
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and optimization of biobased carbon adsorbent monoliths from chitosan-polybenzoxazine for efficient CO2 capture
José E. Mosqueraa,
Liana Delevingnea,
Frédéric Delbecqa,
Elias Daouka,
Audrey Drelicha,
Khashayar Saleha,
Rémi Gautierb and
Mikel Leturia *a
*a
aUniversité de Technologie de Compiègne, ESCOM, TIMR, Compiègne, France. E-mail: mikel.leturia@utc.fr; Tel: +33 6 28 23 87 85
bIMT Nord Europe, Institut Mines-Télécom, CERI Energie et Environnement, F-59508 Douai, France
First published on 3rd March 2025
Abstract
The present study introduces a novel method for the preparation of a CO2 carbon adsorbent derived from biobased precursors. Porous carbon adsorbents were synthesized through the carbonization and thermal activation of biobased chitosan-polybenzoxazine. First, the study explored the influence of varying amounts of the key polymer precursors, lysine (0.05–0.1 g) and chitosan (0.6–0.12 g), on the surface and adsorption characteristics of the obtained carbons. This aimed to identify the most favourable amounts of these precursors that resulted in the highest CO2 adsorption performance. In the subsequent stage, the study investigated the impact of different activation times (1–7 h) to enhance the surface characteristics and CO2 adsorption capacity of the activated carbon. Both carbonization and activation processes were conducted in a tubular furnace at 900 °C under N2 and CO2 atmospheres, respectively. After carbonization, the resulting carbon monoliths exhibited a char yield of approximately 49 wt%, with a BET surface area of up to 541 m2 g−1 and a CO2 uptake of 4.0 mmol g−1 at 0 °C and 1 bar. After activation, the obtained samples displayed a surface area in the range of 650–1000 m2 g−1, with CO2 adsorption capacities at 1 bar ranging from 4.5 to 5.6 mmol g−1 at 0 °C and 3.2 to 4 mmol g−1 at 25 °C. The activated carbons also demonstrated excellent selectivities for CO2/N2 and CO2/CH4 mixtures, along with a stable CO2 adsorption–desorption performance after 10 cycles.
1. Introduction
Over the past few decades, global warming has emerged as a critical concern due to increasing levels of greenhouse gases in the atmosphere, primarily caused by the use of fossil carbon resources in energy, industry and transportation.1 In particular, carbon dioxide (CO2) is widely regarded as the foremost contributor to the global rise in mean temperatures.2 As a result, numerous researchers have focused their efforts on developing efficient methods for CO2 capture and storage. These efforts aim not only to reduce CO2 emissions but also to create strategies for enhancing the selective separation of CO2 from complex gas mixtures. Recently, various CO2 capture technologies have been under investigation, including chemical absorption, adsorption and membrane separation.3–6Among these technologies, CO2 adsorption by solid porous materials has received significant attention, owing to its numerous advantages, including chemical stability, ease of recovery and high adsorption capacity, even under humid conditions.7–9 Recent studies have further demonstrated that carbon adsorbents exhibit excellent CO2 adsorption capacity and selectivity.10–12 Porous carbon materials can be produced through the pyrolysis of various polymeric precursors including polymers of natural origin, as well as synthetic polymers such as polyamide, polyacrylonitrile, phenolic resin and polymer blends.8,13 Despite the development of numerous carbonized materials, there is a strong trend toward creating new adsorbent materials from bioresource-derived precursors with superior adsorption capacity. This trend continues to open up opportunities for further advancements in this type of material.
Renewable resources and bio-based raw materials have been successfully utilized as precursors for producing carbon adsorbents, including wood,14 chitosan,15 polysaccharides16 and lignin,17 among others, showcasing promising results for CO2 capture. Additionally, N-doped carbon frameworks produced from several biomass-derived precursors have shown enhanced CO2 adsorption performance under ambient conditions.18–20
Polybenzoxazines (PBZ) are a class of phenolic thermosetting resins typically synthesized through a cationic ring-opening polymerization of benzoxazine monomers, forming a crosslinked network of tertiary amine bridges. They can be synthesized from cost-effective raw materials, including primary amines, phenol sources and formaldehyde.8,11,20–24 These materials are distinguished from traditional polymers by their exceptional features, such as good chemical and electrical resistance, high thermal stability, good mechanical strength, high char yields, low water adsorption, minimal shrinkage during curing and flame retardancy.18,25–29 These unique qualities extend the utility of PBZ across various applications, from serving as adsorbents for CO2,21,30 water treatment31 and also electronics and aerospace industries.16,32 In recent times, there has been a growing interest in polybenzoxazines derived from renewable bioresource materials.25 Notably, benzoxazine molecules have been synthesized using green solvents, such as water and aqueous solutions.26 In this work, we will explore the development of chitosan-based polybenzoxazine-derived carbon materials for CO2 capture. For comparison, Table 1 summarizes the CO2 adsorption capacity of various polybenzoxazine-derived carbon materials reported in the literature.
Chitosan (2-amino-2-deoxy-D-glucopyranose) is a natural polymer, primarily derived from the exoskeletons of crab and shrimp shells.28 It is soluble in acidic solvents having a pH value lower pH 6.0.36 This biopolymer has attracted enormous interest due to its advantageous characteristics, which include reactive functionality, a high concentration of amine groups, low production costs, widespread availability, low toxicity and environmental friendliness.28,37 Consequently, chitosan finds applications in various domains, ranging from drug delivery systems,38 separation membrane39 to active food packaging.40 Several carbon adsorbents derived from chitosan-based precursors for CO2 capture have been reported. For instance, Witoon et al.41 synthesized a meso–macroporous polyethyleneimine-loaded silica monolith using chitosan as a biotemplate. The resulting carbon product exhibited a BET surface area of 246 m2 g−1 and a CO2 adsorption capacity of 3.8 mmol g−1 at 80 °C. Alhwaige et al.42 fabricated chitosan-graphene oxide hybrid aerogels through a freeze-drying method. The resulting carbon possessed a BET surface area of 415 m2 g−1 and a CO2 adsorption capacity of 4.15 mmol g−1 at 25 °C and 1 bar pressure. In a subsequent study, the authors prepared a carbon aerogel from clay-reinforced biobased chitosan-polybenzoxazine using the same method, yielding a carbon material with high surface areas (up to 710 m2 g−1) and excellent adsorption performance of 5.72 mmol g−1 at 25 °C and 1 bar.19 Kamran et al.43 developed chitosan-based porous carbons through hydrothermal carbonization and chemical activation with KOH and NaOH. The products displayed a superior surface area of 4168 m2 g−1 and a maximum CO2 uptake of 8.36 mmol g−1 at 0 °C and 1 bar. Ghimbeu and Luchnikov15 produced nitrogen-doped carbonized beads from chitosan acetate, cross-linkers and Pluronic F127 co-solution drops. After freeze-drying and carbonization, the resulting carbon achieved a surface area of 433 m2 g−1 and a CO2 uptake of 2.85 mmol g−1 at 0 °C at 1 bar.
Within the preceding scope, the present study reports a simple, scalable and eco-friendly approach for preparing carbon adsorbent monoliths for CO2 adsorption. The formation of carbon monoliths is achieved through the utilization of biobased precursors, wherein chitosan serves as both a bio-templating and nitrogen source, lysine functions as a polymerization agent, and water serves as the exclusive solvent. Subsequently, the resulting polymer monolith undergoes drying at 75 °C, followed by carbonization and thermal activation. Therefore, the aim of this work is to fabricate a porous carbon monolith derived from chitosan-polybenzoxazine, and enhance its properties by thermal activation, including surface characteristics and adsorption capacity. The production yields, CO2/N2 and CO2/CH4 selectivities, as well as adsorption/desorption cyclability are also evaluated. It is noteworthy that this study highlights a method with the following key advantages: it mainly uses sustainable biobased precursors, still offering simplicity and scalability, and includes optimized formulation and fabrication processes, making it well suited for industrial applications.
2. Materials and methods
2.1. Materials
Chitosan (≥75% deacetylated from shrimp), resorcinol (99.0%) and formalin (37 wt% in water), were purchased from Sigma-Aldrich Corp. Lysine (98%) was purchased from Acros Organics, and APG (alkyl polyglycoside C8–C10, trade name Plantacare® 2000 UP) from BASF. All chemicals were used as received.2.2. Preparation of chitosan-PBZ carbon monolith
The chitosan-PBZ monolith was synthesized via a sol–gel process, as shown in Fig. 1. In a typical synthesis process, 0.2 g of APG was dissolved in 13 mL of deionized water, in a 50 mL flask with magnetic stirring under ambient conditions (step 1). Afterward, resorcinol (27.3 mmol, 3 g), lysine (0.1 g) and chitosan (0.9 g) were added to this solution. The mixture was vigorously stirred until complete dissolution of resorcinol and lysine, leaving a uniform suspension of chitosan (step 2). The resulting suspension was poured into a 25 mL plastic syringe and subsequently, formaldehyde (54.5 mmol, 1.63 g, corresponding to 4 mL of formalin at 37 wt% in water) was quickly added to the solution (step 3). Then, the syringe containing the suspension was vigorously agitated and placed vertically in an oven, preheated to 75 °C for 2 hours (step 4). After that, the remaining liquid and the syringe plunger were removed, and the syringe barrel was returned to the oven at the same temperature, for an additional 48 hours to achieve complete drying (step 5). | ||
| Fig. 1 Schematic of the experimental procedure for activated carbon preparation. | ||
The as-prepared polymer monolith was extracted from the syringe and pyrolyzed for 2 hours in a tubular furnace at 900 °C (with a heating rate of 5 °C min−1), under nitrogen atmosphere (with a gas flow rate of 200 mL min−1) (step 6). In this work, the selection of the pyrolysis temperature (900 °C), heating rate (5 °C min−1) and hold time (2 hours) was based on a comprehensive review of existing literature, preliminary experiments, and insights derived from prior research.44
As a first study, three polymer monoliths were prepared, each with different amounts of chitosan and lysine (corresponding to low, medium and high concentrations, as detailed in Table 2), while the mass of the remaining precursors stayed constant. These polymer monoliths were denoted as PCHL-x (polybenzoxazine–chitosan–lysine), where “x” indicates the concentration level of chitosan and lysine in the resulting material (x = 1 for low concentration, x = 2 for medium concentration and x = 3 for high concentration). Subsequently, the three polymer monoliths were pyrolyzed and the CO2 adsorption capacity of the obtained porous carbons was characterized (characterization techniques are detailed in section 2.3).
| Sample name | Concentration level | Chitosan [g] | Lysine [g] |
|---|---|---|---|
| PCHL-1 | Low | 0.6 | 0.05 |
| PCHL-2 | Medium | 0.9 | 0.1 |
| PCHL-3 | High | 1.2 | 0.15 |
As a second study, the pyrolyzed sample displaying the highest CO2 adsorption capacity was chosen for further investigation regarding the subsequent thermal activation step. In order to optimize the surface characteristics and adsorption capacity, different activation times (1, 3, 5 and 7 hours) were applied to the pyrolyzed monolith previously selected. In all these experiments, the carbon monolith was heated from room temperature to 900 °C, with a ramp rate of 20 °C min−1 under nitrogen atmosphere.
The flowing gas was subsequently switched from nitrogen to a gas mixture of CO2/N2 (20/80 molar%), maintained for the selected activation time, and then switched back to nitrogen, all at the same flow rate (200 mL min−1). The resulting activated carbons were designated as ACP-x-y (activated carbon polybenzoxazine), where “x” indicates the concentration level of chitosan and lysine in the polymer monolith, and “y” refers to the activation time (in hours).
2.3. Characterization techniques
Fourier Transform Infrared (FTIR) spectroscopy was used to identify the functional groups of the PBZ materials and the porous carbons, using a Thermo Scientific Nicolet iS5 with iD1 transmission FTIR spectrometer (Thermo 190 Scientific®, USA) at room temperature in the range of 400–4000 cm−1 at a resolution of 4 cm−1, with a KBr pellet. Elemental analysis was carried out on a Thermo Scientific Flash 2000 CHNS/O Analyzer (Thermo Fisher Scientific, USA). The surface topography of porous carbon materials was observed by Scanning Electron Microscopy (SEM), with a FEI Quanta 3D FIB FEG instrument operated at 20 kV. A 3Flex sorption analyser (Micromeritics, Norcross, GA, USA) was used to assess the surface characteristics and gas adsorption properties, by measuring the isotherms for CO2, N2 and CH4. N2 adsorption/desorption isotherms were conducted at −196 °C using nitrogen (99.998% purity), after degassing the porous carbons at 220 °C for 20 h. The specific surface area (SBET) was calculated from the N2 adsorption isotherm by using the Brunauer–Emmett–Teller (BET) method, while the pore size distribution was estimated with the Horvath–Kawazoe (HK) method. The total pore volume (Vtotal) was calculated from the amount of N2 adsorbed at a relative pressure of 0.99. Micropore volume (Vmic) and micropore surface area (Smic) were calculated using the t-plot method. The adsorption isotherms of CO2, N2 and CH4 were conducted at three different temperatures (0, 25 and 50 °C) in the pressure range of 0 to 1 bar. Prior to each adsorption test, the samples were degassed at 220 °C for at least 6 hours.The char yield refers to the amount of carbon obtained after the carbonization process, expressed as a percentage of the initial mass of the precursor material. It was calculated based on the following equation:
 | (1) |
The burn-off reflects the activation progress and is calculated as the percentage of mass lost during the activation of the carbonized material:
 | (2) |
The activation process leads to a higher surface area but also results in a mass loss of the activated product (burn-off). Thus, determining the optimal balance between CO2 adsorption capacity and available mass of activated carbon is crucial. To address this, we introduce the concept of “available CO2 adsorption capacity”, expressed in mmol of CO2 per gram of pyrolyzed material. This requires converting the CO2 adsorbed per unit mass of activated material (denoted as Ac) into CO2 adsorbed per unit mass of pyrolyzed material. Consequently, the available CO2 adsorption capacity is calculated as:
 | (3) |
The CO2/N2 and CO2/CH4 selectivities of the carbon adsorbent were calculated using the Ideal Adsorbed Solution Theory (IAST),5,22 given by:
 | (4) |
3. Results and discussion
3.1. Structural properties of the polymer and porous carbon monoliths
Porous carbon monoliths were successfully prepared from chitosan-PBZ polymers. In Fig. 2a, a representative example of chitosan-PBZ polymer monolith and its carbonized counterpart are presented. Notably, the carbonization process yields a crack-free carbon monolith that retains a uniform shape, similar to the original polymer structure. The resulting carbon monolith typically exhibits dimensions of 31 mm in length and 15 mm in diameter, which corresponds to an overall volume shrinkage of 60%. SEM images (Fig. 2b and c) further illustrate that both the polymer and carbon materials consist of interconnected microspheres and possibly amorphous solid, forming a microporous framework. The FTIR spectra of both the precursor polymer and the porous carbon are presented in Fig. 2d. The FTIR spectrum of chitosan-PBZ resin exhibits characteristic absorption peaks in the range of 3400–3200 cm−1 attributed to the O–H bonds and/or N–H stretching vibrations of chitosan amide groups.39 Additionally, bands at 2922 cm−1 and 2847 cm−1 correspond to C–H bending. The absorption peaks in the range of 1600 cm−1 and 1485 cm−1 can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C within the aromatic rings of the benzoxazine structure.30,45 Furthermore, the bands observed at 1240 and 1160 cm−1 were assigned to asymmetric and symmetric stretching of the oxazine structure C–O–C.27 Moreover, a band around 1110 cm−1 suggests the presence of the C–N stretching vibrations,46 while the weak absorption bands occurring at 926, 874 and 764 cm−1 are indicative of C–H groups.
C and C–C within the aromatic rings of the benzoxazine structure.30,45 Furthermore, the bands observed at 1240 and 1160 cm−1 were assigned to asymmetric and symmetric stretching of the oxazine structure C–O–C.27 Moreover, a band around 1110 cm−1 suggests the presence of the C–N stretching vibrations,46 while the weak absorption bands occurring at 926, 874 and 764 cm−1 are indicative of C–H groups.
 | ||
| Fig. 2 (a) Representative photographs of polymer and carbon monoliths; (b) and (c) SEM images of resin and carbonized materials, respectively; (d) FTIR spectra of resin and carbonized materials. | ||
3.2. Effect of chitosan and lysine concentrations
As noted earlier, three monoliths with varying amounts of lysine and chitosan were synthesized and carbonized (while the mass of the remaining precursors stayed constant). The CO2 adsorption isotherms, measured at 0 °C, are illustrated in Fig. 3. The highest CO2 uptake capacity at 0 °C was achieved with the PCHL-2 sample. To understand this behaviour, Table 3 presents the key characteristics of the three monoliths. The data reveals an almost constant char yield for all the samples of approximately 49 wt%. In addition, elemental analysis demonstrated an increase in nitrogen content with the addition of chitosan and lysine, ranging from 1.7 to 2.3 wt%. This observation aligns with expectations, considering that both precursors can be considered as nitrogen sources. Furthermore, the results (Table 3 and Fig. 3) reveal that the highest BET surface area (541 m2 g−1) and CO2 uptake capacity at 0 °C and 1 bar (4.05 mmol g−1) were achieved with the PCHL-2 sample. These results suggest that the cross-linking of chitosan and lysine at an intermediate concentration level results in an optimized carbon framework structure, with increased surface area and active sites for CO2 adsorption. | ||
| Fig. 3 CO2 adsorption isotherms at 0 °C of chitosan-based porous carbon monoliths. | ||
| Sample | Chitosan and lysine concentration level | Char yield [%] | SBET [m2 g−1] | CO2 uptake at 0 °C and 1 bar [mmol g−1] | Elemental analysis (wt%) | ||
|---|---|---|---|---|---|---|---|
| N | C | H | |||||
| PCHL-1 | Low | 51% | 489 | 3.36 | 1.73 | 93.41 | 0.40 |
| PCHL-2 | Medium | 49% | 541 | 4.05 | 2.07 | 91.47 | 0.37 |
| PCHL-3 | High | 47% | 506 | 3.66 | 2.30 | 93.70 | 0.41 |
According to the work of Sun et al.,47 a structured network made of chitosan and other elements, such as amino acids, could generate nitrogen-enriched substructures, including pyridinic or graphitic moieties, after pyrolysis. These substructures are reported to be important for the stabilization of the CO2 uptake. Consequently, the PCHL-2 formula was selected as the preferred material for the subsequent thermal activation experiments.
3.3. Effect of activation progress
The carbon material obtained after pyrolysis of the chitosan-PBZ polymer was subjected to activation with CO2 at 900 °C, in order to enhance its surface characteristics and CO2 adsorption capacity. The evolution of the textural properties (specific surface area, micropore surface area, total and micropore volumes) with activation progress (i.e., burn-off) is summarized in Table 4, together with the CO2 adsorption capacity and elemental analysis.| Sample | Burn-off [%] | SBETa [m2 g−1] | Smicb [m2 g−1] | Vtotalc [cm3 g−1] | Vmicd [cm3 g−1] | CO2 uptake [mmol g−1] | Elemental analysis [wt%] | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 °C | 25 °C | N | C | H | O | ||||||
| a BET specific surface area obtained from the adsorption data in the P/P0 range from 0.05 to 0.2.b Microporous specific surface area obtained from t-plot method.c Total pore volume at a relative pressure of 0.99.d Micropore volume. The results are shown as mean values (n = 2 replicates). | |||||||||||
| PCHL-2 | 0 | 541 | 501 | 0.28 | 0.25 | 4.05 | 3.25 | 2.07 | 91.47 | 0.37 | 1.53 |
| ACP-2-1 | 5 | 660 | 622 | 0.33 | 0.32 | 4.52 | 2.16 | 91.74 | 0.53 | 1.22 | |
| ACP-2-3 | 12 | 930 | 845 | 0.46 | 0.42 | 5.22 | 3.99 | 1.98 | 92.60 | 0.49 | 1.11 |
| ACP-2-5 | 17 | 995 | 888 | 0.49 | 0.45 | 5.44 | 1.87 | 92.48 | 0.42 | 1.02 | |
| ACP-2-7 | 23 | 998 | 902 | 0.50 | 0.46 | 5.60 | 3.95 | 1.84 | 92.32 | 0.42 | 1.29 |
First, it is evident that the burn-off increases with activation time, ranging from 5% to 23% within the studied time range (1 to 7 hours). This increase is expected due to the extended contact time between CO2 and the carbon material during the activation treatment. However, the resulting activated carbon monoliths did not show any significant modifications in their physical structure during the activation process, as the shrinkage did not exceed 5% under all studied conditions. In the same way, there is no clear trend observed in the elemental composition, particularly in terms of nitrogen content. The samples still contain approximately 2 wt% of nitrogen after activation. These nitrogen content values are of interest due to the potential role that N heteroatoms can play in CO2 capture.8,48
The nitrogen adsorption/desorption isotherms and the corresponding pore-size distributions of the obtained carbon materials are displayed in Fig. 4a and b, respectively. As shown in Fig. 4a, the porous carbon materials exhibit typical type-I isotherms, with a rapid increase of N2 sorption at low relative pressures (<0.05), indicating the predominance of micropores for all of these samples.
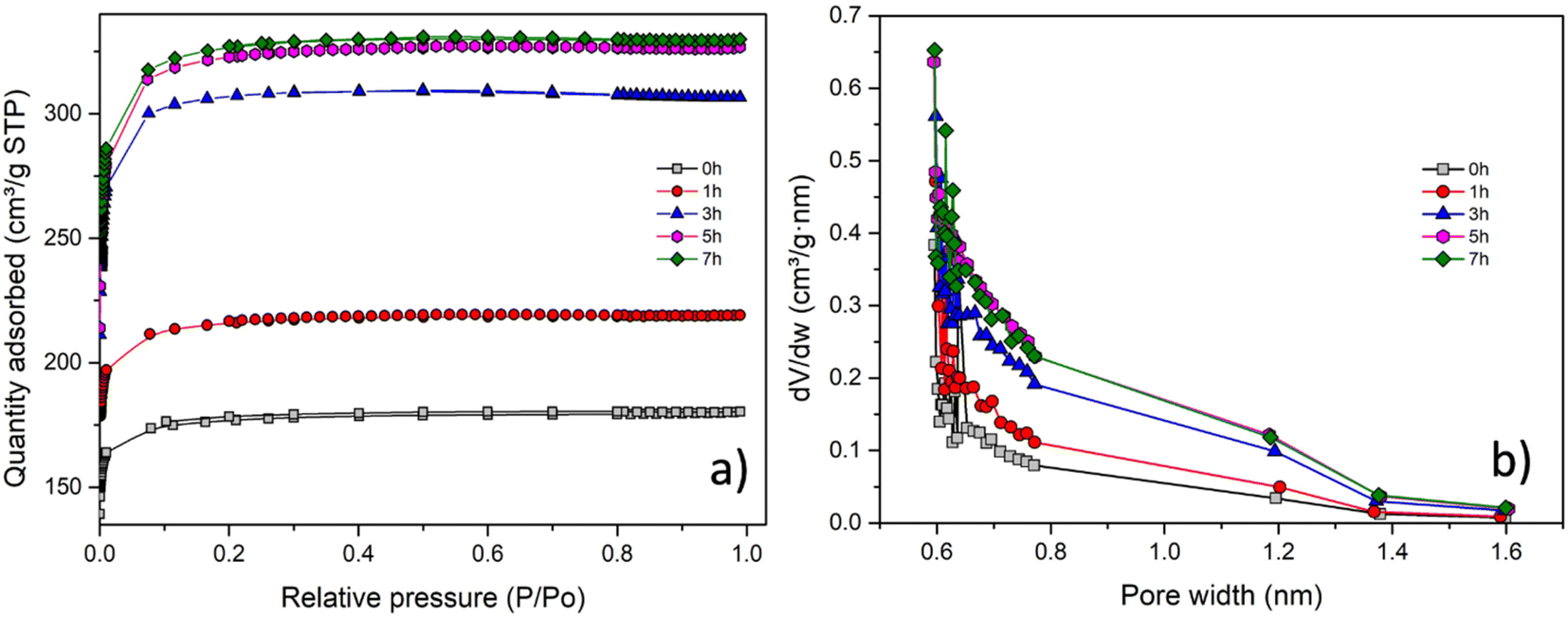 | ||
| Fig. 4 (a) N2 sorption isotherms for different activation times. (b) Pore size distributions (PSDs) calculated from the Horvath–Kawazoe method. | ||
From Table 4, a progressive increase of the main surface characteristics (specific surface area, micropore surface area, total and micropore volumes) can be observed with the activation progress. For instance, SBET increased from 541 to 998 m2 g−1, while Vtotal increased from 0.28 to 0.50 cm3 g−1. This trend is consistent with findings in prior studies on CO2 activation of carbons.49–51 The enhancement of these textural properties can be attributed to the prolonged contact time between CO2 and the char, leading to increased burn-off and greater pore development.52 Also, it should be noted that the increase of the main surface characteristics becomes slower in the later stages of activation. The Horvath–Kawazoe micropore size distributions of the obtained porous carbons are presented in Fig. 4b. The majority of the micropores are concentrated in the 0.6–1.2 nm range, with the cumulative pore volume increasing as activation progresses. It can be concluded that thermal activation significantly improves the surface properties of the carbon material. Notably, through this process, the activated carbon achieves up to twice the specific surface area SBET and pore volume Vtotal, compared to the non-activated carbon.
Finally, the CO2 adsorption capacity of the porous carbon materials was investigated at 0 °C under atmospheric pressure (1 bar). Table 4 provides a summary of the CO2 uptakes and Fig. 5 shows the CO2 adsorption isotherms at 0 °C. From both Table 4 and Fig. 5, it can be noticed that the CO2 adsorption capacity progressively increases with the activation progress.
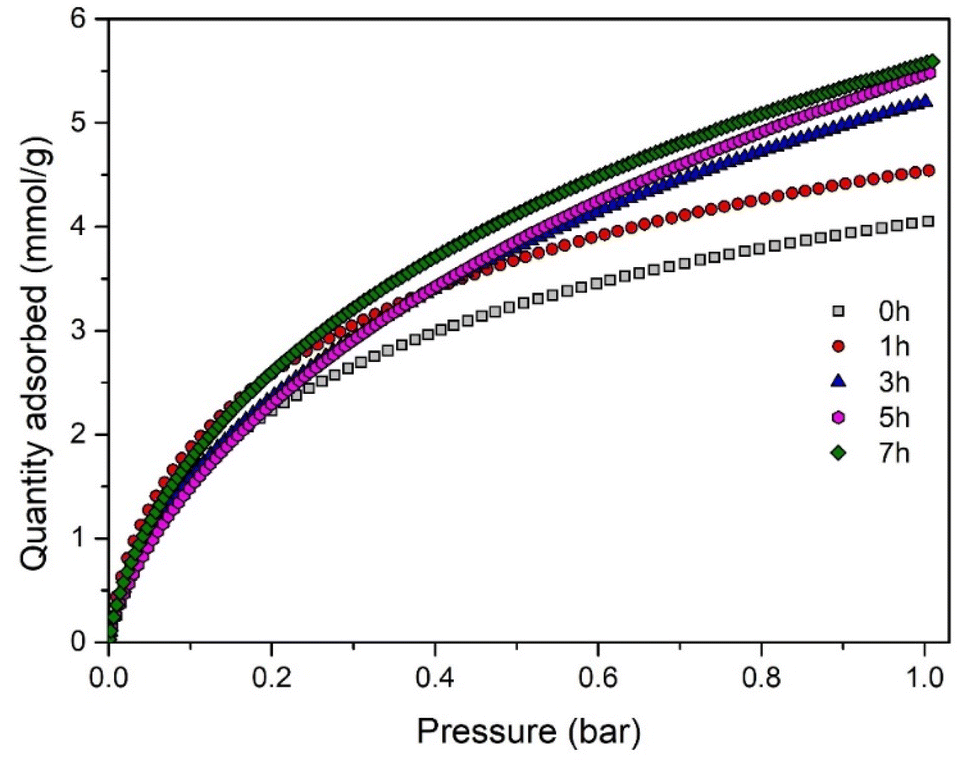 | ||
| Fig. 5 CO2 adsorption isotherms at 0 °C for different activation times. | ||
The highest CO2 adsorption capacity of 5.6 mmol g−1 is achieved after 7 h of activation, corresponding to a burn-off of 23%. Notably, this represents an increase in adsorption capacity of up to 38% compared to non-activated carbon. This enhancement is consistent with the earlier trends observed for the BET surface area and pore volume. Furthermore, as the activation progresses, there is an enlargement of the micropore range, especially in the fraction of micropores smaller than 0.8 nm (Fig. 4b). This factor is likely to contribute to the improvement of CO2 uptake, as the CO2 adsorption capacity at 1 bar is highly dependent on the micropores in this size range.5,34,48,50 It is also noteworthy that the resulting carbon materials exhibit a very high CO2 uptake under ambient conditions, ranging from 3.25 to 4 mmol g−1 at 25 °C and 1 bar.
3.4. Production yields of the carbon adsorbents
Fig. 6 shows the evolution of the BET surface area, CO2 uptake (at 0 °C and 1 bar) and available CO2 adsorption capacity of the activated carbons with burn-off. As explained in the previous section, the activation process leads to an enhancement of textural properties and CO2 adsorption capacity (Fig. 6a and b). However, a higher activation progress also leads to a higher mass loss (burn-off), i.e., a lower quantity of the final adsorbent material. Therefore, in the context of industrial applications, it becomes imperative to identify the best compromise between CO2 adsorption capacity and available mass of activated carbon. To this purpose, the available CO2 adsorption capacity was calculated for all activated carbons, using eqn (3). Fig. 6c displays the CO2 adsorption capacity per unit mass of pyrolyzed carbon as a function of the activation burn-off.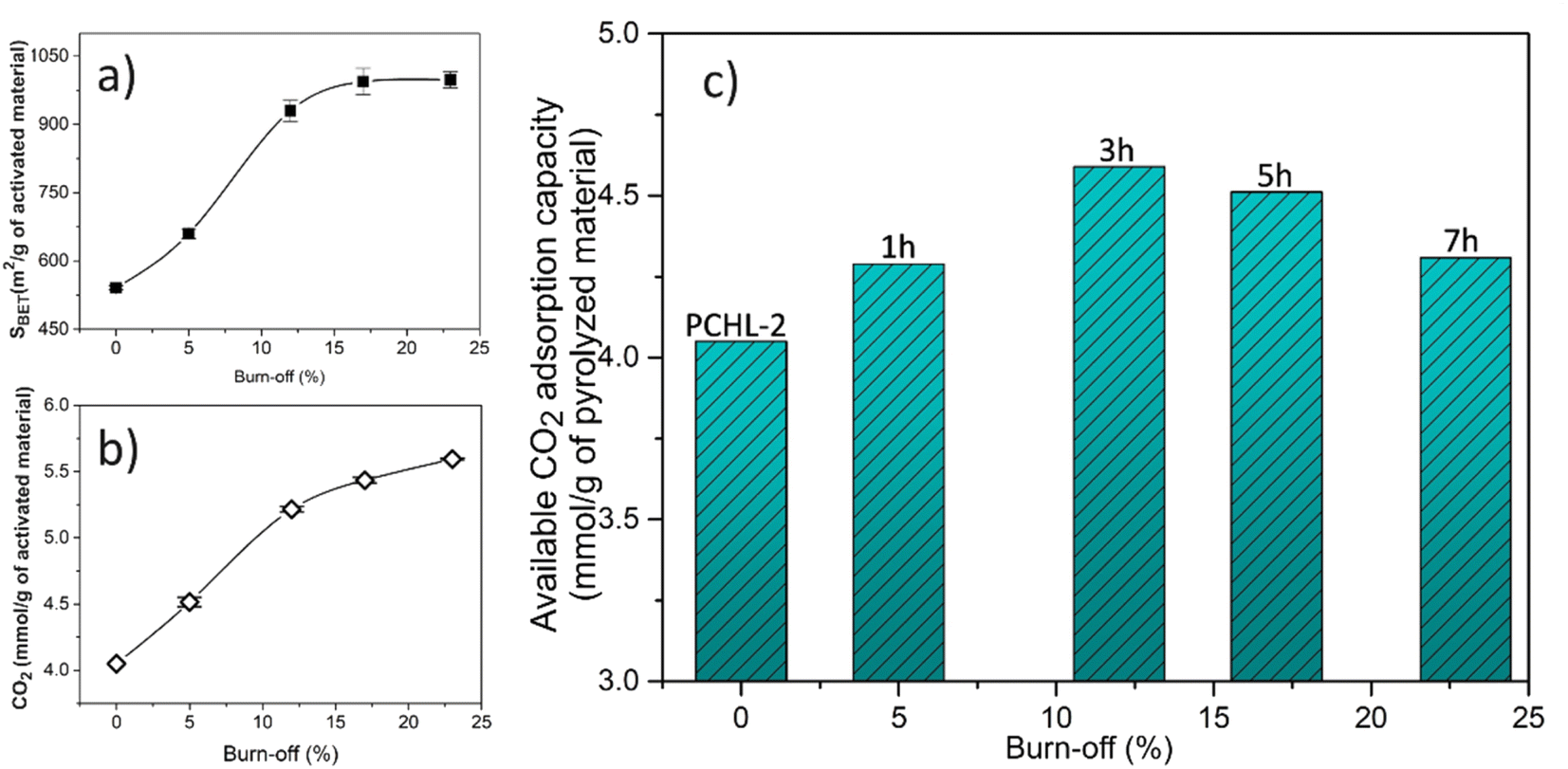 | ||
| Fig. 6 Evolution of the (a) BET surface area, (b) CO2 uptake (at 0 °C and 1 bar), and (c) available CO2 adsorption capacity of the activated carbons with burn-off. | ||
The obtained results show that the carbon adsorbent ACP-2-3, obtained after 3 hours of activation (corresponding to a burn-off of 12%), exhibits the best balance between adsorption capacity and mass loss. Accordingly, a recommended route for producing an optimal carbon adsorbent involves carbonizing at 900 °C the chitosan-PBZ monolith with an intermediate concentration of chitosan and lysine, followed by a 3 hour thermal activation process.
3.5. CO2/N2 and CO2/CH4 selectivities
A crucial aspect in the development of a CO2 adsorbent is its ability to separate CO2 from other gases, particularly N2 (for CO2 capture from flue gas streams) and CH4 (for natural gas sweetening). Therefore, an effective adsorbent material with high adsorption performance should also exhibit a high CO2 selectivity over N2 and/or CH4. As a consequence, the CO2/N2 and CO2/CH4 selectivities of the non-activated porous carbon (PCHL-2) and the chosen activated porous carbon (ACP-2-3) were investigated. To this end, the N2 and CH4 adsorption isotherms were characterized at three temperatures (0, 25 and 50 °C) for pressures ranging from 0 to 1 bar. The obtained results are presented in Fig. 7 and summarized in Table 5. | ||
Fig. 7 CO2 and N2 adsorption isotherms for (a) PCHL-2 and (b) ACP-2-3; CO2 and CH4 adsorption isotherms for (c) PCHL-2 and (d) ACP-2-3; IAST selectivity analyses for (e) (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85) CO2/N2 mixture and (f) (50:50) CO2/CH4 mixture, for PCHL-2 (t = 0 h) and ACP-2-3 (t = 3 h). :85) CO2/N2 mixture and (f) (50:50) CO2/CH4 mixture, for PCHL-2 (t = 0 h) and ACP-2-3 (t = 3 h). | ||
| Sample | CO2 uptake [mmol g−1] | N2 uptake [mmol g−1] | CH4 uptake [mmol g−1] | IAST selectivity (at 1 bar) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO2/N2 (15:85) |
CO2/CH4 (50:50) |
||||||||||||||
| 0 °C | 25 °C | 50 °C | 0 °C | 25 °C | 50 °C | 0 °C | 25 °C | 50 °C | 0 °C | 25 °C | 50 °C | 0 °C | 25 °C | 50 °C | |
| PCHL-2 | 4.0 | 3.2 | 1.9 | 0.79 | 0.43 | 0.27 | 1.0 | 0.8 | 0.6 | 36.6 | 35.4 | 23,3 | 35.3 | 25.0 | 13.8 |
| ACP-2-3 | 5.2 | 4.0 | 2.1 | 0.82 | 0.49 | 0.30 | 2.1 | 1.0 | 1.0 | 33.0 | 28.0 | 16.0 | 6.5 | 5.7 | 3.2 |
The N2 (Fig. 7a and b) and CH4 (Fig. 7c and d) adsorption capacities of PCHL-2 and ACP-2-3 measured at 1 bar pressure and 0 °C, 25 °C and 50 °C, range from 0.27 to 0.82 and 0.6 to 2.1 mmol g−1, respectively. On the other hand, the CO2 adsorption capacity for the same materials under the same conditions ranges from 1.9 to 5.2 mmol g−1. This observation indicates significantly lower N2 and CH4 uptakes compared to that of CO2 under the same conditions, suggesting the preferential adsorption of CO2 over N2 and CH4.
The selectivity (Sads) performance of the binary mixtures of CO2/N2 and CO2/CH4 was estimated using the Ideal Adsorbed Solution Theory (IAST), as defined in section 2.3 (eqn (4)). In the 0–1 bar range, the CO2/N2 selectivity (Fig. 7e) was calculated considering a typical flue gas composition of 15% CO2 and 85% N2, whereas for CO2/CH4 selectivity (Fig. 7f), a 50:50 mixture was considered (natural gas sweetening). As shown in Table 5, PCHL-2 demonstrates CO2/N2 IAST selectivity factors (at 1 bar) of 36, 35 and 23, while ACP-2-3 exhibits factors of 33, 28, and 16 at 0 °C, 25 °C and 50 °C, respectively. These results indicate that the carbon adsorbent is effective in selectively separating CO2 from N2, particularly at the lowest studied temperature (close to 0 °C), where the material demonstrated the highest selectivity. The same IAST selectivity analysis for the CO2/CH4 (50:50) mixture shows that at 1 bar pressure and for temperatures ranging from 0 °C to 50 °C, PCHL-2 displays IAST selectivity factors between 14 and 36, whereas ACP-2-3 shows lower factors ranging from 3 to 6. Similar to the CO2/N2 mixture, it is evident that the carbon material shows a higher selectivity for CO2/CH4 separation at temperatures close to 0 °C.
However, it is noteworthy that the selectivity of the non-activated carbon (PCHL-2) is higher than that of the activated carbon (ACP-2-3), particularly for the separation of a CO2/CH4 (50:50) mixture. In fact, materials with smaller average pore sizes are expected to exhibit higher selectivity for CO2 over CH4.53 CO2, which has a higher quadrupole moment than CH4, interacts more strongly with carbon surfaces within small pores, particularly ultramicropores (<0.7 nm), leading to preferential adsorption.54 As shown in Fig. 4b, activation leads to the formation of wider pores in activated carbons compared to the non-activated carbon. This pore enlargement enhances CH4 adsorption, thereby decreasing CO2 selectivity. From an industrial perspective, these results suggest the activation step could potentially be omitted from the porous carbon fabrication process. However, other factors must be considered, such as the quantity of adsorbent required, contact time, and additional process parameters that could influence the overall performance and efficiency of an industrial process. Breakthrough curves would be of great interest for process optimization, as they would allow adsorption capacity and selectivity assessment under dynamic flow conditions. From a theoretical standpoint, these results demonstrate that selectivity clearly does not solely depend on the specific surface area but is also likely influenced by other factors such as porous structure and heteroatoms, which undergo alterations during activation and may consequently impact selectivity. Further investigation would be required to clearly identify the mechanisms leading to the decrease in selectivity after activation.
Based on all these results, the adsorbent material has demonstrated high CO2 adsorption capacity and good selectivities for CO2/N2 and CO2/CH4 separations. Consequently, the porous carbon material developed in the present study is suitable for industrial applications in gas separation, such as natural gas sweetening (CO2/CH4 separation) or the removal of CO2 from flue gas (CO2/N2 separation).
3.6. Adsorption/desorption cyclability
Another crucial aspect to investigate for industrial applications is the stability of the adsorption capacity after multiple adsorption–desorption cycles. In this study, one cycle consisted of a desorption stage at 130 °C under vacuum to desorb CO2, followed by an adsorption stage at 0 °C and 1 bar to measure the CO2 adsorption capacity at equilibrium. The 3Flex sorption analyser (Micromeritics, Norcross, GA, USA) was used to evaluate the CO2 adsorption capacity under equilibrium conditions. This adsorption–desorption cycle was repeated 10 times consecutively on the same activated carbon (ACP-2-3). The results are presented in Fig. 8 and show that even after 10 cycles, the CO2 adsorption capacity remains stable. This indicates that ACP-2-3 possesses excellent adsorption–desorption cyclability, which is of great interest for industrial applications. | ||
| Fig. 8 Cyclical CO2 adsorption behaviour of the ACP-2-3 activated carbon: adsorption at 0 °C and 1 bar, and desorption at 130 °C under vacuum. | ||
4. Conclusions
This study introduces a straightforward methodology for the fabrication of microporous carbon monoliths from biobased chitosan-PBZ polymers for CO2 separation. The quantity of chitosan and lysine was found to influence the textural and adsorption properties of the resulting carbon, with medium concentrations providing the most favourable characteristics. Conversely, the char yield remained nearly constant (∼49 wt%) for the three concentrations studied.The thermal activation process significantly enhanced the adsorption capacity of the porous carbon monolith. A higher activation progress contributed to greater pore development in the carbonaceous material, resulting in a notable improvement in textural characteristics and adsorption capacity. The activated carbon exhibited a maximum surface area and CO2 uptake of around 1000 m2 g−1 and 5.6 mmol g−1, respectively, with a burn-off of 23%.
Analysis of the available CO2 adsorption capacity revealed that the activated material achieved the most favourable compromise between increased adsorption capacity and mass loss after a 3 hour activation process (corresponding to a burn-off of 12%). This activated carbon also demonstrated good selectivities for CO2/N2 and CO2/CH4 separation, with excellent adsorption–desorption cyclability.
A very important result of this work is that activation may sometimes lead to a decrease in selectivity. This was particularly evident for the separation of the CO2/CH4 (50:50) mixture. From an industrial perspective, this suggests that the activation step could potentially be omitted from the porous carbon fabrication process. Further investigation would be required to clearly identify the mechanisms leading to the decrease in selectivity after activation.
In summary, the advantages of easy preparation, favourable surface and adsorption properties, good selectivity and regeneration capacity, position the obtained adsorbent carbons (non-activated and activated) as promising candidates for industrial CO2 separation applications.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper. Should any raw data files be needed in another format, they are available from the corresponding author upon request. Source data are provided in this paper.Author contributions
José E. Mosquera: data curation, formal analysis, investigation, methodology, resources, validation, visualization, writing – original draft. Liana Delevingne: data curation, investigation, validation, visualization. Frédéric Delbecq: conceptualization, formal analysis, methodology, writing – review & editing. Elias Daouk: conceptualization, formal analysis, methodology, writing – review & editing. Audrey Drelich: conceptualization, formal analysis, methodology, writing – review & editing. Khashayar Saleh: conceptualization, project administration, supervision, writing – review & editing. Rémi Gautier: conceptualization, funding acquisition, methodology, project administration, writing – review & editing. Mikel Leturia: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Région Hauts-de-France (PISCO Project, dispositif STIMulE, volet Partenarial). They also thank Zaira Hernandez for her invaluable contribution during the preliminary experiments conducted as part of her MSc internship.References
- M. W. Jones, G. P. Peters, T. Gasser, R. M. Andrew, C. Schwingshackl, J. Gütschow, R. A. Houghton, P. Friedlingstein, J. Pongratz and C. Le Quéré, Sci. Data, 2023, 10, 1–23 CrossRef.
- J. O. Kaplan and M. New, Clim. Change, 2006, 79, 213–241 CrossRef.
- A. M. Yousef, W. M. El-Maghlany, Y. A. Eldrainy and A. Attia, Energy, 2018, 156, 328–351 CrossRef.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Y. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef.
- X. Ma, Y. Li, M. Cao and C. Hu, J. Mater. Chem. A, 2014, 2, 4819–4826 RSC.
- B. J. Bucior, D. L. Chen, J. Liu and J. K. Johnson, J. Phys. Chem. C, 2012, 116, 25904–25910 CrossRef.
- M. Sai Bhargava Reddy, D. Ponnamma, K. K. Sadasivuni, B. Kumar and A. M. Abdullah, RSC Adv., 2021, 11, 12658–12681 RSC.
- I. Tiwari, P. Sharma and L. Nebhani, Mater. Today Chem., 2022, 23, 100734 CrossRef CAS.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS.
- X. L. Zhou, H. Zhang, L. M. Shao, F. Lü and P. J. He, Waste Biomass Valorization, 2021, 12, 1699–1724 CrossRef CAS.
- G. P. Hao, W. C. Li, S. Wang, G. H. Wang, L. Qi and A. H. Lu, Carbon, 2011, 49, 3762–3772 CrossRef CAS.
- J. Du, W. C. Li, Z. X. Ren, L. P. Guo and A. H. Lu, J. Energy Chem., 2020, 42, 56–61 CrossRef.
- T. H. Le and H. Yoon, Carbon, 2019, 152, 796–817 CrossRef CAS.
- A. C. Lua, F. Y. Lau and J. Guo, J. Anal. Appl. Pyrolysis, 2006, 76, 96–102 Search PubMed.
- C. M. Ghimbeu and V. A. Luchnikov, Microporous Mesoporous Mater., 2018, 263, 42–52 CrossRef.
- B. Kiskan, N. N. Ghosh and Y. Yagci, Polym. Int., 2011, 60, 167–177 CrossRef CAS.
- N. Liu, L. Shao, C. Wang, F. Sun, Z. Wu, P. Zhan, L. Zhang and H. Wan, Int. J. Biol. Macromol., 2022, 221, 25–37 CrossRef CAS PubMed.
- M. M. Samy, M. G. Mohamed and S. W. Kuo, Eur. Polym. J., 2020, 138, 109954 CrossRef CAS.
- A. A. Alhwaige, H. Ishida and S. Qutubuddin, ACS Sustain. Chem. Eng., 2016, 4, 1286–1295 CrossRef CAS.
- G. P. Hao, W. C. Li, D. Qian, G. H. Wang, W. P. Zhang, T. Zhang, A. Q. Wang, F. Schüth, H. J. Bongard and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS.
- S. Wang, W. Li, L. Zhang, Z. Jin and A.-H. Lu, J. Mater. Chem. C, 2015, 3, 4406–4412 Search PubMed.
- L. Hong, S. Ju, X. Liu, Q. Zhuang, G. Zhan and X. Yu, Energy Fuels, 2019, 33, 11454–11464 CrossRef CAS.
- R. Konnola and T. S. Anirudhan, J. Environ. Chem. Eng., 2020, 8, 103614 CrossRef CAS.
- T. Takeichi, T. Kawauchi and T. Agag, Polym. J., 2008, 40, 1121–1131 CrossRef CAS.
- P. Thirukumaran, A. Shakila Parveen, R. Atchudan and S. C. Kim, Eur. Polym. J., 2018, 109, 248–256 CrossRef CAS.
- A. A. Alhwaige, T. Agag, H. Ishida and S. Qutubuddin, Biomacromolecules, 2013, 14, 1806–1815 CrossRef CAS PubMed.
- Y. Zhu, J. Su, R. Lin, Y. Jiang and P. Li, Thermochim. Acta, 2020, 683, 178465 CrossRef CAS.
- N. Ramdani, M. Chrigui, J. Wang, T. T. Feng, X. Y. He, W. Bin Liu and X. S. Zheng, J. Compos. Mater., 2015, 49, 2449–2458 CrossRef CAS.
- N. Thepphankulngarm, T. Chaisuwan, D. Tanangteerapong and P. Kongkachuichay, Mater. Chem. Phys., 2022, 287, 126258 CrossRef CAS.
- Z. E. Jin, J. L. Wang, R. J. Zhao, T. T. Guan, D. D. Zhang and K. X. Li, New Res. Carbon Mater., 2018, 33, 392–401 Search PubMed.
- A. Hariharan, P. Prabunathan, A. Kumaravel, M. Manoj and M. Alagar, Polym. Test., 2020, 86, 106443 CrossRef CAS.
- C. Shaer, L. Oppenheimer, A. Lin and H. Ishida, Polymers, 2021, 13, 3775 CrossRef CAS PubMed.
- L. H. Zhang, W. C. Li, L. Tang, Q. G. Wang, Q. T. Hu, Y. Zhang and A. H. Lu, J. Mater. Chem. A, 2018, 6, 24285–24290 RSC.
- Z. Guo, X. Lu and Z. Xin, Colloids Surf., A, 2022, 646, 128845 CrossRef CAS.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- B. Kancı Bozoğlan, O. Duman and S. Tunç, Colloids Surf., A, 2021, 610, 125600 CrossRef.
- N. Yadav, M. Monisha, R. Niranjan, A. Dubey, S. Patil, R. Priyadarshini and B. Lochab, Carbohydr. Polym., 2021, 254, 117296 CrossRef CAS.
- P. I. Morgado, S. P. Miguel, I. J. Correia and A. Aguiar-Ricardo, Carbohydr. Polym., 2017, 159, 136–145 CrossRef CAS PubMed.
- D. D. Kachhadiya and Z. V. P. Murthy, J. Environ. Chem. Eng., 2023, 11, 109307 CrossRef CAS.
- S. Casalini and M. Giacinti Baschetti, J. Sci. Food Agric., 2023, 103, 1021–1041 CrossRef CAS.
- T. Witoon and M. Chareonpanich, Mater. Lett., 2012, 81, 181–184 CrossRef CAS.
- A. A. Alhwaige, T. Agag, H. Ishida and S. Qutubuddin, RSC Adv., 2013, 3, 16011–16020 RSC.
- U. Kamran and S. J. Park, J. CO2 Util., 2020, 40, 101212 CrossRef CAS.
- J. E. Mosquera, F. Delbecq, E. Daouk, A. Drelich, K. Saleh, R. Gautier and M. Leturia, Processes, 2024, 12, 1604 CrossRef CAS.
- M. G. Mohamed, M. M. Samy, T. H. Mansoure, C. J. Li, W. C. Li, J. H. Chen, K. Zhang and S. W. Kuo, Int. J. Mol. Sci., 2022, 23, 347 CrossRef CAS.
- F. Liu, K. Huang, Q. Wu and S. Dai, Adv. Mater., 2017, 29, 1–8 Search PubMed.
- Y. Sun, Z. He, H. Fan and S. Wang, J. Power Sources, 2024, 614, 235027 CrossRef CAS.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS.
- M. J. Teng, Y. S. Wei, T. G. Hu, Y. Zhang, K. Feng, M. H. Zong and H. Wu, J. Food Eng., 2020, 281, 109993 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 CrossRef CAS PubMed.
- K. Yang, J. Peng, H. Xia, L. Zhang, C. Srinivasakannan and S. Guo, J. Taiwan Inst. Chem. Eng., 2010, 41, 367–372 CrossRef CAS.
- J. F. Kwiatkowski, Activated Carbon : Classifications, Properties and Applications, Nova Science Publishers, New York, 2012 Search PubMed.
- A. Rahimalimamaghani, R. Ramezani, D. A. P. Tanaka and F. Gallucci, Ind. Eng. Chem. Res., 2023, 62, 19116–19132 CAS.
- N. Yang, S. Liu and X. Yang, Appl. Surf. Sci., 2015, 356, 1262–1271 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |