μM against the MCF-7 cell line. The anticancer activity was improved with substitution at the pyrazole ring by a polar group at position 4, while carboxylic or nitro groups at position 5 of the 1H-benzimidazole decreased the activity (Fig. 3).232
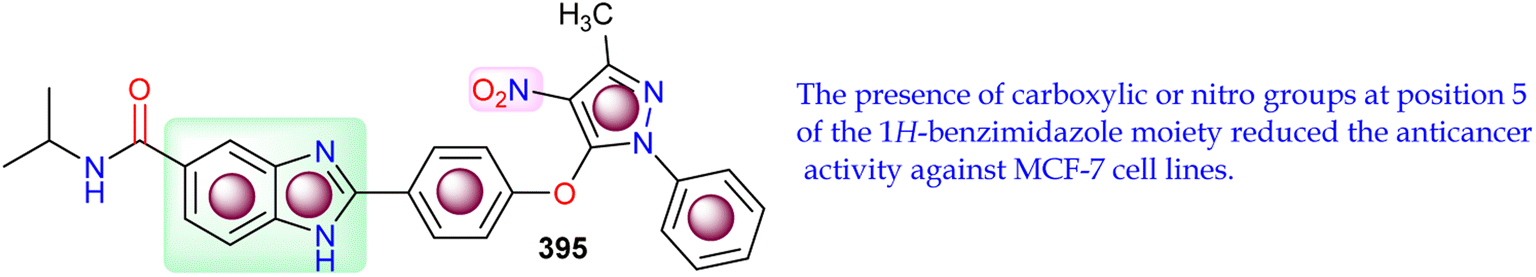 | ||
| Fig. 3 The effects of the benzimidazole 5-position on the anticancer activity. | ||
Novel 1H-benzimidazole-linked β-carbolines 396 were synthesized by Sireesha et al. The compounds tested positive for anticancer activity against several cancer cell lines, including MCF-7 cell lines. The compounds were CLK binders with molecular interactions with kinase. A 5,6-dimethoxy substitution was found to introduce more activity than a molecule without substitution at the 1H-benzimidazole core. The presence of a weak electron-donating group (EDG) and the presence of a dimethyl group on the 1H-benzimidazole entity decreased the activity (Fig. 4).233
 | ||
| Fig. 4 Substitution at positions 5 and 6 favored higher anticancer activity. | ||
Diabetes mellitus resulting from metabolic abnormalities is characterized by elevated blood glucose levels, known as hyperglycemia. Severely diabetic individuals experience a variety of symptoms, including weight loss, refractory infections, recurrent vomiting, dermatological and ocular complications, as well as drug-resistant nausea.234,235 α-Amylase and α-glucosidase are involved in the digestion of carbohydrates. Concerning the antidiabetic structure–activity relationships of benzimidazole derivatives, novel thiazole–benzimidazole 397 analogues have been reported236 as multipotent inhibitors of α-amylase and α-glucosidase. Recently discovered analogs also exhibited inhibition potential against α-amylase and α-glucosidase. SAR studies indicated that the inhibition efficiency is increased by smaller (F and Cl) groups or the presence of groups capable of forming hydrogen bonds (OH) with targeted enzymes. The potency of analogs with fluoro-substitution at the meta-position and homologs with a para-fluoro-substitution towards α-amylase was high, whereas analogs with large substituents, such as Br, or groups that do not form hydrogen bonds, like CH3, showed poor activity (Fig. 5).
 | ||
| Fig. 5 Antidiabetic SAR considerations. | ||
The effectiveness of benzimidazole scaffolds in inhibiting α-amylase and α-glycosidase was evaluated relative to acarbose. Designed products with halogen groups at the para-position of the phenyl part hindered enzyme activity via direct interaction. Molecular docking experiments showed binding affinity with the HPA and HLAG active sites. Aroua et al. proposed a facile method for synthesizing diverse benzoyl-aryl-benzimidazoles 398. This entailed the condensation of 3,4-diaminobenzophenone with an appropriately substituted aryl aldehyde utilizing derivatives, using ammonium chloride or a combination of ammonium chloride and sodium bisulfite as the catalyst (Fig. 6).183
 | ||
| Fig. 6 The SAR for para-substituted halogens on the phenylic part of the benzimidazole. | ||
Benzimidazole derivatives of N-substituted benzimidazoles 399 prevented the growth of methicillin-resistant Staphylococcus aureus MRSA (ATCC4330) under in vitro conditions, presenting MICs of up to 4 μg mL−1, which were better than sultamicillin (MIC = 25 μg mL−1). Also, these compounds exhibited no toxicity toward mammalian Vero cell lines, demonstrating their safe nature as antimicrobials, with IC50 values of 298 μM. The presence of a strong electron-withdrawing group, e.g., nitro, at the 3-position of the phenyl ring at the N-position of the benzimidazole was observed to facilitate efficacy against MRSA (Fig. 7).237
 | ||
| Fig. 7 N-Substitution as a facilitator of the antimicrobial activity of benzimidazole. | ||
Benzimidazole molecules 400 having a short chain alkyl group at the terminal end with mildly hydrophobic character enhanced the antimicrobial activity (Fig. 8).238
 | ||
| Fig. 8 Chain-length effects on bioactivity from SAR studies of benzimidazole derivatives. | ||
A small number of 2-phenylsubstituted benzimidazoles 401 were investigated for their ability to inhibit COX-1 and -2, and 5-lipoxygenase. Fig. 9 showed that the inhibition of COX-1 and -2, and 5-lipoxygenase was best achieved by products with no substitutions at the R2, R3, and R4 positions, while the presence of an amine group at R1 improved the inhibition of all three enzymes. Nonetheless, the inhibition of COX-1 was favored by the presence of a lipophilic group at R5, the inhibition of COX-2 was enhanced by the presence of a hydrophilic group, and the inhibition of 5-lipoxygenase was favored by methoxy substitution. Molecules with 2-aminopyridin-4-yl in the product structures enhanced the inhibition of 5-lipoxygenase (Fig. 9).239
 | ||
| Fig. 9 The SAR of 2-phenyl-substitued benzimidazoles. | ||
4. Recent advances in synthesis and green approaches for benzimidazoles
The search for innovative synthetic procedures for the preparation of prospective lead structures has emerged as a prominent field, with the pursuit of effective and practical synthetic methods for benzimidazole synthesis in continuous focus.240 Concurrently, researchers have sought to develop novel synthetic methodologies,241 with a strong emphasis on green and eco-friendly methodologies.242,243 These green advancements have influenced reaction conditions, reagents, solvents, and a range of other parameters, including the utilization of neat,244 green,240 and ionic solvents;245 the use of material-conserving, cheap, eco-friendly,246 and recyclable solvents and catalysts;247 the incorporation of ionic liquids;248,249 the use of solid supports;250,251 combinatorial and parallel synthetic approaches; and the exploration of energy-efficient protocols, such as the use of microwave irradiation,252 mechanochemical methods,245 and ultrasound energy.240 Shaikh et al.245 reported a novel green synthetic method for the synthesis of benzimidazole derivatives 402 utilizing a catalytic quantity of 1-ethyl-3-methylimidazolium tetrachloroaluminate ([EMIM]AlCl4), the ionic liquid, as a catalyst, ethanol as an eco-friendly solvent, and mechanochemical energy (grinding in a mortar and pestle) at room temperature (Fig. 10). | ||
| Fig. 10 The [EMIM]AlCl4-catalysed synthesis of benzimidazole derivatives. | ||
In addition to its simplicity and the ease of catalyst recovery, the reaction provided good yields in short times. The utilization of catalysts has gained significant attention. The application of Lewis acids as effective catalysts in several transformations has demonstrated more eco-friendly methods for the synthesis of benzimidazole derivatives. In their work on the green synthesis of benzimidazole derivatives, Shaibuna et al.242 employed deep eutectic solvents (DES) as green solvents. The DES consisted of a combination of ZrOCl2·8H2O and urea. After optimization, they determined the optimal ratio of ZrOCl2·8H2O to urea in the DES to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (DES 1). To validate this method, the reaction was performed using 4-chloro-1,2-phenylenediamine and 3,4-diaminotoluene with variously substituted aldehydes (Fig. 11). Notably, the reaction exhibited total selectivity, yielding exclusively mono- or di-substituted benzimidazole derivatives 403 and 404. The di-substituted derivatives were obtained only when thionyl chloride was used as the aldehyde source (Table 2).
:5 (DES 1). To validate this method, the reaction was performed using 4-chloro-1,2-phenylenediamine and 3,4-diaminotoluene with variously substituted aldehydes (Fig. 11). Notably, the reaction exhibited total selectivity, yielding exclusively mono- or di-substituted benzimidazole derivatives 403 and 404. The di-substituted derivatives were obtained only when thionyl chloride was used as the aldehyde source (Table 2).
 | ||
| Fig. 11 The eco-friendly synthesis of benzimidazole derivatives. | ||
| Entry | R1 | R2 | t (min) | Yield (%) | |
|---|---|---|---|---|---|
| 403 | 404 | ||||
| 1 | H | C6H5 | 10 | 97 | 0 |
| 2 | 4-CH3O-C6H4 | 15 | 97 | 0 | |
| 3 | 1-Thienyl | 10 | 0 | 47 | |
| 4 | 3,4-(CH3O)2-C6H3 | 25 | 94 | 0 | |
| 5 | 1-Naphtyl | 20 | 95 | 0 | |
| 6 | 4-Cl-C6H4 | 20 | 90 | 0 | |
| 7 | 4-Br-C6H4 | 25 | 92 | 0 | |
| 8 | 3-NO2-C6H4 | 40 | 85 | 0 | |
| 9 | 4-NO2-C6H4 | 35 | 90 | 0 | |
| 10 | 4-Cl | 1-Thienyl | 15 | 0 | 45 |
| 11 | 1-Naphtyl | 25 | 84 | 0 | |
| 12 | 4-CH3 | 4-CH3-O-C6H4 | 15 | 98 | 0 |
| 13 | 1-Thienyl | 10 | 0 | 49 | |
| 14 | 4-Br-C6H4 | 25 | 89 | 0 | |
Srinivasulu et al.253 achieved the synthesis of benzimidazole derivatives 405 under solvent-free conditions, employing a catalytic amount of the commercially available, inexpensive, and eco-friendly catalyst zinc acetate at room temperature (Fig. 12). The reaction proceeded with high selectivity and excellent yields.
 | ||
| Fig. 12 The synthesis of benzimidazole using an efficient green Zn(OAc)2 catalyst. | ||
Zhang et al.254 achieved the synthesis of a set of benzimidazole derivatives 406 utilizing PhSiH3 and CO2, a greenhouse gas, as reagents. The reaction was carried out under convenient conditions using a B(C6F5)3 catalyst, affording yields of up to 95% (Fig. 13).
 | ||
| Fig. 13 The synthesis of benzimidazole derivatives obtained upon reacting CO2, an atmospheric pollutant. | ||
Interestingly, nearly all of the low molecular weight (LMW), <500 amu, drug molecules contained at least 59% of nitrogen heterocycles in one form or another as part of their structural template. The benzimidazoles, owing to the availability of 1 to 5 substitution site positions, are second only to indoles in the drug design, discovery, and bioactivity fields; a diverse range of bioactivities and structural variations are available, and they show ease of synthesis, as multiple routes and starting materials are available to produce benzimidazole derivatives and structural variations. Moreover, due to the versatility of the benzimidazoles as potent pharmacophores for various medicinal domains, benzimidazole derivatives have been widely reported to exhibit diverse biological and pharmacological activities.255 One of the reasons for the widespread interest in their use to develop newer drugs is their propensity for relatively straightforward preparation with numerous synthetic routes available. The benzimidazole core also exhibits sufficient stability to undergo multiple successive reactions, enabling molecular modifications to achieve the desired target structure without ring cleavage. These properties make them a valuable scaffold for the development of a wide range of compounds with applications in numerous medicinal fields.
A recent search of the Scopus database for derivatives of benzo-fused five-membered heterocycles yielded almost 135000 (134662) publications. The results showed that indole is the most frequently reported moiety, accounting for 69.41% of publications (94858). Benzimidazole follows this, with 20.84% (28897 publications). Other moieties, such as benzothiazole (7.54%), benzotriazole (3.18%), benzoxazole, and benzopyrazole, were less frequently cited, each representing only 0.01% of publications (Chart 1); this confirmed the versatility, interest in, and usability of these categories of compounds and their vital roles in chemical development and from a pharmacological activity standpoint.
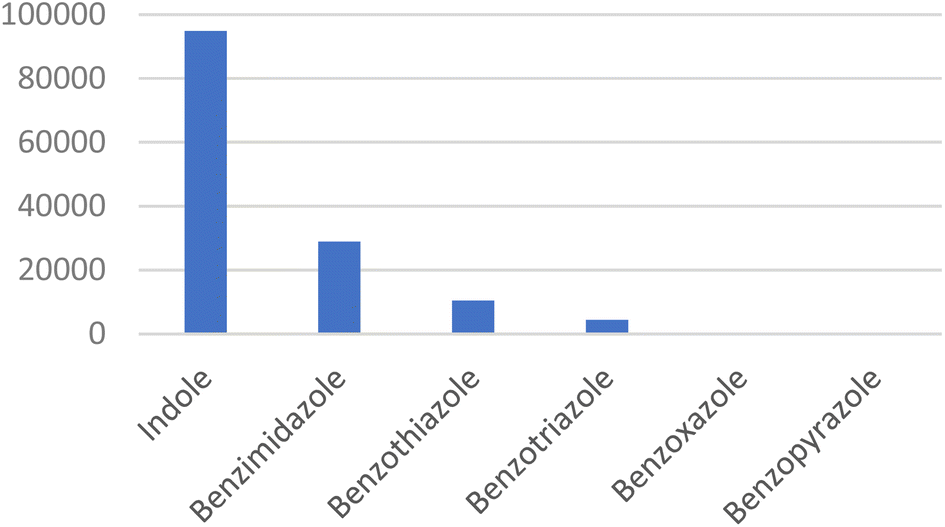 | ||
| Chart 1 The number of publications on benzo-fused five-membered heterocyclic derivatives, according to the Scopus database. | ||
5. Limitations, challenges, and shortcomings related to the benzimidazole structure, preparation techniques, and toxicity
Benzimidazole derivatives have exhibited diverse biological activities, but their development has been accompanied by many constraints. Their pharmacological utility is diminished due to the toxicity of some benzimidazole derivatives. Certain benzimidazoles and their metabolic byproducts cause liver and DNA damage, adverse reactions, and side effects.256 The resistance of parasitic nematodes to benzimidazole anthelmintics is well-documented and typically caused by mutations in the α-tubulin gene, and the situation is similar for certain other benzimidazole-based antiparasitic drugs.257 Also, during synthesis, the use of harsh conditions and toxic raw materials, catalysts and solvents plays a part in eliciting toxicity.258 Additionally, solubility issues connected with certain benzimidazoles have limited their bioavailability and, in turn, their therapeutic efficacy.259 Nonetheless, certain benzimidazoles are rapidly metabolized under in vivo conditions, leading to shorter half-lives and reduced therapeutic efficiency. This necessitates structural optimization during the development of benzimidazole-based drug candidates and related pro-drugs.2606. Benzimidazole-based drugs and drug candidates under clinical trials
The benzimidazole core has gained importance as a pharmacophore and as part of extended pharmacophores in drug discovery exercises owing to its inherent wide-ranging pharmacological properties.261 The rational synthesis of benzimidazole derivatives has become a focal point in medicinal chemistry, enabling the development of novel therapeutics with improved efficiency and reduced side effects and toxicity.262Clinical uses of benzimidazoles include in the management of gastrointestinal disorders, such as gastric ulcers, and as proton pump inhibitors. The role of omeprazole and lansoprazole is one such example. Combating parasitic infections with albendazole and mebendazole and treating nausea and vomiting with droperidol are some common clinical applications now undertaken. Benzimidazole derivatives, such as astemizole and pimozide, are also available as antihistamine and neuroleptic drugs, respectively. The wide range of clinical and therapeutic uses of benzimidazole has shown how important this group of synthetic chemical entities is, in terms of both pharmacodynamics and pharmacokinetics. This has also driven researchers and pharmaceutical companies to design new benzimidazole templates as drug leads, with the aim of developing new drugs and enhancing the safety profile and bioavailability of existing benzimidazoles. Fig. 14 shows known and marketed benzimidazole drugs.
 | ||
| Fig. 14 Marketed benzimidazole drugs. | ||
Table 3 provides a list of the trade and chemical names of benzimidazole-based drugs, their clinical applications as a pharmacological class, and their principal mode of action; all of which are available on the market.
| Benzimidazole drug | Chemical name | Clinical applications | Mechanism of action |
|---|---|---|---|
| Albendazole | Methyl-N-(6-propylsulfanyl-1H-benzimidazol-2-yl)carbamate | Antihelmintic | Interferes with tubulin polymerization |
| Mebendazole | Methyl-N-(6-benzoyl-1H-benzimidazol-2-yl)carbamate | Antihelmintic | Inhibits microtubule production in parasite cells |
| Thiabendazole | 4-(1H-Benzimidazol-2-yl)-1,3-thiazole | Antihelmintic | Helminth-specific enzyme fumarate reductase inhibition |
| Omeprazole | 6-Methoxy-2-[(R)-(4-methoxy-3,5-dimethylpyridin-2-yl)methylsulfinyl]-1H-benzimidazole | Proton-pump inhibitor for gastric ulcers | Inhibits the parietal cell H+/K+ adenosine triphosphate pump |
| Lansoprazole | 2-[[3-Methyl-4-(2,2,2-trifluoroethoxy) pyridin-2-yl]methylsulfinyl]-1H-benzimidazole | Proton-pump inhibitor for gastric ulcers | Inhibits the parietal cell H+/K+ adenosine triphosphate pump |
| Rabeprazole | 2-[[4-(3-Methoxypropoxy)-3-methyl pyridin-2-yl]methylsulfinyl]-1H-benzimidazole | Proton-pump inhibitor for gastric ulcers | Inhibits the parietal cell H+/K+ adenosine triphosphate pump |
| Etodesnitazene | 2-[2-[(4-Ethoxy phenyl) methyl] benzimidazol-1-yl]-N,N-diethylethanamine | Analgesic | Synthetic opioid |
| Metodesnitazene | N,N-Diethyl-2-[(4-methoxyphenyl) methyl]-1H-benzimidazole-1-ethanamine | Analgesic | Synthetic opioid |
| Etodesnitazene | 2-[(4-Ethoxyphenyl)methyl]-N,N-diethyl-1H-benzimidazole-1-ethanamine | Analgesic | Synthetic opioid |
| Bendamustine | 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid | Chemotherapy of chronic lymphocytic leukemia | Alkylating agent |
| Nocodazole | Methyl-N-[6-(thiophene-2-carbonyl)-1H-benzimidazol-2-yl]carbamate | Chemotherapy | Inhibits the self-assembly of tubulin |
| Dovitinib | 4-Amino-5-fluoro-3-[5-(4-methyl-1-piperazinyl)-1,3-dihydrobenzimidazol-2-ylidene]-2-quinolinone | Anticancer | Multi-target FGFR kinase inhibitor |
| Binimetinib | 6-(4-Bromo-2-fluoroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide | Anticancer | Mitogen-activated protein kinase (MEK) inhibitor |
| Selumetinib | 6-(4-Bromo-2-chloroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide | Anticancer | MEK1 and MEK2 inhibitor |
| Veliparib | 2-[(2R)-2-Methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide | Anticancer | PARP inhibitor preventing DNA repair in cancer cells |
| Pracinostat | (E)-3-[2-Butyl-1-[2-(diethylamino) ethyl]benzimidazol-5-yl]-N-hydroxyprop-2-enamide | Anticancer | Histone deacetylase (HDAC) inhibitor |
| Galeterone | (3S,8R,9S,10R,13S,14S)-17-(Benzimidazol-1-yl)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydro-1H-cyclopenta[a]phenanthren-3-ol | Anticancer | Androgen receptor modulator and CYP17 lyase inhibitor |
Also, at the same time, several benzimidazole-derived drug candidates are approaching the final stages of clinical trial and licensing, demonstrating the ongoing interest in this pharmacophore. Researchers are studying benzimidazoles for their potential efficacy in cancer treatment and as antiviral agents. Preliminary research has shown encouraging results, indicating the compound's capacity to suppress tumor proliferation and augment the effectiveness of currently available cancer chemotherapeutics. A list of benzimidazole-based compounds involved in clinical trials is provided in Table 4.
| Drug candidate | Chemical name | Clinical trial ID | Clinical trial area | Clinical application |
|---|---|---|---|---|
| Nazartinib | N-[7-Chloro-1-[(3R)-1-[(E)-4-(dimethylamino)but-2-enoyl]azepan-3-yl]benzimidazol-2-yl]-2-methylpyridine-4-carboxamide | NCT03529084 | Third-generation, mutant-selective epidermal growth factor receptor (EGFR) inhibitor | Anticancer |
| Nazartinib | N-[7-Chloro-1-[(3R)-1-[(E)-4-(dimethylamino)but-2-enoyl]azepan-3-yl]benzimidazol-2-yl]-2-methylpyridine-4-carboxamide | NCT02335944 | The combination of capmatinib and nazartinib for patients with EGFR-mutated non-small-cell lung cancer | Anticancer |
| Nazartinib | N-[7-Chloro-1-[(3R)-1-[(E)-4-(dimethylamino)but-2-enoyl]azepan-3-yl]benzimidazol-2-yl]-2-methylpyridine-4-carboxamide | NCT02108964 | Safety and effectiveness of nazartinib (EGF816) in people with EGFR-mutant non-small-cell lung cancer | Anticancer |
| Binimetinib | 6-(4-Bromo-2-fluoroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide | NCT04965818 | Binimetinib tested in combination with futibatinib in patients with advanced KRASmt tumors | Anticancer |
| Binimetinib | 6-(4-Bromo-2-fluoroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide | NCT03170206 (https://clinicaltrials.gov/show/NCT03170206) | Combination of palbociclib and binimetinib for patients with advanced KRAS mutant non-small-cell lung cancer | Anticancer |
| Bendamustine | 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid | NCT04217317 | Bendamustine tested in combination with CPI-613 in patients with relapsed/refractory T-cell non-Hodgkin lymphoma | Anticancer |
| Bendamustine | 4-[5-[Bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid | NCT04510636 | Study of pembrolizumab with bendamustine against Hodgkin lymphoma | Anticancer |
| Selumetinib | 6-(4-Bromo-2-chloroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide | NCT02768766 | Combination of selumetinib and dacarbazine for patients with metastatic uveal melanomas | Anticancer |
| Abemaciclib | N-[5-[(4-Ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine | NCT04003896 (https://clinicaltrials.gov/show/NCT04003896) | A trial to assess abemaciclib in late biliary tract carcinomas that failed prior first-line therapy | Anticancer |
| Abemaciclib | N-[5-[(4-Ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine | NCT04040205 | A trial to assess abemaciclib activity in the treatment of bone and soft-tissue sarcomas with cyclin-dependent kinase (CDK) pathway alteration | Anticancer |
| Veliparib | 2-[(2R)-2-Methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide | NCT02723864 | A combination of veliparib and cisplatin in patients with refractory solid tumors | Anticancer |
| Veliparib | 2-[(2R)-2-Methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide | NCT01434316 | The combination of veliparib and dinaciclib in treating patients with advanced solid tumors | Anticancer |
| Dovitinib | 4-Amino-5-fluoro-3-[5-(4-methyl-1-piperazinyl)-1,3-dihydrobenzimidazol-2-ylidene]-2-quinolinone | NCT01635907 | A trial conducted to assess the effect of dovitinib in managing cancer in people with certain kinds of neuroendocrine tumors. This research will also assess the safety of this medicine | Anticancer |
| Pracinostat | (E)-3-[2-Butyl-1-[2-(diethylamino)ethyl]benzimidazol-5-yl]-N-hydroxyprop-2-enamide | NCT03848754 | The combination of pracinostat and gemtuzumab ozogamicin (PraGO) in patients with relapsed/refractory acute myeloid leukemia | Anticancer |
| Galeterone | (3S,8R,9S,10R,13S,14S)-17-(Benzimidazol-1-yl)-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15-decahydro-1H-cyclopenta[a]phenanthren-3-ol | NCT04098081 | A combination of galeterone with gemcitabine for patients with metastatic pancreatic adenocarcinomas | Anticancer |
7. Specific areas of research or application that can be the most impactful for benzimidazole-based compounds in the next decade
The versatility of benzimidazole-based compounds has made them a hot topic of research in many different industries. Over the past few years, benzimidazole pharmacophores have found various applications in the pharmaceutical industry, including antibacterial,263–265 antifungal,266,267 antitubercular,268,269 anti-inflammatory,270 antidiabetic,271,272 and anticancer273–275 uses. Research indicates that benzimidazole materials are commonly used as insecticides and fungicides in agricultural settings.276,277 Polymer science278,279 and corrosion inhibition280,281 are two areas where the benzimidazole core has been shown to be effective in materials research.Furthermore, environmental science represents a significant area of application. In this context, several research studies have been conducted, including the prediction of potential risks associated with ten azole and benzimidazole fungicides in relation to their aryl hydrocarbon receptor agonistic activity in aquatic ecosystems; the development of two coordination polymers based on rigid benzimidazole carboxylic acid ligands, focusing on electrode performance and dye adsorption;282 the preparation of molecular-scale hybrid membranes utilizing benzimidazole-based monomers for high-performance hydrogen purification;283 the exploration of benzil-imidazole blue fluorophores and their application in blue/white light-emitting diodes,284 sensing, and anticounterfeiting; and the investigation of tetra-benzimidazoles flanking divinyl-phenothiazine as AIEgens acting as aza-Michael acceptors in concentration-tuned responses to biogenic amine vapors.285
A range of environmentally friendly synthesis methods for benzimidazoles includes the use of a dual-chain metallo-micellar catalyst for aerobic oxidative synthesis in water;286 a sustainable approach utilizing reusable CaAl2O4 nanophosphors as a catalyst for benzimidazole-based Schiff base synthesis, with a focus on metal(II) complexes and DNA interactions;287 an effective microwave-assisted copper-catalyzed aerobic oxidation strategy for quinazolinone and benzimidazole synthesis;288 and the green synthesis of benzimidazole scaffolds employing copper-substituted zinc aluminate via a sol–gel process.289 In addition, benzimidazole materials were utilized in battery technology to improve the safety and performance of lithium metal batteries; a thermally stable poly(aryl ether benzimidazole) separator with 2D-functionalized boron nitride was used for 3000 hours of lithium plating/stripping290 and multi-channeled halloysite nanotube-blended polybenzimidazole separators were used for enhancing lithium-ion battery performance.291 A benzimidazole-linked polymer has also been identified for its application potential in membranes designed for efficient syngas (H2/CO/CO2) separation.292
8. Patent landscape
The patent landscape relating to benzimidazoles is diverse. Benzimidazoles and their derivatives have been patented for a range of uses and applications in diverse industries, including the pharmaceutical industry. A recent search revealed >100000 entries in the Google Patents database; for the European patent office, an Espacenet search of benzimidazole-specific patents returned 10000 entries [Espacenet – results view (https://worldwide.espacenet.com/searchResults?submitted=true%26locale=en_EP%26DB=EPODOC%26ST=singleline%26query=benzimidazole)]; the Japanese patent office showed 71 patents [Simple Search|J-PlatPat [JPP] (https://www.j-platpat.inpit.go.jp/s0100)]; the World Intellectual Property Organization (WIPO) through Patentscope® provided 1805 entries [WIPO – Search International and National Patent Collections (https://patentscope.wipo.int/search/en/result.jsf?_vid=P21-M6O7U9-54819)]; a United States Patents Office site search produced >95000 entries [Patent Public Search Basic|USPTO (https://ppubs.uspto.gov/pubwebapp/static/pages/ppubsbasic.html)]; and Canadian patent entries related to benzimidazole were found to reach around 20000 [Search Results – Canadian Patents Database (https://brevets-patents.ic.gc.ca/opic-cipo/cpd/eng/search/results.html?query=benzimidazole%26start=1%26num=10%26type=basic_search%26newSearch=0)]. The patent activity peaked between 2001 and 2013, with a majority of patents being filed during this period. Benzimidazole bioactivity patents peaked during the period 2016–2019 [bioactivity benzimidazole – Google Patents (https://patents.google.com/?q=(bioactivity+benzimidazole)%26oq=bioactivity+benzimidazole)]. The Takeda, Merck, Pfizer, and Janssen pharmaceutical and chemical companies are worth mentioning. A review compilation of benzimidazole patents in the periods 2010–2012,293 2013–2014,294 and 2021 (ref. 295) demonstrates over 10 years of progress in the area of benzimidazole patents. Compilations of marketed drugs and drugs under development, which are part of clinical trial registries, are provided above.
9. Conclusions
Based on a benzimidazole molecular template, a plethora of structurally diverse compounds has been derived through various functional-group and ring-system substitutions, together with substructure entanglements through the use of a vast spectrum of different synthons, starting materials, and intermediates; in addition, new and novel synthetic approaches have yielded compounds and significant leads that have exhibited a range of different kinds of pharmacological activities. Several categories of biologically promising compounds with different activities, including antimicrobial, antiviral, antiparasitic, anticancer, anti-inflammatory, antioxidant, antihypertensive, and antidiabetic activity, have been overviewed and described in detail with their respective synthetic schemes, highlighting the synthetic protocols and how benzimidazole is the core pharmacophore responsible for the activity. Different structures that were developed based on a benzimidazole core have shown potential for further research to develop bioactive compounds in the form of new chemical entities, lead compounds and drugs, strengthening drug discovery. The benzimidazole molecular template, as a core part of active organo-medicinal products, possesses potential for the expedited development of new drug candidates with various types of bioactivity and pharmacological action. The various structural derivatives and benzimidazole-templated analogues have successfully opened new vistas, utilizing SAR- and QSAR-based drug discovery, with benzimidazole as a central molecular component. The vast number of structures and derivatives produced through benzimidazole intermediaries that are currently available also demonstrates the potential to further explore 3D-QSAR-based approaches in minute detail for intricate drug design and consequential drug discovery; this can be carried out in conjunction with activity prediction, structural mapping, and toxicity studies, using in vivo, in vitro, and in silico approaches. The current status of structural diversification and the range of notable bioactivities have also provided an opening for understanding the mechanisms of action of benzimidazole-derived drugs, both developed and in development, helping to understand the metabolomics and toxicokinetics, to suppress and remove the inherent toxicity, side effects, and biological cross-reactivity in developed products that are harming future drug development plans. The profound versatility of pharmacological activities, the structural diversity, and the utility of the retained benzimidazole pharmacophore are all beneficial; in addition, the biological potential of the would-be and already synthesized pools of diversely substituted benzimidazole structures, which may be produced through combinatorial and parallel synthesis, as well as their bioactivities, which have been confirmed through high-throughput screenings, offer new avenues and unexplored domains for chemical synthesis, drug design and development, and the introduction of new pharmacological activities while working with the benzimidazole template.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Researchers would like to thank the Deanship of Graduate Studies and Scientific Research at Qassim University for financial support (QU-APC-2025).References
- O. Ebenezer, F. Oyetunde-Joshua, O. D. Omotoso and M. Shapi, Results Chem., 2023, 5, 100925 CrossRef CAS.
- N. D. Mahurkar, N. D. Gawhale, M. N. Lokhande, S. J. Uke and M. M. Kodape, Results Chem., 2023, 101139 CAS.
- S. I. Alaqeel, J. Saudi Chem. Soc., 2017, 21, 229–237 CAS.
- M. Shaharyar and A. Mazumder, Arabian J. Chem., 2017, 10, S157–S173 Search PubMed.
- V. V. Fedotov, V. L. Rusinov, E. N. Ulomsky, E. M. Mukhin, E. B. Gorbunov and O. N. Chupakhin, Chem. Heterocycl. Compd., 2021, 57, 383–409 Search PubMed.
- E. M. Dokla, N. S. Abutaleb, S. N. Milik, D. Li, K. El-Baz, M.-A. W. Shalaby, R. Al-Karaki, M. Nasr, C. D. Klein and K. A. Abouzid, Eur. J. Med. Chem., 2020, 186, 111850 CAS.
- V. R. Mishra, C. W. Ghanavatkar, S. N. Mali, S. I. Qureshi, H. K. Chaudhari and N. Sekar, Comput. Biol. Chem., 2019, 78, 330–337 CAS.
- S. Gatadi, Y. Madhavi, S. Chopra and S. Nanduri, Bioorg. Chem., 2019, 92, 103252 CAS.
- A. P. Tayade and R. P. Pawar, Polycyclic Aromat. Compd., 2022, 42, 1474–1478 CAS.
- D. Song and S. Ma, ChemMedChem, 2016, 11, 646–659 CAS.
- L. Aroua, Asian J. Chem., 2020, 32, 1266–1272 CAS.
- A. T. Mavrova, K. K. Anichina, D. I. Vuchev, J. A. Tsenov, P. S. Denkova, M. S. Kondeva and M. K. Micheva, Eur. J. Med. Chem., 2006, 41, 1412–1420 CAS.
- E. K. Francis, A. Antonopoulos, M. E. Westman, J. McKay-Demeler, R. Laing and J. Šlapeta, Int. J. Parasitol., 2024, 54, 55–64 CAS.
- J. Vichapong, Y. Santaladchaiyakit, R. Burakham and S. Srijaranai, Anal. Lett., 2015, 48, 617–631 CAS.
- T. Zhang, Q. Liu and Y. Ren, Tetrahedron, 2020, 76, 131027 Search PubMed.
- A. Spasov, A. Kucheryavenko, V. Sirotenko, V. Anisimova, L. Divaeva, T. Kuzmenko and A. Morkovnik, Bull. Exp. Biol. Med., 2019, 166, 747–750 CrossRef CAS PubMed.
- T.-L. Yen, M.-P. Wu, C.-L. Chung, W.-B. Yang, T. Jayakumar, P. Geraldine, C.-M. Chou, C.-Y. Chang, W.-J. Lu and J.-R. Sheu, J. Biomed. Sci., 2016, 23, 1–13 CrossRef PubMed.
- S. B. Bhalekar and S. N. Shelke, ChemistrySelect, 2019, 4, 12170–12175 CrossRef CAS.
- I. Vazzana, E. Terranova, B. Tasso, M. Tonelli, A. Piana, S. Gastaldi and F. Sparatore, Chem. Biodiversity, 2008, 5, 714–728 CrossRef CAS PubMed.
- H.-L. Kuo, J.-C. Lien, C.-H. Chung, C.-H. Chang, S.-C. Lo, I.-C. Tsai, H.-C. Peng, S.-C. Kuo and T.-F. Huang, Naunyn Schmiedebergs Arch. Pharmacol., 2010, 381, 495–505 CrossRef CAS PubMed.
- S. S. Bharadwaj, B. Poojary, S. K. M. Nandish, J. Kengaiah, M. P. Kirana, M. K. Shankar, A. J. Das, A. Kulal and D. Sannaningaiah, ACS Omega, 2018, 3, 12562–12574 CrossRef CAS PubMed.
- H. Yang, Y. Ren, X. Gao and Y. Gao, Chem. Res. Chin. Univ., 2016, 32, 973–978 Search PubMed.
- W. Ren, Y. Ren, M. Dong and Y. Gao, Helv. Chim. Acta, 2016, 99, 325–332 CrossRef CAS.
- M. Imran, L. T. Al Kury, H. Nadeem, F. A. Shah, M. Abbas, S. Naz, A.-u. Khan and S. Li, Biomolecules, 2020, 10, 108 CrossRef CAS PubMed.
- A. Rathore, R. Sudhakar, M. J. Ahsan, A. Ali, N. Subbarao, S. S. Jadav, S. Umar and M. S. Yar, Bioorg. Chem., 2017, 70, 107–117 CrossRef CAS PubMed.
- A. A. Moneer, K. O. Mohammed and H. B. El-Nassan, Chem. Biol. Drug Design, 2016, 87, 784–793 CrossRef CAS PubMed.
- S. Kankala, R. K. Kankala, P. Gundepaka, N. Thota, S. Nerella, M. R. Gangula, H. Guguloth, M. Kagga, R. Vadde and C. S. Vasam, Bioorg. Med. Chem. Lett., 2013, 23, 1306–1309 CrossRef CAS PubMed.
- M. T.-E. Maghraby, O. M. Abou-Ghadir, S. G. Abdel-Moty, A. Y. Ali and O. I. Salem, Bioorg. Med. Chem., 2020, 28, 115403 CrossRef CAS PubMed.
- A. Noor, N. G. Qazi, H. Nadeem, A.-u. Khan, R. Z. Paracha, F. Ali and A. Saeed, Chem. Cent. J., 2017, 11, 1–13 CrossRef PubMed.
- R. Radhamanalan, M. Alagumuthu and N. Nagaraju, Future Med. Chem., 2018, 10, 1805–1820 CrossRef CAS PubMed.
- N. T. Chandrika, S. K. Shrestha, H. X. Ngo and S. Garneau-Tsodikova, Bioorg. Med. Chem., 2016, 24, 3680–3686 CrossRef CAS PubMed.
- E. Guzel, U. Acar Çevik, A. E. Evren, H. E. Bostancı, U. D. Gul, U. u. Kayış, Y. Ozkay and Z. A. Kaplancıklı, ACS Omega, 2023, 8, 4369–4384 CrossRef CAS PubMed.
- L. Emami, Z. Faghih, K. Zomorodian, M. Behrooz, L. Zamani, A. Rostami, A. Jalilian and S. Khabnadideh, Med. Chem. Res., 2022, 31, 2220–2230 Search PubMed.
- A. Ç. Karaburun, B. Kaya Çavuşoğlu, U. Acar Çevik, D. Osmaniye, B. N. Sağlık, S. Levent, Y. Özkay, Ö. Atlı, A. S. Koparal and Z. A. Kaplancıklı, Molecules, 2019, 24, 191 CrossRef PubMed.
- I. A. Khodja, H. Boulebd, C. Bensouici and A. Belfaitah, J. Mol. Struct., 2020, 1218, 128527 CrossRef.
- Y. Khan, W. Rehman, R. Hussain, S. Khan, A. Malik, M. Khan, A. Liaqat, L. Rasheed, F. Begum and S. Fazil, J. Heterocycl. Chem., 2022, 59, 2225–2239 CrossRef CAS.
- Z. Alım, T. Tunç, N. Demirel, A. Günel and N. Karacan, J. Mol. Struct., 2022, 1268, 133647 Search PubMed.
- F. Doganc, I. Celik, G. Eren, M. Kaiser, R. Brun and H. Goker, Eur. J. Med. Chem., 2021, 221, 113545 Search PubMed.
- S. Choudhary, M. Arora, H. Verma, M. Kumar and O. Silakari, Eur. J. Pharmacol., 2021, 899, 174027 CrossRef CAS PubMed.
- V. M. Patel, N. B. Patel, M. J. Chan-Bacab and G. Rivera, Synth. Commun., 2020, 50, 858–878 CAS.
- R. S. Keri, C. K. Rajappa, S. A. Patil and B. M. Nagaraja, Pharmacol. Rep., 2016, 68, 1254–1265 CAS.
- S. Yadav, S. M. Lim, K. Ramasamy, M. Vasudevan, S. A. A. Shah, A. Mathur and B. Narasimhan, Chem. Cent. J., 2018, 12, 1–14 Search PubMed.
- Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208–6236 CAS.
- M. Azadbakht, A. Davoodi, S. J. Hosseinimehr, M. Keighobadi, M. Fakhar, R. Valadan, R. Faridnia, S. Emami, M. Azadbakht and A. Bakhtiyari, Exp. Parasitol., 2020, 213, 107902 CAS.
- M. Tonelli, E. Gabriele, F. Piazza, N. Basilico, S. Parapini, B. Tasso, R. Loddo, F. Sparatore and A. Sparatore, J. Enzyme Inhib. Med. Chem., 2018, 33, 210–226 CAS.
- R. Nieto-Meneses, R. Castillo, A. Hernández-Campos, B. Nogueda-Torres, E. O. López-Villegas, A. Moreno-Rodríguez, F. Matadamas-Martínez and L. Yépez-Mulia, Int. J. Mol. Sci., 2024, 25, 659 CAS.
- D. A. Bakhotmah and A. A. Al-Ahmadi, Polycyclic Aromat. Compd., 2021, 41, 1459–1471 Search PubMed.
- Y. K. Yoon, M. A. Ali, A. C. Wei, T. S. Choon and R. Ismail, Eur. J. Med. Chem., 2015, 93, 614–624 Search PubMed.
- V. Francesconi, E. Cichero, S. Schenone, L. Naesens and M. Tonelli, Molecules, 2020, 25, 1487 CAS.
- D. Sharma, B. Narasimhan, P. Kumar, V. Judge, R. Narang, E. De Clercq and J. Balzarini, J. Enzyme Inhib. Med. Chem., 2009, 24, 1161–1168 CAS.
- T. Pan, X. He, B. Chen, H. Chen, G. Geng, H. Luo, H. Zhang and C. Bai, Eur. J. Med. Chem., 2015, 95, 500–513 CrossRef CAS PubMed.
- J. F. Miller, E. M. Turner, K. S. Gudmundsson, S. Jenkinson, A. Spaltenstein, M. Thomson and P. Wheelan, Bioorg. Med. Chem. Lett., 2010, 20, 2125–2128 CrossRef CAS PubMed.
- Z. Wang, X. Deng, R. Xiong, S. Xiong, J. Liu, X. Cao, X. Lei, Y. Chen, X. Zheng and G. Tang, MedChemComm, 2018, 9, 305–315 RSC.
- I. Singh, V. Luxami and K. Paul, Org. Biomol. Chem., 2019, 17, 5349–5366 RSC.
- D. Schmit, U. Milewicz, M. A. Boerneke, S. Burley, K. Walsworth, J. Um, D. Hecht, T. Hermann and B. M. Bergdahl, Aust. J. Chem., 2020, 73, 212–221 CrossRef CAS.
- R. Gondru, Y. Li and J. Banothu, Eur. J. Med. Chem., 2022, 227, 113921 CrossRef PubMed.
- M. Nardi, N. C. H. Cano, S. Simeonov, R. Bence, A. Kurutos, R. Scarpelli, D. Wunderlin and A. Procopio, Catalysts, 2023, 13, 392 CrossRef CAS.
- M. Serafini, E. Torre, S. Aprile, E. D. Grosso, A. Gesù, A. Griglio, G. Colombo, C. Travelli, S. Paiella and A. Adamo, J. Med. Chem., 2020, 63, 3047–3065 CAS.
- A. J. Marcus, I. Iezhitsa, R. Agarwal, P. Vassiliev, A. Spasov, O. Zhukovskaya, V. Anisimova and N. M. Ismail, Eur. J. Pharmacol., 2019, 850, 75–87 CrossRef CAS PubMed.
- Z. Wu, M.-B. Xia, D. Bertsetseg, Y.-H. Wang, X.-L. Bao, W.-B. Zhu, P.-R. Chen, H.-S. Tang, Y.-J. Yan and Z.-L. Chen, Bioorg. Chem., 2020, 101, 104042 CrossRef CAS PubMed.
- B. T. B. Hue, P. H. Nguyen, T. Q. De, M. Van Hieu, E. Jo, N. Van Tuan, T. T. Thoa, L. D. Anh, N. H. Son and D. La Duc Thanh, ChemMedChem, 2020, 15, 1453–1463 CAS.
- R. Hussain, S. Iqbal, M. Shah, W. Rehman, S. Khan, L. Rasheed, F. Rahim, A. A. Dera, S. Kehili and E. B. Elkaeed, Molecules, 2022, 27, 6457 CrossRef CAS PubMed.
- F. Rahim, K. Zaman, M. Taha, H. Ullah, M. Ghufran, A. Wadood, W. Rehman, N. Uddin, S. A. A. Shah and M. Sajid, Bioorg. Chem., 2020, 94, 103394 CrossRef CAS PubMed.
- S. Guzelj, M. Gobec, D. Urbančič, I. Mlinarič-Raščan, E. Corsini and Ž. Jakopin, Eur. J. Med. Chem., 2020, 190, 112089 CrossRef PubMed.
- A. Spasov, O. Zhukovskaya, N. Gurova, L. Naumenko, N. Eliseeva, A. Kucheryavenko, V. Kosolapov, D. Yakovlev, V. Muravyeva and V. Babkova, Russ. J. Bioorg. Chem., 2022, 48, 281–291 CrossRef CAS.
- M. Taha, A. Mosaddik, F. Rahim, S. Ali, M. Ibrahim and N. B. Almandil, J. King Saud Univ. Sci., 2020, 32, 191–194 CrossRef.
- M. A. Argirova, M. K. Georgieva, N. G. Hristova-Avakumova, D. I. Vuchev, G. V. Popova-Daskalova, K. K. Anichina and D. Y. Yancheva, RSC Adv., 2021, 11, 39848–39868 RSC.
- A. Baldisserotto, M. Demurtas, I. Lampronti, M. Tacchini, D. Moi, G. Balboni, S. Pacifico, S. Vertuani, S. Manfredini and V. Onnis, Bioorg. Chem., 2020, 94, 103396 CrossRef CAS PubMed.
- S. MC, J. Taibah Univ. Sci., 2016, 10, 122–130 Search PubMed.
- M. A. Shaldam, D. Hendrychová, R. El-Haggar, V. Vojáčková, T. A. Majrashi, E. B. Elkaeed, N. Masurier, V. Kryštof, H. O. Tawfik and W. M. Eldehna, Eur. J. Med. Chem., 2023, 258, 115610 CAS.
- S. Guo, T. Jia, X. Xu, F. Yang, S. Xiao, Z. Hou, H. Xu, S. Ma, X. Liu and C. Luo, Eur. J. Med. Chem., 2023, 248, 115093 CAS.
- B. Ko, Y. Jang, M. H. Kim, T. T. Lam, H. K. Seo, P. Jeong, M. Choi, K. W. Kang, S.-D. Lee and J.-H. Park, Eur. J. Med. Chem., 2023, 262, 115860 CAS.
- G. Surineni, Y. Gao, M. Hussain, Z. Liu, Z. Lu, C. Chhotaray, M. M. Islam, H. A. Hameed and T. Zhang, Medchemcomm, 2019, 10, 49–60 CAS.
- K. Veena, M. S. Raghu, K. Y. Kumar, C. P. Kumar, F. A. Alharti, M. K. Prashanth and B.-H. Jeon, J. Mol. Struct., 2022, 1269, 133822 CAS.
- S. Patil, P. Bhatt, H. Suryawanshi, J. Patil and R. Chaudhari, Chem. Proc., 2021, 8, 55 Search PubMed.
- T. M. Dhameliya, K. I. Patel, R. Tiwari, S. K. Vagolu, D. Panda, D. Sriram and A. K. Chakraborti, Bioorg. Chem., 2021, 107, 104538 CAS.
- P. S. Jadhavar, K. I. Patel, T. M. Dhameliya, N. Saha, M. D. Vaja, V. S. Krishna, D. Sriram and A. K. Chakraborti, Bioorg. Chem., 2020, 99, 103774 CAS.
- F. Scott, A. M. Fala, L. E. Pennicott, T. D. Reuillon, K. B. Massirer, J. M. Elkins and S. E. Ward, Bioorg. Med. Chem. Lett., 2020, 30, 127040 CAS.
- N. S. Goud, S. M. Ghouse, J. Vishnu, D. Komal, V. Talla, R. Alvala, J. Pranay, J. Kumar, I. A. Qureshi and M. Alvala, Bioorg. Chem., 2019, 89, 103016 CAS.
- M. Wang, Y. Wu, C. Xu, R. Zhao, Y. Huang, X. Zeng and T. Chen, Chem. – Asian J., 2019, 14, 2648–2655 CAS.
- R. Sivaramakarthikeyan, S. Iniyaval, V. Saravanan, W.-M. Lim, C.-W. Mai and C. Ramalingan, ACS Omega, 2020, 5, 10089–10098 CAS.
- A. Lotfi, Egypt. J. Chem., 2020, 63, 4757–4767 Search PubMed.
- F. M. Alminderej and A. Lotfi, Egypt. J. Chem., 2021, 64, 3351–3364 Search PubMed.
- P. Naik, P. Murumkar, R. Giridhar and M. R. Yadav, Bioorg. Med. Chem., 2010, 18, 8418–8456 CAS.
- C. S. Law and K. Y. Yeong, ChemMedChem, 2021, 16, 1861–1877 CAS.
- A.-M. Monforte, A. Rao, P. Logoteta, S. Ferro, L. De Luca, M. L. Barreca, N. Iraci, G. Maga, E. De Clercq and C. Pannecouque, Bioorg. Med. Chem., 2008, 16, 7429–7435 CAS.
- S. Ferro, M. R. Buemi, L. De Luca, F. E. Agharbaoui, C. Pannecouque and A.-M. Monforte, Bioorg. Med. Chem., 2017, 25, 3861–3870 CrossRef CAS PubMed.
- A. M. Monforte, L. De Luca, M. R. Buemi, F. E. Agharbaoui, C. Pannecouque and S. Ferro, Bioorg. Med. Chem., 2018, 26, 661–674 CrossRef CAS PubMed.
- A.-M. Monforte, S. Ferro, L. De Luca, G. L. Surdo, F. Morreale, C. Pannecouque, J. Balzarini and A. Chimirri, Bioorg. Med. Chem., 2014, 22, 1459–1467 CrossRef CAS PubMed.
- B. Sahoo and B. Banik, Microwave Assisted Green Synthesis of Benzimidazole Derivatives and Evaluation of Their Anticonvulsant Activity, Curr. Microwave Chem., 2019, 6, 23–29 CrossRef CAS.
- D. Tiglani, S. Salahuddin, A. Mazumder, M. S. Yar, R. Kumar and M. j. Ahsan, Polycyclic Aromat. Compd., 2021, 42, 5044–5066 CrossRef.
- N. Siddiqui, M. S. Alam, R. Ali, M. S. Yar and O. Alam, Med. Chem. Res., 2016, 25, 1390–1402 CrossRef CAS.
- B. Pathare and T. Bansode, Results Chem., 2021, 3, 100200 CrossRef CAS.
- F. Khan and S. Nadeem, Pharmacologyonline, 2011, 2, 1217 Search PubMed.
- K. M. Nagesh, T. Prashanth, H. A. Khamees and S. A. Khanum, J. Mol. Struct., 2022, 1259, 132741 CrossRef CAS.
- A. A. Al Awadh, Saudi Pharm. J., 2023, 101698 Search PubMed.
- L. A. Mohammed, M. A. Farhan, S. A. Dadoosh, M. A. Alheety, A. H. Majeed, A. S. Mahmood and Z. H. Mahmoud, SynOpen, 2023, 7, 652–673 CAS.
- M. C. Sharma, D. V. Kohli and S. Sharma, Int. J. Drug Delivery, 2010, 2, 228–237 CAS.
- T. Roy, N. N. Petersen, G. Gopalan, J. Gising, M. Hallberg and M. Larhed, Bioorg. Med. Chem., 2022, 66, 116804 CAS.
- J. Y. Xu, Q. Ran, W. Y. Hua, X. M. Wu, Q. J. Wang and J. Zhang, Chin. Chem. Lett., 2007, 18, 251–254 CAS.
- N. N. Mrabti and M. Elhallaoui, J. Taibah Univ. Sci., 2017, 11, 18–39 CrossRef.
- Y. Tamura, I. Morita, Y. Hinata, E. Kojima, Y. Sasaki, T. Wada, M. Asano, M. Fujioka, Y. Hayasaki-Kajiwara and T. Iwasaki, Bioorg. Med. Chem. Lett., 2023, 79, 129059 CrossRef CAS PubMed.
- A. Sharma, S. K. Anand, N. Singh, U. N. Dwivedi and P. Kakkar, Exp. Cell Res., 2023, 428, 113614 CrossRef CAS PubMed.
- G. Yadav and S. Ganguly, Eur. J. Med. Chem., 2015, 97, 419–443 CrossRef CAS PubMed.
- Y. T. Lee, Y. J. Tan and C. E. Oon, Acta Pharm. Sin. B, 2023, 13, 478–497 CrossRef CAS PubMed.
- S. Tahlan, S. Kumar and B. Narasimhan, BMC Chem., 2019, 13, 1–27 CrossRef PubMed.
- R. S. Keri, A. Hiremathad, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 19–65 CrossRef PubMed.
- A. T. Mavrova, D. Yancheva, N. Anastassova, K. Anichina, J. Zvezdanovic, A. Djordjevic, D. Markovic and A. Smelcerovic, Bioorg. Med. Chem., 2015, 23, 6317–6326 CrossRef CAS PubMed.
- M. Terreni, M. Taccani and M. Pregnolato, Molecules, 2021, 26, 2671 CrossRef CAS PubMed.
- S. Moghadam Farid, M. Noori, M. Nazari Montazer, M. Khalili Ghomi, M. Mollazadeh, N. Dastyafteh, C. Irajie, K. Zomorodian, S. S. Mirfazli and S. Mojtabavi, Sci. Rep., 2023, 13, 4392 CrossRef CAS PubMed.
- V. S. Padalkar, B. N. Borse, V. D. Gupta, K. R. Phatangare, V. S. Patil, P. G. Umape and N. Sekar, Arabian J. Chem., 2016, 9, S1125–S1130 CrossRef CAS.
- Y. Shi, K. Jiang, R. Zheng, J. Fu, L. Yan, Q. Gu, Y. Zhang and F. Lin, Chem. Biodiversity, 2019, 16, e1800510 CrossRef PubMed.
- K. Mahmood, W. Hashmi, H. Ismail, B. Mirza, B. Twamley, Z. Akhter, I. Rozas and R. J. Baker, Polyhedron, 2019, 157, 326–334 CrossRef CAS.
- O. O. Ajani, O. O. Tolu-Bolaji, S. J. Olorunshola, Y. Zhao and D. V. Aderohunmu, J. Adv. Res., 2017, 8, 703–712 CAS.
- K. Gullapelli, G. Brahmeshwari, M. Ravichander and U. Kusuma, Egypt. J. Basic Appl. Sci., 2017, 4, 303–309 Search PubMed.
- K. V. Chikkula and R. Sundararajan, Med. Chem. Res., 2017, 26, 3026–3037 CAS.
- N. El-Gohary and M. Shaaban, Eur. J. Med. Chem., 2017, 131, 255–262 CAS.
- S. K. Mohanty, A. Khuntia, N. Yellasubbaiah, C. Ayyanna, B. N. Sudha and M. S. Harika, Beni-Suef Univ. J. Basic Appl. Sci., 2018, 7, 646–651 Search PubMed.
- P. Picconi, C. Hind, S. Jamshidi, K. Nahar, M. Clifford, M. E. Wand, J. M. Sutton and K. M. Rahman, J. Med. Chem., 2017, 60, 6045–6059 CAS.
- F. Naaz, R. Srivastava, A. Singh, N. Singh, R. Verma, V. K. Singh and R. K. Singh, Bioorg. Med. Chem., 2018, 26, 3414–3428 CAS.
- C. Shahini, G. Achar, S. Budagumpi, M. Tacke and S. A. Patil, Inorg. Chim. Acta, 2017, 466, 432–441 CAS.
- I. Tumosienė, A. Peleckis, I. Jonuškienė, R. Vaickelionienė, K. Kantminienė, J. Šiugždaitė, Z. J. Beresnevičius and V. Mickevičius, Monatsh. Chem., 2018, 149, 577–594 Search PubMed.
- N. Desai, D. Pandya and D. Vaja, Med. Chem. Res., 2018, 27, 52–60 CAS.
- H. Bektas, C. Albay, B. B. Sökmen, S. Aydın, E. Menteşe, G. Aydın and D. Şen, J. Heterocycl. Chem., 2018, 55, 2400–2407 CAS.
- Y.-M. Huang, N. S. Alharbi, B. Sun, C. Shantharam, K. Rakesh and H.-L. Qin, Eur. J. Med. Chem., 2019, 181, 111566 CAS.
- L. Garuti, M. Roberti and G. Gentilomi, Il Farmaco, 2000, 55, 35–39 CAS.
- T. Fonseca, B. Gigante, M. M. Marques, T. L. Gilchrist and E. De Clercq, Bioorg. Med. Chem., 2004, 12, 103–112 CAS.
- J. Cheng, J. Xie and X. Luo, Bioorg. Med. Chem. Lett., 2005, 15, 267–269 CrossRef CAS PubMed.
- K. Starčević, M. Kralj, K. Ester, I. Sabol, M. Grce, K. Pavelić and G. Karminski-Zamola, Bioorg. Med. Chem., 2007, 15, 4419–4426 CrossRef.
- L. B. Townsend, R. V. Devivar, S. R. Turk, M. R. Nassiri and J. C. Drach, J. Med. Chem., 1995, 38, 4098–4105 CrossRef CAS PubMed.
- R. Zou, K. R. Ayres, J. C. Drach and L. B. Townsend, J. Med. Chem., 1996, 39, 3477–3482 CrossRef CAS.
- K. S. Gudmundsson, J. C. Drach, L. L. Wotring and L. B. Townsend, J. Med. Chem., 1997, 40, 785–793 CrossRef CAS PubMed.
- J. R. Hwu, R. Singha, S. C. Hong, Y. H. Chang, A. R. Das, I. Vliegen, E. De Clercq and J. Neyts, Antiviral Res., 2008, 77, 157–162 CrossRef CAS PubMed.
- T. M. Evans, J. M. Gardiner, N. Mahmood and M. Smis, Bioorg. Med. Chem. Lett., 1997, 7, 409–412 CrossRef CAS.
- A. Ahmadi, E. Mohammadnejadi, P. Karami and N. Razzaghi-Asl, Int. J. Antimicrob. Agents, 2022, 59, 106518 CrossRef CAS PubMed.
- M. M. Teixeira, D. T. Carvalho, E. Sousa and E. Pinto, Pharmaceuticals, 2022, 15, 1427 CrossRef CAS PubMed.
- R. S. Kankate, P. S. Gide and D. P. Belsare, Arabian J. Chem., 2019, 12, 2224–2235 CrossRef CAS.
- S. A. Katti, V. Gupta, S. P. Pingle and A. P. Pingle, Pharmacophore, 2016, 7, 35–40 Search PubMed.
- X. Wang, Y.-F. Chen, W. Yan, L.-L. Cao and Y.-H. Ye, Molecules, 2016, 21, 1574 CrossRef PubMed.
- L. Zamani, Z. Faghih, K. Zomorodian, B. B. F. Mirjalili, A. Jalilian and S. Khabnadideh, Res. Pharm. Sci., 2019, 14, 496–503 CrossRef PubMed.
- N. Ö. Can, U. Acar Çevik, B. N. Sağlık, S. Levent, B. Korkut, Y. Özkay, Z. A. Kaplancıklı and A. S. Koparal, J. Chem., 2017, 2017, 9387102 Search PubMed.
- H. Küçükbay, R. Durmaz, E. Orhan and S. Günal, Il Farmaco, 2003, 58, 431–437 Search PubMed.
- F. Arjmand, B. Mohani and S. Ahmad, Eur. J. Med. Chem., 2005, 40, 1103–1110 CrossRef CAS PubMed.
- U. A. Çevik, D. Osmaniye, B. K. Çavuşoğlu, B. N. Sağlik, S. Levent, S. Ilgin, N. Ö. Can, Y. Özkay and Z. A. Kaplancikli, Med. Chem. Res., 2019, 28, 2252–2261 CrossRef.
- R. Kenchappa, Y. D. Bodke, S. Telkar and M. Aruna Sindhe, ChemBioChem, 2017, 10, 11–23 CAS.
- L.-X. Li, J. Jiao, X.-B. Wang, M. Chen, X.-C. Fu, W.-J. Si and C.-L. Yang, Molecules, 2018, 23, 746 Search PubMed.
- Y.-B. Bai, A.-L. Zhang, J.-J. Tang and J.-M. Gao, J. Agric. Food Chem., 2013, 61, 2789–2795 Search PubMed.
- R. V. Shingalapur, K. M. Hosamani and R. S. Keri, Eur. J. Med. Chem., 2009, 44, 4244–4248 CAS.
- K. Laxmikeshav, A. Himaja and N. Shankaraiah, J. Mol. Struct., 2022, 1253, 132251 Search PubMed.
- A. Bistrović, L. Krstulović, I. Stolić, D. Drenjančević, J. Talapko, M. C. Taylor, J. M. Kelly, M. Bajić and S. Raić-Malić, J. Enzyme Inhib. Med. Chem., 2018, 33, 1323–1334 Search PubMed.
- H. Torres-Gómez, E. Hernández-Núñez, I. León-Rivera, J. Guerrero-Alvarez, R. Cedillo-Rivera, R. Moo-Puc, R. Argotte-Ramos, M. del Carmen Rodríguez-Gutiérrez, M. J. Chan-Bacab and G. Navarrete-Vázquez, Bioorg. Med. Chem. Lett., 2008, 18, 3147–3151 Search PubMed.
- E. Hernández-Núñez, H. Tlahuext, R. Moo-Puc, D. Moreno, M. O. González-Díaz and G. Navarrete Vazquez, Molecules, 2017, 22, 579 Search PubMed.
- M. A. Ismail, R. Brun, T. Wenzler, F. A. Tanious, W. D. Wilson and D. W. Boykin, Bioorg. Med. Chem., 2004, 12, 5405–5413 CrossRef CAS PubMed.
- J. Pérez-Villanueva, A. Hernández-Campos, L. Yépez-Mulia, C. Méndez-Cuesta, O. Méndez-Lucio, F. Hernández-Luis and R. Castillo, Bioorg. Med. Chem. Lett., 2013, 23, 4221–4224 Search PubMed.
- A. T. Mavrova, D. Wesselinova, J. A. Tsenov and L. A. Lubenov, Eur. J. Med. Chem., 2014, 86, 676–683 CAS.
- D. Valdez-Padilla, S. Rodríguez-Morales, A. Hernández-Campos, F. Hernández-Luis, L. Yépez-Mulia, A. Tapia-Contreras and R. Castillo, Bioorg. Med. Chem., 2009, 17, 1724–1730 CAS.
- O. Soria-Arteche, A. Hernández-Campos, L. Yépez-Mulia, R. Castillo and R. Navarrete-Vazquez, Bioorg. Med. Chem. Lett., 2013, 23, 6838–6841 CrossRef CAS PubMed.
- L. Keurulainen, H. Vahokoski, J. Liukkonen, J. Koskinen and P. Koulu, J. Med. Chem., 2015, 58, 4573–4580 CrossRef CAS PubMed.
- P. S. Hameed, M. Chinnapattu, G. Shanbag, S. Chinnappan, M. Srinivasan, G. Ramachandran and M. Arumugham, J. Med. Chem., 2014, 57, 5702–5713 CrossRef PubMed.
- R. Khajuria, S. Rasheed, C. Khajuria, K. K. Kapoor and P. Das, Synthesis, 2018, 50, 2131–2149 CrossRef CAS.
- K. C. Achar, K. M. Hosamani and H. R. Seetharamareddy, Eur. J. Med. Chem., 2010, 45, 2048–2054 CrossRef CAS PubMed.
- A. Gutiérrez, Y. Galván, E. Domínguez-Mendoza, G. García, J. Pacheco, R. Aguilar and F. Rodríguez, J. Chem., 2019, 2019, 1–8 CrossRef.
- K. Y. Yeong, C. W. Ang, M. A. Ali, H. Osman and S. C. Tan, Med. Chem. Res., 2017, 26, 770–778 CrossRef CAS.
- M. R. Anguru, A. K. Taduri, R. D. Bhoomireddy, M. Jojula and S. K. Gunda, Chem. Cent. J., 2017, 11, 1–12 CrossRef PubMed.
- M. I. García-Aranda, R. González-Ruiz, D. Morales, J. A. Moreno and E. Angulo, Bioorg. Med. Chem., 2020, 28, 115427 CrossRef PubMed.
- M. T. E. Maghraby, O. M. F. Abou-Ghadir, S. G. Abdel-Moty, A. Y. Ali and O. I. A. Salem, Bioorg. Med. Chem., 2020, 28, 115403 CrossRef CAS PubMed.
- K. C. S. Achar, K. M. Hosamani and H. R. Seetharamareddy, Eur. J. Med. Chem., 2010, 45, 2048–2054 CrossRef CAS PubMed.
- S. Dixit, P. K. Sharma and N. Kaushik, Med. Chem. Res., 2013, 22, 900–904 CrossRef CAS.
- C. Sridevi, K. Balaji, A. Naidu and R. Suthakaran, J. Chem. Pharm. Sci., 2010, 3, 234–238 Search PubMed.
- K. Lavrador-Erb, J. D. Nelson, H. Tsujimura, M. J. Ford, S. M. Kramer and P. Gavva, Bioorg. Med. Chem. Lett., 2010, 20, 2916–2919 CAS.
- W. Zhu, Y. Da, D. Wu, H. Zheng, L. Zhu, L. Wang, Y. Yan and Z. Chen, Bioorg. Med. Chem., 2014, 22, 2294–2302 CrossRef CAS PubMed.
- Z. Wu, X. L. Bao, W. B. Zhu, H. Liu, Y. Wang, J. Zhan and Y. Zhong, ACS Med. Chem. Lett., 2018, 10, 40–43 Search PubMed.
- G. Navarrete-Vázquez, H. Moreno-Diaz, F. Aguirre-Crespo, I. León-Rivera, R. Villalobos-Molina, O. Muñoz-Muñiz and S. Estrada-Soto, Bioorg. Med. Chem. Lett., 2006, 16, 4169–4173 Search PubMed.
- W. Zhu, X. Bao, H. Ren, J. Wang, Y. Wu, Z. He and J. Zhang, Eur. J. Med. Chem., 2016, 115, 161–178 CrossRef CAS PubMed.
- N. Kaur, A. Kaur, Y. Bansal, D. I. Shah, G. Bansal and M. Singh, Bioorg. Med. Chem., 2008, 16, 10210–10215 CrossRef CAS PubMed.
- Y. Kim, M. R. Kumar, N. Park, Y. Heo and S. Lee, J. Org. Chem., 2011, 76, 9577–9583 CAS.
- Y. El Bakri, E. H. Anouar, I. Marmouzi, K. Karrouchi, A. Abouri, H. Cherrah and S. Alaoui, J. Mol. Model., 2018, 24, 1–11 CAS.
- R. V. Shingalapur, K. M. Hosamani, R. S. Keri and M. H. Hugar, Eur. J. Med. Chem., 2010, 45, 1753–1759 CAS.
- A. Gutierréz-Hernández, Y. Galván-Ciprés, E. A. Domínguez-Mendoza, Y. Aguirre-Vidal, S. Estrada-Soto, J. C. Almanza-Pérez and G. Navarrete-Vázquez, J. Chem., 2019, 2019, 1650145 Search PubMed.
- L. Deswal, V. Verma, D. Kumar, C. P. Kaushik, A. Kumar, Y. Deswal and S. Punia, Arch. Pharm., 2020, 353, 2000090 CAS.
- M. Taha, F. Rahim, K. Zaman, M. Selvaraj, N. Uddin, R. K. Farooq, M. Nawaz, M. Sajid, F. Nawaz, M. Ibrahim and K. M. Khan, Bioorg. Chem., 2020, 95, 103555 CAS.
- M. Taha, N. Uddin, M. Ali, F. Rahim, G. Khan, R. K. Farooq, M. Gollapalli, N. Iqbal, M. Farooq and K. M. Khan, Int. J. Biol. Macromol., 2020, 161, 355–363 CAS.
- L. M. Aroua, H. R. Almuhaylan, F. M. Alminderej, S. Messaoudi, S. Chigurupati, S. Al-Mahmoud and H. A. Mohammed, A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity, Bioorg. Chem., 2021, 114, 105073 CAS.
- R. V. Shingalapur, K. M. Hosamani, R. S. Keri and M. H. Hugar, Eur. J. Med. Chem., 2010, 45, 1753–1759 CrossRef CAS PubMed.
- S.-C. Lee, H. T. Kim, C.-H. Park, D. Y. Lee, H.-J. Chang, S. Park and Y.-G. Suh, Bioorg. Med. Chem. Lett., 2012, 22, 4221–4224 CrossRef CAS PubMed.
- H. J. Kwak, Y. M. Pyun, J. Y. Kim, H. S. Pagire, K. Y. Kim, K. R. Kim, H. T. Kim and J. H. Ahn, Bioorg. Med. Chem. Lett., 2013, 23, 4713–4718 CrossRef CAS PubMed.
- D. A. Babkov, O. N. Zhukowskaya, A. V. Borisov, V. A. Babkova, E. V. Sokolova, A. A. Brigadirova and A. A. Spasov, Bioorg. Med. Chem. Lett., 2019, 29, 5476–5484 Search PubMed.
- X. Wang, N. Ling, Q. T. Che, Y. W. Zhang, H. X. Yang, Y. Ruan and T. T. Zhao, Inorg. Chem. Commun., 2019, 105, 97–101 CAS.
- A. A. Akande, U. Salar, K. M. Khan, U. Ashraf, K. Rasheed, N. Siddiqui, H. Moin and W. Tanveer, ACS Omega, 2021, 6, 22726–22739 CAS.
- A. A. Adegboye, K. M. Khan, U. Salar, K. Rasheed, N. Siddiqui, H. Moin and W. Tanveer, Eur. J. Med. Chem., 2018, 150, 248–260 CrossRef CAS PubMed.
- Y. K. Yoon, M. A. Ali, A. C. Wei, A. N. Shirazi, K. Parang and T. S. Choon, Eur. J. Med. Chem., 2014, 83, 448–454 CAS.
- A. Kanwal, F. A. Saddique, S. Aslam, M. Ahmad, A. F. Zahoor, N. Mohsin and A. ul, Pharm. Chem. J., 2018, 51, 1068–1077 CAS.
- A. Kumar, S. Banerjee, P. Roy, S. M. Sondhi and A. Sharma, Mol. Diversity, 2017, 22, 113–127 Search PubMed.
- H. A. Ghabour, H. Almahli and W. M. Eldehna, Crystals, 2020, 10, 446 Search PubMed.
- E. Üstün, A. Özgür, K. A. Coşkun, S. Demir Düşünceli, İ. Özdemir and Y. Tutar, Transition Met. Chem., 2017, 42, 331–337 Search PubMed.
- M. F. AlAjmi, A. Hussain, M. T. Rehman, A. A. Khan, P. A. Shaikh and R. A. Khan, Int. J. Mol. Sci., 2018, 19, 1492 Search PubMed.
- J. E. Cheong, M. Zaffagni, I. Chung, M. Kim, Y. Jeong, S. Lee and G. Kim, Eur. J. Med. Chem., 2018, 144, 372–385 CAS.
- S. T. Huang, I. J. Hsei and C. Chen, Bioorg. Med. Chem., 2006, 14, 6106–6119 CAS.
- A. Hussain, M. F. AlAjmi, M. T. Rehman, A. A. Khan, P. A. Shaikh and R. A. Khan, Molecules, 2018, 23, 1232 CrossRef PubMed.
- M. J. Akhtar, A. A. Khan, Z. Ali, S. A. Khan, A. Murtaza and M. I. Choudhary, Bioorg. Chem., 2018, 78, 158–169 CrossRef CAS PubMed.
- H. M. Refaat, Eur. J. Med. Chem., 2010, 45, 2949–2956 CrossRef CAS PubMed.
- M. Shaharyar, M. M. Abdullah, M. A. Bakht and J. Majeed, Eur. J. Med. Chem., 2010, 45, 114–119 CrossRef CAS PubMed.
- P. Sharma, T. S. Reddy, N. P. Kumar, K. R. Senwar, S. K. Bhargava and N. Shankaraiah, Eur. J. Med. Chem., 2017, 138, 234–245 CrossRef CAS.
- S. A. Elsayed, S. Harrypersad, H. A. Sahyon, M. A. El-Magd and C. J. Walsby, Molecules, 2020, 25, 4284 CrossRef CAS PubMed.
- A. Al-Hakimi, F. Alminderej, L. Aroua, H. Al-Jahdali, A. Al-Otaibi, A. Al-Tarawneh and N. Al-Mahri, Arabian J. Chem., 2020, 13, 480–496 Search PubMed.
- H. A. Mohammed, G. M. Sulaiman, S. Albukhaty, A. Z. Al-Saffar, F. A. Elshibani and E. A. Ragab, ChemistrySelect, 2023, 8, 202303306 CrossRef.
- H. Mohammed, Published online, 2009.
- H. A. Mohammed and R. A. Khan, Int. J. Mol. Sci., 2022, 23, 2149 CrossRef CAS PubMed.
- F. A. El-Shibani, G. M. Sulaiman, A. S. Abouzied, A. Al Ali, A. K. Abdulkarim, A. D. Alamami, M. Asiri and H. A. Mohammed, ACS Omega, 2023, 8, 48269–48279 CrossRef CAS PubMed.
- S. V. Bhandari, O. G. Nagras, P. V. Kuthe, A. P. Sarkate, K. S. Waghamare, D. N. Pansare, S. Y. Chaudhari, S. N. Mawale and M. C. Belwate, J. Mol. Struct., 2023, 1276, 134747 CrossRef CAS.
- M. Taha, N. Uddin, M. Ali, F. Rahim, K. Zaman, R. K. Farooq, M. Selvaraj, N. Iqbal, M. Nawaz and K. M. Khan, Int. J. Biol. Macromol., 2020, 161, 355–363 CrossRef CAS PubMed.
- A. Kanwal, F. A. Saddique, S. Aslam, M. Ahmad, A. F. Zahoor and N. ul A. Mohsin, Pharm. Chem. J., 2018, 51, 1068–1077 CrossRef CAS.
- A. S. Alpan, G. Sarıkaya, G. Çoban, S. Parlar, G. Armagan and V. Alptüzün, Arch. Pharm., 2017, 350, 1–16 CrossRef PubMed.
- B. B. Kashid, A. A. Ghanwat, V. M. Khedkar, S. R. Bhandare, M. S. Shingate and S. P. Gowda, J. Heterocycl. Chem., 2019, 56, 895–908 CrossRef CAS.
- V. Chithra and A. Reji, Indian J. Chem., 2019, 58, 1279–1283 Search PubMed.
- N. Karaali, S. Aydin, N. Baltas and E. Mentese, J. Heterocycl. Chem., 2020, 1–10 Search PubMed.
- D. Ashok, S. Gundu, V. K. Aamate and M. G. Devulapally, Mol. Diversity, 2018, 22, 769–778 CrossRef CAS PubMed.
- F. Ke, P. Zhang, Y. Xu, L. Zhang, Q. Wang, J. Wu, H. Hu and X. Ma, Synlett, 2018, 29, 2844–2849 CrossRef.
- M. Özil, C. Parlak and N. Baltaş, Bioorg. Chem., 2018, 76, 468–477 CrossRef PubMed.
- N. O. Anastassova, A. T. Mavrova, D. Y. Yancheva, I. V. Ivanova, S. I. Ivanov, E. B. Zvezdanova and I. I. Bogdanov, Arabian J. Chem., 2018, 11, 353–369 CrossRef CAS.
- M. Bellam, M. Gundluru, S. Sarva, K. K. Satish, V. V. Rao and P. Reddy, Chem. Heterocycl. Compd., 2017, 53, 1–6 CrossRef.
- N. Karmaker, D. Lira, B. Das, U. Kumar and A. Rouf, Dhaka Univ. J. Pharm. Sci., 2018, 16, 245 CrossRef.
- H. Wu, J. Yuan, Y. Bai, G. Pan, H. Wang and X. Shu, J. Photochem. Photobiol., B, 2012, 107, 65–72 CrossRef CAS PubMed.
- D. Prabu, S. Lakshmanan, N. Ramalakshmi, K. Thirumurugan, G. Dharman and D. S. Arul Antony, Synth. Commun., 2019, 49, 1–13 Search PubMed.
- X. B. Fu, J. J. Zhang, D. D. Liu, S. Wang, J. S. Li, L. Wang and Z. Q. Liu, J. Inorg. Biochem., 2015, 143, 77–87 CrossRef CAS PubMed.
- R. G. Ingle and D. D. Magar, Heterocyclic chemistry of benzimidazoles and potential activities of derivatives, Int. J. Drug Deliv. Technol., 2011, 1, 26–32 Search PubMed.
- C. Sun, C. Chen, S. Xu, J. Wang, Y. Zhu, D. Kong, H. Tao, M. Jin, P. Zheng and W. Zhu, Synthesis and anticancer activity of novel 4-morpholino-7, 8-dihydro-5H-thiopyrano [4, 3-d] pyrimidine derivatives bearing chromone moiety, Bioorg. Med. Chem., 2016, 24, 3862–3869 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- G. Satija, B. Sharma, A. Madan, A. Iqubal, M. Shaquiquzzaman, M. Akhter, S. Parvez, M. A. Khan and M. M. Alam, Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets, J. Heterocycl. Chem., 2022, 59, 22–66 CrossRef CAS.
- S. Sana, V. G. Reddy, T. S. Reddy, R. Tokala, R. Kumar, S. K. Bhargava and N. Shankaraiah, Cinnamide derived pyrimidine-benzimidazole hybrids as tubulin inhibitors: synthesis, in silico and cell growth inhibition studies, Bioorg. Chem., 2021, 110, 104765 CrossRef CAS PubMed.
- Y. L. Zhang, R. Yang, L. Y. Xia, R. J. Man, Y. C. Chu, A. Q. Jiang, Z. C. Wang and H. L. Zhu, Synthesis, anticancer activity and molecular docking studies on 1, 2-diarylbenzimidazole analogues as anti-tubulin agents, Bioorg. Chem., 2019, 92, 103219 CrossRef CAS PubMed.
- S. A. Galal, A. S. Abdelsamie, S. A. Shouman, Y. M. Attia, H. I. Ali, A. Tabll, R. El-Shenawy, Y. S. El Abd, M. M. Ali, A. E. Mahmoud and A. H. Abdel-Halim, Part I: Design, synthesis and biological evaluation of novel pyrazole-benzimidazole conjugates as checkpoint kinase 2 (Chk2) inhibitors with studying their activities alone and in combination with genotoxic drugs, Eur. J. Med. Chem., 2017, 134, 392–405 CrossRef CAS PubMed.
- R. Sireesha, R. Sreenivasulu, C. Chandrasekhar, S. S. Jadav, Y. Pavani, M. V. Rao and M. Subbarao, Design, synthesis, anti-cancer evaluation and binding mode studies of benzimidazole/benzoxazole linked β-carboline derivatives, J. Mol. Struct., 2021, 1226, 129351 CrossRef CAS.
- D. J. Magliano and E. J. Boyko, IDF Diabetes Atlas, scientific committee, International Diabetes Federation, Brussels 10th edn, 2021 Search PubMed.
- D. Care, Medical care in diabetes 2020, Diabetes Care, 2020, 43, 135 CrossRef PubMed.
- R. Hussain, S. Iqbal, M. Shah, W. Rehman, S. Khan, L. Rasheed, F. Rahim, A. A. Dera, S. Kehili, E. B. Elkaeed and N. S. Awwad, Synthesis of novel benzimidazole-based thiazole derivatives as multipotent inhibitors of α-amylase and α-glucosidase: in vitro evaluation along with molecular docking study, Molecules, 2022, 27, 6457 CrossRef CAS PubMed.
- S. R. Chaudhari, P. N. Patil, U. K. Patil, H. M. Patel, J. D. Rajput, N. S. Pawar and D. B. Patil, Green synthesis of N-substituted benzimidazoles: The promising methicillin resistant Staphylococcus aureus (MRSA) inhibitors, Chem. Data Collect., 2020, 25, 100344 CrossRef CAS.
- N. Ranjan, S. Story, G. Fulcrand, F. Leng, M. Ahmad, A. King, S. Sur, W. Wang, Y. C. Tse-Dinh and D. P. Arya, Selective inhibition of Escherichia coli RNA and DNA topoisomerase I by Hoechst 33258 derived mono-and bisbenzimidazoles, J. Med. Chem., 2017, 60, 4904–4922 CrossRef CAS PubMed.
- S. N. Bukhari, G. Lauro, I. Jantan, C. Fei Chee, M. W. Amjad, G. Bifulco, H. Sher, I. Abdullah and N. A. Rahman, Anti-inflammatory trends of new benzimidazole derivatives, Future Med. Chem., 2016, 8, 1953–1967 CrossRef CAS PubMed.
- N. D. Mahurkar, N. D. Gawhale, M. N. Lokhande, S. J. Uke and M. M. Kodape, Results Chem., 2023, 6, 101139 CrossRef CAS.
- A. Bodkhe, D. Pansare, D. Karpe, V. Sudrik, M. Suryawanshi, R. Shinde and S. Lawande, Results Chem., 2025, 13, 101937 CrossRef CAS.
- M. Shaibuna, K. Hiba, A. M. Shebitha, M. J. K. Kuniyil, P. B. Sherly mole and K. Sreekumar, Curr. Res. Green Sustainable Chem., 2022, 5, 100285 Search PubMed.
- V. P. Sachin and R. N. Deepak, Res. J. Chem. Environ., 2022, 26(4), 99–104 Search PubMed.
- L. Wang, J. Sheng, H. Tian and C. Qian, Synth. Commun., 2004, 34, 4265–4272 CAS.
- S. M. Shaikh, G. M. Ansari, Z. Z. Karbari, A. A. Babre, V. V. Borge, V. M. Bangade, D. N. Kommi and B. B. Popatkar, Results Chem., 2024, 12, 101884 CAS.
- A. P. Sarkate, S. D. Shinde, A. O. Barde and A. P. Sarkate, Int. J. ChemTech Res., 2015, 8, 496–500 Search PubMed.
- N. H. Cano, J. G. Uranga, M. Nardi, A. Procopio, D. A. Wunderlin and A. N. Santiago, Beilstein J. Org. Chem., 2016, 12, 2410–2419 CAS.
- J. Flieger and M. Flieger, Int. J. Mol. Sci., 2020, 21, 6267 Search PubMed.
- I. I. I. Alkhatib, M. L. Ferreira, C. G. Alba, D. l. Bahamon, F. Llovell, A. B. Pereiro, J. M. M. Araújo, M. R. M. Abu-Zahra and L. F. Vega, J. Chem. Eng. Data, 2020, 65, 5844 CAS.
- S. Rahimi and E. Soleimani, Results Chem., 2020, 2, 100060 CAS.
- S. Dadwal, M. Kumar and V. Bhalla, J. Org. Chem., 2020, 85, 13906 CAS.
- M. B. Swami, A. H. Jadhav, S. R. Mathpati, H. G. Ghuge and S. G. Patil, Res. Chem. Intermed., 2017, 43, 2033–2053 CAS.
- R. Srinivasulu, K. R. Kumar and P. V. V. Satyanarayana, Green Sustainable Chem., 2014, 4, 33 CAS.
- Z. Zhang, Q. Sun, C. Xia and W. Sun, Org. Lett., 2016, 18(24), 6316–6319 CAS.
- M. Nardi, N. C. H. Cano, S. Simeonov, R. Bence, A. Kurutos, R. Scarpelli, D. Wunderlin and A. Procopio, Catalysts, 2023, 13, 392 CrossRef CAS.
- K. Błaszczak-Świątkiewicz, J. Sikora, J. Szymański, M. Danilewicz and E. Mikiciuk-Olasik, Biological evaluation of the toxicity and the cell cycle interruption by some benzimidazole derivatives, Tumor Biol., 2016, 37, 11135–11145 Search PubMed.
- F. Martin, P. Halvarsson, N. Delhomme, J. Höglund and E. Tydén, Exploring the β-tubulin gene family in a benzimidazole-resistant Parascaris univalens population, Int. J. Parasitol.: Drug, 2021, 17, 84–91 Search PubMed.
- S. I. Alaqeel, Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review, J. Saudi Chem. Soc., 2017, 21, 229–237 Search PubMed.
- Y. T. Lee, Y. J. Tan and C. E. Oon, Benzimidazole and its derivatives as cancer therapeutics: The potential role from traditional to precision medicine, Acta Pharm. Sin. B, 2023, 13, 478–497 Search PubMed.
- J. Wang, X. Sun, N. Li, R. Sheng and R. Guo, An Overview of Thrombin Inhibitors in the Perspective of Structure activity Relationships, Curr. Med. Chem., 2023, 30, 2864–2930 CrossRef CAS PubMed.
- S. Sharma, M. Gupta and J. Sahu, Significance of Benzimidazole analogues for the creation of novel molecules in drug discovery, Curr. Chem. Lett., 2023, 12, 27–44 CrossRef.
- C. M. Singh, P. Katiyar, K. Kushwaha, A. Srivastava, S. Patel and V. D. Verma, Investigation of Benzimidazole Derivatives in Molecular Targets for Breast Cancer Treatment: A Comprehensive review, Crit. Rev. Oncog., 2025, 30, 43–58 CrossRef PubMed.
- A. Akhtar, A. Ali, K. Azeem, M. Abid, N. A. Mazumdar and A. Inam, 2-Benzimidazolamine-Acetamide Derivatives as Antibacterial Agents: Synthesis, ADMET, Molecular Docking, and Molecular Simulation Studies, Synthesis, 2025, 57, 637–646 Search PubMed.
- G. Wang, Y. C. Wang, J. Sun, W. F. Yan, Y. P. Wang, J. Jin, Z. Y. Li, Y. Z. Dong, J. W. Yu and X. Zhang, A novel cocrystal of the Zn(II) coordination molecule and the benzimidazole for efficient detection of triethylamine and antibacterial property, J. Mol. Struct., 2025, 1321, 140054 CrossRef CAS.
- N. Şahin, M. A. Mosrati, A. Merghni, İ. Özdemir, H. Sellami, K. Bedchiche, S. Krayiem, S. Aifa, D. Abdelmalek and D. Sémeril, Synthesis, antimicrobial and antibiofilm activities of silver (I) complexes with N-alkylbenzimidazole derivatives and their protein interaction modelling study, J. Mol. Struct., 2025, 1322, 140440 Search PubMed.
- M. Li, Y. Long, L. Shao, J. Meng, Z. Zheng, Y. Wu, X. Zhou, L. Liu, Z. Li, Z. Wu and S. Yang, Targeting tubulin protein to combat fungal disease: Design, synthesis, and its new mechanistic insights of benzimidazole hydrazone derivatives, Int. J. Biol. Macromol., 2025, 140226 CAS.
- B. S. Daisylet, S. J. Raphael, P. Kumar, A. Mohan and A. Dasan, Exploring photocatalytic, antimicrobial, and biofilm inhibition properties of cuminaldehyde derived benzimidazole metal complexes, J. Mol. Struct., 2025, 141311 CrossRef CAS.
- V. M. Patel, N. B. Patel, M. J. Chan-Bacab, G. Rivera, T. R. Humal and A. S. Gamit, Synthesis and computational studies of 1, 3, 4-thiadiazole and benzothiazole clubbed benzimidazole analogous as anti-tubercular and anti-protozoal agent, J. Mol. Struct., 2025, 1319, 139326 CAS.
- G. Krishnu, G. Paleti, B. K. Chagaleti, A. Mueen and S. Balakrishnan Design, Synthesis and Evaluation of Substituted Benzimidazoles as Potent Polyketide Synthase 13 Inhibitors for Tuberculosis, J. Young Pharm., 2025, 17, 149 Search PubMed.
- H. Shawky, D. B. Fayed, S. S. Abd El-Karim, H. Rezk, M. A. Esawy and E. K. Farrag, Immunotherapeutic effects of de novo benzimidazole derivative and prebiotic bacterial levan against triple-negative breast tumors by harnessing the immune landscape to intercept the oncogenic transcriptome, Int. J. Biol. Macromol., 2025, 289, 138844 CAS.
- M. Hatamfayazi, M. Mahdavi, S. M. Dehaghi, M. Khoshneviszadeh and A. Iraji, Synthesis and biological assessment of benzimidazole-acrylonitrile-1, 2, 3-triazole derivatives as α-glucosidase inhibitors, Bioorg. Chem., 2025, 154, 108060 CAS.
- A. Shakoor, G. Fareed, I. Ahmad, A. A. Elhenawy, M. Khan, N. Fareed, E. Al-Olayan, M. R. Abukhadra, A. Alam and M. Ibrahim, Exploring the anti-diabetic activity of benzimidazole containing Schiff base derivatives: In vitro α-amylase, α-glucosidase inhibitions and in silico studies, J. Mol. Struct., 2025, 1321, 140136 CAS.
- N. Wagih, I. M. Abdel-Rahman, N. A. El-Koussi and G. E. Abuo-Rahma, Anticancer benzimidazole derivatives as inhibitors of epigenetic targets: a review article, RSC Adv., 2025, 15, 966–1010 CAS.
- M. A. Soliman, H. E. Ahmed, E. H. Eltamany, A. T. Boraei, A. Aljuhani, S. A. Salama, R. Alghamdi, A. K. Aljohani, M. Almaghrabi and M. R. Aouad, Novel bis-benzimidazole-triazole hybrids: anticancer study, in silico approaches, and mechanistic investigation, Future Med. Chem., 2025, 17, 93–107 Search PubMed.
- E. Mashhadi, J. Safaei-Ghomi and M. Yahya, Synthesis and Anticancer Activity of Benzimidazole and Benzothiazole Derivatives Bearing Furan Moiety by CAN as a Catalyst Under Ultrasonic Irradiation and Molecular Docking Studies, ChemistrySelect, 2025, 10, 202404904 Search PubMed.
- C. Shen, X. Ding, W. Rao, J. Hu, T. Lin, X. Z. Zhou, Y. Zheng, F. Dong and G. Fan, Prediction of Potential Risk for Ten Azole and Benzimidazole Fungicides with the Aryl Hydrocarbon Receptor Agonistic Activity to Aquatic Ecosystems, J. Agric. Food Chem., 2025, 73, 1167–1181 CAS.
- F. K. Zandian, H. Z. Rafie and H. Bagheri, Bimodal UVM-7-based periodic mesoporous organosilica containing benzimidazole for thin film microextraction of triazole fungicides in fruiting vegetables prior to gas chromatography-mass spectrometry, Anal. Chim. Acta, 2025, 1335, 343420 Search PubMed.
- C. Liu, S. Zhang, M. Sun, X. Zhang, M. Zhao, C. Song, X. Bai, W. Liu and B. Zhang, Development of Benzimidazole-Containing Thermosetting Imide Oligomers With Enhanced Thermal Stability and Adhesive Properties, J. Appl. Polym. Sci., 2025, 142, 56512 Search PubMed.
- A. Shankhyan, G. C. Sharma, P. N. Bhakuni, M. Kazemi and R. Javahershenas, One-Pot, for the Green Preparation of Benzimidazoles and 2, 3-Dihydroquinazolin-4 (1H)-ones Catalyzed by In2O3 NPs/Glycerol, J. Inorg. Organomet. Polym. Mater., 2025, 24, 1–5 Search PubMed.
- J. Zhang, R. Zhai, J. Li, C. Hong, Y. Xu, M. Chen and C. Zhou, Benzimidazole functionalized waterborne polyurethane with excellent mechanical properties, UV and corrosion resistance, Prog. Org. Coat., 2025, 199, 108945 CAS.
- C. Liao, Y. Chen, C. Luo, B. Guan, Q. Hu, H. Guan, C. Luc and Z. Hu, In situ observation and SKPFM analysis to study the adsorption behavior and corrosion inhibition mechanism of 2-aminobenzimidazole inhibitor on Cu- 5 wt% Fe alloys, Appl. Surf. Sci., 2025, 687, 162232 CAS.
- B. Y. Liu, J. Zhao, T. Zeng, T. C. Yue, L. L. Wang and D. Z. Wang, Two Coordination Polymers Based on Rigid Benzimidazole Carboxylic Acid Ligands: Electrode Performance and Adsorption of Dyes, Appl. Organomet. Chem., 2025, 39, 7982 Search PubMed.
- L. Luan, P. Shi, Z. Guo, S. Cong, S. Tang, B. Zhang, Z. Wang and X. Liu, Molecular-scale hybrid membranes prepared with benzimidazole based monomers for high performance H2 purification, J. Membr. Sci., 2025, 29, 123789 CrossRef.
- B. P. Debata, M. D. Thiyagarajan, R. Prathap, J. D. Girase, S. K. Iyer, S. Patel and S. Vaidyanathan, Benzil-imidazole blue fluorophores and their applications in blue/white light-emitting diodes, sensing and anticounterfeiting, J. Mater. Chem. C, 2025, 13, 2711–2731 RSC.
- S. Singh, K. Mandal and M. Chakravarty, Tetra-benzimidazoles flanking divinyl-phenothiazine: AIEgens as aza-Michael acceptors in concentration-tuned responses to biogenic amine vapors, Chem. Commun., 2025, 61, 728–731 CAS.
- P. Sunani, P. Thiruvengetam and D. K. Chand, A Double Chain Based Metallomicellar Catalyst for Aerobic Oxidative Synthesis of Benzimidazoles in Water, Dalton Trans., 2025, 54, 3704–3713 CAS.
- M. Manjunath, F. H. Sujata, A. H. Shridhara, B. Vinay Kumar, K. Prashantha, K. Yogendra and N. Madhusudhana, Sustainable synthesis of benzimidazole-based Schiff base using reusable CaAl2O4 nanophosphors catalyst: Insights into metal (II) complexes and DNA interactions, Nucleosides, Nucleotides Nucleic Acids, 2025, 1–23 CrossRef CAS PubMed.
- S. K. Bera, M. Irfan and A. Porcheddu, An Efficient Strategy for Quinazolinone and Benzimidazole Synthesis Using Microwave-Assisted Copper-Catalyzed Aerobic Oxidation of Amines, ChemCatChem, 2025, 202401813 CrossRef.
- U. D. Kadam, R. S. Pandav, S. M. Arde, M. S. Hossain, A. Dinesh, K. Radhakrishnan, M. Ayyar, R. P. Patil and B. S. Shirke, Green synthesis of benzimidazole scaffolds using copper-substituted zinc aluminate in a sol-gel process, J. Indian Chem. Soc., 2025, 102, 101494 CrossRef CAS.
- G. Zhang, D. Xian, S. Hussain, B. Zhang, W. Lin, X. Xiang and L. Wang, Enhancing lithium metal battery safety and performance: a thermally stable poly (aryl ether benzimidazole) separator with 2D-functionalized boron nitride for 3000 hours lithium plating/stripping, J. Mater. Chem. A, 2025, 13 10.1039/D4TA08225G.
- Q. Zhou, S. Hussain, J. Peng, B. Zhang and L. Wang, Multi-channeled halloysite nanotube-blended polybenzimidazole separators for enhancing lithium-ion battery performance, J. Membr. Sci. Res., 2025, 714, 123417 CrossRef CAS.
- S. Duan, H. Xu, J. Zhang, M. Shan, S. Zhang, Y. Zhang, X. Wang and F. Kapteijn, Benzimidazole-linked polymer membranes for efficient syngas (H2/CO/CO2) separation, J. Membr. Sci., 2025, 717, 123595 CrossRef CAS.
- F. Fei and Z. Zhou, New substituted benzimidazole derivatives: a patent review (2010–2012), Expert Opin. Ther. Pat., 2013, 23, 1157–1179 CrossRef CAS PubMed.
- M. Wang, X. Han and Z. Zhou, New substituted benzimidazole derivatives: a patent review (2013–2014), Expert Opin. Ther. Pat., 2015, 25, 595–612 CrossRef CAS PubMed.
- C. S. W. Law and K. Y. Yeong, Benzimidazoles in Drug Discovery: A Patent Review, ChemMedChem, 2021, 16, 1861–1877 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08864f |
| This journal is © The Royal Society of Chemistry 2025 |