
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ transformation of Pd to metal–metalloid alloy Pd2B for alkyne semi-hydrogenation†
Shuaiwen Xua,
Lei Wanga,
Pengfei Tianb and
Shenghu Zhou *a
*a
aShanghai Key Laboratory of Multiphase Materials Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China. E-mail: zhoushenghu@ecust.edu.cn
bKey Laboratory of Pressure Systems and Safety, Ministry of Education, School of Mechanical and Power Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, P. R. China
First published on 3rd March 2025
Abstract
In this work, we report alumina-supported metal-metalloid alloy nanoparticles (Pd2B/Al2O3) as highly efficient alkyne semi-hydrogenation catalysts. The mentioned catalysts contain orthorhombically distorted hcp Pd2B nanoparticles and were prepared by in situ transformation of Pd/Al2O3 with borane dimethylamine complex by a solvothermal method. The synthesized Pd2B/Al2O3 demonstrated greatly enhanced semi-hydrogenation performance. Under reaction conditions of 30 °C, 0.1 MPa of H2 and a substrate/Pd molar ratio of 1000/1, the conversions of 3-hexyne-1-ol could reach 99.8% in 15 min with a yield of cis-3-hexen-1-ol of 93.3%. Importantly, the alkene and cis- to trans-alkene selectivity only slightly decreases with an extended reaction time, showing the inhibition of deep hydrogenation. Experimental studies and density functional theory calculations indicate that the catalytic enhancement is originated from the formation of Pd2B alloy, which alters the electronic and geometric properties of surface species, thus suppressing the deep hydrogenation with the enhanced alkene selectivity.
Introduction
Semi-hydrogenation of alkynes to alkenes is one of the model reactions for rational design of selective hydrogenation catalysts,1–5 and is frequently used in real industrial processes to produce various chemicals.6–8 For example, semi-hydrogenation of phenylacetylene9–12 over Pd/CaCO3 with Pb salts and quinoline, the Lindlar catalyst,13 has been used to remove the alkyne in styrene to avoid the deactivation of styrene polymerization catalysts. In general, supported noble metal-based catalysts14–18 are recognized as efficient semi-hydrogenation catalysts. However, their alkene selectivity is relatively low, and keeping high alkene selectivity including cis- to trans-alkene selectivity during an extended reaction time is challenging since hydrogenation of alkynes are tandem and isomerization reactions.Among various noble metal catalysts, Pd-based ones could catalyze hydrogenations of alkynes under mild reactions due to their high activity. Literatures have reported the poisoning19,20 strategy employing S- or N-containing organic ligands21–24 to improve the alkene selectivity for Pd-based catalysts. Such poisoning could alter the electronic/geometric properties of surface Pd species, resulting in the inhibition of alkene adsorption to mitigate their deep hydrogenation. For instance, diphenyl sulfide linkages-containing polymer supported Pd nanoparticles (NPs) improved the alkene selectivity with minimal over-hydrogenation25 while 2, 2′-bipyridine (bipy) and imidazolium-functionalized bipy ligands stabilized Pd NPs26 exhibited enhanced alkene selectivity for hydrogenation of 1-hexyne.
Another strategy to enhance the alkene selectivity is to alloy Pd with other metals. By the interaction between Pd and the second metal, the electron density as well as the geometric properties of surface Pd species is changed, thus avoiding deep-hydrogenation of alkenes.27–33 For example, Pd-Zn/Al2O3 demonstrated better styrene selectivity at high conversions for hydrogenation of phenylacetylene than monometallic Pd/Al2O3,34 and dilute PdxAu1−x NPs embedded in raspberry colloid-templated silica35 showed better performance at high conversions for gas phase semi-hydrogenation of 1-hexyne than individual Pd. Although alloying Pd with transition metal or main group metal has shown enhanced alkene selectivity for hydrogenation of alkynes to more or less degree, maintaining high alkene selectivity over the extended reaction time is challenging. Compared with individual metals, metal–metalloid alloys have different surface electronic structures and geometric characteristics due to the unique electronegativity and atomic radius of metalloid elements, which can affect the catalytic activity, selectivity, and even the crystal structures of the alloys.36 Currently, Pd-metalloid alloys are mainly applied in electrocatalysis,37–39 catalytic oxidation40,41 and reduction,42 the use of Pd-metalloid alloys for semi-hydrogenations is rarely reported.43
In this work, we report alumina-supported metal-metalloid Pd2B alloy NPs (Pd2B/Al2O3) as highly active and selective alkyne semi-hydrogenation catalysts. The mentioned Pd2B/Al2O3 was prepared by in situ transformation of Pd/Al2O3 with borane dimethylamine (DMAB) in tetrahydrofuran (THF) at 160 °C by a solvothermal method.42 Scheme 1 shows the synthetic procedures of Pd2B/Al2O3, where the in situ transformation could preserve the small size of catalytic functionality. Pd2B/Al2O3 demonstrated significantly enhanced alkene selectivity for hydrogenation of 3-hexyne-1-ol (HY). Under 30 °C, a molar ratio of substrate/Pd of 1000/1 and atmospheric H2 pressure, 93.6% of cis-3-hexen-1-ol (cis-HE) selectivity at 99.8% of conversion was achieved over Pd2B/Al2O3 during 15 min for hydrogenation of HY, and the alkene selectivity only exhibited a slight decrease with the reaction proceeding, showing much better performance than Pd/Al2O3. Through the combination of experimental studies and density functional theory (DFT) calculations, the performance enhancement of Pd2B/Al2O3 is ascribed to the alteration of electronic/geometric properties by alloying Pd with B, which inhibits the adsorption and hydrogenation of alkenes, thus improving the alkene selectivity.
 | ||
| Scheme 1 The synthetic procedures of Pd2B/Al2O3. | ||
Results and discussion
Characterizations
Fig. 1a and b present the transmission electron microscope (TEM) images of Pd/Al2O3 and Pd2B/Al2O3, respectively, while their corresponding size distributions are shown in Fig. S1.† It can be seen that Pd and Pd2B NPs illustrate well-faceted shapes with average particle sizes of 7.2 and 7.9 nm (Fig. S1†), respectively. The spacing of lattice fringe in the insert of Fig. 1a is 0.228 nm, which can be attributed to the (111) spacing of fcc Pd while the insert of Fig. 1b shows the lattice spacing of 0.233 nm corresponding to the spacing of (002) of hcp Pd2B. The lattice expansion is presumed to be due to the insertion of B.44–46 Fig. 1c to f show the high angle annular dark field scanning electron microscope (HAADF-STEM) images of Pd2B/Al2O3 and their corresponding energy dispersion spectroscopy (EDS) phase mappings and line-scans of the selected Pd2B NPs. The Pd phase mappings in Fig. 1d clearly demonstrate the well dispersed Pd while the B phase mapping contains much noise due to its small atomic number. Therefore, EDS line-scans of B element were used. As shown in Fig. 1e and f, the highest B concentration aligns with the highest concentration of Pd, confirming the co-presence of Pd and B in Pd2B NPs. | ||
| Fig. 1 TEM images showing: (a), Pd/Al2O3, insert showing the lattice fringe of Pd (111); (b), Pd2B/Al2O3, insert showing the lattice fringe of Pd2B (002). HAADF-STEM images of Pd2B/Al2O3 showing: (c), the selected areas of Pd2B/Al2O3 for EDS phase mapping, the arrow indicating the selected NPs for EDS line-scans; (d), the Pd phase mapping; (e) and (f), the corresponding EDS line-scans. | ||
X-ray diffraction (XRD) patterns of Al2O3, Pd/Al2O3 and Pd2B/Al2O3 are shown in Fig. 2a. Due to the overlap of alumina and Pd diffractions, the distinct Pd diffractions were not observed in Pd/Al2O3. Compared with that of alumina, the relative intensity ratio of the diffraction at 39.3° (2θ degree) to that at 37.4° in Pd/Al2O3 is significantly increased, suggesting that the fcc Pd (111) diffraction at 39.6° is overlapped with that at 39.3° of alumina. In contrast, a set of the diffractions at 41.4°, 54.2°, 70.9° and 78.6° appear in Pd2B/Al2O3, which can be ascribed to (101), (102), (103) and (112) of hcp Pd2B,47 respectively. To further confirm the formation of Pd2B by in situ transformation, silica supports were used for comparison, where the Pd2B/SiO2 was prepared by the similar process to that of Pd2B/Al2O3 except that silica was used. As shown in Fig. 2b, Pd2B/SiO2 shows the distinct hcp Pd2B diffractions of (100) at 36.7°, (002) at 38.2°, (101) at 41.6°, (102) at 54.0°, (110) at 66.2°, (103) at 71.1° and (112) at 78.7°, and Pd diffractions totally disappear, further confirming the complete transformation of Pd into Pd2B by the solvothermal method.
 | ||
| Fig. 2 (a), XRD patterns of Al2O3 (black), Pd/Al2O3 (blue) and Pd2B/Al2O3 (red); (b), XRD patterns of Pd/SiO2 (black) and Pd2B/SiO2 (red). The green vertical dashed lines indicate the diffractions of the reference hcp Pd2B. The synthetic procedures of Pd2B/Al2O3 and Pd2B/SiO2 are same except that different supports were used. | ||
Fig. 3 presents the X-ray photoelectron spectroscopy (XPS) analysis of various samples. The binding energies of 335.0/340.3 and 336.4/341.5 (eV eV−1) in Fig. 3a are attributed to the 3d5/2/3d3/2 of Pd0 and Pd2+ species of Pd/Al2O3,48–50 respectively. However, the binding energies of Pd0 at 334.6/339.9 and Pd2+ at 336.0/341.1 of Pd2B/Al2O3 in Fig. 3b show a shift of 0.4 eV to the lower binding energies relative to those of Pd/Al2O3, suggesting the electron flow from B to Pd. Fig. 3c demonstrates the B 1s peak of Pd2B/Al2O3, where the binding energies at 187.8 and 192.0 eV are attributed to B0 and B–Ox species, respectively. Compared with 186–187.1 eV of B0 species of individual B,51,52 the binding energy of B0 of Pd2B exhibits a shift of 0.7–1.8 eV to the higher binding energies, further confirming the electron flow from B to Pd. The high percentage of B–Ox is attributed to the formation of amorphous B2O3 by decomposition of excess DMAB in the solovothermal process.47,53 Table S1† summarizes the metal loadings of various materials determined by inductively coupled plasma optical emission spectrometer (ICP-OES). Indeed, the B loading is much higher than the theoretic value, confirming the formation of amorphous B2O3 in the solvothermal process.
 | ||
| Fig. 3 XPS spectra: (a), Pd 3d spectra of Pd/Al2O3; (b), Pd 3d spectra of Pd2B/Al2O3; (c), B 1s spectra of Pd2B/Al2O3. | ||
The change of electron density of surface Pd species was further studied by diffuse reflection Fourier transform infrared spectroscopy (DRIFT-IR) with CO probes. As shown in the Fig. 4, CO bands above 2100 cm−1 disappear after 10 min of argon purging, suggesting that these bands are due to free CO. The left weak peak at 2036 cm−1 for Pd/Al2O3 in Fig. 4a is related to the linear CO species coordinated on Pd,54,55 while the major peak at 1944 cm−1 and those shoulders at 1960 and 1920 cm−1 can be attributed to the characteristic bands of bridged CO species on Pd.56–59 However, the major bridged CO peak of Pd2B/Al2O3 in Fig. 4b is found at 1920 cm−1, showing a red shift of 24 cm−1. Such a red shift indicates the electron-sufficiency of surface Pd species by alloying Pd with B, and is consistent with the XPS study. Through the combination of XPS studies and DRIFT-IR studies with CO probes, the surface Pd species of Pd2B are electron-sufficient. It is well known that the change of electron densities of surface species will change the adsorption behavior of reactants, intermediates or products, and further DFT calculation confirms that Pd2B inhibits the adsorption of the intermediate alkene and thus increases the selectivity of alkene for alkyne hydrogenation.
 | ||
| Fig. 4 DRIFT-IR spectra with CO probes of (a) Pd/Al2O3 and (b) Pd2B/Al2O3. | ||
Catalytic hydrogenation of alkyne over Pd2B/Al2O3
Hydrogenation of 3-hexyne-1-ol (HY) was carried out under selected solvents and atmospheric H2. Table S1† shows the Pd metal loadings of different catalysts. To keep the substrate/Pd molar ratio constant, different weights of Pd/Al2O3 and Pd2B/Al2O3 were used. Fig. 5 presents the time courses of HY hydrogenations over Pd/Al2O3 and Pd2B/Al2O3. These two catalysts show basically similar activity with >99.8% of conversions and 95.3% of 3-hexen-1-ol (HE) yields in 15 min. However, the selectivity of HE and cis-HE of Pd/Al2O3 in Fig. 5a rapidly decreases while those over Pd2B/Al2O3 in Fig. 5b only slightly decrease. For example, the HE/cis-HE yields of Pd2B/Al2O3 in Fig. 5b could maintain 85.1/74.2% in 120 min. In sharp contrast, the HE/cis-HE yields of Pd/Al2O3 in Fig. 5a are only 50.8/21.6% at the same time. In addition, the control experiment using individual BxOy/Al2O3 in Fig. S2† didn't show obvious catalytic activity. Therefore, the maintenance of the selectivity of HE and cis-HE for Pd2B/Al2O3 is due to the inhibition of alkene adsorption by alloying Pd with B. | ||
| Fig. 5 Reaction profiles of HY hydrogenation over: (a), Pd/Al2O3; (b), Pd2B/Al2O3. Reaction conditions: HY/Pd molar ratio, 1000/1; HY, 1.0 mmol; MeOH, 10.0 mL; reaction temperature, 30 °C; speed of agitation, 700 rpm; H2 pressure, 0.1 MPa. Red curves are the yields of alkene including cis- and trans-alkene. | ||
To further confirm the inhibition of alkene hydrogenation over Pd2B/Al2O3, the experiments using cis-HE as substrates were performed. As shown in Fig. 6a, individual Pd/Al2O3 exhibits rapid alkene hydrogenation (100% conversion in 25 min) and cis to trans isomerization (a maximum of 43.1% trans-HE yield in 20 min). Moreover, some non-identified products were also found, possibly due to the double bond migration.60,61 In sharp contrast, Pd2B/Al2O3 in Fig. 6b only shows cis to trans isomerization reactions with a trans-HE yield of 7.9% in 60 min (no alkane detected). Although the cis-HE hydrogenation over Pd2B/Al2O3 is greatly inhibited in Fig. 6b, its hydrogenation when HY as substrates in Fig. 5b cannot be totally inhibited. Currently, the reason is not clear, and we speculate that the intermediate cis-HE already on Pd2B when HY as substrates could favor its further hydrogenation.
 | ||
| Fig. 6 Reaction profiles of cis-HE hydrogenations over: (a), Pd/Al2O3; (b), Pd2B/Al2O3. Reaction conditions: cis-HE/Pd molar ratio, 1000/1; cis-HE, 1.0 mmol; MeOH, 10.0 mL; reaction temperature, 30 °C; speed of agitation, 700 rpm; H2 pressure, 0.1 MPa. Others in (a) may be due to double bond migration. | ||
The effects of solvents on the catalytic performance of Pd2B/Al2O3 are shown in Table 1. MeOH and EtOH as solvents exhibit basically similar performance with >95% of conversions and >93% of HE yields in 15 min while propan-2-ol and THF show lower activity and alkene selectivity. In this study, the polarities of solvents decrease in the following order: MeOH > EtOH > propan-2-ol and THF, which is aligned with the activity decreasing order of Pd2B/Al2O3 in these solvents, suggesting an important role of the solvent polarity. Moreover, the competitive adsorption of solvents may also influence the catalytic active sites,62 which further affects the alkene hydrogenation.
| Solventsa | Time (min) | Conv. (%) | Yield (%) | ||
|---|---|---|---|---|---|
| HE | cis-HE | trans-HE | |||
| a Reaction conditions: HY/Pd molar ratio, 1000/1; HY, 1.0 mmol; solvent, 10.0 mL; reaction temperature, 30 °C; speed of agitation, 700 rpm; H2 pressure, 0.1 MPa. | |||||
| MeOH | 15 | 99.8 | 95.9 | 93.3 | 2.6 |
| 120 | 100.0 | 85.1 | 74.2 | 10.9 | |
| EtOH | 15 | 95.9 | 93.4 | 91.3 | 2.0 |
| 120 | 100.0 | 85.0 | 75.0 | 10.0 | |
| Propan-2-ol | 15 | 79.7 | 79.7 | 78.1 | 1.6 |
| 120 | 100.0 | 77.7 | 67.1 | 10.5 | |
| THF | 15 | 44.3 | 44.3 | 43.6 | 0.7 |
| 120 | 100.0 | 77.1 | 57.8 | 19.3 | |
Fig. 7 presents the effect of reaction temperatures on catalytic performance. As shown in Fig. 7 and 5b, no significant difference of catalytic activity was observed for HY hydrogenations over Pd2B/Al2O3 in the temperature range 20–40 °C, and the alkene selectivity is only slightly decreased with an extended reaction time in this temperature range, showing the robust inhibition of alkene adsorption and deep-hydrogenation on Pd2B NPs. More hydrogenations of other alkyne substrates were also investigated. As shown in Fig. S3,† Pd2B/Al2O3 shows higher activities as well as better alkene selectivity for hydrogenation of 2-butyn-1-ol (BY) than Pd/Al2O3 while Pd2B/Al2O3 shows higher activities for diphenylacetylene (DPA) hydrogenation although Pd/Al2O3 and Pd2B/Al2O3 all demonstrate good alkene selectivity during the extended reaction time.
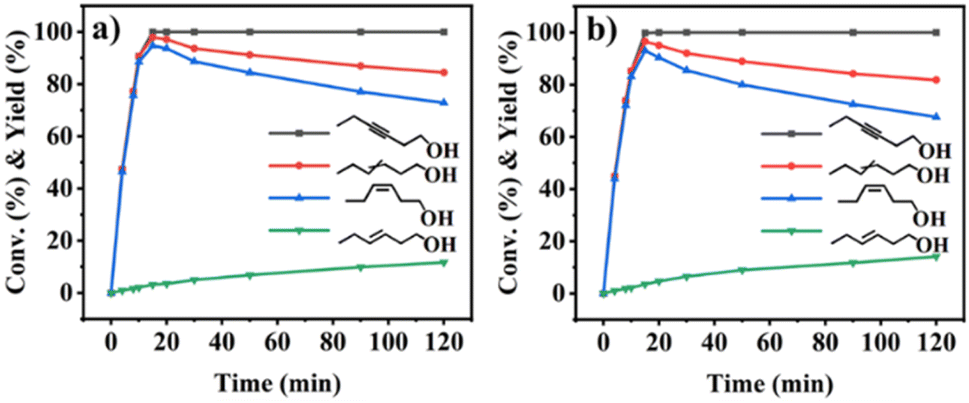 | ||
| Fig. 7 Reaction profiles of HY hydrogenation over Pd2B/Al2O3 at atmospheric H2 pressure and various reaction temperatures: (a), 20 °C; (b), 40 °C. Reaction conditions: HY/Pd ratio, 1000/1; HY, 1.0 mmol; MeOH, 10.0 mL; speed of agitation, 700 rpm. | ||
Fig. 8 shows the stability investigation of the Pd2B/Al2O3 catalysts. The experiment was first performed at a HY/Pd ratio of 5000/1 (five times higher). After treating five times more reactants, the catalysts were recovered, thoroughly washed and dried, and then were tested at a HY/Pd ratio of 1000/1. As shown in the high ratio experiment (Fig. 8a), the conversion of HY reached 99.9% at 100 min with a yield of cis-HE of 94.6% (the alkene yield of 97.3%). At 600 min, the yield of HE could be maintained at 84.0% with a cis-HE yield of 72.1%, showing good selectivity stability at a high ratio of substrate/Pd. Moreover, the recovered catalysts in Fig. 8b obtained a complete conversion and the HE/cis-HE yields of 95.7%/92.1% at 15 min, showing the similar performance to the fresh catalysts. The HE/cis-HE yields of the recovered catalysts during the extended reaction time only slightly decrease to 78.6%/64.7% at 120 min, showing a quite good catalytic stability. Fig. S4† presents the TEM images, size distribution and XRD pattern of the recovered catalysts. Except the Pd2B particle size increasing from 7.9 to 8.9 nm, no other obvious changes were found after recycling experiments, further confirming their structural stability. Table S2† summarizes the comparison between this work and the reported ones. As indicated in Table S2,† the Pd2B/Al2O3 is comparable to state-of-the-art Pd-based catalysts for liquid phase 3-hexyne-1-ol semi-hydrogenation.
 | ||
| Fig. 8 (a) Reaction profiles of HY hydrogenation over Pd2B/Al2O3 at a HY/Pd ratio of 5000/1 (five times higher); (b) HY hydrogenation at a HY/Pd ratio of 1000/1 over the recycled Pd2B/Al2O3. Reaction conditions: HY, 15.0 mmol for (a) and 1.0 mmol for (b); MeOH, 30.0 mL for (a) and 10.0 mL for (b); reaction temperature, 30 °C; speed of agitation, 700 rpm; H2 pressure, 0.1 MPa. | ||
The origin of the enhanced alkene selectivity of Pd2B/Al2O3
DFT calculations were performed to gain further mechanistic understanding of HY hydrogenation on the Pd/Al2O3 and Pd2B/Al2O3 catalysts. Pd (111) and Pd2B (001)47 surfaces were employed to model the Pd and Pd2B catalysts, respectively (Fig. 9 and S5†). Since the decrease in HE selectivity primarily stems from the HE hydrogenation reaction, the two-step hydrogenation pathway for HE conversion to hexanol was considered in the calculations. | ||
| Fig. 9 DFT calculation results. (a) Pd (111) and (b) Pd2B (001) models, Pd-blue and B-pink; (c) potential energy diagram for hydrogenation of cis-HE to hexanol on Pd (111) and Pd2B (001). TS refers to transition state, * indicating the surfaces of Pd (111) and P2B (001). | ||
As shown in Fig. S5,† the binding energy of −1.57 eV of cis-HE on Pd2B (001) is obviously higher than −1.90 eV of cis-HE on Pd (111), indicating that the adsorption of HE on the Pd2B alloy is suppressed. Fig. 9c presents the potential energy diagrams for cis-HE hydrogenation on Pd (111) and Pd2B (001). Despite the two catalysts show similar activation barrier for the first hydrogenation step, the hydrogenation barrier of C6H13O* intermediate to C6H14O* is markedly increased from 0.44 eV to 0.89 eV with adding B to construct Pd2B alloy, suggesting that Pd2B is more unfavorable for HE hydrogenation. Thus, the DFT calculations confirm that the enhanced HE selectivity over Pd2B/Al2O3 is due to the suppression of alkene adsorption and its over-hydrogenation.
Conclusion
In this work, alumina supported Pd2B NPs were prepared by in situ transformation of Pd/Al2O3 with borane dimethylamine complex by a solvothermal method. At mild reaction conditions, the obtained Pd2B/Al2O3 exhibited enhanced alkene/cis-alkene selectivity for hydrogenation of 3-hexyne-1-ol during an extended reaction time. The experimental studies and theoretical calculations indicate that alloying Pd with B alters the electronic/geometric properties of surface species of catalytic functionality, where alloying B with Pd makes the Pd electron-sufficient and the binding energy of cis-HE on Pd2B (001) is obviously higher than that of cis-HE on Pd (111), resulting in inhibition of the adsorption of alkene and suppression of their deep hydrogenation with enhanced alkene selectivity. We believe that the in situ transformation by a solvothermal method could extend to other metals, and such metal–metalloid alloy NPs may find more applications in heterogeneous catalysis.Experimental section
Synthesis of Pd/Al2O3
A portion of PdCl2 (15.0 mg), PVP-K30 (28.7 mg) and EG (6.5 mL) were charged into a three-necked round-bottomed flask under vigorous magnetic stirring. The resultant mixture was heated to and maintained at 60 °C for 30 min under nitrogen to fully dissolve the chemicals, and then subjected to heating at 90 °C for 30 min, followed by an additional heat treatment at 150 °C for 120 min. The resultant dark colloid was cooled to room temperature and subsequently centrifuged with acetone to collect Pd NPs. The above mentioned Pd NPs were then re-dispersed in 7.5 mL of EtOH to form Pd colloid, and then transferred into a three-necked round-bottomed flask. A portion of 240 mg of Al2O3 calcined at 500 °C for 180 min was added into the system under vigorous magnetic stirring. EtOH was then removed at 60 °C by a N2 flow to obtain gray powder, which was further dried at 60 °C in an oven for 8.0 h, and then subjected to washing with acetone and EtOH to remove PVP-K30. Finally, the collected solids were dried in an oven at 60 °C for 8.0 h to obtain Pd/Al2O3 catalysts.Synthesis of Pd2B/Al2O3
Typically, Pd/Al2O3 (20.0 mg) and DMAB (600 mg) were mixed in 10.0 mL of THF, and then subjected to ultrasonic treatment for 60 min. Next, the mixture was transferred into a 20 mL Teflon-lined hydrothermal reactor. The system was heated to and maintained at 160 °C for 5.0 h. After cooling the system to room temperatures, the solids were collected, washed with THF and EtOH, and then dried in an oven at 60 °C for 8.0 h to obtain Pd2B/Al2O3 catalysts.Catalytic hydrogenations of HY
In a typical experiment, 1.0 mmol of HY, a desired amount of catalysts (HY/Pd molar ratio of 1000/1), and 10.0 mL of solvents were charged into a three-necked round-bottomed flask. The reaction was performed at the defined temperature and atmospheric H2 with magnetic stirring. During the defined time intervals, the reaction mixture was sampled, filtered, and analyzed by a gas chromatography (GC) with a flame ionization detector. The detailed GC analysis method can be found in ESI.†The stability experiment of Pd2B/Al2O3 was first performed using 15.0 mmol of HY and 30.0 mL of MeOH (a HY/Pd molar radio of 5000/1) under 30 °C and atmospheric H2. After the reaction, the catalysts were collected by centrifugation, washing with EtOH, and drying at 60 °C for 8 h. Then, the HY hydrogenation over the recovered Pd2B/Al2O3 was carried out using 1.0 mmol of HY and 10.0 mL of MeOH (a HY/Pd ratio of 1000/1) under 30 °C and atmospheric H2.
Computational models and methods
The detailed theoretical calculations can be found in ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conception and design of the research: Shuaiwen Xu, Shenghu Zhou. Acquisition of data: Shuaiwen Xu, Lei Wang. Analysis and interpretation of the data: Shuaiwen Xu, Lei Wang. Experimental: Shuaiwen Xu. Calculation: Peifei Tian. Funding acquisition: Shenghu Zhou, Pengfei Tian. Writing of the manuscript: Shuaiwen Xu. Critical revision of the manuscript for intellectual content: Shenghu Zhou.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (Grant No. 22078099, 22178110 and 22378125) and the Basic Research Program of Science and Technology Commission of Shanghai Municipality (Grant No. 22JC1400600). The authors thank these organizations for providing these kind financial supports.References
- M. K. Karunananda and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 14598–14601 CrossRef CAS PubMed.
- M. Li, N. Zhang, R. Long, W. Ye, C. Wang and Y. Xiong, Small, 2017, 13, 1604173 CrossRef PubMed.
- H. Alawisi, H. D. Arman and Z. J. Tonzetich, Organometallics, 2021, 40, 1062–1070 Search PubMed.
- J. Ballesteros-Soberanas, J. A. Carrasco and A. Leyva-Perez, J. Org. Chem., 2023, 88, 18–26 Search PubMed.
- H. Liu, L. Zhang, K. Wang, L. Wang, L. Zhang, B. Yu and F. Yang, J. Phys. Chem. C, 2023, 127, 7211–7219 CrossRef CAS.
- M. Tejeda-Serrano, M. Mon, B. Ross, F. Gonell, J. Ferrando-Soria, A. Corma, A. Leyva-Perez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2018, 140, 8827–8832 Search PubMed.
- M. Cordoba, F. Coloma-Pascual, M. E. Quiroga and C. R. Lederhos, Ind. Eng. Chem. Res., 2019, 58, 17182–17194 CrossRef CAS.
- P. McNeice, M.-A. Müller, J. Medlock, W. Bonrath, N. Rockstroh, S. Bartling, H. Lund, K. Junge and M. Beller, ACS Sustainable Chem. Eng., 2022, 10, 9787–9797 Search PubMed.
- L. Yang, Y. Jin, X. Fang, Z. Cheng and Z. Zhou, Ind. Eng. Chem. Res., 2017, 56, 14182–14191 CrossRef CAS.
- Y. Liu, W. Guo, X. Li, P. Jiang, N. Zhang and M. Liang, ACS Appl. Nano Mater., 2021, 4, 5292–5300 CrossRef CAS.
- C. Shen, Y. Ji, P. Wang, S. Bai, M. Wang, Y. Li, X. Huang and Q. Shao, ACS Catal., 2021, 11, 5231–5239 CrossRef CAS.
- Z. Wang, C. Xu, Y. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2023, 15, 10292–10301 Search PubMed.
- Z. S. Wang, C. L. Yang, S. L. Xu, H. Nan, S. C. Shen and H. W. Liang, Inorg. Chem., 2020, 59, 5694–5701 CrossRef CAS PubMed.
- X. Song, F. Shao, Z. Zhao, X. Li, Z. Wei and J. Wang, ACS Catal., 2022, 12, 14846–14855 Search PubMed.
- C.-T. Kuo, Y. Lu, L. Kovarik, M. Engelhard and A. M. Karim, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- Z. Li, Q. Ren, X. Wang, W. Chen, L. Leng, M. Zhang, J. H. Horton, B. Liu, Q. Xu, W. Wu and J. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 2530–2537 Search PubMed.
- H. Zhou, B. Li, Y. Zhang, X. Yan, W. Lv, X. Wang, B. Yuan, Y. Liu, Z. Yang and X. Lou, ACS Appl. Mater. Interfaces, 2021, 13, 40429–40440 CrossRef CAS PubMed.
- F. Huang, M. Peng, Y. Chen, X. Cai, X. Qin, N. Wang, D. Xiao, L. Jin, G. Wang, X. D. Wen, H. Liu and D. Ma, J. Am. Chem. Soc., 2022, 144, 18485–18493 CrossRef CAS PubMed.
- T. Yoshii, D. Umemoto, Y. Kuwahara, K. Mori and H. Yamashita, ACS Appl. Mater. Interfaces, 2019, 11, 37708–37719 Search PubMed.
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Catal., 2019, 9, 1993–2006 Search PubMed.
- E. Karakhanov, A. Maximov, Y. Kardasheva, V. Semernina, A. Zolotukhina, A. Ivanov, G. Abbott, E. Rosenberg and V. Vinokurov, ACS Appl. Mater. Interfaces, 2014, 6, 8807–8816 CrossRef CAS PubMed.
- W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 Search PubMed.
- Y. Park, S. Lee, K. Hyun, J. Lee, J. Y. Park, R. Ryoo and M. Choi, J. Catal., 2021, 404, 716–725 Search PubMed.
- X. Zhao, L. Zhou, W. Zhang, C. Hu, L. Dai, L. Ren, B. Wu, G. Fu and N. Zheng, Chem, 2018, 4, 1080–1091 Search PubMed.
- S. Yun, S. Lee, S. Yook, H. A. Patel, C. T. Yavuz and M. Choi, ACS Catal., 2016, 6, 2435–2442 Search PubMed.
- M. Crespo-Quesada, R. R. Dykeman, G. Laurenczy, P. J. Dyson and L. Kiwi-Minsker, J. Catal., 2011, 279, 66–74 Search PubMed.
- W. Huang, J. McCormick, R. Lobo and J. Chen, J. Catal., 2007, 246, 40–51 CrossRef CAS.
- P. V. Markov, G. O. Bragina, A. V. Rassolov, G. N. Baeva, I. S. Mashkovsky, V. Y. Murzin, Y. V. Zubavichus and A. Y. Stakheev, Mendeleev Commun., 2016, 26, 502–504 CrossRef CAS.
- C. M. Kruppe, J. D. Krooswyk and M. Trenary, ACS Catal., 2017, 7, 8042–8049 Search PubMed.
- J. Liu, J. Shan, F. R. Lucci, S. Cao, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Catal. Sci. Technol., 2017, 7, 4276–4284 RSC.
- J. Zhang, W. Xu, L. Xu, Q. Shao and X. Huang, Chem. Mater., 2018, 30, 6338–6345 CrossRef CAS.
- A. Gonzalez-Fernandez, A. Berenguer-Murcia, D. Cazorla-Amoros and F. Cardenas-Lizana, ACS Appl. Mater. Interfaces, 2020, 12, 28158–28168 Search PubMed.
- X. Wang, M. Chu, M. Wang, Q. Zhong, J. Chen, Z. Wang, M. Cao, H. Yang, T. Cheng, J. Chen, T. K. Sham and Q. Zhang, ACS Nano, 2022, 16, 16869–16879 CrossRef CAS PubMed.
- Z. Wang, L. Yang, R. Zhang, L. Li, Z. Cheng and Z. Zhou, Catal. Today, 2016, 264, 37–43 Search PubMed.
- M. Luneau, T. Shirman, A. C. Foucher, K. Duanmu, D. M. A. Verbart, P. Sautet, E. A. Stach, J. Aizenberg, R. J. Madix and C. M. Friend, ACS Catal., 2019, 10, 441–450 Search PubMed.
- Y. Wang, H. Lv, L. Sun and B. Liu, ACS Nano, 2021, 15, 18661–18670 CrossRef CAS PubMed.
- X. Ai, X. Zou, H. Chen, Y. Su, X. Feng, Q. Li, Y. Liu, Y. Zhang and X. Zou, Angew. Chem., Int. Ed., 2020, 59, 3961–3965 CrossRef CAS PubMed.
- L. Sun, H. Lv, Y. Wang, D. Xu and B. Liu, J. Phys. Chem. Lett., 2020, 11, 6632–6639 CrossRef CAS PubMed.
- Y. Wang, H. Lv, L. Sun, X. Guo, D. Xu and B. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 17599–17607 CrossRef CAS PubMed.
- N. I. Kuznetsova, V. N. Zudin, L. I. Kuznetsova, V. I. Zaikovskii, H. Kajitani, M. Utsunomiya and K. Takahashi, Appl. Catal., A, 2016, 513, 30–38 CrossRef CAS.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y.-F. Han, Chin. J. Catal., 2018, 39, 673–681 CrossRef CAS.
- H. Lv, D. Xu, C. Kong, Z. Liang, H. Zheng, Z. Huang and B. Liu, ACS Cent. Sci., 2020, 6, 2347–2353 CrossRef CAS PubMed.
- C. W. Chan, A. H. Mahadi, M. M. Li, E. C. Corbos, C. Tang, G. Jones, W. C. Kuo, J. Cookson, C. M. Brown, P. T. Bishop and S. C. Tsang, Nat. Commun., 2014, 5, 5787 Search PubMed.
- K. Jiang, J. Chang, H. Wang, S. Brimaud, W. Xing, R. J. Behm and W. B. Cai, ACS Appl. Mater. Interfaces, 2016, 8, 7133–7138 Search PubMed.
- T. Chen, I. Ellis, T. J. N. Hooper, E. Liberti, L. Ye, B. T. W. Lo, C. O'Leary, A. A. Sheader, G. T. Martinez, L. Jones, P. L. Ho, P. Zhao, J. Cookson, P. T. Bishop, P. Chater, J. V. Hanna, P. Nellist and S. C. E. Tsang, J. Am. Chem. Soc., 2019, 141, 19616–19624 Search PubMed.
- Y. M. Liu, B. Q. Miao, H. Y. Yang, X. Ai, T. J. Wang, F. Shi, P. Chen and Y. Chen, Adv. Funct. Mater., 2024, 34, 2402485 Search PubMed.
- L. Chen, L.-R. Zhang, L.-Y. Yao, Y.-H. Fang, L. He, G.-F. Wei and Z.-P. Liu, Energy Environ. Sci., 2019, 12, 3099–3105 Search PubMed.
- H. Lv, L. Sun, D. Xu, J. Henzie, Y. Yamauchi and B. Liu, J. Mater. Chem. A, 2019, 7, 24877–24883 Search PubMed.
- A. González-Fernández, C. Pischetola and F. Cárdenas-Lizana, J. Phys. Chem. C, 2021, 125, 2454–2463 Search PubMed.
- S. Chen, T. Yang, H. Lu, Y. Liu, Y. He, Q. Li, J. Gao, J. Feng, H. Yan, J. T. Miller and D. Li, ACS Catal., 2022, 12, 15696–15706 Search PubMed.
- J. Li, J. Chen, Q. Wang, W.-B. Cai and S. Chen, Chem. Mater., 2017, 29, 10060–10067 CrossRef CAS.
- F. Li, B. Cao, R. Ma, H. Song and H. Song, Can. J. Chem. Eng., 2015, 94, 89–93 Search PubMed.
- L. Artiglia, D. Lazzari, S. Agnoli, G. A. Rizzi and G. Granozzi, J. Phys. Chem. C, 2013, 117, 13163–13172 Search PubMed.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799–2812 Search PubMed.
- H. Li, M. Shen, J. Wang, H. Wang and J. Wang, Ind. Eng. Chem. Res., 2020, 59, 1477–1486 Search PubMed.
- P. Zhang, Y. Hu, B. Li, Q. Zhang, C. Zhou, H. Yu, X. Zhang, L. Chen, B. Eichhorn and S. Zhou, ACS Catal., 2015, 5, 1335–1343 Search PubMed.
- X. Chen, X. Shi, P. Chen, B. Liu, M. Liu, L. Chen, D. Ye, X. Tu, W. Fan and J. Wu, ACS Environ. Au, 2023, 3, 223–232 Search PubMed.
- V. Marchionni, M. Nachtegaal and D. Ferri, ACS Catal., 2020, 10, 4791–4804 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
- F. Costantino, M. Nocchetti, M. Bastianini, A. Lavacchi, M. Caporali and F. Liguori, ACS Appl. Nano Mater., 2018, 1, 1750–1757 CrossRef CAS.
- W. Oberhauser, M. Frediani, I. Mohammadi Dehcheshmeh, C. Evangelisti, L. Poggini, L. Capozzoli and P. Najafi Moghadam, ChemCatChem, 2022, 14, e202101910 Search PubMed.
- R. L. Augustine, R. W. Warner and M. J. Melnick, J. Org. Chem., 1984, 49, 4853–4856 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00302d |
| This journal is © The Royal Society of Chemistry 2025 |