
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning tools for the characterization of bioactive metabolites derived from different parts of Ochrosia elliptica Labill. for the management of Alzheimer's disease†
Mohamed A. Salem‡
a,
Essam Abdel-Sattar *b,
Asmaa A. Mandour*c and
Riham A. El-Shiekhb
*b,
Asmaa A. Mandour*c and
Riham A. El-Shiekhb
aDepartment of Pharmacognosy and Natural Products, Faculty of Pharmacy, Menoufia University, Gamal Abd El Nasr St, Shibin Elkom, 32511, Menoufia, Egypt. E-mail: mohamed.salem@phrm.menofia.edu.eg
bPharmacognosy Department, Faculty of Pharmacy, Cairo University, Kasr El Aini St, P. B. 11562, Cairo, Egypt. E-mail: Riham.adel@pharma.cu.edu.eg; essam.abdelsattar@pharma.cu.edu.eg
cPharmaceutical Chemistry Department, Faculty of Pharmacy, Future University in Egypt (FUE), Cairo 11835, Egypt. E-mail: asmaa.abdelkereim@fue.edu.eg
First published on 7th April 2025
Abstract
Currently, natural products are one of the most valuable resources for discovering novel chemical medicinal entities. A total of 41 compounds were tentatively identified from the stems, barks, roots, and fruits of Ochrosia elliptica Labill. using UPLC-MS/MS analysis. The binding affinities of these biomarkers for the active sites of acetyl- and butyryl-cholinesterase enzymes were further validated using molecular docking studies, which showed good results with (–)C-Docker interaction energy ranges of 30.17–86.73 and 26.81–72.42 (kcal mol−1), respectively. The most active predicted compound was a quercetin derivative [quercetin-3-O-rhamnosyl-(1-3)-rhamnosyl-(1-6)-hexoside, E = −86.73, −72.42 kcal mol−1], which was subjected to dynamic simulation studies against the two enzymes to investigate the stability of the docked conformation. Root-mean-square fluctuations (RMSFs) showed values of 0.25–4.0 and 0.50–4.75 compared to free-state protein RMSF values of 0.25–4.5 and 0.5–7.5, revealing stable fluctuations over time after docking of this compound to AChE and BChE active pockets, respectively. AI in pharmacology can significantly improve patient outcomes and advance healthcare. Ligand binding or catalytic sites for AzrBmH21, AzrBmH22/3, and AzrBmH24/5 were predicted using a machine learning algorithm based on the Prank Web and DeepSite chemoinformatics tools. These findings will establish a scientific foundation for further investigations into the Ochrosia genus, particularly in relation to Alzheimer's disease.
1 Introduction
Alzheimer's disease (AD) is a neurological condition that impairs judgement, thinking, orientation, and cognitive functions.1–4 One in every 85 people worldwide is predicted to have AD by 2050 owing to the disease's increasing prevalence in the elderly population.5,6 The disease's pathogenic features include the absence of the essential neurotransmitter acetylcholine (ACh), the build-up of amyloid plaques (Aβ), the formation of neurofibrillary tangles (NFTs), and nerve damage due to oxidative stress.7 Research is now focused on the development of cholinesterase inhibitors that can reinstate ACh activity, antioxidants, anti-amyloid, and anti-inflammatory medications.8–10Regretfully, the FDA has only approved few medications, including galantamine, donepezil, and rivastigmine for the treatment of AD. These medications work by raising the brain's ACh levels. However, these medications exhibit a poor efficacy rate and serious adverse effects, including hepatotoxicity.11,12 According to published research, there are about 189 medications that are licensed for use in clinical settings that are derived from plant sources,12–15 reducing the risk of certain chronic illnesses such as neurodegenerative conditions, especially AD.16
The genus Ochrosia (Apocynaceae) has approximately 39 species of trees that grow naturally in tropical or subtropical Malaysia and west of the Pacific Islands.17,18 Phytochemical investigations on plants in this genus resulted in the identification of distinct skeletal types of indole alkaloids with anticancer properties, which piqued the interest of pharmacologists.19 O. elliptica Labill. is primarily recognized for the ellipticine series of indole alkaloids, as well as lignans, coumarins, and phenolic acids.19–22 Previously studied biological activities were primarily involved in the cytotoxic attributes of ellipticine. Ellipticine and its derivatives largely impede cell cycle progression and trigger apoptotic pathways in various cancer cell types.23
The integration of machine learning (ML) into natural product research is revolutionizing drug discovery by addressing traditional challenges and unlocking new opportunities.24,25 The increasing trend of using ML in natural product research and drug discovery is reshaping the pharmaceutical landscape. ML algorithms can analyze large datasets swiftly, identify promising compounds, predict drug–target interactions, and optimize drug development processes. This integration is making drug discovery more efficient, accurate, and cost-effective, ultimately accelerating the development of new and innovative treatments. It is an exciting advancement that holds great potential for the future of medicine and healthcare.
Metabolomics is a valuable method for thoroughly analyzing and comparing metabolites in biological systems.26 Metabolomics has proven to be a useful method, especially for plants, where thousands of undiscovered metabolites occur in substantial variety and numbers.27 The untargeted approach helps researchers better understand the complexity of these mixtures by offering an objective technique for comparing metabolite profiles between groups.28 It may also aid in understanding which metabolites are most important for specific pharmacological activities.29 Previous metabolomic techniques, particularly liquid chromatography-tandem mass spectroscopy (LC-MS/MS)-based metabolomics, focused on specific groups of substances such as alkaloids from the Apocynaceae family.30–32
Molecular docking and molecular dynamics simulation can improve our understanding of protein–ligand interactions. Furthermore, it has proven to be quite precise in screening bioactive components against a wide range of targets. It saves scientists' time, effort, and budget during the lengthy drug discovery process.33,34 Docking, in particular, can be utilized to anticipate new therapeutic drugs capable of treating a variety of chronic diseases.35,36 AI-based prediction models are being developed to address these challenges, and they may play a role in future screening, diagnosis, treatment selection, and salvage therapy decisions.37
The cholinesterases, which include acetylcholinesterase (AChE, EC 3.1.1.7) and butyrylcholinesterase (BChE, EC 3.1.1.8), are a family of esterases that catalyze the hydrolysis of cholinergic neurotransmitters into choline and their corresponding acids, resulting in the resumption of the activated cholinergic neuron to its state of rest.38 AChE is considered a high-performance cholinesterase with highly specific catalytic activity towards ACh (80%), whereas BChE, a replacement for AChE, is a non-selective cholinesterase that can breakdown both ACh and butyrylcholine.39 Previous research has shown that ACh depletion can produce a variety of neurological abnormalities in the brain's cortical cholinergic locations in AD patients. Several pharmacotherapeutics acting as acetylcholinesterase inhibitors (AChEIs) have been found to increase the concentration of Ach, and hence, contribute to the relief of the symptoms of AD.40
As a part of our ongoing research into phytochemically and biologically interesting medicinal plants,41–45 this study aimed to use a metabolite profiling strategy for understanding the chemical profiles of the stems, barks, roots, and fruits of O. elliptica Labill., correlating them with their anti-cholinesterase activities (AChE and BChE inhibitory activities) for the first time. Our study will be the first metabolite profiling record of O. elliptica Labill. identifying all metabolites in a single run. Furthermore, molecular docking simulations were utilized to investigate the potential binding mechanisms and intermolecular interactions of the discovered compounds with AChE and BChE active sites. These findings may be the first to show that this genus and its identified metabolites have multi-targeted potential against AD.
2 Material and methods
2.1. Plant materials
The stems, barks, roots, and fruits of Ochrosia elliptica Labill. were collected in December 2022 from the Experimental Station of the Faculty of Pharmacy, Cairo University, Giza, Egypt. The plant was authenticated by Mrs Therese Labib, Consultant of Plant Taxonomy at the Ministry of Agriculture and the Former Director of El-Orman Botanical Garden. A voucher with specimen no. (28.12.2012) was deposited at the herbarium of the Pharmacognosy Department, Faculty of Pharmacy, Cairo University.2.2. Preparation of the samples
The air-dried samples (200 g) were extracted separately with 70% ethanol (3× 350 mL) till exhaustion. Solvents were removed using a rotary evaporator at 60 °C to obtain the extracts, after that 10 mg from each extract was moved to an LC vial, dissolved in 1.5 mL methanol, and kept at −20 °C till further LC-MS analysis. For each sample, three replicates were extracted in parallel under the same conditions. To conduct biological analysis, 5 mg of dried methanolic extracts were dissolved in methanol.2.3. Chemicals
Tris–HCl buffer, dithio-bis-(2-nitrobenzoic acid) (DTNB), butyrylthiocholine iodide, acetylthiocholine, acetylcholinesterase (AChE) from electric eel, and butyrylcholinesterase (BChE) from horse serum were supplied by Sigma Chemical Co. (St. Louis, MO, USA).2.4. UPLC-MS/MS chemical profiling and metabolite annotations
Aliquots of the samples (1 mg of the methanol extract) were resuspended in 1 mL of LC-grade 50% methanol in water, samples were quickly vortexed, sonicated for 3 min and finally centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min adopting the conditions mentioned previously.46 The metabolites resulting from the tested extracts were separated using an RP High Strength Silica (HSS) T3 C18 column (100 mm × 2.1 mm containing 1.8 μm diameter particles) in a Waters UPLC system (Acquity, Waters Corporation, USA), with an injection volume of 3 μL and a flow rate of 0.3 mL min−1. The chromatographic conditions are as follows: mobile phase A (water containing 0.1% formic acid) and mobile phase B (acetonitrile containing 0.1% formic acid). The separation gradient is as follows: 0–1 min at 1% mobile phase B; 1–11 min mobile phase B was increased linearly from 1 to 40%; 11–13 min, mobile phase B was increased linearly from 40 to 70%; 13–15 min, mobile phase B was increased linearly from 70 to 99%; 15–16 min, mobile phase B was maintained at 99%; 16.0–17.0 min, mobile phase B was linearly decreased from 99 to 1%; 17.0–20.0 min, mobile phase B was kept at 1%. The column temperature was 40 °C. Mass spectra were attained using a high-resolution Orbitrap mass analyzer (Q Exactive system, Thermo Fisher Scientific, Waltham, MA, USA). Data acquisition was completed in data-independent acquisition (DIA), which was obtained in the positive mode, with the scanning range from 100 to 1500 m/z. The ion spray voltage and positive ion voltage were both 3500 V, the negative ion voltage was 3000 V, ion source was Heated Electrospray Ionization (HESI), the sheath gas pressure was 40 psi, the auxiliary heating gas pressure was 15 psi, and the ion source heating temperature was 300 °C. The Thermo X-calibur software (Thermo Scientific, Germany) was used for instrument control, data acquisition and quality checking of the MS/MS data. The raw files were subsequently brought into MSDial for subsequent processing.47
000 rpm for 5 min adopting the conditions mentioned previously.46 The metabolites resulting from the tested extracts were separated using an RP High Strength Silica (HSS) T3 C18 column (100 mm × 2.1 mm containing 1.8 μm diameter particles) in a Waters UPLC system (Acquity, Waters Corporation, USA), with an injection volume of 3 μL and a flow rate of 0.3 mL min−1. The chromatographic conditions are as follows: mobile phase A (water containing 0.1% formic acid) and mobile phase B (acetonitrile containing 0.1% formic acid). The separation gradient is as follows: 0–1 min at 1% mobile phase B; 1–11 min mobile phase B was increased linearly from 1 to 40%; 11–13 min, mobile phase B was increased linearly from 40 to 70%; 13–15 min, mobile phase B was increased linearly from 70 to 99%; 15–16 min, mobile phase B was maintained at 99%; 16.0–17.0 min, mobile phase B was linearly decreased from 99 to 1%; 17.0–20.0 min, mobile phase B was kept at 1%. The column temperature was 40 °C. Mass spectra were attained using a high-resolution Orbitrap mass analyzer (Q Exactive system, Thermo Fisher Scientific, Waltham, MA, USA). Data acquisition was completed in data-independent acquisition (DIA), which was obtained in the positive mode, with the scanning range from 100 to 1500 m/z. The ion spray voltage and positive ion voltage were both 3500 V, the negative ion voltage was 3000 V, ion source was Heated Electrospray Ionization (HESI), the sheath gas pressure was 40 psi, the auxiliary heating gas pressure was 15 psi, and the ion source heating temperature was 300 °C. The Thermo X-calibur software (Thermo Scientific, Germany) was used for instrument control, data acquisition and quality checking of the MS/MS data. The raw files were subsequently brought into MSDial for subsequent processing.47
2.5. Cholinesterase inhibitory activities determination
The cholinesterase inhibition assays were performed by the Ellman colorimetric method with slight modifications.4,48 In a total volume of 170 μL of Tris–HCl buffer (0.1 M, pH 8), 20 μL of various extracts in methanol with different concentrations, and 20 μL of enzyme solution containing 1 U mL−1 were incubated for 10 min at room temperature. After adding 20 μL of substrate solution and 40 μL of DTNB, the mixture was incubated at room temperature for 15 minutes. The absorbance of the combination was measured at 410 nm using a plate reader (Techan, USA). Donepezil was employed as a positive control. The percentage inhibition of enzyme activity was calculated by comparing with the negative controlwhere A0 is the absorbance of the negative control (enzyme without samples) and A1 is the absorbance of the enzyme with the tested samples. Assays were carried out in triplicate. To calculate the IC50 values, each sample was assayed at 5 concentrations (200, 100, 50, 10, and 5 μg mL−1). The IC50 values were obtained from the dose–effect curves by linear regression.

2.6. Molecular modeling
Further, the 2D chemical structures of forty-one compounds were first sketched using ChemBioDraw Ultra 14.00 (Cambridge Soft Corp., Cambridge, MA) and then converted into three-dimensional (3D) structures using Discovery Studio (DS 4.0 Biovia Discovery Studio 2016 64-bit Client). Simulation for the ligands was carried out by applying CHARMm Forcefield with partial charge MMFF94 for ligand preparation.
To reconfirm the previously predicted catalytic amino acids of the active pockets of the two mentioned proteins, CAVER Web 1.0 tools (https://loschmidt.chemi.muni.cz/caverweb/) was employed for quantum tunneling that played an important role in ligands' dimension determination to investigate the fitting level of the ligand and predict its biological effect.54
3 Results and discussion
3.1. UPLC-MS/MS chemical profiling and metabolite annotations
The LC-ESI-MS/MS approach is presented for profiling phytocompounds in the tested plant extracts. Furthermore, the fragmentation patterns found in the mass spectrum helped the annotation of these molecules. Compounds were identified by comparing their retention times and distinctive MS spectrum data to those from the reference literature. The combination of UHPLC's superior chromatographic resolution and separation capabilities with HR-MS allows for structural annotations based on accurate mass measurement in both MS and MS–MS investigations. These approaches provide a substantial advantage for screening target chemicals from complicated matrices. O. elliptica contains a high concentration of alkaloids.18 Alkaloid-bearing plants were detected in positive ion mode, with 39 chemicals found in various organs. In addition, two quercetin derivatives were identified, bringing the total number of 41 compounds detected in positive ionization mode (see Table 1, Fig. 1, and S6†). Representative compounds with fragmentation patterns are illustrated in the respective paragraphs.| Comp no. | RT (min) | Compound name | Exact mass | [M + H]+ | Error (ppm) | O. elliptica L. Area (×106) | Molecular formula | Mass fragments | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stems | Barks | Roots | Fruits | |||||||||
| 1 | 4.93 | 10-Methoxyyohimbine | 384.2049 | 385.2122 | −0.14 | 25200 |
18500 |
26900 |
18900 |
C22H28O4N2 | 383 (M-l), 369 (M-15), 214, 199, 192 (M++), 186 | 59 |
| 2 | 4.99 | Ajmaline | 326.1994 | 327.2065 | 0.169 | 69.076736 | 76.689184 | 17.30335 | 42.032544 | C20H26O2N2 | 239, 220, 210, 194, 182, 158, 144 | 60 |
| 3 | 5.23 | 10-Hydroxydihydrocorynantheol | 314.1994 | 315.2067 | 0.081 | 0.120714 | 0 | 5.281832 | 0 | C19H26O2N2 | 313, 285, 283 (–2OH), 269, 267, 257, 241, 200, 186, 161, 185172 |
61 |
| 4 | 5.25 | 18-Hydroxyepialloyohimbine | 370.1892 | 371.1967 | 0.421 | 4790 | 10000 |
9625.18528 | 3890 | C21H26N2O4 | 339, 353, 240, 222, 158, 144 | 62 |
| 5 | 5.34 | 10,11-Dimethoxyajmalicinine | 430.2103 | 431.2178 | 0.248 | 1.613632 | 1.86388 | 4.428456 | 2.493541 | C23H30N2O6 | 355, 353, 252, 222, 212, 117, 178, 144 | Natural products dictionary |
| 6 | 5.49 | 10-Methoxydihydrocorynantheol | 328.2150 | 329.2222 | −0.47 | 111 | 99.329912 | 188.719536 | 26.58068 | C20H28N2O2 | 313, 299, 297, 283, 281, 255, 214, 200, 199, 186, 168 | 61 |
| 7 | 5.86 | 16,22-Dihydro-16-hydroxyapparicine | 282.1732 | 283.1806 | 0.495 | 0.524963 | 0.045075 | 9.128824 | 0.171156 | C18H22N2O | 264 (M-18), 172, 158, 130 | 63 |
| 8 | 5.93 | Corynantheal | 294.1732 | 295.1807 | 0.678 | 308 | 65.234364 | 1380 | 663 | C19H22N2O | 281, 255, 214, 200, 199, 186, 168 | 61 |
| 9 | 6.09 | Reserpinine | 382.1892 | 383.1965 | 0.068 | 254 | 111 | 169 | 77.758112 | C22H26N2O4 | 351, 222, 159, 188, 174 | 62 |
| 10 | 6.11 | Reserpiline | 412.1998 | 413.2076 | 1.165 | 77.830664 | 80.647272 | 110 | 32.254536 | C23H28N2O5 | 397, 323, 222, 218, 204 | 62 |
| 11 | 6.17 | Norajmaline | 312.1837 | 313.1907 | −1.228 | 38.796436 | 4.932806 | 6.774066 | 564 | C19H24N2O2 | 295, 239, 220, 210, 194, 182, 158, 144, 130 | 62 |
| 12 | 6.35 | Quercetin-3-O-rhamnosyl-(1-3)-rhamnosyl-(1-6)-hexoside | 756.2112 | 757.2184 | −0.171 | 10500 |
32500 |
20400 |
6720 | C33H40O20 | 609, 465, 303 | Natural products dictionary |
| 13 | 6.70 | Rutin | 610.1533 | 611.1616 | 1.569 | 29.008288 | 2.795345 | 4.912448 | 676 | C27H30O16 | 465, 303 | Natural products dictionary |
| 14 | 6.71 | Ochropposinine | 358.2256 | 359.2329 | 0.141 | 651 | 1760 | 1570 | 496 | C21H30N2O3 | 329, 219, 204, 169, 154 (lo), 140, 125 | 61 |
| 15 | 6.72 | Dihydro-reserpiline | 414.2154 | 415.2231 | 0.871 | 10.607523 | 12.781067 | 10.589717 | 10.413892 | C23H30O5N2 | 383, 397, 254, 236, 222, 188, 174, 160 | 62 |
| 16 | 6.81 | 10,11-Dimethoxy-19,20-dihydro-16S-sitsirikine | 416.2311 | 417.2385 | 1.47 | 10.774761 | 20.76162 | 18.364846 | 7.31043 | C23H32N2O5 | 415, 401, 313, 311, 285, 230 | 61 |
| 17 | 7.05 | 3,14-Dihydroellipticine | 248.1313 | 249.1122 | 0.058 | 44.865748 | 1.232219 | 10.745313 | 11.298486 | C17H16N2 | 247 | Sirus |
| 18 | 7.12 | Ochropamine | 366.1943 | 367.2019 | 0.846 | 400 | 317 | 579.685248 | 71.348168 | C22H26N2O3 | 144, 138, 218, 219, 168 | Sirus |
| 19 | 7.27 | Reserpic acid | 400.1998 | 401.2075 | 0.976 | 970 | 775 | 1640 | 291 | C22H28N2O5 | 383, 369, 321, 240, 222, 188, 174 | 62 |
| 20 | 7.3 | Dihydrocorynantheol | 298.2045 | 299.2122 | 1.237 | 2440 | 2490 | 5580 | 643 | C19H26N2O | 283, 281, 255, 214, 200, 199, 186, 168 | 61 |
| 21 | 7.39 | Reserpiline-N-oxide | 428.1947 | 429.2027 | 1.6 | 874 | 1500 | 2340 | 871 | C23H28N2O6 | 397, 369, 339, 220, 210, 205, 189, 178, 150 | 62 |
| 22 | 7.47 | Corynantheol | 296.1888 | 297.1964 | 0.909 | 113 | 27.411926 | 81.495312 | 15.870396 | C19H24N2O | 283, 281, 255, 214, 200, 199, 186, 168 | 61 |
| 23 | 7.48 | O-Acetylyohimbine | 396.2049 | 397.2111 | 0.551 | 328 | 594 | 350.388512 | 171 | C23H28N2O4 | 338 (M-59, COOCH3), 184, 170, 169, 156 | 59 |
| 24 | 7.5 | O-Methyl-vobasinol | 368.2099 | 369.2173 | 0.137 | 12200 |
16500 |
18000 |
1600 | C22H28N2O3 | 353, 326, 297, 265, 281, 222, 174, 144 | Natural products dictionary |
| 25 | 7.57 | 3-Dihydrocorynantheol N-methosalt | 312.2201 | 313.2274 | 0.127 | 77.830664 | 80.647272 | 110 | 32.254536 | C20H28N2O | 283, 281, 255, 214, 200, 199, 186, 168 | 61 |
| 26 | 7.59 | Yohimbine | 354.1943 | 355.2018 | 0.622 | 386 | 383 | 285 | 596 | C21H26N2O3 | 117, 144, 162, 180, 194, 212, 224 | 59 |
| 27 | 7.66 | Ochroborine B | 444.1896 | 445.1971 | 0.41 | 26.964956 | 54.577132 | 157.585968 | 129 | C23H28N2O7 | 365, 339, 337, 294, 248, 154, 144 | 64 |
| 28 | 7.93 | 10,11-Dimethoxypicraphylline | 442.2103 | 443.2180 | 0.805 | 205 | 252 | 312.399168 | 99.130208 | C24H30N2O6 | 413, 368, 317, 281, 279, 240, 204, 154 | 65 |
| 29 | 8.02 | Polyneuridine aldehyde | 350.1630 | 351.1707 | 1.142 | 23.164102 | 217 | 12.213688 | 17.48936 | C21H22N2O3 | 352, 336, 322, 263, 249, 168, 142 | 66 |
| 30 | 8.19 | Holeinine (4-methylreserpiline) | 426.2154 | 427.2230 | 0.635 | 51600 |
111000 |
95000 |
32800 |
C24H30N2O5 | 397, 323, 222, 204, 218 | 62 |
| 31 | 8.42 | Apparicine | 264.1626 | 265.1700 | 0.32 | 9830 | 8100 | 14800 |
1660 | C18H20N2 | 249, 235, 222, 208, 194, 180, 167, 145, 130, 128 | 67 |
| 32 | 8.43 | 1,2,3,4-Tetrahydro-9-methoxyellipticine | 280.1575 | 281.1651 | 0.819 | 29.7627 | 76.93712 | 59.162264 | 19.478692 | C18H20N2O | 267, 246 | Natural products dictionary |
| 33 | 8.6 | 1,2,3,4-Tetrahydroellipticine | 250.1469 | 251.1545 | 0.816 | 0.304959 | 0.649391 | 209.45456 | 0.223243 | C17H18N2 | 244 | Natural products dictionary |
| 34 | 8.74 | Ochrosamine A | 322.1681 | 323.1756 | 0.698 | 2450 | 3430 | 2640 | 455 | C20H22N2O2 | 255, 219, 149 | 68 |
| 35 | 8.98 | Ellipticine/olivacine | 246.1156 | 247.1229 | −0.142 | 11200 |
18900 |
6930 | 5140 | C17H14N2 | 247, 246, 232 | Natural products dictionary |
| 36 | 9.1 | 9-Methoxyellipticine | 276.1262 | 277.1334 | −0.432 | 23.95127 | 72.96672 | 7250 | 43.883868 | C18H16N2O | 246, 245 | Natural products dictionary |
| 37 | 9.4 | 10,11-Dimethoxyalstonine | 408.1685 | 409.1759 | 0.322 | 0.379272 | 0.116939 | 7.528142 | 0 | C23H24N2O5 | 379, 349 | Natural products dictionary |
| 38 | 9.45 | 1,2-Dihydro-9-methoxyellipticine | 278.1419 | 279.1857 | 0.554 | 7.014448 | 7.858734 | 2.745834 | 3.771581 | C18H18N2O | 246, 245 | Natural products dictionary |
| 39 | 9.69 | Serpenticine | 378.1579 | 379.1659 | 9.69 | 0.017724 | 0.055235 | 3.216962 | 0.012426 | C22H22N2O4 | 347, 293, 144 | 60 |
| 40 | 11.84 | Dehydro-reserpine | 606.2577 | 607.2658 | 1.223 | 1.40914 | 1.230805 | 34.67404 | 2.777899 | C33H38O9N2 | 367, 335, 236, 221, 206, 190, 144 | 62 |
| 41 | 11.88 | Reserpine | 608.2733 | 609.2818 | 1.876 | 625 | 1990 | 6790 | 265 | C33H40O9 N2 | 577, 448, 436, 397, 365, 236, 195, 174 | 62 |
 | ||
| Fig. 1 Structures of the tentatively identified compounds from different organs of O. elliptica Labill. using UPLC-MS/MS. | ||
Compounds 1, 4, 23, and 26 are types of indole alkaloids identified as yohimbine and its derivatives. The MS spectra of yohimbine showed a protonated ion at m/z 355.2018 [M + H]+ and MS/MS showed ions at m/z 117, 144, 162, 180, 194, 212, and 224, and the most abundant ion was found at m/z 144 and ions at m/z 214 and m/z 224 are the other ions showed up with considerable abundance. Ring cleavage at C-2,3 and C-4,5 resulted in the formation of indole derivative with m/z 144 [C10H10N+] and m/z 212 [C11H18NO3+]. Similarly, ring cleavage at C-2,3 and C-5,6 resulted in the formation of m/z 224 [C12H18NO3+]. These three peaks are characteristic peaks for identifying the similar yohimbine type of compound like 10-methoxyyohimbine (1), which showed peaks at m/z 384 (M+), 383 (M-l), 369 (M-15), 355, 354, 353, 324, 214, 201, 200, 199, 192 (M++), 186, 174, 173, and 153.5. The peaks at m/z 214, 200, 199, and 186 are the typical yohimbine peaks moved to a value 30 mass units higher because of the presence of the aromatic methoxyl group. 18-Hydroxyepialloyohmbine (4) and O-acetylyohimbine (23) are identified in the same pattern.
Compounds 2 and 11: ajmaline and norajmaline are other types of indole alkaloids, showing protonated ions at m/z 327.2065, and 313.1907 [M + H]+, respectively, and ions at m/z 131, 144, 158, 182, 194, 210, 220, and 239. N-Methyl indole derivative (m/z 158) is formed by ring cleavage at C-2,3 and C-4,5, and further loss of methyl group from nitrogen atom yields the base peak at m/z 144 [C10H10N+]. The loss of C8H7NO from protonated ions gives m/z 194 [C12H20NO+] and cleavage at C-2,3 and C-4,5 from m/z 194 yields m/z 108 [C7H10N+].
Compound 5: 10,11-dimethoxyajmalicinine showed protonated ions at m/z 431.2178 and ions at m/z 117, 144, 178, 212, 222, and 252. The fragmentation behavior of ajmalicine is similar to that of yohimbine, except the presence of double bond (C-16, 17), oxygen atom in ring D, and methyl group at C-19 position.
Compounds 32, 33, 35, 36, and 38 are types of indole alkaloids identified as ellipticine and its derivatives showed molecular ion peaks at m/z 281.16507, 251.1545, 247.1229, 277.1334, 409.17593, and 279.1857, respectively, and characteristic fragment ions at 246 and 247.
Compounds 40 and 41: reserpine and dehydro-reserpine are trimethoxybenzoic acid-substituted alkaloids, with protonated ions [M + H]+ at m/z 609.2818 and 607.2658, respectively, with fragment ions at m/z 174, 195, 236, 365, 397, 448, and 577. Neutral loss trimethoxybenzoic acid of [M + H]+ resulted in the formation of m/z 397 [C23H29N2O4+]. The loss of a hydroxyl group from trimethoxybenzoic acid resulted in the generation of the most abundant peak at m/z 195 [C10H11O4+]. The loss of CH3OH from m/z 397 yields an ion at m/z 365 [C22H25N2O3+]. Ring cleaving at C-2, 3, C-4, 5, and C-5, 6 resulted in the formation of m/z 174 [C11H12NO+] and m/z 448 [C23H29NO8+]. Neutral loss of trimethoxybenzoic acid from m/z 448 generates m/z 236 [C13H18NO3+].
Compounds 12 and 13: quercetin-3-O-rhamnosyl-(1-3)-rhamnosyl-(1-6)-hexoside and rutin were identified in fruits only with molecular ion peaks at m/z 757.2184, and 611.16162, respectively. The compounds showed characteristic fragment ions at m/z 465, and 303 due to loss of sugar moieties.69,70
3.2. Biological activities of different parts of O. elliptica Labill.
The activity of different samples against Alzheimer activity is summarized in Table 2. Acetyl- and butyrylcholinesterase revealed that the samples investigated had more selectivity index to AChE than BChE. Carbazole alkaloids possess the acetylcholinesterase inhibitory potential by the most widely used method, i.e. Ellman's method and also are efficient inhibitors of beta amyloid fibril formation.71 From the results, it can be concluded that the hydroalcoholic extract of O. elliptica fruits exhibited the highest biological activity among the samples studied. This enhanced activity is likely attributed to its rich flavonoid content, as indicated by the UPLC/MS profiling conducted during the research. Phenolics, i.e., flavonoids, are known for their significant therapeutic potential, particularly in nutraceutical applications for AD patients.72,73 These compounds can be integrated into daily diets to help increase acetylcholine levels and mitigate brain inflammation.74,75 Incorporating a diet rich in antioxidant and anti-inflammatory flavonoids has been shown to interact beneficially with tau receptors, which may lead to a reduction in gliosis and a decrease in neuroinflammatory markers, as observed in various animal model studies. Such dietary strategies could represent a promising avenue for supporting cognitive health and managing symptoms associated with neurodegenerative conditions.76 The fruit extract is predominantly characterized by the presence of quercetin derivatives. This specific profile may play a crucial role in the extract's activity as inhibitors of acetylcholinesterase and butyrylcholinesterase. The strong effects of quercetin derivatives suggest that they may be valuable components in developing nutraceuticals due to their antioxidant properties, antibacterial activity, and antiproliferative effects.69,70,77 These benefits are particularly relevant for promoting cognitive health and enhancing cholinergic function.78| Hydroalcoholic extract of Ochrosia elliptica Labill. | IC50 (μg mL−1) | |
|---|---|---|
| AChE | BChE | |
| Stems | 25.37 ± 0.69 | 48.01 ± 0.12 |
| Barks | 18.24 ± 1.66 | 35.77 ± 1.34 |
| Roots | 16.11 ± 0.79 | 35.39 ± 0.52 |
| Fruits | 12.77 ± 0.24 | 29.98 ± 0.51 |
| Donepezil | 0.23 ± 0.03 | 0.31 ± 0.0033 |
3.3. Molecular modeling studies
Table 3 presents the Prank Web prediction results for AChE protein (PDB ID: 7E3H) showing 13 binding pockets; only pockets ranked 1 and 2 showed the highest results regarding score (30.22, 28.81), probability (0.918, 0.910), residue number (27, 28) and conservation (0.743, 0.717) for pockets 1 and 2, respectively. Visualization showed the high-confident regions of the structure (score >0.070) colored as per pocket ranking. Pocket 1 was suspected to be the most predictable binding pocket for small molecules binding via interaction with the identified amino acid residues (Fig. 2).
 |
 | ||
| Fig. 2 PrankWeb predictions of small-molecule binding pockets of (A) AChE (PDB ID: 7E3H) (pockets 1–13) and (B) BChE (PDB ID: 4TPK) (pockets 1–6) with the ranking color code highlighting mainly pockets 1 (red) and 2 (yellow). | ||
Furthermore, Prank Web prediction results for BChE (PDB ID: 4TPK) showed 6 binding pockets, from which pockets that ranked 1 and 2 were the best based on score results (29.97, 27.62), probability (0.916, 0.903), residue number (25, 26) and conservation average (0.869, 0.813), respectively (Table 4). Visualization results suggested pocket 1 to be the most predictable ligand binding pocket based on the essential amino acid residues' interactions (Fig. 2).
 |
The DeepSite Web server prediction results revealed only four predicted pockets for AChE protein (PDB ID: 7E3H) for either chain A or B. However, the prediction results of BChE (PDB ID: 4TPK) showed 3 binding pockets when testing chain A and 4 binding pockets for chain B prediction. The visualization of the results showed that pocket 1 in either 7E3H or 4TPK proteins confirmed the best results, and although chain A confirmed some binding interactions with the key amino acid residues, the number of binding interactions with most of the essential amino acid residues for chain B exceeded that of chain A in both targets. In AChE pocket 1, interactions with TYR 72 and PHE 338 were observed during chain A prediction, and interactions with Phe295, Phe297, Trp86, Ser293, Ser125 and Tyr72 were observed after testing chain B (Fig. 3).
 | ||
| Fig. 3 DeepSite predictions showing binding pocket 1 (sited as red sphere) of (A) AChE (PDB ID: 7E3H) chain A prediction, (B) AChE (PDB ID: 7E3H) chain B prediction and (C) BChE (PDB ID: 4TPK) chain A prediction and (D) BChE (PDB ID: 4TPK) chain B prediction. | ||
Moreover, the visualization of 4TPK chain A DeepSite Web server prediction showed that pocket 1 confirmed binding interactions with Trp82, Phe329, His438, and Ser287, and chain B showed Trp82, Asp70, Ser72, Tyr332, Phe329, and Ser287 interactions (Fig. 3).
Findings revealed that PrankWeb predictions showed more possible regions in both tested targets compared to DeepSite, where 7E3H showed 13 predicted binding pockets by PrankWeb compared to only 4 by DeepSite and 4TPK showed 6 by PrankWeb compared to only 3 predicted binding regions by DeepSite, with common predicted amino acid residues by both servers. These analyses confirmed that the PrankWeb results are more likely to be more valid giving more accurately identified predictions, as reported previously54 (Fig. 4, Tables 3 and 4).
 | ||
| Fig. 4 Best score predicted protein pockets for (A) AChE (PDB ID: 7E3H) (pockets 1–6 out of 32 pockets: magenta, blue, red, yellow, orange, and cyan) and (B) BChE (PDB ID: 4TPK) (pockets 1–6 out of 14 pockets: red, magenta, yellow, blue, orange, and cyan) using CAVER Web 1.0 tools, viewed using PyMol. | ||
CAVER Web 1.0 tool (https://loschmidt.chemi.muni.cz/caverweb/) was used for further confirmation of the binding sites of the two mentioned targeted proteins. The CAVER Web tool was used for quantum tunneling of the best pose of each of the two active binding sites; from the 32 and 14 pockets predicted by CAVER for AChE (7E3H) and BChE (4TPK), respectively, the best six score ranked pockets are illustrated in Fig. 4. For quantum tunneling of the best pose, the results were measured and ranked according to the investigated priority scores for the four tunnels of AChE: (0.0920–0.900), with radius and length dimensions of the best tunnel priority, tunnel 1 of 2.249 Å and 3.896 6 Å, respectively. Furthermore, the results of BChE showed 11 tunnels (0.138–0.930) and tunnel 1 showing the best score revealed a radius and length of 3.646 Å and 8.733 Å, respectively (Fig. 5 and 6). The results showed that tunnel 1 was of the highest radius during the investigation of both enzymes. This plays an important role in predicting the ligand dimensions such as width, height or length as fitting requirements within the binding pocket.54
 | ||
| Fig. 5 Four predicted protein tunnels for 7E3H using CAVER Web 1.0 tools: (A) tunnel 1 (priority: 0.90), (B) tunnel 2 (priority: 0.50), (C) tunnel 3 (priority: 0.27) and (D) tunnel 4 (priority: 0.09). | ||
 | ||
| Fig. 6 The best 2 predicted protein tunnels (2 out of 11) for 4TPK using CAVER Web 1.0 tools: (A) tunnel 1 (priority: 0.93) and (B) tunnel 2 (priority: 0.83). | ||
3.4. Molecular docking study
All the compounds were docked into the two target proteins AChE (PDB ID: 7E3H) and BChE (PDB ID: 4TPK) using C-Docker protocol. Fig. 7 shows the 2D-diagram of the docking results of the two ligands, with a (–)C-Docker interaction energy of donepezil of 57.72 (kcal mol−1), and of N-((1-(2,3-dihydro-1H-inden-2-yl)piperidin-3-yl)methyl)-N-(2-methoxyethyl)-2-naphthamide as 47.71 (kcal mol−1) on AChE and BChE, respectively, while (–)C-Docker interaction of the tested compounds lied within the range of 30.17–86.73 (kcal mol−1) for AChE and 26.81–72.42 (kcal mol−1) for BChE. | ||
| Fig. 7 2D diagram showing docking interactions of the ligands with the key amino acid residues at binding pockets: (A) donepezil with AChE (PDB ID: 7E3H), (E = −57.72 kcal mol−1): hydrogen bond with Phe295; hydrophobic bond with Phe338, Tyr337, Tyr341, Trp86, Trp286 and Ser293. (B) N-((1-(2,3-Dihydro-1H-inden-2-yl)piperidin-3-yl)methyl)-N-(2-methoxyethyl)-2-naphthamide with BChE (PDB ID: 4TPK), (E = −47.71 kcal mol−1): hydrophobic bonds with Phe329, His438, Trp82, Trp231, Asn68 and Leu286. Color code of dotted lines: green for H bonds, pink for Pi–alkyl bonds, purple for Pi–Pi bonding, and pale greenish for van der Waals interactions. | ||
All the docked compounds (41 compounds) showed good interaction results with AChE (PDB ID: 7E3H), where most of the compounds showed comparable binding interactions at the active pocket to that of donepezil with stable (–)C-Docker interaction energy values, where 17 tested compounds revealed (–)C-Docker interaction energy values exceeding 50 kcal mol−1 compared to donepezil (E = −57.72 kcal mol−1). Upon further filtration, it was observed that five compounds showed better interaction energy than that of the ligand as follows: compound 12 showed the best binding results (E = −86.73 kcal mol−1), showing one hydrogen bond (H bond) with Phe295, Phe338, Gln291, Gly120, Asp74, Thr75, Tyr124, Tyr133, and Trp86 and two H bonds with Ser293, a hydrophobic bond with Phe338, Tyr337, Tyr341 and Trp286. Compound 13 (E = −63.22 kcal mol−1) showed a H bond with Phe338, Asn87, Tyr341, and His447 and 2H bonds with Glu202. Moreover, carbon–hydrogen (C–H) interaction with Asp74 and van der Waals interaction with Ty72. Compound 16 (E = −59.64 kcal mol−1) showed a H bond with His447, Gly122, and Val294 and a hydrophobic bond with Phe338, Tyr124, Tyr337, Tyr341 and Trp86. Compound 30 (E = −58.31 kcal mol−1) showed a hydrophobic bond with Phe338, Phe297, Tyr341 and Trp86 and a C–H bond with Trp86, Ser293, and Ser125. In addition to Pi carbon interaction with Tyr337, compound 40 (E = −67.22 kcal mol−1) showed a hydrophobic bond with Phe338, Tyr124, Tyr337, Tyr331, Tyr34, Trp86 and Trp286 and a C–H bond with Gly448, Glu202, His447, Ser125, and Tyr337. In addition, there was a Pi carbon interaction with Tyr337 (Fig. 8).
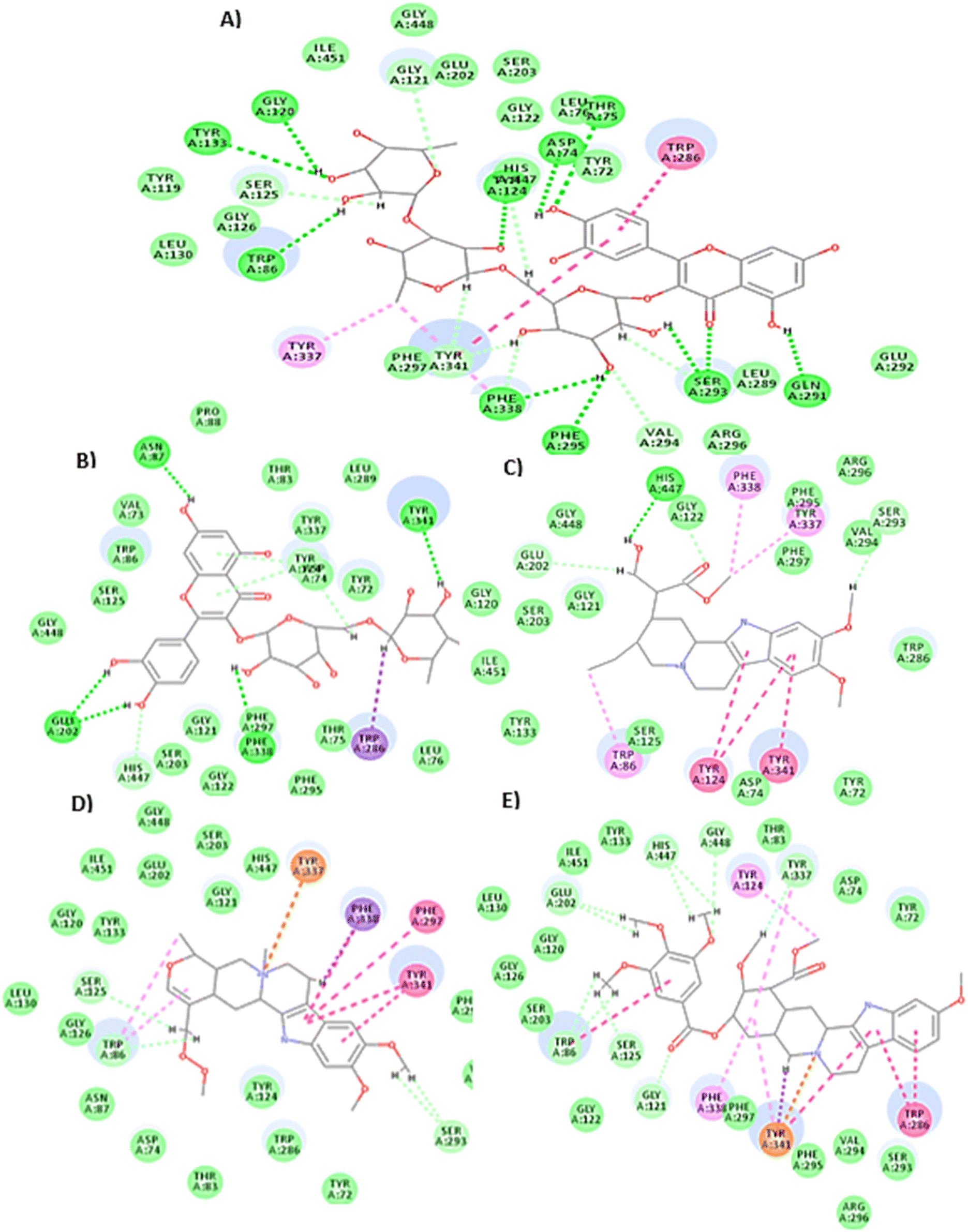 | ||
| Fig. 8 2D interaction diagram of the best ligands after docking on AChE (PDB ID: 7E3H): (A) compound (12) (E = −86.73 kcal mol−1), (B) compound (13) (E = −63.22 kcal mol−1), (C) compound (16) (E = −59.64 kcal mol−1), (D) compound (30) (E = −58.31 kcal mol−1), and (E) compound (40) (E = −67.22 kcal mol−1). | ||
Furthermore, only six compounds showed (–)C-Docker interaction energy values less than 40 kcal mol−1. Although these compounds revealed lower stability interaction energy values, their visual inspection confirmed good interactions at the binding site with the essential amino acid residues as follows: compound 17 (E = −37.93 kcal mol−1), compound 31 (E = −38.10 kcal mol−1), and compound 34 (E = −36.69 kcal mol−1) showing a H bond with Phe 338, compound 35 (E = −30.17 kcal mol−1) confirmed a H bond with Phe 295, compound 36 (E = −32.88 kcal mol−1), compound 37 (E = −38.67 kcal mol−1) and compound 38 (E = −37.38 kcal mol−1). Fig. 9 presents compound 35 which showed the least stability results, yet with comparable binding interactions at the binding pockets of both AChE (PDB ID: 7E3H) and BChE (PDB ID: 4TPK).
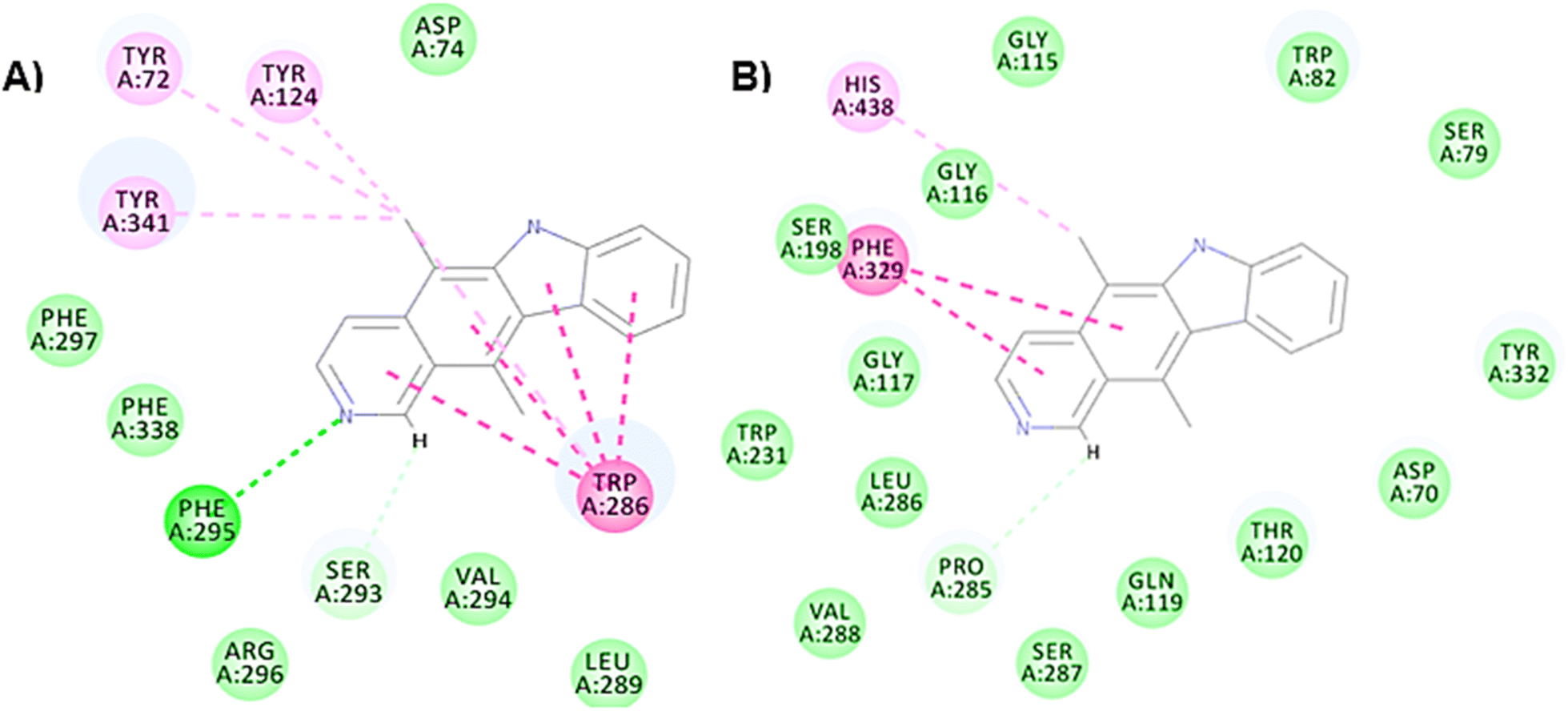 | ||
| Fig. 9 2D interaction diagram of the least stable compound (compound 35) after docking on (A) AChE (PDB ID: 7E3H) (E = −30.17 kcal mol−1) and (B) BChE (PDB ID: 4TPK) (E = −26.81 kcal mol−1), showing good interactions on both targets. | ||
While checking the BChE target (PDB ID: 4TPK) docking results, it was observed that most of the compounds showed promising results. Ten compounds out of 41 docked compounds showed more stable C-Docker interaction energy than the re-docked ligand (E = −47.71 kcal mol−1), as follows: compound 10 (E = −53.41 kcal mol−1), compound 12 (E = −72.42 kcal mol−1), compound 13 (E = −68.66 kcal mol−1), compound 14 (E = −49.58 kcal mol−1), compound 15 (E = – 59.78 kcal mol−1), compound 16 (E = −50.79 kcal mol−1), compound 24 (E = −49.01 kcal mol−1), compound 27 (E = −51.32 kcal mol−1), compound 30 (E = −50.34 kcal mol−1), compound 40 (E = −69.02 kcal mol−1), and compound 41 (E = −66.03 kcal mol−1), which were visualized, revealing good interactions at the binding site. It was observed that the five best docked compounds (compounds 12, 13, 16, 30 and 40) against AChE (PDB ID: 7E3H) were also superior while targeting BChE (PDB ID: 4TPK), confirming the essential binding interactions with amino acid residues in the active pockets (Fig. 10).
 | ||
| Fig. 10 2D interaction diagram of the five most promising ligands after docking on BChE (PDB ID: 4TPK): (A) compound (12) (E = −72.42 kcal mol−1), (B) compound (13) (E = −68.66 kcal mol−1), (C) compound (16) (E = −50.79 kcal mol−1), (D) compound (30) (E = −50.34 kcal mol−1), and (E) compound (40) (E = −69.02 kcal mol−1). | ||
Based on the docking results, it was observed that compound 12 could be considered as the most promising compound, confirming the essential binding interactions on the two studied targets with the best docking interaction energy compared to all the docked compounds and the ligands. The overlay visualization of compound 12 with each of the two ligands in the two active sites of the two studied enzymes showed good fitting with the neighboring amino acid residues, which suggested promising biological interactions at the binding sites (Fig. 11). These results encouraged further study applying dynamic simulation studies to determine the stability of the two studied enzymes AChE and BChE after docking.
 | ||
| Fig. 11 Overlay of the ligand and compound 12 in the active sites of (A) AChE protein (7E3H) (gray), ligand (red), compound 12 (magenta), Phe295 (blue), Phe338 (green), Trp286 (magenta), His447 (green), tyr341 (yellow). (B) BChE protein (4TPK) (bluish grey), ligand (blue), compound 12 (magenta), Trp82 (yellow), Phe329 (pink), His438 (orange) visualized using Pymol. | ||
Moreover, the visualization of the molecular fitting of compound 12 on both enzymes after using the CAVER Web 1.0 tool showed good fitting within the best score-ranked 4 tunnels. It is worth noting that the tunnel dimensions hinge on variables either being dependent or independent, and the protein radius is essential to calculate the needed ligand's fitting dimensions in order to achieve the best biological results (Fig. 12).
 | ||
| Fig. 12 The best 4 predicted protein tunnels with compound 12 (magenta) using CAVER Web 1.0 tools: (A) 7E3H (yellow) and tunnels 1–4 (blue, green, red and cyan, respectively); (B) 4TPK (orange) and tunnels 1–4 (blue, green, red and yellow, respectively), viewed using PyMol. | ||
3.5. Standard dynamic simulations
Discovery Studio 4.0 was used to investigate the stability of both proteins: AChE and BChE, and the produced conformations after their docking via standard dynamic simulation were analyzed through trajectory studies. The stability of the most promising docked compound (12) was compared to the free states of the two enzymes AChE and BChE. The stability was determined as the total energy plot against the time and calculation of root mean square fluctuations (RMSFs) against the produced conformations. The free state of AChE (7E3H) and BChE (4TPK) showed a decrease in total energy versus time; it is obvious that, among the same time frame, the docked enzymes with compound 12 also showed a total energy decrease (Fig. 13), which could be explained as similar behaviors conducted after docking by compound 12 being of high stability compared to the two enzymes in their free states. | ||
| Fig. 13 Dynamic simulation study results represented by the total energy versus time plot in the production step: (A) AChE (PDB ID: 7E3H) in its free state before docking, (B) AChE after docking with compound 12, (C) BChE (PDB ID: 4TPK) in its free state before docking and (D) BChE after docking with compound 12. | ||
The values of RMSFs of AChE (7E3H) in its free state lied in the 0.25–4.5 range, while after being docked with compound 12, the RMSF values were: 0.25–4.0, revealing stable fluctuations over time. The same for BChE (4TPK): the RMSF values of the free protein showed a range of 0.5–7.5, and for the docked state with compound 12, the range was 0.50–4.75, showing high stability (Fig. 14).
 | ||
| Fig. 14 Analyzed trajectory study results represented by the RMSF versus residue index plot of: (A) AChE (PDB ID: 7E3H) in its free state before docking, (B) AChE after docking with compound 12, (C) BChE (PDB ID: 4TPK) in its free state before docking and (D) BChE after docking with compound 12. | ||
3.6. Biological activity prediction
The predicted biological activity of the most promising compound (12) targeting different proteins was performed using PASS Online Way2Drug https://www.way2drug.com/passtargets/, by uploading the 2D ChemBioDraw chemical structure of the compound. The results indicated the activity against muscarinic acetylcholine receptor M1 (ChEMBL id: CHEMBL216) with 0.2819 confidence value, and activity against acetylcholinesterase (ChEMBL id: CHEMBL220) with a maximum confidence of 0.1483.56 The cellular and molecular mechanisms of the quercetin and its derivatives involved in the protection against AD were highly documented in previous works, which are mainly due to their anti-inflammatory and antioxidant actions.79 Furthermore, quercetin derivatives have relevance to the Alzheimer's disease (AD) pathogenesis cascade and demonstrate the ability to protect neuronal cells from oxidative stress-induced damage.77,80 In addition, flavonoids such as quercetin of 12 and 13 exhibited multiple mechanisms of action that are involved in the pathogenesis of AD including AChE inhibition, antioxidant, neuroprotection, and anti-Aβ aggregation. In addition, quercitrin exhibited antioxidant and anti-hyaluronidase properties.81 Consequently, flavonoid-rich nutraceuticals may represent a cost-effective treatment option for AD, but additional well-designed, randomized, and placebo-controlled clinical trials are necessary to evaluate their optimal dosages, effectiveness, and long-term safety in humans.824 Conclusions
A total of 39 Apocynaceae alkaloids were tentatively identified based on the UPLC-MS/MS analysis of different parts of O. elliptica. The major bioactive metabolites were docked against both AChE and BChE target enzymes and reflected a good potential as anti-Alzheimer therapies with good binding scores and comparable binding modes at the binding pockets compared to the downloaded ligands; furthermore, a dynamic simulation study was conducted on the most predicted active compound quercetin-3-O-rhamnosyl-(1-3)-rhamnosyl-(1-6)-hexoside and confirmed good stability results compared to the free state proteins. Future work could progress towards implementing this strategy to identify novel derivatives isolated from Ochrosia plants as targets for AD.List of abbreviations
| AChE | Acetylcholinesterase |
| AD | Alzheimer's disease |
| Aβ | Amyloid plaques |
| BChE | Butyrylcholinesterase |
| DTNB | Dithio-bis-(2-nitrobenzoic acid) |
| ESI | Electrospray ionization |
| FDA | Food and drug administration |
| NFTs | Neurofibrillary tangles |
| RMSFs | Root-mean-square fluctuations |
| UPLC-MS/MS | Ultra-performance liquid chromatography coupled with tandem mass spectrometry |
Data availability
The data supporting the findings of this study are available within the article.Author contributions
Conceptualization: M. A. S., R. A. E., and E. A. S.; data curation: A. A. M., M. A. S., and R. A. E.; resources: M. A. S., A. A. M., and R. A. E.; investigation: A. A. M., M. A. S., and R. A. E.; analysis: A. A. M., M. A. S., and R. A. E.; writing – original draft; writing, review, and editing: all the authors. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research received no external funding.References
- M. Citron, Alzheimer's disease: strategies for disease modification, Nat. Rev. Drug Discovery, 2010, 9, 387–398 CrossRef CAS PubMed.
- A. F. Kassem, M. A. Omar, A. Temirak, R. A. El-Shiekh and A. M. Srour, Barbiturate–sulfonate hybrids as potent cholinesterase inhibitors: design, synthesis and molecular modeling studies, Future Med. Chem., 2024, 1–17 Search PubMed.
- R. A. El-Shiekh, H. A. Kassem, A. E. Khaleel and M. M. Abd El-Mageed, Anticholinesterases activity of Murraya koenigii (L.) Spreng. and Murraya paniculata (L.) Jacq. essential oils with GC/MS analysis and molecular docking, Nat. Prod. Res., 2024, 38, 2155–2159 CrossRef CAS PubMed.
- R. M. Ibrahim, R. A. El-Shiekh, O. G. Mohamed, A. A. Al-Karmalawy, A. Tripathi and P. M. Abdel-Baki, LC/MS-Based Metabolomics Reveals Chemical Variations of Two Broccoli Varieties in Relation to Their Anticholinesterase Activity: In vitro and In silico Studies, Plant Foods Hum. Nutr., 2024, 1–8 Search PubMed.
- P. Scheltens, K. Blennow, M. M. Breteler, B. De Strooper, G. B. Frisoni and S. Salloway, et al., Alzheimer's disease, Lancet, 2016, 388, 505–517 CrossRef CAS PubMed.
- M. A. Omar, R. A. El-Shiekh, D. H. Dawood, A. Temirak and A. M. Srour, Hydrazone–sulfonate hybrids as potential cholinesterase inhibitors: design, synthesis and molecular modeling simulation, Future Med. Chem., 2023, 15, 2269–2287 Search PubMed.
- M. H. Mahnashi, M. A. Alshahrani, M. H. Nahari, S. Su Hassan, M. S. Jan and M. Ayaz, et al., In-vitro, in-vivo, molecular docking and ADMET studies of 2-substituted 3, 7-dihydroxy-4H-chromen-4-one for oxidative stress, inflammation and Alzheimer's disease, Metabolites, 2022, 12, 1055 CrossRef CAS PubMed.
- J. D. Grill and J. L. Cummings, Current therapeutic targets for the treatment of Alzheimer's disease, Expert Rev. Neurother., 2010, 10, 711–728 CrossRef CAS PubMed.
- D. H. Dawood, A. M. Srour, M. A. Omar, T. A. Farghaly and R. A. El-Shiekh, Synthesis and molecular docking simulation of new benzimidazole–thiazole hybrids as cholinesterase inhibitors, Arch. Pharm., 2024, 357, 2300201 CrossRef CAS PubMed.
- M. A. Youssef, S. S. Panda, R. A. El-Shiekh, E. M. Shalaby, D. R. Aboshouk and W. Fayad, et al., Synthesis and molecular modeling studies of cholinesterase inhibitor dispiro [indoline-3, 2′-pyrrolidine-3′, 3′′-pyrrolidines], RSC Adv., 2020, 10, 21830–21838 RSC.
- A. A. Abduljawad, M. A. Elawad, M. E. M. Elkhalifa, A. Ahmed, A. A. E. Hamdoon and L. H. M. Salim, et al., Alzheimer's disease as a major public health concern: Role of dietary saponins in mitigating neurodegenerative disorders and their underlying mechanisms, Molecules, 2022, 27, 6804 CrossRef CAS PubMed.
- M. H. Mahnashi, M. Ashraf, A. H. Alhasaniah, H. Ullah, A. Zeb and M. Ghufran, et al., Polyphenol-enriched Desmodium elegans DC. ameliorate scopolamine-induced amnesia in animal model of Alzheimer's disease: In Vitro, In Vivo and In Silico approaches, Biomed. Pharmacother., 2023, 165, 115144 CrossRef CAS PubMed.
- H. M. Hussein, E. Mohsen, A. R. Abdelmonem, M. A. Abdel Kawy, A. A. Al-Karmalawy and R. A. El-Shiekh, New Insight into the Anticholinesterase Potential of Cordia dichotoma G. Forst. and Cordia sebestena L. Leaves, Phenolic Characterization of their Active Extracts by HPLC-DAD and Molecular Modeling, Egypt. J. Chem., 2024, 67, 225–237 Search PubMed.
- A. A. Hamed, R. A. El-Shiekh, O. G. Mohamed, E. A. Aboutabl, F. I. Fathy and G. A. Fawzy, et al., Cholinesterase inhibitors from an endophytic fungus Aspergillus niveus Fv-er401: metabolomics, isolation and molecular docking, Molecules, 2023, 28, 2559 CrossRef CAS PubMed.
- R. A. El-Shiekh, A. A. Shalabi, O. S. Al-Hawshabi, M. A. Salkini and E. Abdel-Sattar, Anticholinesterase and anti-inflammatory constituents from Caralluma awdeliana, a medicinal plant from Yemen, Steroids, 2023, 193, 109198 CrossRef CAS PubMed.
- A. Agnihotri and O. I. Aruoma, Alzheimer's disease and Parkinson's disease: a nutritional toxicology perspective of the impact of oxidative stress, mitochondrial dysfunction, nutrigenomics and environmental chemicals, J. Am. Coll. Nutr., 2020, 39, 16–27 CrossRef CAS PubMed.
- R. A. El-Shiekh, D. A. Al-Mahdy, M. S. Hifnawy and E. A. Abdel-Sattar, Pharmacognostical study of Ochrosia elliptica Labill.(Apocynaceae), J. Appl. Pharm. Sci., 2019, 9, 049–057 CrossRef CAS.
- E. Abdel-Sattar and R. A. El-Shiekh, Chemical composition and biological activities of genus Ochrosia: a mini review, Phytochem. Lett., 2024, 62, 92–111 CAS.
- Y.-H. Fua, Structurally diverse indole alkaloids from Ochrosia elliptica, Heterocycles, 2017, 94, 743–749 Search PubMed.
- R. A. El-shiekh, D. A. Al-Mahdy, M. S. Hifnawy, T. Tzanova, E. Evain-Bana and S. Philippot, et al., Chemical and biological investigation of Ochrosia elliptica Labill. cultivated in Egypt, Rec. Nat. Prod., 2017, 11, 552–557 CAS.
- R. A. El-Shiekh, D. A. Al-Mahdy, M. S. Hifnawy and E. Abdel-Sattar, Biological and chemical assessment of Ochrosia elliptica Labill leaves, Arabian J. Sci. Eng., 2021, 46, 5247–5255 CAS.
- R. Elshimy, W. Y. Khawagi, I. A. Naguib, S. I. Bukhari and R. A. El-Shiekh, 9-Methoxyellipticine: Antibacterial Bioactive Compound Isolated from Ochrosia elliptica Labill. Roots, Metabolites, 2023, 13, 643 CAS.
- R. M. Labib, F. Zulfiqar, M. A. Ibrahim, P. Balachandran, J. Zhang and S. A. Ross, FOXO signal activating alkaloids isolated from Ochrosia elliptica leaf cultivated in Egypt, Med. Chem. Res., 2019, 28, 1628–1632 CAS.
- S. Mikutis, S. Lawrinowitz, C. Kretzer, L. Dunsmore, L. Sketeris and T. Rodrigues, et al., Machine Learning Uncovers Natural Product Modulators of the 5-Lipoxygenase Pathway and Facilitates the Elucidation of Their Biological Mechanisms, ACS Chem. Biol., 2023, 19, 217–229 Search PubMed.
- F. Noor, M. Junaid, A. H. Almalki, M. Almaghrabi, S. Ghazanfar and M. Tahir ul Qamar, Deep learning pipeline for accelerating virtual screening in drug discovery, Sci. Rep., 2024, 14, 28321 CAS.
- Y.-Q. Wang, L.-P. Hu, G.-M. Liu, D.-S. Zhang and H.-J. He, Evaluation of the nutritional quality of Chinese kale (Brassica alboglabra Bailey) using UHPLC-Quadrupole-Orbitrap MS/MS-based metabolomics, Molecules, 2017, 22, 1262 Search PubMed.
- R. C. De Vos, S. Moco, A. Lommen, J. J. Keurentjes, R. J. Bino and R. D. Hall, Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry, Nat. Protoc., 2007, 2, 778–791 CAS.
- M. Maldini, F. Natella, S. Baima, G. Morelli, C. Scaccini and J. Langridge, et al., Untargeted metabolomics reveals predominant alterations in lipid metabolism following light exposure in broccoli sprouts, Int. J. Mol. Sci., 2015, 16, 13678–13691 Search PubMed.
- D. Lučić, I. Pavlović, L. Brkljačić, S. Bogdanović, V. Farkaš and A. Cedilak, et al., Antioxidant and Antiproliferative Activities of Kale (Brassica oleracea L. Var. acephala DC.) and Wild Cabbage (Brassica incana Ten.) Polyphenolic Extracts, Molecules, 2023, 28, 1840 Search PubMed.
- L. A. Barny, J. A. Tasca, H. A. Sanchez, C. R. Smith, S. Koptur and T. Livshultz, et al., Chemotaxonomic investigation of Apocynaceae for retronecine-type pyrrolizidine alkaloids using HPLC-MS/MS, Phytochemistry, 2021, 185, 112662 CAS.
- P. Kumar, A. Bhushan, P. Gupta and S. Gairola, Comparative morpho-anatomical standardization and chemical profiling of root drugs for distinction of fourteen species of family Apocynaceae, Bot. Stud., 2022, 63, 12 CAS.
- J. Liu, Y. Liu, Y.-j Pan, Y.-G. Zu and Z.-H. Tang, Determination of alkaloids in Catharanthus roseus and Vinca minor by high-performance liquid chromatography–tandem mass spectrometry, Anal. Lett., 2016, 49, 1143–1153 CrossRef CAS.
- A. A. Al-Karmalawy, R. Alnajjar, A. A. Elmaaty, F. A. Binjubair, S. T. Al-Rashood and B. S. Mansour, et al., Investigating the promising SARS-CoV-2 main protease inhibitory activity of secoiridoids isolated from Jasminum humile; in silico and in virto assessments with structure-activity relationship, J. Biomol. Struct. Dyn., 2023, 1–13 Search PubMed.
- K. I. Eissa, M. M. Kamel, L. W. Mohamed, A. S. Doghish, R. Alnajjar and A. A. Al-Karmalawy, et al., Design, synthesis, and biological evaluation of thienopyrimidine derivatives as multifunctional agents against Alzheimer’s disease, Drug Dev. Res., 2023, 937–961 CrossRef CAS PubMed.
- A. Tripathi and K. Misra, Molecular docking: A structure-based drug designing approach, JSM Chem., 2017, 5, 1042–1047 Search PubMed.
- R. Nisar, S. Ahmad, K.-u-R. Khan, A. E. Sherif, F. Alasmari and A. F. Almuqati, et al., Metabolic profiling by GC-MS, in vitro biological potential, and in silico molecular docking studies of Verbena officinalis, Molecules, 2022, 27, 6685 CrossRef CAS PubMed.
- J. R. Christie, P. Lang, L. M. Zelko, D. A. Palma, M. Abdelrazek and S. A. Mattonen, Artificial intelligence in lung cancer: bridging the gap between computational power and clinical decision-making, Can. Assoc. Radiol., J., 2021, 72, 86–97 CrossRef PubMed.
- Y. Wu, X. Su, J. Lu, M. Wu, S. Y. Yang and Y. Mai, et al., In Vitro and in silico analysis of phytochemicals from fallopia dentatoalata as dual functional cholinesterase inhibitors for the treatment of Alzheimer's disease, Front. Pharmacol, 2022, 13, 905708 CrossRef CAS PubMed.
- Y. Nicolet, O. Lockridge, P. Masson, J. C. Fontecilla-Camps and F. Nachon, Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products, J. Biol. Chem., 2003, 278, 41141–41147 Search PubMed.
- A. Martinez and A. Castro, Novel cholinesterase inhibitors as future effective drugs for the treatment of Alzheimer's disease, Expert Opin. Invest. Drugs, 2006, 15, 1–12 Search PubMed.
- R. A. El-Shiekh, M. M. Okba, A. A. Mandour, O. Kutkat, R. Elshimy and H. A. Nagaty, et al., Eucalyptus Oils Phytochemical Composition in Correlation with Their Newly Explored Anti-SARS-CoV-2 Potential: in Vitro and in Silico Approaches, Plant Foods Hum. Nutr., 2024, 1–7 Search PubMed.
- R. A. El-Shiekh, R. Elshimy, A. A. Mandour, H. A. Kassem, A. E. Khaleel and S. Alseekh, et al., Murraya koenigii (L.) Sprengel seeds and pericarps in relation to their chemical profiles: new approach for multidrug resistant Acinetobacter baumannii ventilator-associated pneumonia, Appl. Biol. Chem., 2024, 67, 1–19 Search PubMed.
- S. M. Mohamed, M. A. Shalaby, R. A. El-Shiekh, A. F. Bakr, M. M. Rashad and S. R. Emam, et al., Vitis vinifera L. seed standardized extract; a promising therapeutic against metabolic syndrome induced by high-fat/high-carbohydrate diet and streptozotocin in rats, S. Afr. J. Bot., 2024, 167, 476–486 CrossRef CAS.
- R. A. El-Shiekh, R. Elshimy, A. A. Mandour, H. A. Kassem, A. E. Khaleel and S. Alseekh, et al., Phytochemical characterisation of leaves and stems of Murraya koenigii (L.) Sprengel and Murraya paniculata (L.) Jack and their antibacterial activity against multidrug-resistant Acinetobacter baumannii bacterial infection, Int. J. Food Sci. Technol., 2024, 59(10), 7998–8010 CrossRef CAS.
- E. Abdel-Sattar, O. Kutkat, R. A. El-Shiekh, M. K. El-Ashrey and A. M. El Kerdawy, In Silico and In Vitro Screening of Some Pregnane Glycosides Isolated from Certain Caralluma Species as SARS-COV-2 Main Protease Inhibitors, Chem. Biodiversity, 2024, e202301786 CrossRef CAS PubMed.
- R. M. Merghany, M. A. Salem, S. M. Ezzat, S. F. Moustafa, S. A. El-Sawi and M. R. Meselhy, A comparative UPLC-orbitrap-MS-based metabolite profiling of three Pelargonium species cultivated in Egypt, Sci. Rep., 2024, 14, 22765 CrossRef CAS PubMed.
- H. Tsugawa, T. Cajka, T. Kind, Y. Ma, B. Higgins and K. Ikeda, et al., MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis, Nat. Methods, 2015, 12, 523–526 CrossRef CAS PubMed.
- H. M. Hussein, E. Mohsen, A. R. Abdelmonem, M. A. Abdel Kawy, A. A. Al-Karmalawy and R. A. El-Shiekh, New Insight into the Anticholinesterase Potential of Cordia dichotoma G. Forst. and Cordia sebestena L. Leaves, Phenolic Characterization of their Active Extracts by HPLC-DAD and Molecular Modeling, Egypt. J. Chem., 2024, 67(8), 225–237 Search PubMed.
- K. Dileep, K. Ihara, C. Mishima-Tsumagari, M. Kukimoto-Niino, M. Yonemochi and K. Hanada, et al., Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery, Int. J. Biol. Macromol., 2022, 210, 172–181 CrossRef CAS PubMed.
- B. Brus, U. Kosak, S. Turk, A. Pislar, N. Coquelle and J. Kos, et al., Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor, J. Med. Chem., 2014, 57, 8167–8179 CrossRef CAS PubMed.
- K. Dileep, K. Ihara, C. Mishima-Tsumagari, M. Kukimoto-Niino, M. Yonemochi and K. Hanada, et al., Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery, Int. J. Biol. Macromol., 2022, 210, 172–181 CrossRef CAS PubMed.
- B. Brus, U. Kosak, S. Turk, A. Pislar, N. Coquelle and J. Kos, et al., Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor, J. Med. Chem., 2014, 57, 8167–8179 CrossRef CAS PubMed.
- R. Krivák and D. Hoksza, P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure, J. Cheminf., 2018, 10, 1–12 Search PubMed.
- H. A. Oyewusi, R. A. Wahab, K. A. Akinyede, G. M. Albadrani, M. Q. Al-Ghadi and M. M. Abdel-Daim, et al., Bioinformatics analysis and molecular dynamics simulations of azoreductases (AzrBmH2) from Bacillus megaterium H2 for the decolorization of commercial dyes, Environ. Sci. Eur., 2024, 36, 31 CrossRef CAS.
- A. A. Mandour, E. B. Elkaeed, M. Hagras, H. M. Refaat and N. S. Ismail, Virtual screening approach for the discovery of selective 5α-reductase type II inhibitors for benign prostatic hyperplasia treatment, Future Med. Chem., 2023, 15, 2149–2163 Search PubMed.
- Y. A. Elkhawas, H. A. Gad, M. M. AbdelRazek, A. A. Mandour, M. M. Bishr and N. M. Al Musayeib, et al., LC-ESI-MS/MS-Based Comparative Metabolomic Study, Antioxidant and Antidiabetic Activities of Three Lobelia Species: Molecular Modeling and ADMET Study, ACS Omega, 2024, 9(18), 20477–20487 CrossRef CAS PubMed.
- E. M. Azmy, I. F. Nassar, M. Hagras, I. M. Fawzy, M. Hegazy and M. M. Mokhtar, et al., New indole derivatives as multitarget anti-Alzheimer's agents: synthesis, biological evaluation and molecular dynamics, Future Med. Chem., 2023, 15, 473–495 CrossRef CAS PubMed.
- E. M. Azmy, I. F. Nassar, M. Hagras, I. M. Fawzy, M. Hegazy and M. M. Mokhtar, et al., New indole derivatives as multitarget anti-Alzheimer's agents: synthesis, biological evaluation and molecular dynamics, Future Med. Chem., 2023, 15, 473–495 CrossRef CAS PubMed.
- P. Benoin, R. Burnell and J. Medina, Alkaloids of Aspidosperma excelsum Benth, Can. J. Chem., 1967, 45, 725–730 CrossRef CAS.
- S. Sagi, B. Avula, Y.-H. Wang and I. A. Khan, Quantification and characterization of alkaloids from roots of Rauwolfia serpentina using ultra-high performance liquid chromatography-photo diode array-mass spectrometry, Anal. Bioanal. Chem., 2016, 408, 177–190 Search PubMed.
- A. Ahond, H. Fernandez, J. Moore, C. Poupat, V. Sanchez and P. Potier, et al., Contribution to the study of Ochrosiineae: alkaloids from Ochrosia moorei, J. Nat. Prod., 1981, 45, 725–730 Search PubMed.
- S. Kumar, A. Singh, V. Bajpai, M. Srivastava, B. P. Singh and B. Kumar, Structural characterization of monoterpene indole alkaloids in ethanolic extracts of Rauwolfia species by liquid chromatography with quadrupole time-of-flight mass spectrometry, J. Pharm. Anal., 2016, 6, 363–373 Search PubMed.
- P. Perera, T. Van Beek and R. Verpoorte, 16 (S)-Hydroxy-16, 22-dihydroapparicine, a new alkaloid from the leaves of Tabernaemontana dichotoma, J. Nat. Prod., 1984, 47, 835–838 CrossRef CAS.
- B. J. Zhang, J. M. Yan, Z. K. Wu, Y. P. Liu, M. F. Bao and G. G. Cheng, et al., Alkaloids from Ochrosia borbonica, Helv. Chim. Acta, 2013, 96, 2288–2298 CrossRef CAS.
- J. Bruneton, T. Sevenet and A. Cavé, Alcaloïdes des ecorces d'ochrosia vieillardii, Phytochemistry, 1972, 11, 3073–3075 CrossRef CAS.
- W. Yin, M. S. Kabir, Z. Wang, S. K. Rallapalli, J. Ma and J. M. Cook, Enantiospecific total synthesis of the important biogenetic intermediates along the ajmaline pathway,(+)-polyneuridine and (+)-polyneuridine aldehyde, as well as 16-epivellosimine and macusine A, J. Org. Chem., 2010, 75, 3339–3349 CrossRef CAS PubMed.
- P. Crooks and B. Robinson, The isolation and identification of (−)-apparicine from Tabernamontana cumminsii, J. Pharm. Pharmacol., 1970, 22, 799–800 CAS.
- A. R. Carroll, R. Addepalli, G. Fechner, J. Smith, G. P. Guymer and P. I. Forster, et al., Alkaloids from the Australian rainforest tree Ochrosia moorei, J. Nat. Prod., 2008, 71, 1063–1065 CrossRef CAS PubMed.
- R. R. Cardoso, R. O. Neto, C. T. dos Santos D'Almeida, T. P. do Nascimento, C. G. Pressete and L. Azevedo, et al., Kombuchas from green and black teas have different phenolic profile, which impacts their antioxidant capacities, antibacterial and antiproliferative activities, Food Res. Int., 2020, 128, 108782 CrossRef CAS PubMed.
- X.-Q. Zheng, Y. Nie, Y. Gao, B. Huang, J.-H. Ye and J.-L. Lu, et al., Screening the cultivar and processing factors based on the flavonoid profiles of dry teas using principal component analysis, J. Food Compos. Anal., 2018, 67, 29–37 CrossRef CAS.
- B. Sungthong, K. Sithon, P. Panyatip, S. Tadtong, N. Nunthaboot and P. Puthongking, Quantitative analysis and in silico molecular docking screening for acetylcholinesterase inhibitor and ADME prediction of coumarins and carbazole alkaloids from Clausena harmandiana, Nat. Prod., 2022, 16, 358–369 CAS.
- P. Sadhukhan, S. Saha, S. Dutta, S. Mahalanobish and P. C. Sil, Nutraceuticals: An emerging therapeutic approach against the pathogenesis of Alzheimer's disease, Pharmacol. Res., 2018, 129, 100–114 Search PubMed.
- Y. R. Hassan, R. A. El-Shiekh, H. M. El Hefnawy and C. G. Michael, Irvingia gabonensis baill.(African Mango): A comprehensive review of its ethnopharmacological significance, unveiling its long-standing history and therapeutic potential, J. Ethnopharmacol., 2024, 117942 CrossRef CAS PubMed.
- M. I. Morales-Rivera, R. Alemón-Medina, A. Martínez-Hernández, C. Contreras-Cubas, N. F. Altamirano-Bustamante and J. Gómez-Garduño, et al., Exome Sequence Data of Eight SLC Transporters Reveal That SLC22A1 and SLC22A3 Variants Alter Metformin Pharmacokinetics and Glycemic Control, Pharmaceuticals, 2024, 17, 1385 CAS.
- J. Melrose, The potential of flavonoids and flavonoid metabolites in the treatment of neurodegenerative pathology in disorders of cognitive decline, Antioxidants, 2023, 12, 663 CAS.
- M. Al Amin, Z. Dehbia, M. H. Nafady, M. Zehravi, K. P. Kumar and M. A. Haque, et al., Flavonoids and Alzheimer's disease: reviewing the evidence for neuroprotective potential, Mol. Cell. Biochem., 2024, 1–31 Search PubMed.
- S. Kamiloglu, M. Tomas and E. Capanoglu, Dietary flavonols and O-glycosides, Handbook of dietary phytochemicals, 2019, pp. 1–40 Search PubMed.
- H. Hassan, Neuroprotective and anti-inflammatory potentials of rutin in an in vitro model of Alzheimer's disease 2024 Search PubMed.
- E. Zaplatic, M. Bule, S. Z. A. Shah, M. S. Uddin and K. Niaz, Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer's disease, Life Sci., 2019, 224, 109–119 CrossRef CAS PubMed.
- M. A. Mirza, S. Mahmood, A. R. Hilles, A. Ali, M. Z. Khan and S. A. A. Zaidi, et al., Quercetin as a therapeutic product: evaluation of its pharmacological action and clinical applications—a review, Pharmaceuticals, 2023, 16, 1631 CAS.
- K. Plekratoke, P. Waiwut, C. Yenjai, O. Monthakantirat, P. Takomthong and N. Nualkaew, et al., Multi-Target Actions of Flavonoid Derivatives from Mesua ferrea Linn Flower against Alzheimer's disease Pathogenesis, Biomed. Sci. Clin. Med., 2023, 62, 169–180 Search PubMed.
- S. M. Kothawade, H. S. Buttar, H. S. Tuli and G. Kaur, Therapeutic potential of flavonoids in the management of obesity-induced Alzheimer's disease: an overview of preclinical and clinical studies, Naunyn-Schmiedeberg's Arch. Pharmacol., 2023, 396, 2813–2830 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00021a |
| ‡ Current address: The BioActives Lab, Biological and Environmental Science and Engineering Division (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2025 |