
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing HER performance via nitrogen defects: a comparative DFT study of Fe and Ru single-atom catalysts on graphene†
Hongyu Yanga,
Xin Xia *a,
Shijie Ana,
Moujie Huangb,
Hao Maa,
Feng Yea,
Chuang Pengb and
Chao Xua
*a,
Shijie Ana,
Moujie Huangb,
Hao Maa,
Feng Yea,
Chuang Pengb and
Chao Xua
aBeijing Laboratory of New Energy Storage Technology, North China Electric Power University, Beijing 102206, China. E-mail: xxia@ncepu.edu.cn
bSchool of Resource and Environmental Science, Wuhan University, Wuhan 430072, China
First published on 10th March 2025
Abstract
Exploring high-performance catalysts for the hydrogen evolution reaction (HER) is essential for the development of clean hydrogen energy. Single atom catalysts (SACs) have garnered significant attention due to their maximum atomic efficiency, high catalytic performance and excellent selectivity. In this work, we systematically investigated the HER activity of Ru and Fe SACs on nitrogen-doped graphene using density functional theory (DFT) calculations. Various nitrogen defect configurations (N to 4N) were examined to access their impact on structural stability and catalytic performance. The results indicate that lower N-coordinated moieties, particularly N_pyrrolic and 2N, exhibit superior HER activity, while high N-coordinated moieties (4N) demonstrate greater stability. Volcano plot analysis reveals that catalytic performance is highly sensitive to metal–support interactions, which can be effectively described using binding energy and metal charge state. Ru SACs@N_pyrrolic and Fe SACs@2N achieve the most favorable performance, with additional active sites and low overpotentials of approximately 0.26 V and 0.23 V, respectively. Bader charge analysis further confirms that moderate positive charge states enhance electronic metal–support interactions, optimizing hydrogen adsorption and desorption. These findings highlight the critical role of nitrogen coordination in tuning the electronic and energetic properties of SACs on N doped graphene, providing valuable insights into the rational design of Pt-free SACs for highly efficient HER catalysis.
1. Introduction
The hydrogen evolution reaction (HER) via water electrolysis has emerged as an attractive and sustainable approach for hydrogen production.1–3 Electrocatalysts are essential to improve the efficiency of HER. However, due to the scarcity and high cost of precious metal catalyst, developing cost-effective, highly efficient and stable electrocatalysts remains a significant challenge for the large-scale utilization of hydrogen energy. Single atom catalysts (SACs), which exhibit maximum atomic efficiency, high selectivity, and high activity towards a variety of chemical reactions, have opened up a new frontier in the field of catalysis.4–7 The development of SACs composed of inexpensive, earth-abundant metals presents an opportunity to accelerate the hydrogen economy.In 2011, Qiao and coworkers pioneered single atom catalysis by employing a co-precipitation method to prepare Pt SACs supported on iron oxides for CO oxidation.8 This milestone set the stage for SACs as a significant research direction in heterogeneous catalysis. Cheng et al. demonstrated that single platinum atom catalysts synthesized using the atomic layer deposition exhibited significantly enhanced HER catalytic activity (up to 37 times) and high stability compared to commercial Pt/C catalysts.9 Fang et al. uncovered that the single Pt atom tends to dynamically release from the nitrogen–carbon substrate, with the geometric structure less coordinated to support and electronic property closer to zero valence, during the reaction. Theoretical simulations support that the Pt sites with weakened Pt–support interaction and more 5d density are the real active centers. The single-atom Pt catalyst exhibits very high hydrogen evolution activity and long-term durability in wide-pH electrolytes.10 The unique electronic structures and unsaturated coordination environment of SACs contribute to enhanced metal–support interaction and intrinsic catalytic activity, yet the high surface energy of isolated single atoms can lead to aggregation, reducing their efficiency.11 Using substrate materials to anchor single atoms are emerging as an important solution to stabilize SACs. Various types of substrate materials, including metal oxides, carbon-based materials and metal–organic frameworks have been used to anchor metal SACs.12
With the increasing application in catalytic aspects, graphene-supported metal atoms have also been identified as suitable catalysts.13–15 Graphene, a two-dimensional (2D) materials with large specific surface area, good stability and high electrical conductivity, provides an ideal substrate for to dispersing nanoparticles and SACs which can greatly enhance electrocatalytic performance.16–18 The seminal work by Sun et al. prepared the first graphene-based metal SAC using the atomic layer deposition (ALD) method.19 Extensive studies have shown that the graphene-based carbon materials are a robust candidate as SACs' supports. The introduction of metal SAs can make the inert graphene a highly active catalyst for many reactions with remarkable activity and selectivity, such as selective hydrogenation, oxygen reduction reaction (ORR) and hydrogen evolution reaction (HER).20 Additionally, N-doping has proven particularly effective in stabilizing SACs on graphene. Hossain et al. and Jung et al. demonstrated the excellent HER activity of single-atom Co on nitrogen-doped graphene through theoretical design and experimental verification.21–23 Zang et al. confirmed that atomically-dispersed Ni with triple nitrogen coordination (Ni–3N) can achieve efficient hydrogen evolution reaction (HER) performance in alkaline media, offering a low cost and corrosion resistant alternative for electrocatalytic water splitting.24 The introduction of nitrogen atoms into carbon lattice offers relatively higher electronegativity and slightly larger radius compared to carbon, making them ideal dopants to enhance electron transfer and catalytic performance in graphene-based materials.25–28 The N-doped carbon-based nanomaterials have been extensively studied as efficient catalysts for electrocatalysis.29–32 Among them, B–N, Fe–N and S–N co-doped carbons are outstanding ORR electrocatalysts, and P–N co-doped carbons are remarkable OER electrocatalyst, while Co–N co-doped carbons are typical HER electrocatalyst.33–37 Several recent experimental and theoretical studies further highlighted different M–Nx moieties are responsible for the superior electrocatalytic activity of various metal based SACs. Liu et al.'s work on the C3N4 supported Co SACs suggests that the low coordination Co–N structure significantly enhances HER performance due to its unique electronic structure and low free energy barrier.38 Similarly, Feng et al. identified that unsaturated Ni–N2 are the active structures for HER in Ni SAC embedded on nanoporous carbon nanotubes (CNT). The average chemical state of Ni in Ni–N–C/Ni@CNT-H was between metallic Ni (Ni0) and oxidized Ni (NiII).39 In addition, Levell et al. compared various Fe–N–C site models and revealed that a hybrid coordination environment between pyridinic and poryphyrinic Fe–Nx moieties plays a key role in driving outstanding ORR activity.40 The importance of N-doping in enhancing catalytic activity has driven our investigation into the effects of N dopants on the HER performance of SACs.
In our previous work,41,42 we have investigated Ru nanocluster supported on O and N co-doped graphene, achieving high intrinsic HER activity. N-doped graphene provides a versatile substrate for stabilizing metal atoms while improving electron transfer and catalytic performance. In this work, we extend this approach by using density functional theory (DFT) to explore N-doped graphene structures with 1N to 4N defect centers as SAC supports. Two potential HER catalyst alternatives, Ru and Fe SACs, have been studied and compared in terms of their catalytic active sites and interactions with various nitrogen defect centers. This theoretical study highlights the tunability of catalytic properties and structural stability through N doping, providing valuable insights into the design of Pt free SACs catalysts based on N-doped graphene.
2. Computational methods
All density functional theory (DFT) calculations were performed using the Vienna ab inito Simulation Package (VASP).43,44 The electron–ion interactions were described using the projector-augmented wave (PAW) pseudopotentials, and the electronic exchange–correlation effects were treated with the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA).45–47 To account for long-range van der Waals interactions, the D3 Grimme correction (DFT + D3) was employed.43 The convergences of the geometric optimization were achieved with a SCF energy tolerance of 1.0 × 10−6 eV per atom and a maximum force tolerance of 0.01 eV Å−1. The energy cut-off was chosen as 500 eV. A 6 × 6 graphene supercell was used as the substrate model, with a vacuum layer layer of 15 Å. Monkhorst–Pack scheme K-points grid sampling was set to 3 × 3 × 1 in the reciprocal space.The defect formation energy (EDEF) of N-doped graphene was calculated using the equation:
| EDEF = EN − EC − xμN + yμC | (1) |
The binding energy (Ebind) serves as an energetic descriptor for the metal–support interaction (MSI) between metal SACs and the N-doped graphene surface. It is calculated as
| Ebind = Etot − Esub − EM | (2) |
The Gibbs free energy of hydrogen adsorption (ΔGH*) on graphene substrate with various configurations of N defects was evaluated as a key descriptor for HER activity, The HER catalytic performance was assessed using the following equation for ΔGH*.
| ΔGH* = ΔEH* + ΔEZPE − TΔSH | (3) |
| ΔEH* = Etot − Esub − 1/2E[H2]. | (4) |
Additionally, electronic structure studies including charge density difference calculations and Bader charge analysis were performed to understand the interaction between the SACs and the N-doped graphene substrate. Crystal Orbital Hamilton Population (COHP) analysis was also used to examine the bonding characteristics of hydrogen on various active sites between the transition metals and the N-doped graphene substrate.49
3. Results and discussion
3.1 Defect configurations of N doped graphene
In order to study the stability and activity of N-doped graphene, we constructed various N defect configurations (N to 4N) by extracting carbon atoms form the graphene lattice, as shown in Fig. 1 and 2. According to the energetic calculation results, creating a vacancy by directly extracting a single carbon atom from undoped graphene lattice (Vac_C) requires a high vacancy formation energy of 7.89 eV, which is unlikely to occur under standard conditions. Substituting nitrogen atoms into the graphene lattice can significantly lower the defect formation energy indicating that nitrogen doping makes structural modifications more feasible. We examined the defect formation energies for various N-doped configurations, with values summarized in Table S1.† The EDEF values for three N defect models, i.e. N_graphitic, N_pyridinic and N_pyrrolic are 0.91 eV, 6.19 eV and 8.64 eV, respectively. Among these, N_graphitic configuration shows the lowest defect formation energy of 0.91 eV, indicating it as the most stable configuration for N defect. Considering vacancy containing models of graphene is crucial, since the unsaturated carbon sites can act as active sites for the HER process. Our calculation results suggest that one nitrogen substitution into the graphene vacancy structure lowers the total defect formation energy to a less positive value of 6.19 eV for the N_pyridinic configuration. This reduction of EDEF indicates that the pyridinic site N substitution at unsaturated carbon sites is energetically favorable. In contrast, the N_pyrrolic substitution increases the defect formation energy, making it thermodynamically unfavorable. | ||
| Fig. 1 Structure models of (a) N_graphitic, (b) N_pyridinic, (c) N_pyrrolic, (d) Vac_C, (e) Vac2_C, and (f) N_Vac2_C. Dark brown and blue represent C and N, respectively. | ||
When we extended the study to two nitrogen substitutions (2N) in the vacancy-containing graphene, the 2N_Vac defect structure shown in Fig. 2(a) has a defect formation energy of 4.73 eV, which is significantly lower than N_pyridinic and Vac_C. As the number of N dopants increased to three shown in Fig. 2(b), the close proximity of the doped nitrogen atoms reduced the bond breaking energy, resulting in a favorable defect formation energy of 3.67 eV. For the 4N defect center, as shown in Fig. 2(f), the defect formation energy further decreased to 3.51 eV, which is more favorable than 3N and 2N. This suggests that increasing the number of nitrogen substitution at the two-fold edge sites in the vacancy containing graphene tends to reduce the defect formation energy and enhance structural stability.
 | ||
| Fig. 2 Structure models of (a) 2N_Vac, (b) 3N_Vac, (c) 4N_Vac, (d) 2N_Vac2, (e) 3N_Vac2 and (f) 4N_Vac2. Dark brown and blue represent C and N, respectively. | ||
We also examined the effect of nitrogen substitution in the two-carbon vacancy-containing graphene model (Vac2_C). Extracting two carbon atoms from graphene can form a vacancy structure with 8 and 5 carbon rings at the edges, as shown in Fig. 1(e). The formation energy of Vac2_C (5.47 eV) is significantly lower than that of Vac_C. However, due to the strong lattice relaxation stabilizing the Vac2_C configuration, nitrogen doping at these three-fold edge sites is less likely to occur (5.75 eV). A further comparison of the defect formation energies for N_Vac and N_Vac2 suggests that the N_Vac2 structure model is less stable (Table S1†). Nevertheless, configurations with multiple nitrogen dopants such as 3N and 4N shown in Fig. 2(e) and (f) continue to show relatively low defect formation energies than Vac2_C. Therefore, nitrogen doping effectively stabilizes under-coordinated edge sites in graphene, enhancing the overall stability of the defective graphene structures.
In summary, the structural stability of the N-doped graphene configurations can be estimated as follows: 4N_Vac > 3N_Vac > 2N_Vac > N_pyridinic > Vac_C > N_pyrrolic. Due to this stabilization effect of N doping, single metal atoms were adsorbed on the N-doped graphene for further study.
3.2 Metal–support interaction between Ru/Fe SAs and N-doped graphene
In this section, we studied the interaction between Ru and Fe single atoms and N-doped graphene structures, as depicted in Fig. S1(a)–(h).† The most stable 1N to 4N structures in each case have been selected for incorporation with metal SACs for further HER studies. The metal atom binding energies, bond lengths and charge states are summarized in Tables S2 and S3.† The calculation results indicate that certain defect-containing structures, such as Vac_C and N_pyrrolic, which require high defect formation energies, also tend to anchor metal single atoms (SAs) more strongly, thereby serving as active sites for Ru and Fe deposition. Additionally, Ru SAs generally exhibit higher binding energies than Fe SAs on the same substrate, indicating a stronger metal–support interaction between Fe and N doped graphene. For instance, the N_pyridinic site offers a relatively low binding energy −0.07 eV for Ru, whereas the binding energy for Fe SAs on the same site is as low as −1.99 eV. Thus, Fe atoms bind more strongly to these N-doped defect sites, potentially impacting their catalytic performance in specific reactions. Furthermore, the energetic results indicate a preference for N_pyridinic over N_pyrrolic sites for metal SA binding, consistent XPS characterizations in previous studies.41 Comparing electronic structures of the three N doping configurations, the sp2-bonded graphitic nitrogen introduces a charge deficiency effect on the C5N ring, which attracts Ru/Fe atoms at the hollow sites and favors Ru–C bonds formation over Ru–N bonds. In contrast, for N_pyridinic and N_pyrrolic structures, more charge density is transferred from Ru to neighboring carbon atoms, leaving less charge distribute to N atoms as shown in Fig. 3 (Fe SACs in Fig. S6†). This effect is further evidenced by lower charge states on C and less negative charge on N after Ru deposition. In N_pyridinic doped graphene, the calculated Ru–N and Ru–C bond lengths are 1.91 Å and 1.87 Å, respectively. In N_pyrrolic doped graphene, the neighboring carbon atoms in the C4N ring lose less charge to the N defect than that in the C5N ring of N_pyridinic, and the charge transfer from Ru to N_pyrrolic is enhanced. Fe atoms show even more substantial charge transfer to the N defect center, as suggested by Bader charge analysis, which indicates Ru and Fe lose 0.49e and 0.75e to N_pyridinic, respectively. Fe–N and Fe–C bond lengths at the defect center of N_pyridinic are 1.77 Å and 1.76 Å, reflecting a much stronger Fe–N/C interaction than Ru–N/C.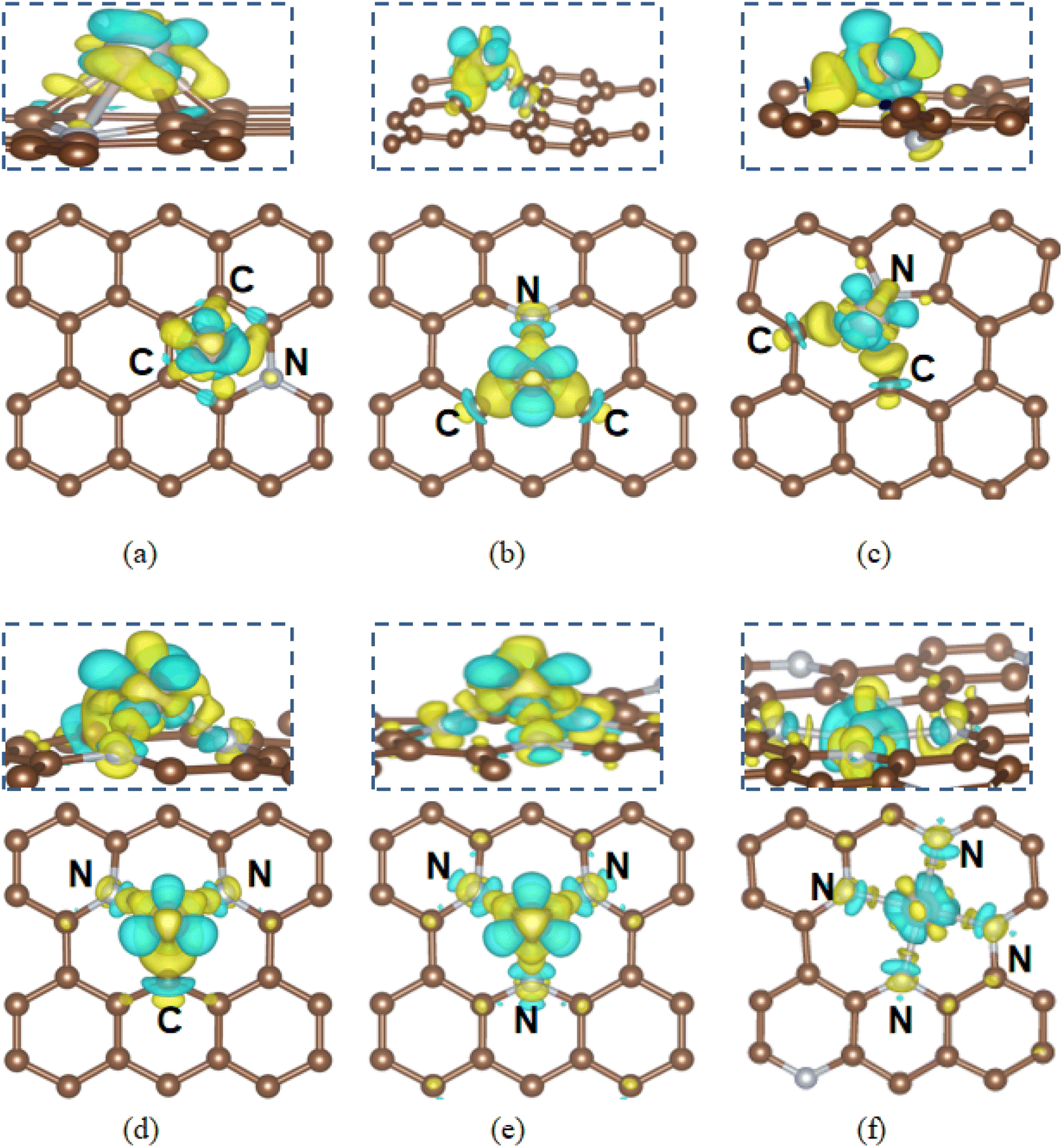 | ||
| Fig. 3 Electron charge density difference for Ru deposition on the substrates of (a) N_graphitic, (b) N_pyridinic, (c) N_pyrrolic, (d) 2N_Vac, (e) 3N_Vac and (f) 4N_Vac2 graphene. The yellow and blue areas denote electron accumulation and depletion. | ||
With increased N substitution, the binding energy for Ru/Fe SAs decreases from 2N to 4N defects. Notably, Fe SA has a very low binding energy on the 4N defect center (−2.09 eV), indicating strong metal–support interaction. The lowest formation energy (ΔE) among all the calculated models has been found for the Fe@4N moiety (1.42 eV), suggesting the energetically preference of this 4N-coordinated SAC structure. The binding energy of Ru on the 4N defect center is relatively low (0.25 eV) with promoted charge transfer of 0.96e from Ru to the 4N defect (0.99e for Fe). The formation of Ru SACs@4N is also energetically favorable. Bond lengths for Ru–N and Fe–N in the 4N defect center are slightly stretched to 1.94 Å and 1.88 Å, respectively. The COHP analysis for metal binding on the 4N defect center shows that the Fe–N antibonding orbital shifts below the Fermi level shown in Fig. S2(d),† indicating partial occupancy of the antibonding state, which destabilized the Fe–N bond. This is also reflected by the elongation of Fe–N bond length (1.88 Å) and a smaller ICOHP values compared to the bonds formed with N to 3N defect centers. Similarly, COHP analysis for Ru–N bonds suggests a weakening trend for the 4N configurations, although this effect is more pronounced in the Fe–N bond. As shown in Fig. 4, the 3N defect center exhibits relatively higher antibonding orbitals, while N_pyrrolic and 2N defect centers show highly localized antibonding peaks close to the Fermi level. These accessible antibonding states suggest enhanced charge transfer and potential bond dissociation during the catalytic reaction. Therefore, the general trend is that the higher N coordinated defect sites on graphene (i.e. 4N) shows energetically preference to anchor Ru and Fe SACs. Meanwhile, the lower N coordinated moieties (i.e. N_pyrrolic and 2N) may offer particularly active sites for HER reaction, as their accessible antibonding states facilitate electron transfer and potential bond dissociation. The dominance of Fe@4N motifs is in alignment with several early theoretical studies50,51 and supported by recent experimental results,52,53 which demonstrate that converting the pyrrolic N-coordinated FeN4 sites into highly stable pyridinic N-coordinated FeN4 sites to improve the long-term stability of the Fe SACs for ORR application.53
 | ||
| Fig. 4 Calculated PDOS and COHP analysis for Ru SACs on the (a) N_pyrrolic, (b) 2N, (c) 3N and (d) 4N-doped graphene substrates. | ||
3.3 HER activity and active sites of Ru/Fe@N-doped graphene
Based on the most stable defect configurations of N to 4N moieties, the hydrogen adsorption behavior on Ru and Fe single atoms anchored on N-doped graphene has been further explored in this section. Various adsorption sites, including edging sites C/N atoms and metal atoms were examined. The Gibbs free energy of the hydrogen adsorption (ΔGH*) was calculated under standard conditions and plotted in Fig. 5, serving as a key descriptor for HER performance activity.54 | ||
| Fig. 5 Calculated HER profile for Ru and Fe SACs on the three different configurations of N-doped graphene substrates (a) Ru@N and (c) Fe@N, and on the 2N to 4N moieties substrates (b) Ru@Nx and (d) Fe@Nx. | ||
As presented in Fig. 5(a), the calculated ΔGH* values for H adsorption on Ru SACs@N_graphitic, N_pyridinic and N_pyrrolic-doped graphene are 0.3 eV, 0.34 eV and 0.01 eV, respectively. While for Fe SAs on these three N moieties, the ΔGH* values are −0.39 eV, −0.14 eV and 0.02 eV respectively. For an ideal HER catalyst, ΔGH* should be close to zero. Thus N_pyrrolic defect centers offer moderate hydrogen adsorption energy, making them suitable for HER. In case of Ru SAC@N_pyrrolic, the edge sites carbon atoms adjacent to Ru SACs@N_pyrrolic are the most active sites for HER as shown in Fig. 6(a). For Fe SAC@N_pyrrolic, both edge site carbon atoms and Fe sites exhibit ΔGH* values close to 0. In the Ru SAC@N_pyrrolic, the C–H bond length is 1.20 Å with an ICOHP value of −5.45 eV. While in the Fe SAC@N_pyrrolic, the C–H bond length is 1.17 Å and ICOHP value is −5.86 eV, indicating stronger C–H interaction. Compared to H adsorption on the undoped Vac2_C, where ΔGH* is 0.17 eV shown in Fig. S4(a),† with additionally, in Ru SAC@N_pyrrolic, the adsorbed hydrogen is negatively charged (−0.65e), and the bonded edge site carbon atom is positively charged (+1.16e), whereas in the in Fe system, the H atom is negatively charged (−0.27e) and the C atom positively charged (+1.14e). This result indicates the stronger C–H interaction in the Fe SACs model, consistent with the with the higher ICOHP. While for the H atom adsorb on Fe atom, the Fe–H bond length is 1.55 Å and ICOHP value is −2.84 eV. For a good HER catalyst, the absolute value of ΔGH* should be less than 0.20 eV. For Fe SAC@N_pyridinic, the ΔGH* value of −0.14 eV suggests a moderate adsorption of hydrogen on the C atom adjacent to Fe adatom shown in Fig. S4(b),† making it suitable for HER. The C–H bond length is to be 1.11 Å and with an ICOHP of −5.95 eV. Therefore, the most active site for HER is the under-coordinated edge site carbon atom in the N doped graphene structures.
 | ||
| Fig. 6 The optimized hydrogen adsorption configurations at the most active sites on the (a) Ru SAC@N_pyrrolic, (b) Ru SAC@2N, (c) Ru SAC@3N, (d) Ru SACs@4N, (e) Fe SAC @N_pyrrolic, (f) Fe SAC@2N, (g) Fe SAC@3N and (h) Fe SAC@4N doped graphene. Dark brown, blue, gray, yellow and white represent C, N, Ru, Fe and H, respectively (side views are given in Fig. S7†). | ||
For substrates with 2N moieties, the calculated ΔGH* values of −0.1 eV (Ru SAC@2N-doped graphene) and −0.01 eV (Fe SAC@2N-doped graphene) or and, respectively, suggesting excellent HER activity, where hydrogen adsorption occurs directly on the metal atoms. The best ΔGH* is achieved when H atoms adsorb directly onto the metal single atoms as shown in Fig. 6 and COHP is in Fig. S5.† The Ru–H bond length is 1.68 Å, with an ICOHP of −2.93 eV, while the Fe–H bond length is 1.57 Å and the ICOHP is −2.70 eV. Ru and Fe atoms are positively charged (+1.11e and +1.18e, respectively), while H atoms are negatively charged (−0.52e and −0.47e, respectively), indicating enhanced charge transfer and promoting HER activity.
With 3N moieties in the substrate, the optimal adsorption sites are carbon atoms adjacent to the nitrogen dopants for both Ru and Fe SAC@3N-doped graphene as shown in Fig. 6. The ΔGH* values are 0.19 eV for Ru SAC@3N-doped graphene and 0.26 eV for Fe SAC@3N-doped graphene, making both metal SACs suitable for HER catalysis. The C–H bond lengths in both Ru and Fe systems are 1.12 Å with ICOHP values of −5.77 eV from Fig. S3.† The stronger charge transfer between Ru adatoms and 3N moieties results in a lower ΔGH* (−0.41 eV) than that for Fe (−0.28 eV). Ru lose more charges to neighboring atoms and slightly outperforms Fe.
Regarding to 4N defective substrate, the best ΔGH* is achieved when H atoms adsorb directly onto the metal single atoms as shown in Fig. 6 (COHP in Fig. S5†). Fe SAC@4N-doped graphene shows a ΔGH* of −0.18 eV, close to the ideal range for HER, with a Fe–H bond length of 1.63 Å and an ICOHP of −2.08 eV. In contrast, Ru SAC@4N-doped graphene exhibits stronger hydrogen adsorption, with a ΔGH* of −0.64 eV and a shorter Ru–H bond length of 1.58 Å, suggesting that hydrogen release is hindered, leading to inferior HER performance. This stronger Ru–H interaction (ICOHP of −2.93 eV) is due to excessive electron density extraction by the 4N defects, weakening HER efficiency.
To summarize, for graphene doped with a single nitrogen atom, N_pyrrolic defect centers offer moderate hydrogen adsorption energy, with edge carbon atoms adjacent to metal-pyrrolic sites serving as the most active sites for HER. This behavior is also observed in 3N defects, where edge carbon sites remain optimal adsorption sites. However, increased charge transfer in 3N configurations causes slightly more negative adsorption energies. For 2N-doped substrate, Ru and Fe adatoms act as active sites, providing ideal ΔGH* values. In contrast, the 4N defect shows a marked difference between Ru and Fe, with Fe SAC@4N-doped graphene having more moderate hydrogen adsorption, promoting efficient H2 release and superior HER performance. However, the Ru–N bond in Ru SAC@4N-doped graphene is overly strong, inhibiting H2 release and reducing HER efficiency. Overall, the modelling results suggest that Fe SACs on N-doped graphene could achieve superior HER activity, potentially surpassing platinum group metals like Pt and Ru with fine-tuning of nitrogen configurations. Our finding for Fe and Ru SACs on N doped graphene is also consistent with recent experimental studies on other transition metals. e.g. Co SACs on C3N4 substrate, the EXAFS characterization suggests higher N coordinated Co SAC structure are dominate on the dominant on the CNG substrate, whereas lower N-coordinated sites exhibit superior HER activity.38
3.4 The relationship between nitrogen coordination environments and HER activity
To further explore the relationship between N coordination and catalytic activity, Fig. 7 presents the catalytic activity descriptor, the minimum absolute adsorption free energy (min|ΔGH*|), as a function of metal SAC binding energy. A lower binding energy indicates stronger metal–support interactions, min|ΔGH*| identifies the best catalytic sites. The data reveal that an optimal HER activity is achieved when the binding energy of metal SACs on N doped substrate falls within a moderate range. Specifically, the N_pyrrolic and 2N configurations appear at the volcano plot peak, confirming their superior HER activity. Notably, the zigzag edges of N_pyrrolic provide additional active sites for H adsorption/desorption, with ΔGH* values as small as 0.01 eV. For 2N configurations, both Ru SAC@2N and Fe SAC@2N are positioned near the volcano peak, when hydrogen atoms adsorb directly onto the metal atoms. These two lower N-coordinated structures effectively tailor the interaction strength between the metal SACs and the graphene substrate, thereby enhancing HER activity. In contrast, the weak interaction between N_graphitic substrate and Ru/Fe SACs result in relatively poorer activity, while the 4N coordinated sites tend to bond too strongly with Ru, which also hinders HER activity. | ||
| Fig. 7 Volcano plots of catalytic activities as a function of the metal SAC's binding energy on the N-doped graphene substrates: (a) Ru SACs and (b) Fe SACs. | ||
Further comparisons of the overpotentials (max|ΔGH*|) for 1N to 4N structures are provided in Fig. S8.† The most unfavorable adsorption case is considered the rate-limiting step for HER reaction under acidic conditions. The 1N coordinated Ru SAC@N_pyrrolic exhibits the lowest overpotential for Ru, with an approximate values of 0.26 V. For Fe@2N moieties, the overpotential is as low as 0.23 V, positioning this structure at the peak of the overpotential plot for Fe. In contrast, high N-coordinated Fe SAC@4N moieties show a large max|ΔGH*| value due to increased deprotonation tendency at the bridging site between N and Fe (+0.81 eV), caused by excessive charge transfer from Fe to proximal N atoms. Meanwhile, for Ru SAC@4N, the high max|ΔGH*| is primarily attributed to H* desorption of at Ru site (−0.64 eV). Therefore, Ru SAC@N_pyrrolic and Fe SAC@2N emerge as the most favorable structures for HER, balancing adsorption and desorption properties.
To further investigate the impact of coordination environments, we performed Bader charge analysis to estimate the charge states of metal atoms in each configuration. Both Ru and Fe SACs exhibit positive charge states ranging from 0 to +2, with adsorption free energy (ΔGH*) varies accordingly shown in Fig. 8. The high catalytic activities of Ru/Fe SACs on N_pyrrolic and 2N correlates with their moderate charge states, e.g. with +0.84 for Fe in Fe SAC@2N and +0.70 for Ru in Ru SAC@2N. The tunable charge transfer between the metal center and substrate indicates that N doped graphene can actively modulate the valence electron distribution through electronic metal–support interactions.
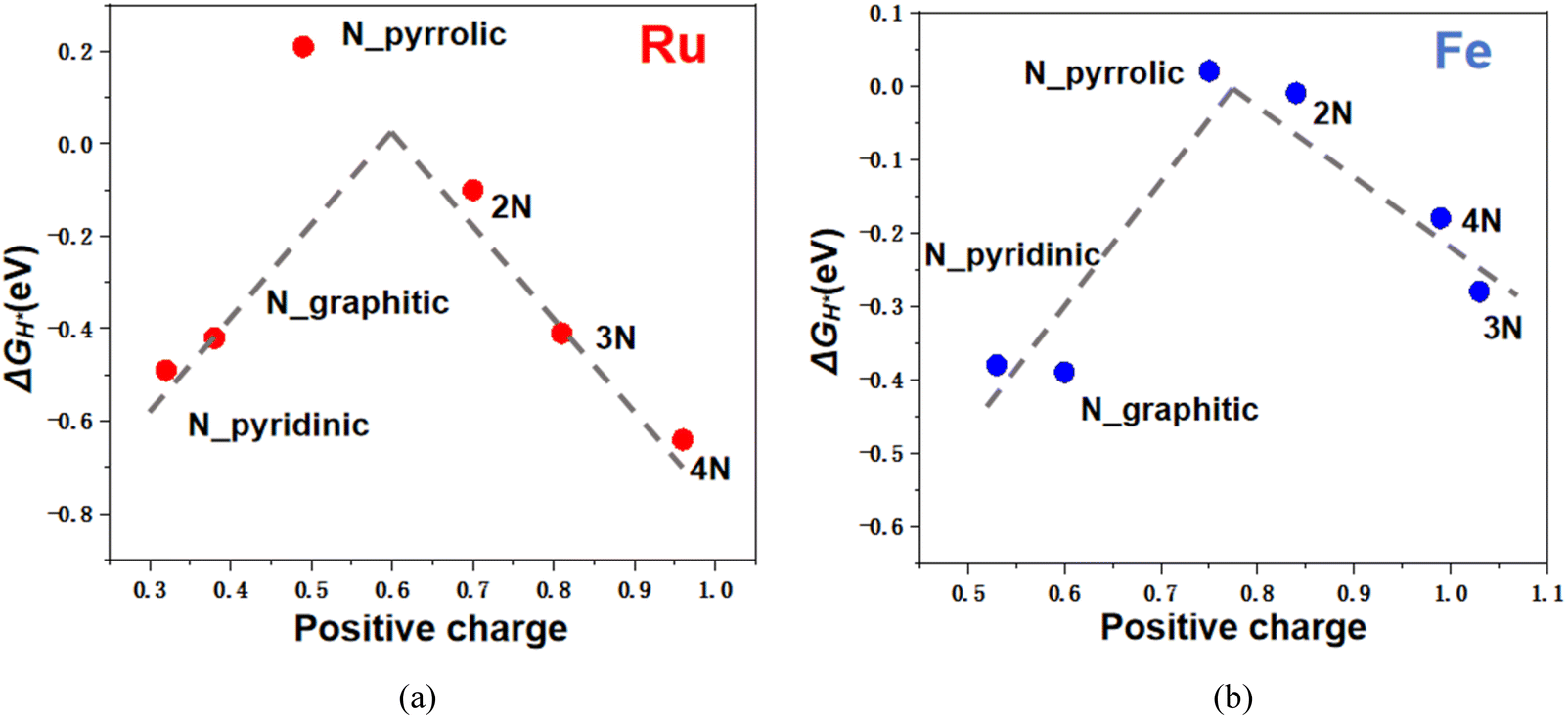 | ||
| Fig. 8 Volcano plots of ΔGH* varies with the positive charges of supported (a) Fe and (b) Ru SACs. | ||
Therefore, both binding energy and metal charge state can serve as key descriptors for energetic and electronic metal–support interactions. When comparing Ru with Fe SACs, moderate charge transfer from the metal to the substrate favors H* adsorption. This effect is consistent with our previous study on support design for catalytic methane conversion.55 Thus, the enhanced activity of metal SACs is primarily due to their moderate charge states, which are tuned by N coordination environments.
4. Conclusion
The search for efficient and cost-effective catalysts for hydrogen evolution reaction has led to the exploration of nitrogen-doped graphene supported single-atom catalysts, which offer tunable catalytic properties and enhanced stability. In this work, we systematically explored the stability of N to 4N defect substitution in graphene and highlighted the effect of nitrogen coordination environments on the HER activity of Ru and Fe SACs supported on N-doped substrate.• Direct extraction of a carbon atom from the graphene lattice requires a high vacancy formation energy making it is unlikely to occur. Nitrogen substitution at the unsaturated carbon sites would significantly decrease the defect formation energy. However, N-doped carbon divacancy structures generally remain less stable than N-doped carbon monovacancy structures.
• Among the invested configurations, 4N moieties provide the most stable anchoring sites for SACs. N defect centers interact more strongly with than Ru SAC. The Ru/Fe–N bonds in N_pyrrolic and 2N doped graphene provide active sites for HER reaction, as their accessible antibonding states facilitate electron transfer and potential bond dissociation.
• HER activity is highly tunable by the local coordination environments of metal SACs. The edge sites carbon atoms adjacent to Ru/Fe–N_pyrrolic moieties serve to offer moderate ΔGH* in 2N-coordinated structures, Ru and Fe adatoms act as primary active sites for HER. 3N and 4N moieties withdraw excessive electron density from Ru SAs, strengthening Ru–H bonds and inferior HER performance.
• Moderate metal–support interactions between metal SACs and supports is crucial for achieving superior HER performance. In particular, the N_pyrrolic and 2N configurations emerge as the most favorable, as they not only reside at the apex of the catalytic volcano plots but also exhibit low overpotentials. The charge state of metal SACs and binding energy have been proposed as key descriptors for the prediction of HER activity, which provide a framework that can be extend to other SAC and supports.
Overall, our study provides valuable insights into the design of efficient HER catalysts, suggesting that Fe SACs on 2N-coordinated graphene substrates hold significant promise for surpassing traditional platinum-group catalysts.
Data availability
The data supporting the findings of this study are available within the article and its ESI.† Additional datasets generated and analyzed during the current study are available from the corresponding author upon request. All computational input files, output data, and scripts used for the material modeling simulations have been archived and can be accessed by contacting email E-mail: xxia@ncepu.edu.cn.Author contributions
X. X. supervised the project. X. X., H. M. and C. P. conceived the idea. H. Y., S. A. and H. M. carried out the DFT calculations. H. M., C. P., Y. F. and C. X. contributed to the result discussion. H. Y. and X. X. drafted the manuscript. C. P., H. M. and F. Y. finalized the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from China National Nature Science Foundation (No. 22005095).References
- R. Akram, F. Chen and F. Khalid, et al., Heterogeneous effects of energy efficiency and renewable energy on carbon emissions: evidence from developing countries, TIDEE: TERI Information Digest on Energy and Environment, 2021, 20(3), 339–340 Search PubMed.
- M. G. Walter, E. L. Warren and J. R. McKone, et al., Solar water splitting cells, Chem. Rev., 2010, 110(11), 6446–6473 CrossRef CAS PubMed.
- J. Luo, J. H. Im and M. T. Mayer, et al., Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalysts, Science, 2014, 345(6204), 1593–1596 CrossRef CAS PubMed.
- L. Liu and A. Corma, Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles, Chem. Rev., 2018, 118(10), 4981–5079 CrossRef CAS PubMed.
- L. Yang, X. Wang and J. Wang, et al., Graphite carbon nitride/boron-doped graphene hybrid for efficient hydrogen generation reaction, Nanotechnology, 2018, 29(34), 345705 CrossRef PubMed.
- H. Jing, P. Zhu and X. Zheng, et al., Theory-oriented screening and discovery of advanced energy transformation materials in electrocatalysis, Adv. Powder Mater., 2022, 1(1), 100013 CrossRef.
- J. Yang, W. H. Li and S. Tan, et al., The electronic metal–support interaction directing the design of single atomic site catalysts: achieving high efficiency towards hydrogen evolution, Angew. Chem., 2021, 133(35), 19233–19239 CrossRef.
- B. Qiao, A. Wang and X. Yang, et al., Single-atom catalysis of CO oxidation using Pt1/FeO x, Nat. Chem., 2011, 3(8), 634–641 CrossRef CAS PubMed.
- N. Cheng, S. Stambula and D. Wang, et al., Platinum single-atom and cluster catalysis of the hydrogen evolution reaction, Nat. Commun., 2016, 7(1), 13638 CrossRef CAS PubMed.
- S. Fang, X. Zhu and X. Liu, et al., Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction, Nat. Commun., 2020, 11(1), 1029 CrossRef CAS PubMed.
- H. Zhang, G. Liu and L. Shi, et al., Single-atom catalysts: emerging multifunctional materials in heterogeneous catalysis, Adv. Energy Mater., 2018, 8(1), 1701343 CrossRef.
- Y. Wang, J. Mao and X. Meng, et al., Catalysis with two-dimensional materials confining single atoms: concept, design, and applications, Chem. Rev., 2018, 119(3), 1806–1854 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Heterogeneous single-atom catalysis, Nat. Rev. Chem, 2018, 2(6), 65–81 CrossRef CAS.
- C. Rivera-Cárcamo and P. Serp, Single atom catalysts on carbon-based materials, ChemCatChem, 2018, 10(22), 5058–5091 CrossRef.
- H. Li, H. Zhang and X. Yan, et al., Carbon-supported metal single atom catalysts, New Carbon Mater., 2018, 33(1), 1–11 CrossRef CAS.
- Y. Liang, Y. Li and H. Wang, et al., Strongly coupled inorganic/nanocarbon hybrid materials for advanced electrocatalysis, J. Am. Chem. Soc., 2013, 135(6), 2013–2036 CrossRef CAS PubMed.
- Y. Li, H. Wang and L. Xie, et al., MoS2 nanoparticles grown on graphene: an advanced catalyst for the hydrogen evolution reaction, J. Am. Chem. Soc., 2011, 133(19), 7296–7299 CrossRef CAS PubMed.
- Q. Liu, J. Tian and W. Cui, et al., Carbon nanotubes decorated with CoP nanocrystals: a highly active non-noble-metal nanohybrid electrocatalyst for hydrogen evolution, Angew. Chem., Int. Ed., 2014, 53(26), 6710–6714 CrossRef CAS PubMed.
- S. Sun, G. Zhang and N. Gauquelin, et al., Single-atom catalysis using Pt/graphene achieved through atomic layer deposition, Sci. Rep., 2013, 3(1), 1–9 Search PubMed.
- H. Y. Zhuo, X. Zhang and J. X. Liang, et al., Theoretical understandings of graphene-based metal single-atom catalysts: stability and catalytic performance, Chem. Rev., 2020, 120(21), 12315–12341 CrossRef CAS PubMed.
- D. W. Ma, T. Li and Q. Wang, et al., Graphyne as a promising substrate for the noble-metal single-atom catalysts, Carbon, 2015, 95, 756–765 CrossRef CAS.
- M. D. Hossain, Z. Liu and M. Zhuang, et al., Rational design of graphene-supported single atom catalysts for hydrogen evolution reaction, Adv. Energy Mater., 2019, 9(10), 1803689 Search PubMed.
- E. Jung, H. Shin and B. H. Lee, et al., Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production, Nat. Mater., 2020, 19(4), 436–442 CrossRef CAS PubMed.
- W. Zang, T. Sun and T. Yang, et al., Efficient hydrogen evolution of oxidized Ni-N3 defective sites for alkaline freshwater and seawater electrolysis, Adv. Mater., 2021, 33(8), 2003846 CrossRef CAS PubMed.
- X. Kong, Y. Huang and Q. Liu, Two-dimensional boron-doped graphyne nanosheet: A new metal-free catalyst for oxygen evolution reaction, Carbon, 2017, 123, 558–564 CrossRef CAS.
- T. He, S. K. Matta and A. Du, Single tungsten atom supported on N-doped graphyne as a high-performance electrocatalyst for nitrogen fixation under ambient conditions, Phys. Chem. Chem. Phys., 2019, 21(3), 1546–1551 RSC.
- B. Bhattacharya and U. Sarkar, The effect of boron and nitrogen doping in electronic, magnetic, and optical properties of graphyne, J. Phys. Chem. C, 2016, 120(47), 26793–26806 CrossRef CAS.
- B. Bhattacharya, N. B. Singh and U. Sarkar, Pristine and BN doped graphyne derivatives for UV light protection, Int. J. Quantum Chem., 2015, 115(13), 820–829 CrossRef CAS.
- Z. L. Wang, X. F. Hao and Z. Jiang, et al., C and N hybrid coordination derived Co–C–N complex as a highly efficient electrocatalyst for hydrogen evolution reaction, J. Am. Chem. Soc., 2015, 137(48), 15070–15073 CrossRef CAS PubMed.
- Z. Xing, Q. Liu and W. Xing, et al., Interconnected Co-Entrapped, N-Doped Carbon Nanotube Film as Active Hydrogen Evolution Cathode over the Whole pH Range, ChemSusChem, 2015, 8(11), 1850–1855 CrossRef CAS PubMed.
- X. Zhang, Z. Yang and Z. Lu, et al., Bifunctional CoNx embedded graphene electrocatalysts for OER and ORR: a theoretical evaluation, Carbon, 2018, 130, 112–119 CrossRef CAS.
- W. Liu, L. Zhang and W. Yan, et al., Single-atom dispersed Co–N–C catalyst: structure identification and performance for hydrogenative coupling of nitroarenes, Chem. Sci., 2016, 7(9), 5758–5764 RSC.
- Y. Zhao, L. Yang and S. Chen, et al., Can boron and nitrogen co-doping improve oxygen reduction reaction activity of carbon nanotubes?, J. Am. Chem. Soc., 2013, 135(4), 1201–1204 CrossRef CAS PubMed.
- S. Chao, Z. Bai and Q. Cui, et al., Hollowed-out octahedral Co/N-codoped carbon as a highly efficient non-precious metal catalyst for oxygen reduction reaction, Carbon, 2015, 82, 77–86 CrossRef CAS.
- G. Zhuang, J. Bai and X. Tao, et al., Synergistic effect of S, N-co-doped mesoporous carbon materials with high performance for oxygen-reduction reaction and Li-ion batteries, J. Mater. Chem. A, 2015, 3(40), 20244–20253 RSC.
- S. Gao, G. D. Li and Y. Liu, et al., Electrocatalytic H2 production from seawater over Co, N-codoped nanocarbons, Nanoscale, 2015, 7(6), 2306–2316 RSC.
- Z. L. Wang, X. F. Hao and Z. Jiang, et al., C and N hybrid coordination derived Co–C–N complex as a highly efficient electrocatalyst for hydrogen evolution reaction, J. Am. Chem. Soc., 2015, 137(48), 15070–15073 CrossRef CAS PubMed.
- X. Liu, Y. Deng and L. Zheng, et al., Engineering low-coordination single-atom cobalt on graphitic carbon nitride catalyst for hydrogen evolution, ACS Catal., 2022, 12(9), 5517–5526 CrossRef CAS.
- L. Feng, C. Fu and D. Li, et al., High-density low-coordination Ni single atoms anchored on Ni-embedded nanoporous carbon nanotubes for boosted alkaline hydrogen evolution, Dalton Trans., 2023, 52(28), 9684–9693 RSC.
- Z. Levell and S. Yu, et al., What is the “Other” site in M-N-C?, ACS Catal., 2025, 147(1), 603–609 Search PubMed.
- M. Huang, Y. Ma and J. Yang, et al., Ultrafine Ru nanoparticles on nitrogen-doped CNT arrays for HER: A CVD-based protocol achieving microstructure design and strong catalyst-support interaction, Appl. Surf. Sci., 2024, 645, 158834 CrossRef CAS.
- M. Huang, H. Yang and X. Xia, et al., Highly active and robust Ir-Ru electrocatalyst for alkaline HER/HOR: Combined electronic and oxophilic effect, Appl. Catal., B, 2024, 358, 124422 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113(18), 7756–7764 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed.
- J. Greeley, T. F. Jaramillo and J. Bonde, et al., Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5(11), 909–913 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Blöchl, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97(33), 8617–8624 CrossRef CAS.
- W. I. Choi, S. H. Jhi and K. Kim, et al., Divacancy-nitrogen-assisted transition metal dispersion and hydrogen adsorption in defective graphene: A first-principles study, Phys. Rev. B:Condens. Matter Mater. Phys., 2010, 81(8), 085441 CrossRef.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions, Phys. Chem. Chem. Phys., 2011, 13(34), 15639–15643 RSC.
- H. T. Chung, D. A. Cullen and D. Higgins, et al., Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357(6350), 479–484 CrossRef CAS PubMed.
- S. Liu, C. Li and M. J. Zachman, et al., Atomically dispersed iron sites with a nitrogen–carbon coating as highly active and durable oxygen reduction catalysts for fuel cells, Nat. Energy, 2022, 7(7), 652–663 CrossRef CAS.
- J. Greeley, T. F. Jaramillo and J. Bonde, et al., Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5(11), 909–913 CrossRef CAS PubMed.
- J. Xing, X. Xia and F. Jin, et al., Density functional theory study on Support-Promoted CH4 reforming in Ni-Based oxygen carriers for chemical-looping conversion, Appl. Surf. Sci., 2024, 656, 159691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra09113b |
| This journal is © The Royal Society of Chemistry 2025 |