
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Semi-synthesis and biological activities of heterocyclic compounds containing camphor
Zeinab S. Hamzaa,
Wael N. Hozzeinb,
Khaled El-Adlcd,
Mohammad K. Oklae,
Gehad AbdElgayedf,
Wael A. A. Fadalyg and
Hussein S. Mohamed *a
*a
aChemistry of Medicinal and Aromatic Plants Department, Research Institute of Medicinal and Aromatic Plants (RIMAB), Beni-Suef University, Beni-Suef 62511, Egypt. E-mail: h_gendy_2010@yahoo.com; Husseinshaban@rimp.bsu.edu.eg; Tel: +201000800296
bBotany and Microbiology Department, Faculty of Science, Beni-Suef University, Beni-Suef, Egypt
cChemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
dPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City 11884, Cairo, Egypt
eBotany and Microbiology Department, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
fIntegrated Molecular Plant Physiology Research, Department of Biology, University of Antwerp, Antwerp, 2020, Belgium
gPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt
First published on 25th April 2025
Abstract
Breast cancer and lung cancer are two of the most prevalent and deadly malignancies worldwide. Both cancers present significant challenges in terms of effective treatment and management, highlighting the urgent need for novel therapeutic strategies that can improve patient outcomes. This study focuses on the synthesis of novel heterocyclic compounds derived from the naturally formed camphor, aimed at evaluating their cytotoxicity. The research addresses the need for effective cancer treatments by presenting compounds that demonstrate significant inhibitory effects against MCF-7 breast carcinoma cells. Among these, compound 20 exhibited remarkable potency, with an IC50 value of 0.78 μM, surpassing the efficacy of standard chemotherapeutics, dasatinib (IC50 = 7.99 μM) and doxorubicin (IC50 = 3.10 μM). In the context of A549 lung cancer cells, compound 20 also showed strong inhibitory activity (IC50 = 1.69 μM), again outperforming dasatinib (IC50 = 11.8 μM) and doxorubicin (IC50 = 2.43 μM). To further elucidate the biological activities of these compounds, molecular docking studies were performed, revealing that compound 20 exhibited the highest binding energy among the tested compounds, supporting the experimental findings. These results indicate that the synthesized camphor-derived heterocycles, particularly compound 20, have significant potential as potent anticancer agents against breast and lung cancer cell lines.
1. Introduction
Breast cancer and lung cancer are two prevalent types of cancer that significantly impact the health and well-being of individuals worldwide. Breast cancer is a type of cancer that arises inside the tissue of the breast, typically originating in the milk ducts or lobules of the breast.1,2 It is the most prevalent cancer among women worldwide, although it can also affect men.3,4 Breast cancer treatment options rely on multiple factors, involving the cancer stage, tumor characteristics, and one's health in general.5–7 Standard treatment options encompass surgical procedures (such as lumpectomy or mastectomy), chemotherapy, radiation, targeted therapy, and hormonal therapy.8–11 With advancements in early detection and treatment strategies, the outlook for carcinoma of the breast has markedly improved in recent years.12,13A lung tumor is a malignancy that arises in the lungs, typically in the cells that line the airways.14 It affects both sexes and is the main reason for cancer-related fatalities globally.15,16 The primary cause of lung cancer is tobacco smoke, including second-hand smoke inhalation and cigarettes, cigars, and tobacco pipe smoking.17–20 Surgery, therapy with radiation, treatment with chemotherapy, targeted therapy, and immunotherapy constitute typical forms of treatment.21,22 Targeted therapies for lung cancer often focus on specific genetic mutations, such as EGFR, ALK, ROS1, and others, which can drive the growth of cancer cells. It is noteworthy that the prognosis for both breast cancer and lung cancer is improved by early detection and prompt medical care.23
EGFR (Epidermal Growth Factor Receptor) enzyme inhibition refers to the suppression or blocking of the activity of the EGFR enzyme.24 The receptor tyrosine kinase EGFR is important for cell proliferation,25 proliferation, and survival.26 Abnormal activation or overexpression of EGFR The development and progression of certain cancer types, including as colorectal, lung, and cancers of the neck and head, are frequently linked to it.27 Inhibition of EGFR activity can be achieved through different approaches, primarily using targeted therapies known as EGFR inhibitors.28 EGFR inhibitors have shown clinical efficacy in certain cancers that exhibit EGFR abnormalities. They are commonly used in EGFR mutation therapy for non-small cell lung cancer (NSCLC) and metastatic colorectal cancer with EGFR overexpression.29 These targeted therapies can be used as monotherapy or in conjunction with additional treatment approaches, such radiation therapy or chemotherapy, based on the particular malignancy and its features.
Camphor is a waxy, white, or transparent solid produced from turpentine oil or from the bark that comes from the plant Cinnamomum camphora.30 It has a distinct, aromatic odor and a cooling sensation when applied to the skin.31 C. camphora has a long history of use in various cultures and is utilized for its medicinal, aromatic, and pest-repellent properties. In traditional medicine, C. camphora has been employed for its potential therapeutic effects.32 Cinnamomum camphora is a species endemic to China and is highly valued as a natural spice plant in the country.33,34 The leaves of it contain a significant amount of essential oil (3.8–4.5%), consisting of more than 40 components, including 1,8-cineole, γ-terpinene, terpinene-4-ol, α-terpineol, and safrole.35 Research has shown that the essential oil derived from the leaves of it exhibits various biological activities, including antimicrobial36–38 effects, antitumor actions,39–41 and analgesic properties.42 In addition to its medicinal uses, C. camphora has been utilized as an aromatic ingredient in perfumes, soaps, and other personal care products.43 Overall, C. camphora is a natural plant with a rich history of traditional use.30
Thiazoles are an important group of compounds due to their diverse chemical and biological properties.44 Their chemical structure and reactivity make them versatile building blocks for the synthesis of complex organic molecules.45,46 Thiazole derivatives often exhibit interesting biological activities, such as antimicrobial, antiviral, anticancer, and anti-inflammatory properties.47 Studies have shown that certain thiazole derivatives exhibit promising anticancer activity against breast and lung cancer cells in vitro (in cell cultures) and in vivo (in animal models).48
Prymidinone and pyrimidinthione are synthetic compounds that belong to the class of pyrimidine derivatives.49,50 Several derivatives are being explored for their ability to inhibit specific kinases involved in cancer progression, such as The receptors for vascular endothelial growth factor (VEGFR) and epidermal growth factor (EGFR).51,52 These inhibitors may enhance targeted cancer therapies.
Pyrazole derivatives have emerged as a promising class of compounds in cancer research due to their diverse biological activities, including anti-cancer properties.53,54 Many pyrazole derivatives can trigger cancer cells that undergo programmed cell death, promoting apoptosis and thereby reducing tumor size and preventing metastasis.52,55 Some pyrazole derivatives function as kinase inhibitors, targeting specific enzymes involved in tumor growth and survival, such as EGFR, VEGFR, and CDK. This makes them valuable in targeted cancer therapies.56
Dasatinib is a potent and selective tyrosine kinase inhibitor (TKI) primarily utilized to treat some cancers, including acute lymphoblastic leukemia (ALL) and chronic myeloid leukemia (CML).57 The U.S. Food and Drug Administration (FDA) gave its approval in 2006,58 Dasatinib was designed to suppress the BCR-ABL fusion protein's activity, which is a characteristic of leukemias with the Philadelphia chromosome.59
Doxorubicin, also known as Adriamycin, is a widely used anthracycline antibiotic and chemotherapeutic agent that is essential to the treatment of many types of cancer.60 First isolated from the bacterium Streptomyces peucetius in the 1960s,61 Doxorubicin has since become a cornerstone in oncology due to its broad-spectrum efficacy against solid tumors and hematological malignancies.62
In summary, the pressing challenges posed by breast and lung cancer necessitate the continued exploration of novel compounds, such as camphor-derived heterocycles, to enhance treatment efficacy and improve patient outcomes. This study aims to contribute to this effort by investigating the potential cytotoxicity of newly synthesized compounds (Fig. 1).
 | ||
| Fig. 1 Structure of compounds has anticancer activities, (1–3) are thiazole derivatives, (4–6) are pyrimidinone derivatives and (7–10) are pyrazole derivatives. | ||
2. Rationale and structure design
Our structures exhibited the necessary requirements of EGFR inhibitors (Fig. 2 and 3). The bio-isosteric replacements were at four points of erlotinib as EGFR inhibitor: (1) a hydrophobic tail (occupy hydrophobic region II), (2) HBD NH group and/or HBA, (3) a hydrophobic domain (occupy hydrophobic area I), and (4) a flat heteroaromatic ring which inhabits the adenine site (Fig. 2 and 3)62–64 | ||
| Fig. 2 Essential necessities for erlotinib as reference TK inhibitor. | ||
 | ||
| Fig. 3 Essential necessities for EGFR inhibitors. | ||
3. Results and discussion
3.1. Chemistry
The synthetic pathway for derivatives 6–20 is illustrated in Scheme 1–4. Starting with a mixture of camphor 1 with thiourea 2 in the presence of potassium carbonate which heated under reflux with the appropriate malononitrile, ethyl cyanoacetate and/or ethyl acetoacetate to give the corresponding derivatives 6, 7 and 8 respectively (Scheme 1). Moreover, cyclocondensation of the same components but using urea instead of thiourea resulted in the corresponding derivatives 10, 11 and 12 respectively (Scheme 2). In addition, condensation of camphor with thiosemicarbazide 13 yielded the corresponding thiosemicarbazone 14 that underwent cyclocondensation with 2-bromo-1-(substituted) ethan-1-one to give the consistent derivatives 16a–c correspondingly (Scheme 3). Finally, the condensation reaction of camphor with hydrazine hydrate 17 produced the corresponding Schiff's base 18 which also condensed with the aldehyde derivative 19 to afford the corresponding derivative 20 respectively (Scheme 4). | ||
| Scheme 1 Synthesis of compounds 6–8. | ||
 | ||
| Scheme 2 Synthesis of compounds 10–12. | ||
 | ||
| Scheme 3 Synthesis of compounds 16a–c. | ||
 | ||
| Scheme 4 Synthesis of compound 20. | ||
3.2. Biological activities
MCF-7 Adenocarcinoma of the breast in humans and A549 small lung cancer lines of cells (purchased from Labco's Sigma-Aldrich) were employed to evaluate the newly synthesized compounds' anti-cancer potential. The IC50 values of these compounds were assessed using the MTT test for cell viability as shown in Table 1. The compounds demonstrated significant inhibitory activity against MCF-7, with IC50 values of 0.78 and 19.92 μM, compared to dasatinib (IC50 = 7.99 μM), erlotinib (IC50 = 13.91 μM) and doxorubicin (IC50 = 3.10 μM) as shown in Fig. 4a. Notably, compound 20 exhibited excellent inhibitory activity (IC50 = 0.78 μM), proving to be more potent than standard drugs, dasatinib (IC50 = 7.99 μM), erlotinib (IC50 = 13.91 μM) and doxorubicin (IC50 = 3.10 μM). In contrast, the compounds showed moderate inhibitory effects against A549 cells, with IC50 values ranging from 1.69 to 56.6 μM, compared to dasatinib (IC50 = 11.8 μM), erlotinib (IC50 = 5.49 μM) and doxorubicin (IC50 = 2.43 μM). Similarly, compound 20 displayed strong inhibitory activity (IC50 = 1.69 μM), again surpassing the standard drugs, dasatinib (IC50 = 11.8 μM), erlotinib (IC50 = 5.49 μM) and doxorubicin (IC50 = 2.43 μM) as shown in Fig. 4.| Compound | Cytotoxicity IC50 (μM)a | |
|---|---|---|
| MCF-7 | A549 | |
| a IC50 values are the mean ± SD of three experiments separately. | ||
| 6 | 19.92 ± 0.73*** | 28.47 ± 0.99**** |
| 7 | 10.11 ± 0.39**** | 22.24 ± 0.77**** |
| 8 | 6.15 ± 0.23 | 3.34 ± 0.12 |
| 10 | 12.66 ± 0.47**** | 23.27 ± 0.81**** |
| 11 | 35 ± 1.32**** | 56.6 ± 2.01**** |
| 12 | 3.20 ± 0.12**** | 14.38 ± 0.5**** |
| 16a | 2.16 ± 0.08**** | 4.13 ± 0.14 |
| 16b | 8.29 ± 0.3 | 15.25 ± 0.53**** |
| 16c | 2.84 ± 0.1**** | 1.29 ± 0.04 |
| 20 | 0.78 ± 0.03**** | 1.69 ± 0.06 |
| Dasatinib | 7.99 ± 0.29 | 11.81 ± 0.41 |
| Erlotinib | 13.91 ± 1.3 | 5.49 ± 0.45 |
| Doxorubicin | 3.12 ± 0.11 | 2.43 ± 0.08 |
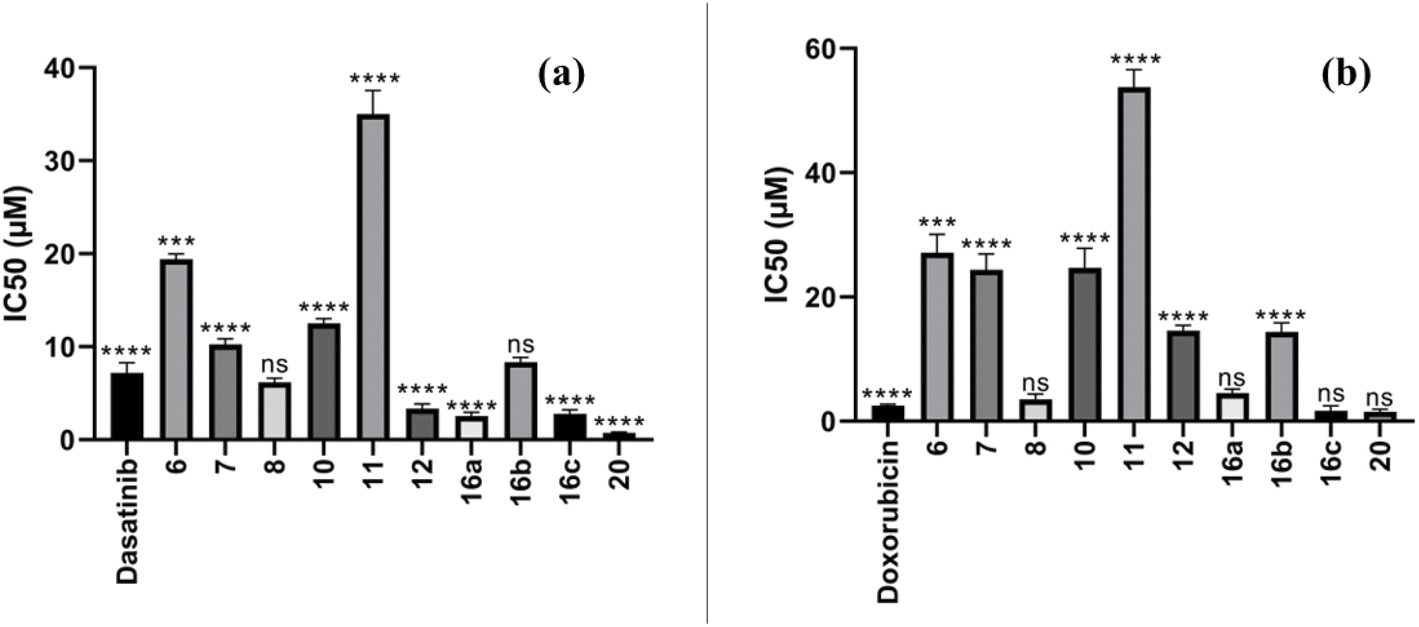 | ||
| Fig. 4 IC50 of the synthesized compounds (a) against of MCF-7 and (b) against of A549 cell lines and reference drugs dasatinib, erlotinib & doxorubicin. Values are mean ± SD. Asterisks indicate significant differences from the respective control (dasatinib for MCF-7 and doxorubicin for A549) as revealed by one-way ANOVA test, *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. | ||
3.3. EGFR kinase inhibitory activity
The EGFR (Catalog # 40321) Kinase Assay Kit was utilized to assess the EGFR kinase inhibitory activity of compound 20 as shown in Fig. 5. Five different concentrations were evaluated in this assay, with erlotinib and dasatinib serving as reference drugs for determining the IC50. The results indicated that compound 20 exhibited enhanced inhibitory activity against EGFRWT, with an IC50 of 0.198 μM, which is comparable to erlotinib and dasatinib's IC50 of 0.15 and 0.041 μM respectively. | ||
| Fig. 5 IC50 values of compound (20) compared to dasatinib on EGFR. | ||
3.4. Molecular docking
For docking studies, Molsoft program was used. The ID of the PDB for EGFRWT wild-type (PDB ID 4HJO) 65 was used.Erlotinib in its normal form shows binding energy = −92.64 kcal mol−1. Two hydrogen bonds were formed with Cys773 (2.95 Å) and Met769 (2.00 Å). The 3-ethynylphenyl head occupied the hydrophobic region I, which was produced by Val702, Asp831, Lys721, Ile765, Thr766, Ala719, and Thr830. Additionally, the hydrophobic furrow II formed by Leu694, Asp776, Cys773, Leu820, Gly772, Phe771, Met769, and Pro770 were occupied by the 2-methoxyethoxy side chains as shown in (Fig. 6).
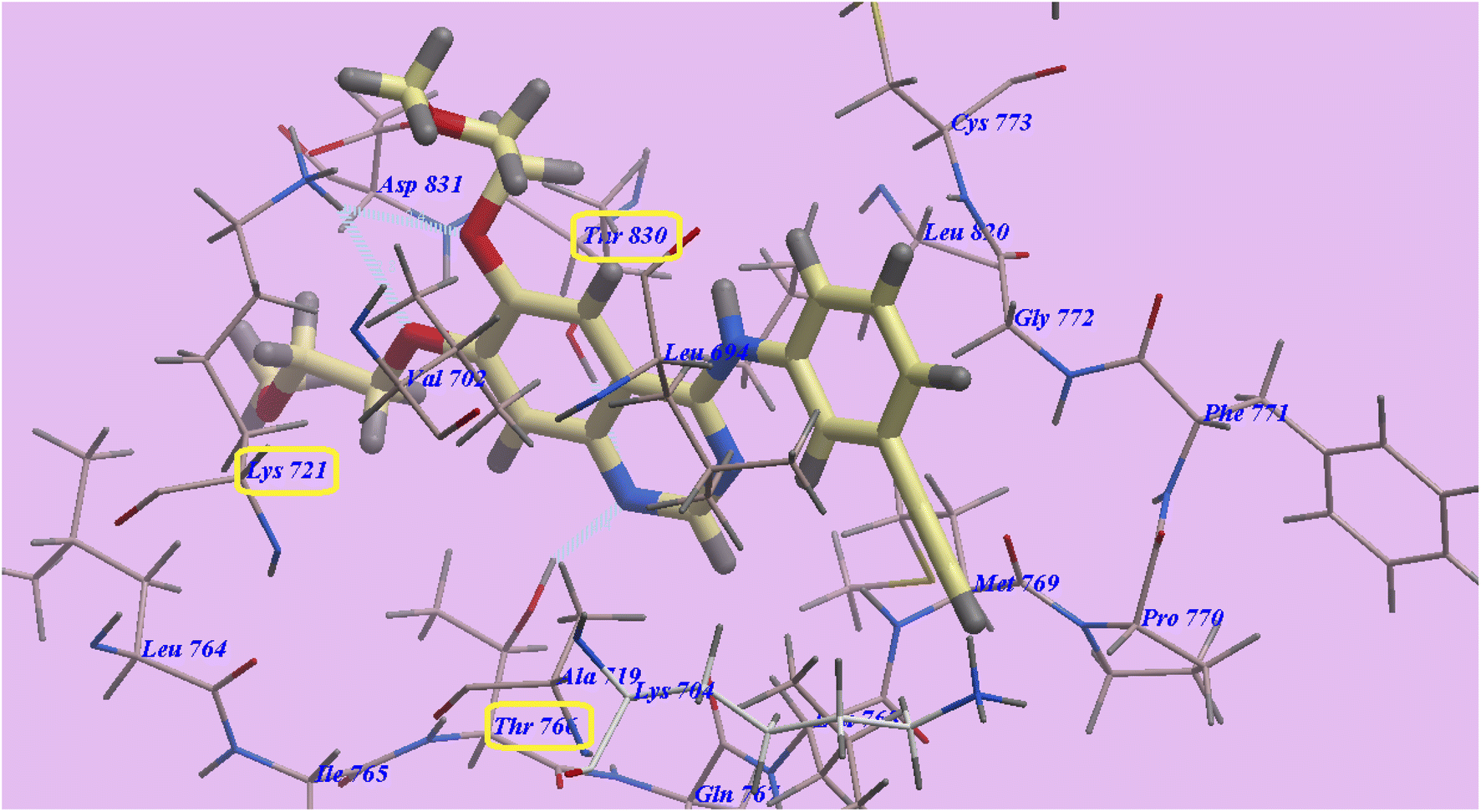 | ||
| Fig. 6 Docking of erlotinib in 4HJO active site. | ||
Compound 20 exhibited nearly the same pattern as that of erlotinib with affinity of −150.65 kcal mol−1 and made 6 H-bonds with Asp837 (2.08 Å), Thr854 (2.97 Å), Met793 (1.74 Å), Gln791 (2.56 Å), Glu762 (2.22 Å) and Lys875 (2.06 Å) (Fig. 7).
 | ||
| Fig. 7 Docking of compound 20 in 4HJO. | ||
3.5. Validation of the accuracy of docking
The re-docked erlotinib seems exactly superimposed on the co-crystallized native bound one (Fig. 8). These results indicated the high accuracy of the docking simulation in comparison with the biological methods.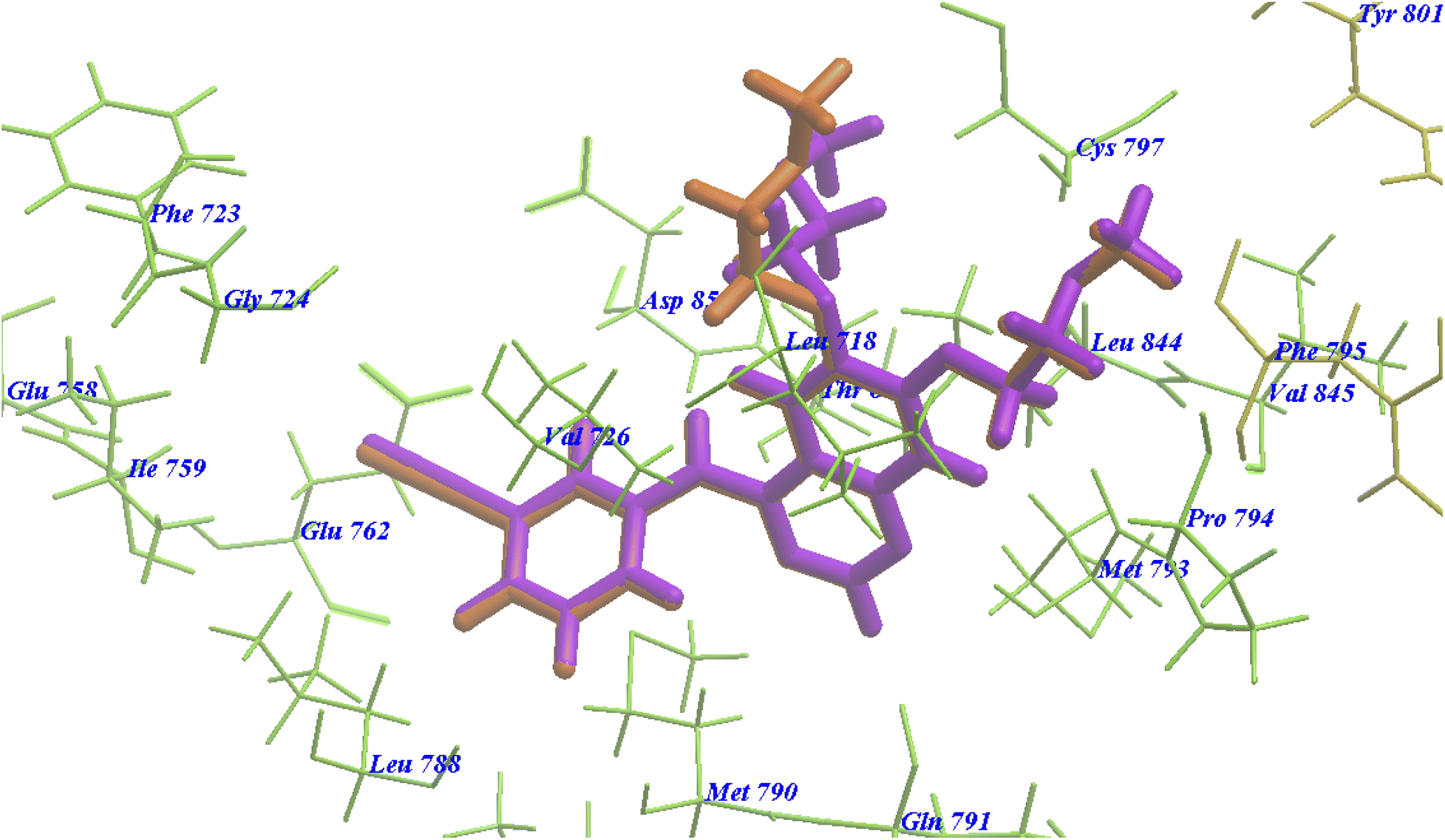 | ||
| Fig. 8 Superimposition of erlotinib on the co-crystallized native bound one with 4HJO. | ||
3.6. Pharmacokinetics and ADME analysis
In drug development, bioavailability is a factor that must be considered in clinical applications. It is affected by the different physicochemical properties of compounds.66The most active compounds and the standard dasatinib and Erlotinib were subjected to view physicochemical properties and drug likeness using the SwissADME site free web tool, developed by the Molecular Modelling Group of the Swiss Institute of Bioinformatics.
In particular, the compliance of compounds to traditional Lipinski's “rule of five” was calculated to determine if the compound can be orally active in human.67 This simple rule states that orally active drug has no more than one violation of the following criteria: molecular weight (MW) less than 500 Da; no more than 10 hydrogen bond acceptors (HBA); no more than five hydrogen bond donors (HBD); and calculated octanol–water partition coefficient (i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP) not greater than 573. Also, the topological polar surface area (TPSA) and the number of rotatable bonds is other critical properties that have been linked to drug bioavailability.68 The reports suggested that compounds with a TPSA of more than 140 Å2 and more than 10 rotatable bonds are thought to have low pharmacological flexibility and permeability, respectively.69 All of the tested compounds have a number of hydrogen bond donor atoms of 1–3 and hydrogen bond acceptor atoms of 2–7 compared to 3 hydrogen bond donors and 7 hydrogen bond acceptors in the standard dasatinib. The number of rotatable bonds of the tested compounds ranged from 0–5 bonds, while dasatinib contains 8 bonds. The molar refractivity (MR) value should be between 40 and 130 for good absorption and oral bioavailability. Acceptable molar refractivity values, in combination with the number of rotatable bonds, indicate that substances have adequate intestinal absorption and oral bioavailability.69 The designed compound's MR values range from 76.56 to 143.35 m3 mol−1, compared to the standard dasatinib (MR 133.62 m3 mol−1). This indicates that most of the proposed compounds (except compound 6 and the standard dasatinib) may have low gastrointestinal absorption and oral bioavailability. Also, the proposed derivatives' TPSA values were range from 68.75 Å2 to 111.08 Å2 compared to TPSA value of dasatinib 134.75 Å2. However, compounds showed TPSA values lower than the standard range. It is worth mentioning that compounds with TPSA values lower than 60 Å2 can penetrate the BBB easily.69 That's why all the proposed derivatives and the standard dasatinib cannot penetrate the blood–brain barrier well, as evidenced by the BBB determination. In addition, the implicit logP (IlogP) n-octanol/water partition coefficients were known to play a critical role in medication absorption in the mouth, as well as facilitating drug interactions with their biological targets.67 The estimated values of IlogP were found to be less than five (1.47–4.00), as recommended by Lipinski's rule of five and compared to dasatinib (3.15). Moreover, the estimated aqueous solubility of the tested compounds Log S (ESOL) ranged from −2.51 to −50.90 compared to −4.84 of dasatinb, indicating their low aqueous solubility (soluble compounds have logS < −4). Furthermore, studying the inhibitory activity of proposed derivatives against a certain CYP isoform becomes a critical factor.67 While all of the target compounds and dasatinib were found not to inhibit CYP1A2. The derivatives 16b, 16c, 20 and the standard dasatinib were anticipated to inhibit CYP2C19 and The derivatives 7, 8, 16b, 16c, 20 and the standard dasatinib were anticipated to inhibit CYP2C9 enzymes which means they are lower exposed to inhibitory drug metabolism.
logP) not greater than 573. Also, the topological polar surface area (TPSA) and the number of rotatable bonds is other critical properties that have been linked to drug bioavailability.68 The reports suggested that compounds with a TPSA of more than 140 Å2 and more than 10 rotatable bonds are thought to have low pharmacological flexibility and permeability, respectively.69 All of the tested compounds have a number of hydrogen bond donor atoms of 1–3 and hydrogen bond acceptor atoms of 2–7 compared to 3 hydrogen bond donors and 7 hydrogen bond acceptors in the standard dasatinib. The number of rotatable bonds of the tested compounds ranged from 0–5 bonds, while dasatinib contains 8 bonds. The molar refractivity (MR) value should be between 40 and 130 for good absorption and oral bioavailability. Acceptable molar refractivity values, in combination with the number of rotatable bonds, indicate that substances have adequate intestinal absorption and oral bioavailability.69 The designed compound's MR values range from 76.56 to 143.35 m3 mol−1, compared to the standard dasatinib (MR 133.62 m3 mol−1). This indicates that most of the proposed compounds (except compound 6 and the standard dasatinib) may have low gastrointestinal absorption and oral bioavailability. Also, the proposed derivatives' TPSA values were range from 68.75 Å2 to 111.08 Å2 compared to TPSA value of dasatinib 134.75 Å2. However, compounds showed TPSA values lower than the standard range. It is worth mentioning that compounds with TPSA values lower than 60 Å2 can penetrate the BBB easily.69 That's why all the proposed derivatives and the standard dasatinib cannot penetrate the blood–brain barrier well, as evidenced by the BBB determination. In addition, the implicit logP (IlogP) n-octanol/water partition coefficients were known to play a critical role in medication absorption in the mouth, as well as facilitating drug interactions with their biological targets.67 The estimated values of IlogP were found to be less than five (1.47–4.00), as recommended by Lipinski's rule of five and compared to dasatinib (3.15). Moreover, the estimated aqueous solubility of the tested compounds Log S (ESOL) ranged from −2.51 to −50.90 compared to −4.84 of dasatinb, indicating their low aqueous solubility (soluble compounds have logS < −4). Furthermore, studying the inhibitory activity of proposed derivatives against a certain CYP isoform becomes a critical factor.67 While all of the target compounds and dasatinib were found not to inhibit CYP1A2. The derivatives 16b, 16c, 20 and the standard dasatinib were anticipated to inhibit CYP2C19 and The derivatives 7, 8, 16b, 16c, 20 and the standard dasatinib were anticipated to inhibit CYP2C9 enzymes which means they are lower exposed to inhibitory drug metabolism.
4. Materials and methods
4.1. Chemistry
4.2. Experimental procedures and spectral data for compounds (6–12)
A reaction mixture consisting of camphor (0.1 mole), potassium bicarbonate (0.01 mole), the relevant malononitrile, ethyl cyanoacetate and/or ethyl acetoacetate (0.01 mole), and thiourea and/or urea (0.01 mole) was prepared in 30 ml of absolute ethanol and subjected to reflux for 4 hours. The resulting precipitate was collected by filtration and subsequently dried upon cooling. Physical and spectral data for them are listed below.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1540 (CN), 1114 (CS), 611 (S); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, 3CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H20N4S (276.14): C, 60.84; H, 7.29; N, 20.27; S, 11.60. Found C = 60.85; H = 7.30; N, 20.25; S = 11.62.O), 1401 (CN), 1114 (CS), 612 (S) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H19N3OS (277.12): C, 60.62; H, 6.90; N, 15.15; O, 5.77; S, 11.56. Found C = 60.62; H = 6.90; N= 15.15; O = 5.77; S = 11.56.O), 1141 (CS), 610 (S) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H22N2OS (278.15): C, 64.71; H, 7.96; N, 10.06; O, 5.75; S, 11.52. Found C = 64.71; H = 7.96; N= 10.06; O = 5.75; S = 11.52.O), 1402 (CN), 1114 (CO) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H20N4O (272.16): C, 66.15; H, 7.40; N, 20.57; O, 5.87. Found C = 66.15; H = 7.40; N= 20.57; O = 5.87.O), 1362 (CN) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H19N3O2 (261.15): C, 64.35; H, 7.33; N, 16.08; O, 12.25. Found C = 64.35; H = 7.33; N= 16.08; O = 12.25.O) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H22N2O3 (278.16): C, 64.73; H, 7.97; N, 10.06; O, 17.24. Found C = 64.73; H = 7.97; N= 10.06; O = 17.24.
N), 1540 (CN), 1114 (CS), 611 (S); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, 3CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H20N4S (276.14): C, 60.84; H, 7.29; N, 20.27; S, 11.60. Found C = 60.85; H = 7.30; N, 20.25; S = 11.62.O), 1401 (CN), 1114 (CS), 612 (S) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H19N3OS (277.12): C, 60.62; H, 6.90; N, 15.15; O, 5.77; S, 11.56. Found C = 60.62; H = 6.90; N= 15.15; O = 5.77; S = 11.56.O), 1141 (CS), 610 (S) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H22N2OS (278.15): C, 64.71; H, 7.96; N, 10.06; O, 5.75; S, 11.52. Found C = 64.71; H = 7.96; N= 10.06; O = 5.75; S = 11.52.O), 1402 (CN), 1114 (CO) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H20N4O (272.16): C, 66.15; H, 7.40; N, 20.57; O, 5.87. Found C = 66.15; H = 7.40; N= 20.57; O = 5.87.O), 1362 (CN) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C14H19N3O2 (261.15): C, 64.35; H, 7.33; N, 16.08; O, 12.25. Found C = 64.35; H = 7.33; N= 16.08; O = 12.25.O) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C15H22N2O3 (278.16): C, 64.73; H, 7.97; N, 10.06; O, 17.24. Found C = 64.73; H = 7.97; N= 10.06; O = 17.24.4.3. Experimental procedures and spectral data for compounds (16a–c)
Camphor-thiosemicarbazone was synthesized following the method described by Brousse BN et al.,70 This process entails the reaction of camphor with thiosemicarbazide in heated ethanol, accompanied by a few drops of sulfuric acid. A mixture of camphor-thiosemicarbazone (0.1 mole) and phenacyl bromide derivatives (0.1 mole) in 30 ml of absolute ethanol, in the presence of sodium acetate, was refluxed for 24 hours. The resulting precipitate was collected by filtration and dried after cooling. Physical and spectral data for them are listed below.C), 1404 (CN), 614 (S) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C20H25N3S (339.18): C, 70.76; H, 7.42; N, 12.38; S, 9.44. Found C = 70.76; H = 7.42; N= 12.38; S = 9.44.C), 1560 (CN), 613 (Br) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C19H22ClN3S (359.12): C, 63.40; H, 6.16; Cl, 9.85; N, 11.67; S, 8.91. Found C = 63.40; H = 6.16; Cl = 9.85; N= 11.67; S = 8.91.N), 1540 (CN), 613 (Cl) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C19H22BrN3S (403.07): C, 56.43; H, 5.48; Br, 19.76; N, 10.39; S, 7.93 Found C = 56.43; H = 5.48; Br = 19.76; N = 10.39; S = 7.93.4.4. Experimental procedures and spectral data for compound (20)
Camphor (0.1 mole) and hydrazine hydrate (0.1 mole) were dissolved in 20 ml of ethanol and transferred to a round-bottom flask. The reaction mixture was acidified with glacial acetic acid and subsequently refluxed in a water bath for 5 hours. Upon cooling, the residue was isolated, filtered, dried, and recrystallized from a 1:1 mixture of benzene and hexane, yielding a colorless crystalline powder.71 A mixture of camphor-hydrazone (0.1 mole) and brominated aldehyde (0.1 mole) in 30 ml of absolute ethanol was refluxed for 20 hours. The precipitate formed upon cooling was then filtered and dried at room temperature, resulting in the production of compound 20. Physical and spectral data for them are listed below.
O), 1402 (CN), 1129 (C–N), 616 (Br) cm−1); 1H NMR (DMSO-d6) at δ = 0.9–1.5 (s, 9H, CH3), 1.6–2.4 (m, 6H, aliphatic protons), 7.45 (s, 1H, NH), 9.2 (s, 1H, CNH), 10.32 (s, 1H, NH), 13C-NMR (100 MHz) (DMSO-d6) at δ = 18.02, 18.23, 18.96, 21.43, 31.9, 35.2, 37.08, 39.9, 41.12, 41,45, 47.33, 118.22, 156.3, 182.1 anal. calcd. For C26H28BrN5O2S (553.11): C, 56.32; H, 5.09; Br, 14.41; N, 12.63; O, 5.77; S, 5.78. Found C = 56.32; H = 5.09; Br = 14.41; N= 12.63; O = 5.77; S = 5.78.4.5. Biological activities
For the MTT assay, cells were plated at a density of 1.2 to 1.8 × 10000 cells per well in a total volume of 200 μl, comprising 100 μl of a full growing medium and 100 μl of the substance that was evaluated in each well. The cells were then incubated for 24 hours.
4.6. Cell culture steps
The culture medium should be transferred to a centrifuge tube. To get rid of serum, quickly rinse the cell layer with a solution of 0.53 mM EDTA and 0.25% (w/v) trypsin, which contains trypsin inhibitors. When the cell layer is fully scattered, which usually happens in 5 to 15 minutes, look at the cells under an inverted microscope after adding 2.0 to 3.0 ml of the Trypsin–EDTA solution to the flask.Note: To prevent clumping, avoid agitating the flask while the cells detach. If detachment proves difficult, incubate the flask at 37 °C to facilitate the process.
Aspirate the cells gently using a pipette after adding 6.0 to 8.0 ml of full growth media. Put the cell suspension and the first step's medium in the centrifuge tube, then centrifuge for five to ten minutes at about 125 × g. Transfer the proper aliquots to new culture vessels, discard the supernatant, and resuspend the cell pellet in fresh growth media. For a full day, incubate the cultures at 37 °C. After the chemical has been treated with repeated concentrations, incubate for a further 48 hours at 37 °C. To perform the MTT assay, examine the plates using an inverted microscope.
4.7. MTT – cytotoxicity assay protocol
The MTT method is highly effective for evaluating in vitro cytotoxicity in multi-well plates. For best results, utilize cells in the logarithmic phase of growth, ensuring that the final cell density does not surpass 106 cells per cm2. Additionally, incorporate a blank control containing complete medium without any cells. Remove the cultures from the incubator and transfer them to a laminar flow hood or sterile workspace. Reconstitute each vial of MTT [M-5655] with 3 ml of medium or balanced salt solution that is free from phenol red and serum. Add the reconstituted MTT in an amount equal to 10% of the total volume of the culture medium. Return the cultures to the incubator for 2 to 4 hours, depending on the cell type and maximum cell density; typically, 2 hours is adequate, though longer incubation may be required for low-density or less metabolically active cells. It is important to maintain consistent incubation times for accurate comparisons.After incubation, remove the cultures from the incubator and dissolve the formed formazan crystals by adding an amount of MTT solution [M-8910] equivalent to the original culture medium volume. Gentle mixing with a gyratory shaker will facilitate the dissolution of the crystals. In denser cultures, pipetting up and down may be necessary to ensure complete dissolution of the MTT formazan crystals. Measure the absorbance spectrophotometrically at 570 nm and assess the background absorbance of the multi-well plates at 690 nm, which should be subtracted from the 570 nm measurement. The tests in multi-well plates can be read using an appropriate plate reader, or the contents can be transferred to cuvettes for measurement.
4.8. EGFR kinase inhibitory activity protocol
The EGFR (Catalog # 40321) Kinase Assay Kit was employed to assess the EGFR kinase inhibitory activity of compound 20. Begin by thawing ATP, Poly (Glu:Tyr 4:1), and PTK substrate (10 mg ml−1) in 5× Kinase Buffer 1. Optionally, to achieve a 10 mM concentration, add DTT to the 5× Kinase Buffer 1 (for example, by adding 10 μl of 1 M DTT to 1 ml of 5× Kinase Buffer 1).
Prepare the primary mixture (25 μl per well) by combining 6 μl of 5× Kinase Buffer 1, 1 μl of ATP (500 μM), 1 μl of PTK substrate Poly (Glu:Tyr 4:1) (10 mg ml−1), and 17 μl of water. Distribute this mixture into N wells, adding 25 μl to each well. Then, introduce 5 μl of the “Test Inhibitor” solution into the appropriate well. For the “Positive Control” and “Blank” samples, add 5 μl of the same solution without the inhibitor (Inhibitor buffer).
To prepare 3 ml of 1× Kinase Buffer 1, combine 2400 μl of water with 600 μl of 5× Kinase Buffer 1. This 3 ml of 1× Kinase Buffer 1 is sufficient for 100 reactions. For the “Blank” wells, add 20 μl of 1× Kinase Buffer 1. Thaw the EGFR enzyme on ice, and once the tube is fully defrosted, briefly spin it to collect all contents. Determine the required amount of EGFR for the assay, then dilute the enzyme in 1× Kinase Buffer 1 to achieve a concentration of 1 ng μl−1. Any remaining undiluted enzyme should be aliquoted and stored at −80 °C. Note that the EGFR enzyme is sensitive to freeze-thaw cycles, so these should be minimized. Reusing thawed aliquots or diluted enzymes is not recommended.
To initiate the reaction, add 20 μl of the diluted EGFR enzyme to both the “Positive Control” and “Test Inhibitor Control” wells. Incubate the mixture for 40 minutes at 30 °C. After this incubation, thaw the Kinase-Glo Max solution. Following the 40-minutes reaction, add 50 μl of the Kinase-Glo Max reagent to each well. Cover the plate with aluminum foil and incubate for an additional 15 minutes at room temperature. Finally, measure luminescence using a microplate reader.
4.9. Molecular docking
Molsoft program was used for docking studies. The protein of EGFRWT wild-type (PDB ID 4HJO) 65 was used. The protein was obtained from the Brookhaven Protein Databank. It was prepared for docking by adding polar hydrogens to its atoms. The protein binding site was defined by overlaying a grid on the center of the co-crystallized ligand. Before conducting docking simulations, all requisite grid maps were calculated. The synthesized compounds were constructed as 3D structures, and their energies were minimized. The co-crystallized ligand was removed from the binding sites, allowing for the docking of the compounds discussed herein into those sites.4.10. Pharmacokinetics and ADME analysis
ADME studies were performed utilizing the Swiss ADME platform, adhering to previously documented methodologies.72 These investigations were designed to evaluate physicochemical parameters67 to assess the drug-likeness and medicinal chemistry of the synthesized compounds, while also establishing correlations between biological activity and these physicochemical characteristics.5. Statistics
GraphPad Prism 7 for Windows (Intuitive Software for Science, San Diego, CA, USA) was used to perform statistical analyses. Analysis of variance (ANOVA), a test for multiple comparisons, was employed to compare the experimental groups. A significant level of P < 0.05 was regarded as the cutoff point for identifying significant differences.6. Conclusion
The findings from this study underscore the promising potential of novel camphor-derived heterocyclic compounds as effective anticancer agents, specifically targeting breast (MCF-7) and lung (A549) cancer cell lines. Notably, compound 20 emerged as a standout candidate, exhibiting exceptional inhibitory activity with an IC50 of 0.78 μM against MCF-7 cells and 1.69 μM against A549 cells. These values not only demonstrate its potency but also surpass the efficacy of established chemotherapeutics, including dasatinib (IC50 = 7.99 μM for MCF-7 and 11.8 μM for A549) and doxorubicin (IC50 = 3.10 μM for MCF-7 and 2.43 μM for A549).Furthermore, the strong correlation between the high binding energy observed in molecular docking studies and the potent biological activity of compound 20 emphasizes the critical role of structural optimization in the design of new anticancer therapies. These results highlight the need for further exploration into the mechanisms of action and in vivo efficacy of compound 20, paving the way for its potential development as a therapeutic agent in cancer treatment. Continued research in this area could lead to effective new strategies for combating breast and lung cancers, ultimately improving patient outcomes.
Ethical approval
This article does not include any studies that use human or animal tissues.Data availability
All data is available within the article.Author contribution
Z. H. and H. S., methodology, design experiments and writing draft manuscript. W. F. and W. H., supervision and editing manuscript. K. E., M. O. and G. H., molecular docking and biology.Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSP2025R374) King Saud University, Riyadh, Saud Arabia.References
- K. Shaikh, et al., An Introduction to Breast Cancer. Artificial Intelligence in Breast Cancer Early Detection and Diagnosis, 2021, pp. 1–20 Search PubMed.
- T. Swathi, S. Krishna, and M. V. Ramesh, A survey on breast cancer diagnosis methods and modalities, in 2019 International Conference on Wireless Communications Signal Processing and Networking (WiSPNET, IEEE, 2019 Search PubMed.
- K. Shaikh, S. Krishnan, and R. M. Thanki, Artificial Intelligence in Breast Cancer Early Detection and Diagnosis, Springer. 2021 Search PubMed.
- O. T. Nemr, et al., Investigating the Anticancer and Antioxidant Potentials of a Polymer-Grafted Sodium Alginate Composite Embedded with CuO and TiO2 Nanoparticles, J. Polym. Environ., 2024, 1–16 Search PubMed.
- S. Łukasiewicz, et al., Breast cancer—epidemiology, risk factors, classification, prognostic markers, and current treatment strategies—an updated review, Cancers, 2021, 13(17), 4287 CrossRef PubMed.
- A. N. Giaquinto, et al., Breast cancer statistics, 2022, Ca-Cancer J. Clin., 2022, 72(6), 524–541 CrossRef PubMed.
- H. J. Burstein, et al., Estimating the benefits of therapy for early-stage breast cancer: the St. Gallen International Consensus Guidelines for the primary therapy of early breast cancer 2019, Ann. Oncol., 2019, 30(10), 1541–1557 CrossRef CAS PubMed.
- N. L. Henry, et al., Cancer of the breast, in Abeloff's Clinical Oncology, Elsevier, 2020, pp, 1560–1603. e12 Search PubMed.
- K. J. Ruddy and P. A. Ganz, Treatment of nonmetastatic breast cancer, Jama, 2019, 321(17), 1716–1717 CrossRef PubMed.
- A. G. Waks and E. P. Winer, Breast cancer treatment: a review, Jama, 2019, 321(3), 288–300 CrossRef CAS PubMed.
- M. Mutebi, et al., Breast cancer treatment: A phased approach to implementation, Cancer, 2020, 126, 2365–2378 CrossRef PubMed.
- M. Zubair, S. Wang and N. Ali, Advanced approaches to breast cancer classification and diagnosis, Front. Pharmacol, 2021, 11, 632079 CrossRef PubMed.
- J. Li, et al., Non-invasive biomarkers for early detection of breast cancer, Cancers, 2020, 12(10), 2767 CrossRef CAS PubMed.
- R. Anupozu and M. Vanamala, Non-small Cell Lung Cancer (Nsclc)-a Comprehensive, 2020 Search PubMed.
- J. Domagala-Kulawik and A. Trojnar, Lung cancer in women in 21th century, J. Thorac. Dis., 2020, 12(8), 4398 CrossRef PubMed.
- A. Siddique, F. M. Otaibi, and S. F. Khan, Lung Cancer in Developing Countries, in Handbook of Medical and Health Sciences in Developing Countries: Education, Practice, and Research, Springer, 2023. pp. 1–28 Search PubMed.
- M. B. Schabath and M. L. Cote, Cancer progress and priorities: lung cancer, Cancer Epidemiol., Biomarkers Prev., 2019, 28(10), 1563–1579 CrossRef PubMed.
- E. S. Cheng, et al., Lung cancer risk in never-smokers: An overview of environmental and genetic factors, Chin. J. Cancer Res., 2021, 33(5), 548 CAS.
- P. Krawczyk, et al., Risk factors and primary prevention of lung cancer. Cessation of cigarette addiction, Oncol. Clin. Pract., 2021, 17(3), 112–124 CrossRef CAS.
- H. S. Mohamed, et al., Computational and molecular docking studies of new benzene sulfonamide drugs with anticancer and antioxidant effects, Curr. Org. Synth., 2023, 20(3), 339–350 CrossRef CAS PubMed.
- N. Duma, R. Santana-Davila, and J. R. Molina, Non–small cell lung cancer: epidemiology, screening, diagnosis, and treatment, in Mayo Clinic Proceedings, Elsevier, 2019 Search PubMed.
- W. D. Travis, et al., IASLC multidisciplinary recommendations for pathologic assessment of lung cancer resection specimens after neoadjuvant therapy, J. Thorac. Oncol., 2020, 15(5), 709–740 CrossRef CAS PubMed.
- Z. Zhang, et al., Low prognostic nutrition index predicts poorer quality of life in late-stage lung cancer, Ann Palliat Med., 2020, 9(6), 3976–3984 CrossRef PubMed.
- P. Harari, Epidermal growth factor receptor inhibition strategies in oncology, Endocr.-Relat. Cancer, 2004, 11(4), 689–708 CAS.
- A. Ayati, et al., A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy, Bioorg. Chem., 2020, 99, 103811 CrossRef CAS PubMed.
- A. Unnisa, et al., Recent advances in epidermal growth factor receptor inhibitors (EGFRIs) and their role in the treatment of cancer: a review, Anticancer Agents Med Chem., 2022, 22(20), 3370–3381 CrossRef CAS PubMed.
- W. A. Fadaly, M. T. Nemr and N. M. Kahk, Discovery of novel pyrazole based Urea/Thiourea derivatives as multiple targeting VEGFR-2, EGFRWT, EGFRT790M tyrosine kinases and COX-2 Inhibitors, with anti-cancer and anti-inflammatory activities, Bioorg. Chem., 2024, 147, 107403 CrossRef CAS PubMed.
- W. A. Fadaly, et al., New 1, 2, 3-triazole/1, 2, 4-triazole hybrids linked to oxime moiety as nitric oxide donor selective COX-2, aromatase, B-RAFV600E and EGFR inhibitors celecoxib analogs: design, synthesis, anti-inflammatory/anti-proliferative activities, apoptosis and molecular modeling study, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2290461 CrossRef PubMed.
- W. A. Fadaly, et al., New pyrazolyl-thiazolidinone/thiazole derivatives as celecoxib/dasatinib analogues with selective COX-2, HER-2 and EGFR inhibitory effects: design, synthesis, anti-inflammatory/anti-proliferative activities, apoptosis, molecular modelling and ADME studies, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2281262 CrossRef PubMed.
- R. B. Malabadi, et al., An age old botanical weapon for herbal therapy: Camphor tree, Cinnamomum camphora, Int. J. Innov. Sci. Res, 2021, 3, 1518–1523 Search PubMed.
- A. Duda-Madej, et al., Is Camphor the Future in Supporting Therapy for Skin Infections?, Pharmaceuticals, 2024, 17(6), 715 CrossRef CAS PubMed.
- J. C. Corzo-Gómez, et al., A Review of Botanical Extracts with Repellent and Insecticidal Activity and Their Suitability for Managing Mosquito-Borne Disease Risk in Mexico, Pathogens, 2024, 13(9), 737 CrossRef.
- C. Xia, et al., Developing long-term conservation priority planning for medicinal plants in China by combining conservation status with diversity hotspot analyses and climate change prediction, BMC Biol., 2022, 20(1), 1–20 CrossRef PubMed.
- W.-B. Xu, et al., Human activities have opposing effects on distributions of narrow-ranged and widespread plant species in China, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(52), 26674–26681 CrossRef CAS PubMed.
- T. Guangfu, D. Jingkai and S. Handong, The chemical constituents of the essential oil from leaves of Cinnamomum longepaniculatum in Hubei, China, Wuhan Bot. Res., 2002, 20(1), 75–77 Search PubMed.
- Q. Wei, et al., Inhibitive Activity of Cinnamomum oil against bacteria, Chin. J. Trop. Agric., 2009, 1, 5–7 Search PubMed.
- C. Tao, et al., Antifungal activity of the essential oil from Cinnamomum longepaniculatum leaves against three species of fungi, Chin. Vet. Sci., 2011, 41(1), 89–93 Search PubMed.
- L. Li, et al., Antibacterial activity of leaf essential oil and its constituents from Cinnamomum longepaniculatum, Int. J. Clin. Exp. Med., 2014, 7(7), 1721 Search PubMed.
- K. Ye, et al., Anticancer activity of the essential oil from Cinnamomumlongepaniculatum leaves and its major component against human BEL-7402, Acta. Anat. Sin, 2012, 43, 381–386 CAS.
- X. Song, et al., Anti-hepatoma effect of safrole from Cinnamomum longepaniculatum leaf essential oil in vitro, Int. J. Clin. Exp. Pathol., 2014, 7(5), 2265 Search PubMed.
- Z.-Q. Yin, et al., γ-terpineol inhibits cell growth and induces apoptosis in human liver cancer BEL-7402 cells in vitro, Int. J. Clin. Exp. Pathol., 2014, 7(10), 6524 Search PubMed.
- M. Cao, et al., Analgesic effects of essential oils purified from Cinnamomum longepaniculatum, Guihaia, 2013, 33(4), 552–555 CAS.
- C. Emmanouil, et al., An insight into ingredients of toxicological interest in personal care products and a small–scale sampling survey of the Greek market: Delineating a potential contamination source for water resources, Water, 2019, 11(12), 2501 CrossRef CAS.
- P. M. Jadhav, et al., A review on biological and medicinal significance of thiazoles, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196(10), 879–895 CrossRef CAS.
- S. A. Ibrahim and H. F. Rizk, Synthesis and biological evaluation of thiazole derivatives, in Azoles-Synthesis, Properties, Applications and Perspectives. 2020, IntechOpen London, UK Search PubMed.
- Y. H. Zaki, et al., Synthesis of 1, 3, 4-thiadiazole derivatives using hydrazonoyl bromide: Molecular docking and computational studies, Polycyclic Aromat. Compd., 2023, 43(2), 1364–1377 CrossRef CAS.
- M. F. Arshad, et al., Thiazole: A versatile standalone moiety contributing to the development of various drugs and biologically active agents, Molecules, 2022, 27(13), 3994 CrossRef CAS PubMed.
- A. Petrou, M. Fesatidou and A. Geronikaki, Thiazole ring—A biologically active scaffold, Molecules, 2021, 26(11), 3166 CrossRef CAS PubMed.
- P. P. Devidas, Design and Synthesis of Thiazines and Triazoles for Antitubercular Activity, Rajiv Gandhi University of Health Sciences, 2007, (India) Search PubMed.
- Y. H. Zaki, et al., Utility of Pyrimidine Thione Derivatives in the Synthesis of Biologically Active Heterocyclic Compounds, Polycyclic Aromat. Compd., 2023, 43(3), 2618–2643 CrossRef CAS.
- F. Musumeci, et al., Vascular endothelial growth factor (VEGF) receptors: drugs and new inhibitors, J. Med. Chem., 2012, 55(24), 10797–10822 CrossRef CAS PubMed.
- F. Yaccoubi, et al., A Simple and Efficient Approach to the Synthesis of 4-Aryl-2-dialkylphosphonomethyl-4-oxobutanenitrile, Molbank, 2022, 2022(1), M1342 CrossRef.
- Y. Zhang, et al., Recent advances in the development of pyrazole derivatives as anticancer agents, Int. J. Mol. Sci., 2023, 24(16), 12724 CrossRef CAS PubMed.
- E. Azzama, et al., Removal of iron (II) from wastewater in oil field using 3-(p-methyl) phenyl-5-thionyl-1, 2, 4-triazoline assembled on silver nanoparticles, Desalin. Water Treat., 2019, 142, 244–251 CrossRef.
- M. F Mohamed, et al., Chalcones incorporated pyrazole ring inhibit proliferation, cell cycle progression, angiogenesis and induce apoptosis of MCF7 cell line, Anticancer Agents Med Chem., 2014, 14(9), 1282–1292 CrossRef PubMed.
- J. Ceramella, et al., Carbazole derivatives as kinase-targeting inhibitors for cancer treatment, Mini-Rev. Med. Chem., 2020, 20(6), 444–465 CrossRef CAS PubMed.
- C. Cerchione, F. Locatelli and G. Martinelli, Dasatinib in the management of pediatric patients with Philadelphia chromosome-positive acute lymphoblastic leukemia, Front. Oncol., 2021, 11, 632231 CrossRef CAS PubMed.
- Y. Wang, et al., Pharmacokinetics and safety of dasatinib and its generic: a phase I bioequivalence study in healthy Chinese subjects, Expert Opin. Invest. Drugs, 2023, 32(3), 263–270 CrossRef CAS PubMed.
- R. Roskoski Jr, Targeting BCR-Abl in the treatment of Philadelphia-chromosome positive chronic myelogenous leukemia, Pharmacol. Res., 2022, 178, 106156 CrossRef PubMed.
- S. Sritharan and N. Sivalingam, A comprehensive review on time-tested anticancer drug doxorubicin, Life Sci., 2021, 278, 119527 CrossRef CAS PubMed.
- C. Ajaykumar, Overview on the side effects of doxorubicin, Advances in Precision Medicine Oncology, IntechOpen, 2020 Search PubMed.
- S. Sritharan and N. Sivalingam, A comprehensive review on time-tested anticancer drug doxorubicin, Life Sci., 2021, 278, 119527 CrossRef CAS PubMed.
- W. Lee, et al., The mutation spectrum revealed by paired genome sequences from a lung cancer patient, Nature, 2010, 465(7297), 473–477 CrossRef CAS PubMed.
- M.-L. Tseng, et al., A literature review on green supply chain management: Trends and future challenges, Resour., Conserv. Recycl., 2019, 141, 145–162 CrossRef.
- K. Aljohani, et al., Oral potentially malignant disorders and oral cancer in Saudi Arabia: an epidemiological review of the literature, J. Clin. Med., 2024, 13(5), 1376 CrossRef PubMed.
- L. S. Muniz and S. S. da Rocha Pita, In silico studies revealed interaction mechanisms of benzylidene–acrylohydrazide derivatives and nsP2 CHIKV, New J. Chem., 2022, 46(14), 6414–6423 RSC.
- C. A. Lipinski, et al., Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- K. R. Abdellatif, et al., Synthesis of novel halogenated triarylpyrazoles as selective COX-2 inhibitors: anti-inflammatory activity, histopatholgical profile and in-silico studies, Bioorg. Chem., 2020, 105, 104418 CrossRef CAS PubMed.
- Z. Y. u. Ibrahim, et al., Pharmacokinetic predictions and docking studies of substituted aryl amine-based triazolopyrimidine designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH), J. Pharm. Sci., 2021, 7, 1–10 Search PubMed.
- B. N. Brousse, et al., Behavior of thiosemicarbazones derived from some terpenones under acetylating conditions, Arkivoc, 2002, 2002(10), 14–23 Search PubMed.
- S. Agrawal, et al., Synthesis molecular modeling and anticonvulsant activity of some hydrazone, semicarbazone, and thiosemicarbazone derivatives of benzylidene camphor, Res. Rep. Med. Chem., 2014, 47–58 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7(1), 42717 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |