
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of nitrate and nitrite on the UV/PDS process: performance and byproduct formation from the perspective of substituents†
Yawei Xie,
Chenda Wu,
Yue Wang,
Shijie Wu,
Yaozu Jin,
Mingdi Yang and
Hongyuan Liu *
*
College of Civil Engineering, Zhejiang University of Technology, Hangzhou, 310023, China. E-mail: lhyzyy@zjut.edu.cn
First published on 10th March 2025
Abstract
Nitrate and nitrite are ubiquitous ions in wastewater that affect the performance of advanced oxidation processes such as UV-activated persulfate (UV/PDS) and lead to the formation of by-products. Three structurally similar compounds with different substituent compounds, namely phenol (Ph), benzoic acid (BA) and salicylic acid (SA), were selected as target pollutants in this study, to explore these issues from a new perspective: the effect of substituents on contaminants. The results showed that both NO3− and NO2− inhibited the removal of the three pollutants in the UV/PDS system. However, the varying substituents on the compounds influenced the electron density of their molecular structures, causing different responses to NO3− and NO2− during treatment. Reactive nitrogen species (RNS) played a more significant role in the oxidation of these compounds in the UV/PDS/NO2− system than in UV/PDS/NO3−. Additionally, NO3− and NO2− seemed to affect the types of RNS that are most active in the process. The different substituents also influenced which positions on the molecule were attacked by reactive species, ultimately impacting the formation of N-containing byproducts. Although oxidation products were theoretically predicted and identified, many potential products remained undetected according to results from Fourier transform ion cyclotron resonance mass spectrometry. This study offers a new perspective by focusing on the characteristics of pollutants to evaluate the impact of NO3− and NO2− when UV/PDS is applied as an advanced wastewater treatment method.
1 Introduction
Sulfate radical (SO4˙−)-based advanced oxidation processes (AOPs) are extensively investigated for the treatment of organic pollutants in wastewater due to the strong oxidizing ability of the sulfate radical (E0 = 1.8–2.7 V). Various activation techniques have been employed to generate sulfate radicals from precursors, including chemical activation (homogeneous1 and/or heterogeneous2–4), thermal activation under moderate temperatures (typically 60–80 °C) and UV irradiation, which is widely adopted due to its operational simplicity and reliability. These methods collectively contribute to advanced oxidation processes and environmental remediation applications.5–7 In UV-based AOPs, the performance is influenced not only by operational conditions,8 but also by the composition of the wastewater matrix. Nitrate (NO3−) and nitrite (NO2−) are commonly found in wastewater, particularly in the effluent from wastewater treatment plants, which often serves as the influent for AOPs used in tertiary treatment.9 The impact of these ions on the performance of UV/PDS has garnered substantial research interest and engineering application over the past several decades. These ions can generate in situ photochemically produced reactive species and/or screen the light,10 thereby exerting complex, positive or negative, effects on the degradation of organic pollutants. The effects of NO3− on the oxidation process remain ambiguous. For instance, Zhao et al.11 investigated the removal of florfenicol and ciprofloxacin by UV/PDS, and they found that NO3− significantly enhanced the removal of ciprofloxacin, while markedly inhibited the removal of florfenicol. Antoine Ghauch et al.12 also found that a specific nitrate concentration interval promotes chloramphenicol degradation. Conversely, Yang et al.13 observed that NO3− inhibits the removal of 2,4-dinitroanisole in a UV/PDS system, while Sbardella et al.14 found that NO3− concentration did not influence the degradation rate of nine pharmaceutically active compounds by UV/PDS. For NO2−, it has generally been found to inhibit the oxidation of organic pollutants, such as aniline,10 dexamethasone15 and 2,4-dichlorophenol.16In addition to the effect on removal efficiency, the presence of NO3− and NO2− in SO4˙−-based AOPs can lead to the formation of toxic nitro-byproducts. Chen et al.17 reported that the input of NO2− resulted in the production of toxic nitrification byproducts, such as 2-nitrophenol and 4-nitrophenol. Similarly, NO3− can absorb sunlight or artificial ultraviolet rays and decompose into ˙OH and NO2˙.9 The NO2˙ can then react with organic compounds, particularly aromatic ones, to produce nitro-byproducts.18 This phenomenon is not limited to specific individual pollutants. Toxic nitro-byproducts such as 4-nitrophenol, 4-hydroxy-3-nitrobenzoic acid and 2,4-dinitrophenol, have been detected even in the treatment of natural organic matter by UV/PDS in the presence of NO3−.19
Although the SO4˙− is generally considered a less selective oxidizing species, varying reaction rates have been observed when UV/PDS was applied to pollutants with different structural characteristics.5,20 Furthermore, as mentioned earlier, NO3− and NO2− exhibit differing effects on the performance of UV/PDS depending on the specific pollutants being treated. The inconsistent results across various studies highlight the significance of the molecular structure of target pollutants in UV/PDS processes. The properties of substituent groups in organic compounds play a crucial role in these outcomes, with electron-withdrawing groups (EWGs) and electron-donating groups (EDGs) being especially common in organic molecules.21 These substituent properties also influence the formation of toxic byproducts during UV/PDS treatment. For instance, aromatic carboxyl groups in natural organic matter (NOM) can undergo decarboxylation upon reaction with SO4˙−, leading to the formation of phenolic intermediates, which contribute to the generation of nitro-containing by-products.19 However, there has been limited discussion on how the structural features of different pollutants affect their response to UV/PDS treatment, particularly in relation to the varying impacts of nitrate and nitrite based on the nature of the substituents present in the target pollutants.
In this study, phenol (Ph), benzoic acid (BA), and salicylic acid (SA) were selected as target pollutants, representing compounds with an electron-donating group (EDG, –OH), an electron-withdrawing group (EWG, –COOH), and both groups, respectively. The aim is to clarify the effects of NO3− and NO2− on the performance of UV/PDS when treating pollutants with different substituents and to examine the characteristics of by-products formed during the process.
2 Materials and methods
2.1 Chemicals
All chemicals were utilized as received, without additional purification. Detailed information is provided in Text S1.†2.2 Experiment steps
The photochemical reaction is carried out in a borosilicate glass made cylindrical reactor as we described in previous report,22 with a 10 W low-pressure mercury lamp emitting at 254 nm, 1.85 cm from the water. The UV intensity was 1.16 mW cm−2 which was measured by a UV radiometer (LS125, Linshang, China). Solution containing certain concentration of NaNO3/NaNO2 and target pollutants solved in the deionized water, the pH was adjusted to designed value with H2SO4 (1 mol L−1) and NaOH (1 mol L−1). Then predetermined amount of PDS was added to the reactor. The reaction was carried out under magnetic stirring and water cooling (25 °C). Samples were collected at predetermined time intervals, and were quenched with sodium thiosulfate (10 mmol L−1).The reaction stoichiometric efficiency (RSE) is defined as the number of moles of the organic contaminants degraded versus the number of mole of PDS consumed. Detailed calculate information is provided in Text S2.†
In the experiment investigating the contribution of various radicals, tert-butyl alcohol and nitrobenzene are used to as quencher of different radicals,23 detailed information are shown in Text S3.†
All tests were repeated at least twice and the averaged results were used.
2.3 Analytical methods
The concentrations of Ph, BA and SA were determined by HPLC (Shimadzu DGU-20A, Japan). The oxidation byproducts during the reaction were identified by LC-TOF-MS (Agilent 6210, USA) in ESI− mode. Detailed information of the methods is described in Text S4.†The FTIR test was conducted using an FTIR spectrometer (NICOLET Avatar 370, Thermo fisher, America) with a scanning range of 4000 to 500 cm−1. The water samples were freeze-dried using a freeze dryer prior to measurement.
Characteristic of oxidation byproducts were analyzed by a 7.0 T Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR-MS)(SolariX, Bruker). Molecular formulas of products were calculated using Data Analysis software. Elemental combinations were limited to molecular formulas containing 12C0–100, 1H0–200, 14N0–4, 16O0–30, and mass peaks with Signal-to-Noise Ratio greater than 6 were considered during molecule assignment. The errors between measured MW and the theoretical one was set to <1. More detailed information and sample pretreatment methods is described in Text S5.†
The concentrations of total organic carbon (TOC) were determined using a TOC analyzer (Shimadzu TOC-LCPH/CPN, Japan). The TOC measurements were triplicated, consuming 5 ml of sample each time.
2.4 Theoretical calculation
The density functional theory (DFT) was used to predict the degradation mechanism conversion of compounds. The Gaussian 09W programs24 were used to conduct all the calculations at the B3LYP/6-31+G (d) level,25 and the Solvation Model Based on Density (SMD) model was employed to account for the solvent effects.26 All the complexes and molecules were optimized to a stationary point and frequency calculation was carried out. Molecular visualization was performed on Gaussian View 5.0. The reactive sites of nucleophilic (f+), electrophilic (f−) and free radical (f0) attacks were predicted by calculating the Condensed Fukui Functions (CFF). The enthalpy change ΔH (kcal mol−1) and the Gibbs free energy change ΔG (kcal mol−1) of the reaction paths were calculated.3 Results and discussion
3.1 Effect of NO3−/NO2− on pollutants removal in UV/PDS
The removal of Ph, BA and SA in UV irradiation process in 60 min are not effective, and the reaction rates are low (Fig. 1 and S1†). UV/PDS significantly accelerates their removal by 41, 13 and 20 times for Ph, BA and SA, respectively, which are understandable and ascribed to the radicals generated in the system. | ||
| Fig. 1 Removal rate of the 3 compounds in various processes (conditions: [Ph] = [BA] = [SA] = 0.1 mM, [PDS] = 0.5 mM, [NO3−] = 2 mM, [NO2−] = 2 mM, pH = 3, T = 60 min). | ||
NO3− accelerated the photolysis of Ph from 0.0056 min−1 to 0.044 min−1 (data not shown here). The acceleration of NO3− on UV photolysis of Ph may result from the high absorbance between 200-400 nm of NO3−, as well as its high photosensitivity, which facilitates the generation of oxidative radicals.
However, the NO3− was found to inhibit the removal of Ph in UV/PDS under pre optimized conditions of NO3− as shown in Fig. 1 and S2.† Even though NO3− suppressed the Ph removal from 0.231 min−1 to 0.141 min−1 in UV/PDS process as shown in Fig. 1, the removal rate is still significantly faster than that under UV irradiation alone. It suggests that the acceleration from NO3− photolysis is not important for Ph removal when NO3− is present in the UV/PDS system. The inhibitory effect of NO3− on Ph removal in UV/PDS can be attributed to its UV filtering effect. NO3− competes with PDS for photons, which reduces the generation of reactive radicals in the UV/PDS process.13 In previous study, nitrate demonstrated a significant increase in Kobs of 37% at the optimal concentration of 10 mg L−1. However, at concentrations of 2.5 mg L−1 and 50 mg L−1, Kobs decreased by approximately 9% and 37%, respectively. This decrease was attributed to the production of highly oxidizing species NO3˙, which can lead to the formation of nitrite and oxygen radicals.12 However, in this study, the concentration of NO3− which may be the reason for the different observation.
NO3− does not significantly affect the removal of BA and SA (Fig. S2†). This observation probably relates to the formation of reactive nitrogenous species (RNSs). NO3− can significantly influence the absorption characteristics of three compounds (Fig. S3†), and it can be photolyzed with lights <280 nm and can slowly react with ˙OH and SO4˙−.27,28 These processes lead to the formation of RNSs such as ONOO˙, NO˙ and small amount of NO2˙ (reactions (1)–(3)).29 The reaction of ONOO− with ˙OH is favored under acidic condition (reactions (3)), and ONOO˙ is likely the main RNSs in the system. These RNSs have weaker redox potentials (E0) compared with that of ˙OH and SO4˙− (e.g. E0(ONOO−, 2H+/NO2−) = 1.2 V).30,31 However, they tend to react more readily with EWG-containing compounds,32,33 such as BA and SA in this study, rather than Ph which contains an EDG. The contribution of these RNSs helps counterbalance the light screening effect of NO3− in the UV/PDS process, which explains why the impact of NO3− on the removal of benzoic acid (BA) and salicylic acid (SA) was not significant. Our findings are consistent with previous studies on the removal of 2,4-dinitroanisole by UV/PDS, where high concentrations of NO3− alleviated the inhibitory effects.13
| NO3− → NO2˙ + O˙− | (1) |
| NO3− + hv → [NO3−]* → ONOO− | (2) |
| ONOO− + ˙OH → ONOO˙ + OH− | (3) |
All three compounds were inhibited by NO2− in UV/PDS process (Fig. 1). In addition to the strong screening effect on the lights (Fig. S3†), NO2− reacts rapidly with SO4˙− and ˙OH (reactions (4) and (5)) forming RNSs (mainly NO2˙). Consequently, in systems containing NO2−, pollutants and NO2− compete for radicals,18 resulting in a reduction in the removal rate of pollutants. The NO2˙ is an electrophilic oxidizer that tends to react with organic molecules containing rich-electron molecular groups.9,34 Ren et al.35 also concluded that the stronger the EDG on the molecular, the faster it reacts with NO2˙. Therefore, Ph, which contains an EDG, is removed more quickly, even in the presence of NO2−.
| NO2− + ˙OH → NO2˙ | (4) |
| NO2− + SO4˙− → NO2˙ | (5) |
In addition, we determined the RSE of the process for removing all three compounds across different systems and compared it with other systems (Table S1†). It is evident that the substituent properties of the compounds, as well as the addition of NO3−/NO2−, significantly influence the RSE. Ph, which contains only EDG, results in a significantly high number of RSE, nearly matching the RSE observed with heat/PDS, unlike other compounds. The reduction in RSE was more pronounced for NO2− than for NO3−, and the effect varied among different compounds.
3.2 Effect of NO3−/NO2− under various pH conditions
pH of a solution affects the form of organic compounds and the concentration of radicals in AOPs.21 As shown in Fig. 2, the removal rate of Ph in UV/PDS system decreased from 0.231 min−1 to 0.056 min−1, as the initial pH increased from 3 to 11. The better performances of UV/PDS under acidic condition can be ascribed to the SO4˙− generation. Under the basic condition, however, the principal radical in the system shifts from SO4˙− to ˙OH36 and Ph mainly exists as phenate.37 The phenate reacts with ˙OH at a much slower rate (1.2 × 106 M−1 s−1) than the rate of reaction between Ph and SO4˙− (6.6 × 109 M−1 s−1).17,38 This disparity in reaction rates explains the inhibition on Ph removal in UV/PDS under basic conditions. | ||
| Fig. 2 Effect of pH on reaction rate of the 3 compounds (conditions: [pollutant] = 0.1 mM, [PDS] = 0.5 mM, [NO3−] = 2 mM, [NO2−] = 2 mM, T = 60 min). | ||
When NO3− was introduced into the UV/PDS system, the Ph removal rate was significantly decreased in acidic condition, while was almost unaffected under basic condition. Very similar tendency was observed when NO2− was added in to the system. The inhibition on radical formation is one of the key reactions in all systems. The reason for the inhibition in acidic condition has been discussed above. Under basic condition, NO2˙ becomes important in both UV/PDS/NO3− and UV/PDS/NO2−. In UV/PDS/NO3−, NO2− is likely formed because HOONO is involved in the reaction mechanism, it reacts more readily to form NO2− when it deprotonated at high pH.39,40 These NO2− are converted to NO2˙ which then undergoes single electron transfer reactions with phenate at a relatively high rate.41 In UV/PDS/NO2−, the large amount of NO2− react with SO4˙− and/or ˙OH in the system forming NO2˙ directly. NO2˙ prefer to react with Ph as discussed before. In addition, the basic condition (0.0086 min−1) slightly favors the photolysis of Ph compared to acidic condition (0.0056 min−1). Therefore, the removal of Ph was not influenced by NO3− and NO2− in basic condition. The performance is even slightly promoted at higher pH level.
BA reacts very quickly with both ˙OH and SO4˙− (5.9 × 109 M−1 s−1 and 1.2 × 109 M−1 s−1, respectively).42 Consequently, it demonstrates similar removal performance under different pH conditions, with only a 7% difference between the highest and the slowest rates in UV/PDS (Fig. 2). When compared to UV/PDS alone, the addition of NO3− does not significantly affect the removal of BA, whereas NO2− dramatically inhibits its removal under various pH conditions. As previously mentioned, the primary conversion product of NO2− in UV/PDS is NO2˙, which tends to react with more readily with Ph rather than BA. Therefore, the observed difference in the effect of NO2− and NO3− on the BA removal indicates that NO2˙ may not be the main RNSs types in UV/PDS/NO3−. Although previous reports have suggested that NO2˙ is also the dominant RNSs in UV-based processes influenced by NO3−,19,43 these conclusions do not align with our observations.
In addition to above reasons, pH also leads to the conversion of the substituents. Under basic condition, –OH can readily be converted to –O−, leading to a decrease in its electron-donating ability. The –COOH group can be converted to –COO−, which exhibits a relatively higher electron-withdrawing ability.44 These conversion in substituents result in the complex response of SA to pH.
3.3 Contribution of reactive species
As shown in Table 1, in the absence of NO3− and NO2−, ˙OH and SO4˙− play comparable roles in the removal of Ph, while ˙OH primarily contributes to the removal of SA and BA. Upon the addition of NO3−, the role of ˙OH is significantly suppressed, indicating strong inhibition of the generation and action of ˙OH, which is consistent with the findings of Wang et al.45 Because of this inhibition on ˙OH, the contribution of SO4˙− becomes more important. Additionally, the formation of RNSs leads to the degradation of pollutants, but the contribution of RNSs is relatively low compared with the other radicals. This is likely because the reaction rates of NO3− with ˙OH and SO4˙− are relatively slow. Moreover, the NO3− can also be photolyzed to ONOO− through the excited [NO3−]*, which can further convert to O2˙−, NO˙. These species are then consumed by inter-reactions in the system.46| Ph | BA | SA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ˙OH (%) | SO4˙− (%) | RNSs (%) | UV (%) | ˙OH (%) | SO4˙− (%) | RNSs (%) | UV (%) | ˙OH (%) | SO4˙− (%) | RNSs (%) | UV (%) | |
| a “—” means no this species. “—” means almost no contribution according to the calculation. | ||||||||||||
| UV/PDS | 42.90 | 53.99 | — | 3.11 | >92 | — | — | 7.69 | 95.05 | — | — | 4.95 |
| UV/PDS-NO3− | 3.73 | 66.37 | 28.20 | 1.70 | 18.97 | 63.02 | 10.08 | 7.94 | 35.23 | 31.84 | 23.66 | 9.28 |
| UV/PDS-NO2− | 2.01 | 19.31 | 69.61 | 9.06 | 4.32 | 7.75 | 70.07 | 17.86 | 4.02 | 1.96 | 83.56 | 10.46 |
With the addition of NO2−, the removal of all compounds was inhibited, but RNSs played a similar and very important role (over 60%) in the removal of the three compounds. The NO2− can easily be converted to NO2˙, leading to a higher contribution of RNSs to Ph. For BA and SA, RNSs do not have priority in reacting with them because of –COOH, the EWG, on their structure. However, RNSs still contribute similarly to the removal of these compounds. These observations can likely be explained by two factors. First, there is competition between NO2− and pollutants for ˙OH, as the reaction rate of ˙OH and NO2− (12 × 109 M−1 s−1) is comparable to that with Ph (6.1 × 109 M−1 s−1), BA (5.9 × 109 M−1 s−1) and SA (1.2 × 1010 M−1 s−1).10,47 Second, the NO2− can be converted to [NO2−]* under UV irradiation, which forms O˙− in acidic condition (reactions (6) and (7)), and protonation of O˙−produces the ˙OH radical (reaction (8)). This formed ˙OH can further react with pollutants. The contribution of this generated ˙OH is likely misattributed to RNSs, making it difficult to distinguish between them using the current experimental methods. These factors likely explain the similar observed contributions of RNSs to the removal of different pollutants.
| NO2− + hv → [NO2−]* | (6) |
| [NO2−]* → NO˙ + O˙− | (7) |
| O˙− + H2O → ˙OH + OH− | (8) |
3.4 TOC removal
The TOC removal efficiencies of the three compounds by UV alone in different situations are generally low (5% to 15%). The results of this study are consistent with those of other studies (approximately 10% to 15%).48,49 The mineralization of the three compounds by UV/PDS are not effective as well, indicating the formation of oxidation byproducts. Performance on TOC removal is better for Ph compared to BA and SA. This can be attributed to the fact that compounds containing the –COOH group have to undergo the process of electrophilic substitution by ˙OH to form phenolic intermediates, which are then mineralized.The presence of NO3− has little effect on TOC removal efficiency compared with the case without NO3−, while the NO2− remarkably inhibits the TOC removal (Fig. 3). Nitro-byproducts are expected to form under the influence of RNSs (mainly NO2˙). Additionally, nitro- is an EWG which decrease the electron density of benzene ring, making the nitro-byproducts more resistant to further mineralization.44
 | ||
| Fig. 3 Effect of NO3−/NO2− on TOC removal in UV/PDS with Ph (a), BA (b) and SA (c) as the target pollutant (condition: [pollutant] = 0.1 mM, [PDS] = 0.5 mM, [NO3−] = 2 mM, [NO2−] = 2 mM,pH = 3). | ||
3.5 Theoretical prediction for the products
Different substituents significantly affect the electron distribution of these molecules (Fig. S5†). In Ph and SA, the electrons in the HOMO orbitals are mainly distributed on the aromatic ring and the –OH, while in BA, the electrons are mainly distributed on the aromatic ring and carbonyl group. This electron enrichment at these sites leads to dipole effects and proximity effects,50 , making these sites more susceptible to electrophilic radical attack.The –OH increases the electron density on para-C of Ph. The p-π conjugation of the lone electron pairs between hydroxyl oxygen and carbonyl π bonds leads to the stabilization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.51 Similarly, the electrostatic potential of Ph and SA shows that the hydrogen on their hydroxyl group is more reactive and more susceptible to attack by electrophiles.52
O bond.51 Similarly, the electrostatic potential of Ph and SA shows that the hydrogen on their hydroxyl group is more reactive and more susceptible to attack by electrophiles.52
According to Fukui function theory, higher values of f−, f+ and f0 indicate that the site is more susceptible to electrophilic, nucleophilic and free radical attack, respectively.53 Radical attack may occur at the neighboring and para-position of the –OH on Ph such as C4, C2 and C6 (Table S1†). The hydroxyl substituent leads to a high electron density center, which will be highly susceptible to ˙OH attack, forming phenolic compounds or poly-hydroxyl substitution structures. In addition, phenoxy structure could be formed through single electron transfer of SO4˙− at C6, O12 and C3. Similarly, C3 and C6 on BA are vulnerable to electrophilic and radical attack (Table S2†). These sites and the formed phenoxy structure are probably attacked by RNSs, forming nitro-byproducts.19 However, the situation of SA is more complex. Similar to Ph, the para-C in SA molecular (C5) is susceptible to radical attack under the influence of –OH. It seems that the influence of –OH obscures the influence of –COOH. Because the para-C of –COOH hydroxyl group (C6) is vulnerable to electrophilic attack under the influence of –COOH just like in BA structure, but C6 is prevented from the electrophilic attack (Table S3†).
3.6 Products identification
We identified the products in UV/PDS/NO3− and UV/PDS/NO2−, and the results are shown in Table 2. Some unique N-containing products were found in the UV/PDS/NO3− and UV/PDS/NO2− systems, respectively. For example, product (m/z = 123) was only formed in UV/PDS/NO2− treating Ph, product (m/z = 139) was only formed in UV/PDS/NO2−. These unique compounds further prove the different RNSs in UV/PDS/NO3− and UV/PDS/NO3−.| UV/PDS/NO2− | UV/PDS/NO3− | |||
|---|---|---|---|---|
| Ph | Pp1 (m/z 139) |  |
Pp1 (m/z 139) |  |
| Pp2 (m/z 123) |  |
Pp3 (m/z 184) | 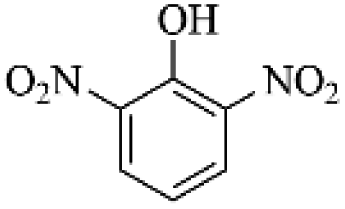 |
|
| Pp3 (m/z 184) |  |
|||
| BA | PB1 (m/z 169) |  |
PB1 (m/z 169) |  |
| PB2 (m/z 139) |  |
|||
| SA | PS1 (m/z 139) |  |
PS1 (m/z 139) | 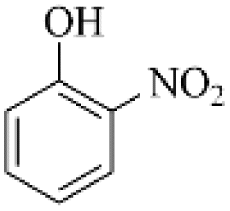 |
| PS2 (m/z 183) | 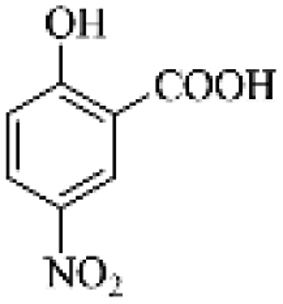 |
PS2 (m/z 183) |  |
|
The detected products match the predicted results well. The unpaired electrons on the phenoxy intermediate product were relocated to its ortho-position and para-position of –OH on Ph (mainly C4), which react with RNSs forming nitrophenol (Pp1), then poly-substituted nitrophenol (Pp3) was formed. Under the influence of the withdrawing property of nitro group and the donating property of –OH, the substitution sites are mainly located in the para-position of –OH and the nitro meta-position.
Benzoic acid radicals and benzene radical are formed via decarboxylation (C6) and attack para-position (C3) of carboxyl group of BA. Then they combine with radicals forming phenol, hydroxy benzoic acid and nitrobenzoic acid (PB1). The phenol then be converted to nitrophenol through the above-mentioned pathway.
Nitrosalicylic acids (Ps1) was found as the products of SA oxidation which imply that –OH and –COOH almost independently affect the pathway of oxidation under the influence of RNSs. SA is electrophilic attacked by SO4˙− forming phenoxy-like radicals, then transfer the unpaired electron to C1 and/or C5, finally form the nitrosalicylic acids.
The FT-IR results also demonstrated the role of RNS (Fig. S6†). For hydroxyl-containing compounds such as Ph and SA, the hydroxyl peaks after UV/PDS-NO2− were significantly lower than those observed after UV/PDS-NO3−. In contrast, BA exhibited hydroxyl peaks that were more similar after both treatments, indicating the selectivity of RNS for EDGs. The carboxyl group absorptions of BA and SA in the range of 2700–2500 cm−1 showed a decrease in intensity, confirming the removal of the carboxyl group. The range of 1800 cm−1 also supports this finding.
In addition, we selected 3 pathways to analyze the energy profiles during the reaction. It shows that SO4˙− and RNSs played important roles in different stages (Fig. 4). Ph is more likely to overcome the energy barrier and form nitrogen-containing byproducts than SA, because ΔG of forming transition state (TS) is smaller for Ph compared with SA. The high ΔG may relate to the withdrawing property of –COOH in SA. For BA, the situation is different. The –COOH decrease the electron density on the BA molecule, it is relatively hard to form its intermediate (ΔG > 0). However, we speculate that the nitration process is easier for BA than for Ph and SA because the ΔG of nitration process is smaller than that of BA.
 | ||
| Fig. 4 Energy change in the reaction of the target pollutants and radicals at/B3LYP/6-31+G(d) level under SMD model (IM for intermediate, TS for transition state, and PC for principal component). | ||
3.7 Characteristics of the oxidation byproducts
Carbon (C), hydrogen (H), oxygen (O), and nitrogen (N) are essential elements of the formed organic products; hence, these byproducts can generally be categorized into CHO and CHON groups based on the absence/presence of N in their structure. The Van Krevelen diagram is used to evaluate the alkylation, hydrogenation, hydration, and oxidation of mixed compounds in the samples.54 Fig. S7† shows that in the presence of NO3− or NO2−, more N-containing products could potentially be formed in the UV/PDS process compared with the formation of CHO-byproducts. For all three compounds, NO3− leads to more byproducts than NO2−, but NO2− result in more N-containing byproducts (Fig. S8† and 5). | ||
| Fig. 5 Van Krevelen diagrams of byproducts from oxidation of Ph, BA and SA under the influence of NO3− (left) and NO2− (right). | ||
The Van Krevelen diagram can be segmented into distinct regions corresponding to various compound classes based on their O/C and H/C values. These classes include aliphatic, protein-like, carbohydrates, unsaturated hydrocarbons, lignin, tannin, and condensed hydrocarbons55,56 corresponding to a–g regions in Fig. 5, respectively. As shown in Fig. 5, under the influence of NO3−, the UV/PDS oxidation products of Ph and SA predominantly consist of N-containing products, specifically lignin and tannins, with a minor presence of condensed hydrocarbons. For BA, the N-containing products are generally tannins. In contrast, under the influence of NO2−, the product distribution is similar for all three compounds, with the primary products being N-containing tannins and lignin. These results further suggest that different RNSs are involved in the UV/PDS processes with NO3− and NO2−, leading to distinct product profiles. Double bond equivalents (DBE) and aromaticity index (AI) indicate the degree of unsaturation, but AI focus on the aromatic.57 Two threshold values, AI > 0.5 and AI > 0.67, are used as unequivocal criteria for the existence of either aromatic or condensed aromatic structures, respectively.58 What interesting is that although the three compounds all consist of carbon of less than 7, the results show many products with a carbon number of >15 were formed. This probably related to the oxidative coupling and polymerization pathways of pollutants during the advanced oxidation processes.59,60 The oxidation products of Ph and SA that with high carbon content (C > 15) are primarily non-aromatic (AI < 0.57). This observation differs from the conclusion of Min et al., who speculated that phenols with polybenzene rings are predominant in these regions.61 Moreover, the NO3−-affected products are more aromatic than those affected by NO2− (Fig. 6). However, the situation is a little different for BA. Almost no compounds with DBE > 12 are formed in the oxidation products of BA in the presence of NO3− or NO2−. The majority of oxidation products are non-aromatic, and the few aromatic products are mainly derived from the influence of NO2−, as indicated by the AI distribution. This observation is corroborated by the region with low O/C and high H/C ratios in Fig. 5.
 | ||
| Fig. 6 DBE and AI distribution of oxidation byproducts of Ph, BA and SA. | ||
4 Conclusions
In this study, three structurally similar compounds were selected, differing only in their substituents. We demonstrated that the nature of the substituent groups significantly impacts not only the efficiency of UV/PDS in the presence of NO2− and NO3−, but also the formation of toxic nitrogen-containing products. NO2− and NO3− negatively affect the UV/PDS process by competing with pollutants for radicals and generating various RNSs.The substituent groups influence the electron density distribution; for instance, EDGs increase electron density on aromatic rings, while EWGs reduce it. The substituent properties of the probe, along with the presence or absence of NO3−/NO2−, influence the RSE of UV/PDS. Additionally, in compounds containing both EDGs and EWGs (e.g., SA in this study), these groups independently influence nitro-product formation.
Further studies with a broader range of model compounds featuring different substituents are needed to deepen our understanding of the effects of substituents on UV/PDS performance under the influence of NO3− and NO2−. While some products were identified in this study, many remained undetected according to FT-ICR-MS results, highlighting the need for additional characterization and evaluation.
Data availability
Most data are provided in support materials and others can be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2025C02217), by National Natural Science Foundation of China (No. 52170093, 52070111), and by the Zhejiang Provincial Natural Science Foundation (No. LY22E080010).References
- M. Amasha, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2018, 350, 395–410 CrossRef CAS.
- S. Naim and A. Ghauch, Chem. Eng. J., 2016, 288, 276–288 CrossRef CAS.
- Z. Abou Khalil, A. Baalbaki, A. Bejjani and A. Ghauch, Environ. Sci.: Adv., 2022, 1, 797–813 Search PubMed.
- R. El Asmar, A. Baalbaki, Z. Abou Khalil, S. Naim, A. Bejjani and A. Ghauch, Chem. Eng. J., 2021, 405, 126701 CrossRef CAS.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- M. Amasha, A. Baalbaki, S. Al Hakim, R. El Asmar and A. Ghauch, J. Adv. Oxid. Technol., 2018, 21, 261–273 CrossRef.
- W. B. Karroum, A. Baalbaki, A. Nasreddine, N. Oueidat and A. Ghauch, Environ. Sci.: Adv., 2024, 3, 1244–1258 Search PubMed.
- R. Yuan, S. N. Ramjaun, Z. Wang and J. Liu, Chem. Eng. J., 2012, 192, 171–178 CrossRef CAS.
- R. C. Scholes, C. Prasse and D. L. Sedlak, Environ. Sci. Technol., 2019, 53, 6483–6491 CrossRef CAS PubMed.
- C. Chen, Y. Lu, J. Liang, L. Wang and J. Fang, Chem. Eng. J., 2023, 451, 138755 CrossRef CAS.
- M. Zhao, Y. Liu, M. Feng, X. Yu and L. Wang, Chem. Eng. J., 2024, 489, 151371 CrossRef CAS.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CrossRef CAS.
- Y. Yang, Y. Ji, P. Yang, L. Wang, J. Lu, C. Ferronato and J.-M. Chovelon, J. Photochem. Photobiol., A, 2018, 360, 188–195 CrossRef CAS.
- L. Sbardella, I. Velo-Gala, J. Comas, I. Rodriguez-Roda Layret, A. Fenu and W. Gernjak, J. Hazard. Mater., 2019, 380, 120869 CrossRef CAS PubMed.
- K. Arman, M. Baghdadi and A. Pardakhti, Int. J. Environ. Anal. Chem., 2022, 1–19, DOI:10.1080/03067319.2022.2059360.
- X. Zhao, J. Gui, P. Yang, D. Kong, J. Lu, J.-M. Chovelon and Y. Ji, ACS ES&T Eng., 2023, 3, 2008–2015 Search PubMed.
- C. Chen, Z. Wu, S. Zheng, L. Wang, X. Niu and J. Fang, Environ. Sci. Technol., 2020, 54, 8455–8463 CrossRef CAS PubMed.
- J. Dzengel, J. Theurich and D. W. Bahnemann, Environ. Sci. Technol., 1998, 33, 294–300 CrossRef.
- X. Gao, Q. Zhang, Z. Yang, Y. Ji, J. Chen and J. Lu, ACS ES&T Eng., 2022, 2, 222–231 Search PubMed.
- M. Mahdi-Ahmed and S. Chiron, J. Hazard. Mater., 2014, 265, 41–46 CrossRef CAS PubMed.
- Y. Xue, Z. Wang, R. Naidu, R. Bush, F. Yang, J. Liu and M. Huang, Chem. Eng. J., 2022, 433, 134546 CrossRef CAS.
- Y. Xie, R. Xu, R. Liu, H. Liu, J. Tian and L. Chen, Chem. Eng. J., 2020, 389, 124457 CrossRef CAS.
- W. Liu, Y. Lu, Y. Dong, Q. Jin and H. Lin, Chem. Eng. J., 2023, 466, 143161 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- X. Ao, X. Zhang, W. Sun, K. G. Linden, E. M. Payne, T. Mao and Z. Li, Water Res., 2024, 253, 121259 CrossRef CAS PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- J. Mack and J. R. Bolton, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS.
- D. M. Stanbury, in Advances in Inorganic Chemistry, ed. A. G. Sykes, Academic Press, 1989, vol. 33, pp. 69–138 Search PubMed.
- G. Merényi and J. Lind, Chem. Res. Toxicol., 1997, 10, 1216–1220 Search PubMed.
- R. C. Scholes, Environ. Sci.: Processes Impacts, 2022, 24, 851–869 RSC.
- Y. Li, H. Qin, Y. Li, J. Lu, L. Zhou, J.-M. Chovelon and Y. Ji, Water Res., 2021, 200, 117275 CrossRef CAS PubMed.
- C. Wang, N. Moore, K. Bircher, S. Andrews and R. Hofmann, Water Res., 2019, 161, 448–458 CrossRef CAS PubMed.
- M. Ren, S. Sun, Y. Wu, Y. Shi, Z.-j. Wang, H. Cao and Y. Xie, Chemosphere, 2022, 296, 134071 CrossRef CAS PubMed.
- T. Olmez-Hanci and I. Arslan-Alaton, Chem. Eng. J., 2013, 224, 10–16 CrossRef CAS.
- R. M. Uppu, J.-N. Lemercier, G. L. Squadrito, H. Zhang, R. M. Bolzan and W. A. Pryor, Arch. Biochem. Biophys., 1998, 358, 1–16 CrossRef CAS PubMed.
- S. Canonica, T. Kohn, M. Mac, F. J. Real, J. Wirz and U. von Gunten, Environ. Sci. Technol., 2005, 39, 9182–9188 CrossRef CAS PubMed.
- G. Mark, H.-G. Korth, H.-P. Schuchmann and C. von Sonntag, J. Photochem. Photobiol., A, 1996, 101, 89–103 CrossRef CAS.
- Y. Cao, Q. Ma, B. Chu and H. He, Front. Environ. Sci. Eng., 2022, 17, 48 CrossRef.
- Y. Ji, L. Wang, M. Jiang, J. Lu, C. Ferronato and J.-M. Chovelon, Water Res., 2017, 123, 249–257 CrossRef CAS PubMed.
- Y. Yang, J. J. Pignatello, J. Ma and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 2344–2351 CrossRef CAS PubMed.
- Y. Wu, L. Bu, X. Duan, S. Zhu, M. Kong, N. Zhu and S. Zhou, J. Cleaner Prod., 2020, 273, 123065 CrossRef CAS.
- M. Ren, S. Sun, Y. Wu, Y. Shi, Z.-j. Wang, H. Cao and Y. Xie, Chemosphere, 2022, 296 Search PubMed.
- P. Wang, L. Bu, Y. Wu, W. Ma, S. Zhu and S. Zhou, Chemosphere, 2021, 267 Search PubMed.
- J. Mack and J. R. Bolton, J. Photochem. Photobiol., A, 1999, 128, 1–13 CrossRef CAS.
- A. Wang, Z. Hua, C. Chen, W. Wei, B. Huang, S. Hou, X. Li and J. Fang, Chem. Eng. J., 2021, 426, 131276 CrossRef CAS.
- A. Balaska, M. E. Samar and A. Grid, Desalin. Water Treat., 2015, 54, 382–392 CrossRef CAS.
- V. E. Sugihartono, N. N. N. Mahasti, Y. J. Shih and Y. H. Huang, Chemosphere, 2022, 296, 133663 CrossRef CAS PubMed.
- J. B. Wang, D. Zhi, H. Zhou, X. W. He and D. Y. Zhang, Water Res., 2018, 137, 324–334 CrossRef CAS PubMed.
- Z.-B. Sun, S.-Y. Li, Z.-Q. Liu and C.-H. Zhao, Chin. Chem. Lett., 2016, 27, 1131–1138 CrossRef CAS.
- S. Liu and L. G. Pedersen, J. Phys. Chem. A, 2009, 113, 3648–3655 CrossRef CAS PubMed.
- J. Guo, H. Sun, X. Yuan, L. Jiang, Z. Wu, H. Yu, N. Tang, M. Yu, M. Yan and J. Liang, Water Res., 2022, 219, 118558 CrossRef CAS PubMed.
- R. L. Sleighter and P. G. Hatcher, J. Mass Spectrom., 2007, 42, 559–574 CrossRef CAS PubMed.
- E. C. Minor, C. J. Steinbring, K. Longnecker and E. B. Kujawinski, Org. Geochem., 2012, 43, 1–11 CrossRef CAS.
- A. C. Maizel and C. K. Remucal, Water Res., 2017, 122, 42–52 CrossRef CAS PubMed.
- A. C. Stenson, A. G. Marshall and W. T. Cooper, Anal. Chem., 2003, 75, 1275–1284 CrossRef CAS PubMed.
- B. P. Koch and T. Dittmar, Rapid Commun. Mass Spectrom., 2006, 20, 926–932 CrossRef CAS.
- Y. Chen, W. Ren, T. Ma, N. Ren, S. Wang and X. Duan, Environ. Sci. Technol., 2024, 58, 4844–4851 CrossRef CAS PubMed.
- Y.-J. Zhang and H.-Q. Yu, Environ. Sci. Technol., 2024, 58, 11205–11208 CrossRef CAS PubMed.
- C. Min, Y. Han, C. Yan, Z. Fan, L. Jun and Y. Chen, Atmos. Environ., 2024, 319, 120319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08989h |
| This journal is © The Royal Society of Chemistry 2025 |