
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePX-MDsim: a rapid and efficient platform for large-scale construction of polyamide membranes via automated molecular dynamics simulations
Yiran Peng a,
Chi Zhangb,
Ming Wua,
Guangle Bub,
Kai Fana,
Xingren Chena,
Lijun Liang*a and
Lin Zhangbc
a,
Chi Zhangb,
Ming Wua,
Guangle Bub,
Kai Fana,
Xingren Chena,
Lijun Liang*a and
Lin Zhangbc
aCollege of Automation, Hangzhou Dianzi University, Hangzhou 310018, P.R. China. E-mail: llj@hdu.edu.cn
bEngineering Research Center of Membrane and Water Treatment of MOE, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, P.R. China
cFuture Environment Laboratory, Innovation Center of Yangtze River Delta, Zhejiang University, Jiaxing 314100, P.R. China
First published on 21st February 2025
Abstract
Polyamide (PA) membranes have attracted extensive attention due to their excellent separation performance in water treatment through reverse osmosis and nanofiltration processes. Although numerous molecular simulation studies attempt to explore their advantages from the microstructure, large-scale construction and simulation of PA membranes remain challenging, mainly due to the complexity and computational intensity of cross-linking reactions of polymers in molecular dynamics simulations. This paper introduces an automated platform called PX-MDsim for modeling and simulation of PA membranes. PX-MDsim is based on the PXLink framework and extends its applicability to any monomer with amino (–NH2) and carboxyl (–COOH) groups. The platform, combined with the PXLink program, realizes the full-process automated cross-linking simulation from input preparation, initial system construction, force field generation, functional group identification, and charge distribution update. Moreover, the software was used to cross-link m-phenylenediamine and 1,4-bis(3-aminopropyl)piperazine with trimesic acid, respectively, and multiple membrane structures with different cross-linking degrees were obtained. Furthermore, the generated membrane microstructure was analyzed using methods such as pore size distribution and order parameter, and the obtained results verified the applicability and accuracy of PX-MDsim in constructing PA membrane structures. The platform is user-friendly and accessible to researchers without prior expertise in molecular dynamics simulation, and it offers new possibilities for polymer research and applications.
1. Introduction
In recent years, polyamide (PA) membranes have become the focus of scientific research due to their outstanding separation performance in water treatment,1 such as seawater desalination, wastewater recycling, and drinking water purification.2 Its excellent selectivity and chemical resistance make it a mainstream technology in the water treatment industry, especially in reverse osmosis (RO) and nanofiltration (NF) processes.3–5In the RO process, the PA membrane works by applying high pressure to salt water, causing water molecules to pass through the membrane, while salt and other impurities are retained on the other side of the membrane.4–6 The NF process is mainly used to separate larger molecules and multivalent ions, and usually operates at lower operating pressures.1 PA nanofiltration membranes can efficiently separate small molecule organic matter, ions and multivalent metals, and are particularly suitable for water softening, wastewater treatment, and concentration and purification processes in the food and pharmaceutical industries.7
At present, there are hundreds of PA membranes formed by cross-linking of different ligands that have been noticed by researchers.3 One of the most complex and critical steps in simulating PA membranes is modeling. The challenges faced by researchers in large-scale construction of PA membranes mainly stem from the complexity and resource requirements of the polymer cross-linking process.8 Simulating the cross-linking reaction at the molecular level is not only technically complex, but also is computer consuming.9,10 There is currently no universal software tool that can flexibly and quickly construct PA membranes based on different monomers and in a personalized manner.
In molecular dynamics (MD) simulations, the construction of PA membranes involves cross-linking reactions of multiple monomer molecules. The cross-linking reaction process is not a simple chemical reaction. It involves non-covalent interactions between molecules, the formation and breaking of chemical bonds, and the accompanying energy and charge changes.9 After this process, these monomers are finally connected together through amide bonds to form a highly cross-linked network structure.11 Therefore, in order to ensure that the structure of the final membrane is consistent with the experimental results, all aspects of this cross-linking process must be accurately handled in the simulation. This requires accurate modeling of force fields, molecular structures, and reaction mechanisms.
Many existing MD simulation software (such as GROMACS, LAMMPS, etc.) require users to have an in-depth understanding of each step of the simulation, from force field selection, initial structure generation to the definition of the reaction process, all of which require manual settings by users.12–15 To simulate cross-linking reactions, users must not only accurately define the chemical reactions in the system but also configure force field parameters for different molecules, which typically requires extensive experience in molecular simulations.16 For most membrane material researchers, this highly specialized operating threshold greatly limits the popularity and application of the tools. At the same time, existing tools are relatively inefficient when dealing with large-scale simulations. Most tools are not optimized for polymer cross-linking reactions, resulting in slow calculation speeds when dealing with complex molecular systems.17
The core function of PXLink is to simulate the cross-linking process of PA membranes, especially to construct polymer networks by calculating the formation of amide bonds between adjacent carboxyl and amino groups. It can effectively generate cross-linked polymer membranes by combining GROMACS for energy minimization and MD simulation. This function has been verified to accurately simulate the structure and dynamic behavior of cross-linked PA membranes of trimesic acid (TMA) and m-phenylenediamine (MPD) in water treatment.9
PX-MDsim is based on the principle of PXLink and further expands its adaptability so that it is no longer limited to specific monomers. PX-MDsim allows users to simulate cross-linking reaction process between carboxyl and amino groups, enabling to construct various polymer cross-linking systems through amino-carboxyl condensation reaction.
In this paper, we first present the design concept and software framework of PX-MDsim. By automating the steps of input preparation, initial system construction, force field generation, functional group identification, and charge distribution calculation, the repetitive tasks in traditional simulations are simplified, significantly improving simulation efficiency and ease of operation. Furthermore, by simulating the cross-linking reactions of different monomers (m-xylylenediamine and TMA, 1,4-bis(3-aminopropyl)piperazine and TMA), and analyzing their properties and structures, the application and reliability of this platform in the construction of polyamide membrane structures are demonstrated, providing a new and efficient tool for the design and optimization of membrane materials.
2. Software description
2.1 Software overview
PX-MDsim is a highly automated MD simulation platform specifically designed to simulate polymer cross-linking reactions with amino (–NH2) and carboxyl (–COOH) monomers, especially suitable for the construction of PA membranes and simulation of the cross-linking process. The software integrates a variety of functional modules and integrates multiple programs such as GROMACS,12,13 Packmol,18 CGenff,19,20 and PXLink.9 The entire process from input data preparation, force field parameter generation to cross-linking reaction simulation has been automated. PX-MDsim is designed to simplify user operation, reduce human intervention, and significantly increase the efficiency of large-scale simulations.2.2 Software architecture
 | ||
| Fig. 1 Flowchart for obtaining the structure of a cross-linked polymer membrane using PX-MDsim. Optional step is marked with dashed boxes. | ||
Afterwards, PX-MDsim uses the ‘pdb2gmx’ command of GROMACS to generate force field topology information for the entire system. PX-MDsim is pre-configured with the CHARMM36 force field, automatically selects the force field, and calls the ‘pdb2gmx’ command of GROMACS based on the force field information of the small molecule we added earlier, to match the atoms, bonds, angles and other information in the initial system with the CHARMM36 force field. At the same time, PX-MDsim will automatically prepare an atomic index file for the system according to the residue name.
 | ||
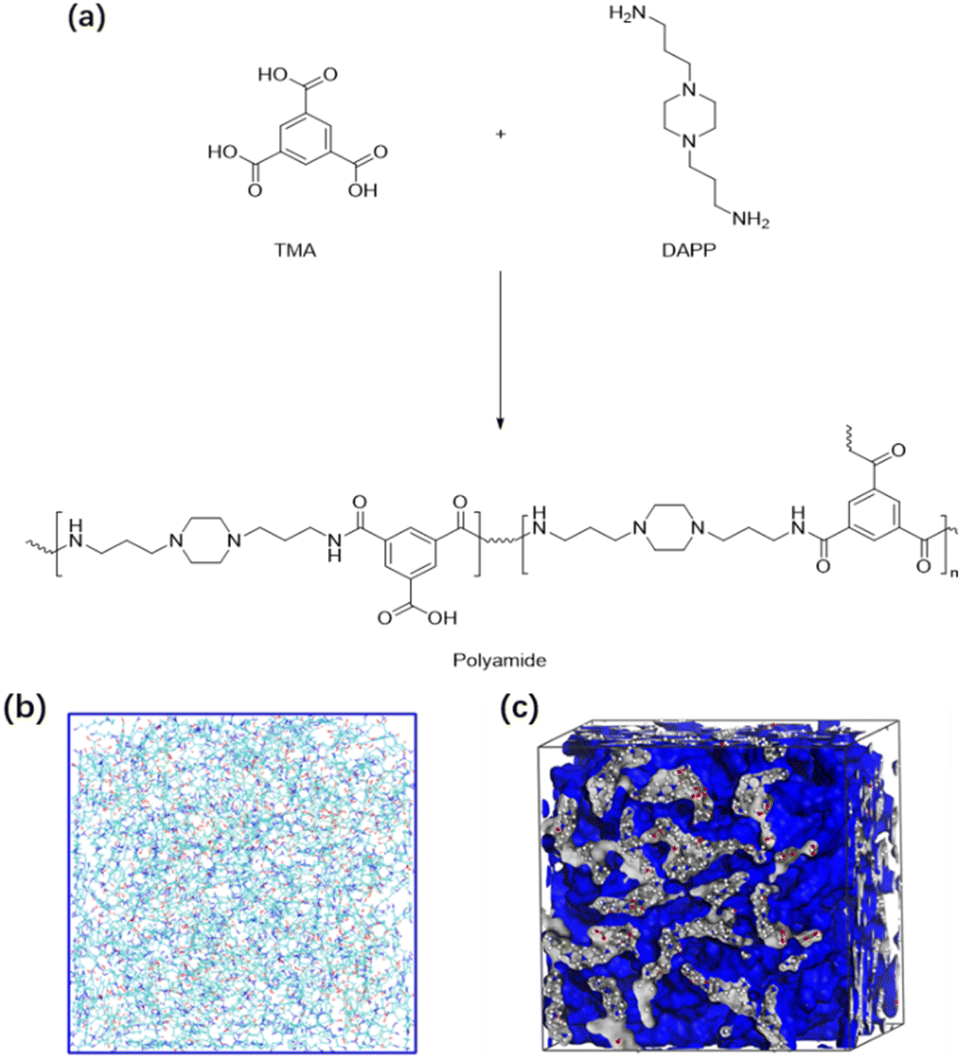
| Fig. 2 Structure formula of XLN and TMA monomers and the crosslinked polyamide. | ||
For example, in the cross-linking process of TMA and XLN, the charges of carbonyl carbon, carbonyl oxygen, amide nitrogen, and amide hydrogen after amide bond formation are 0.327, −0.464, −0.416, and 0.313, respectively. Before bonding, the charges of these atoms are 0.456, −0.432, −0.818, and 0.338. In addition, the charges of the carbon atoms connected to the carbonyl carbon and amide nitrogen have also changed. Our job is to calculate the charge difference of the relevant atoms before and after bonding, and update its value in the specified function in the PXLink program. In this way, after each new amide bond is formed during the cross-linking process, the correct charge change of the atom will be ensured. The automation of this step ensures the rationality of the charge distribution after the cross-linking reaction, thereby improving the accuracy and credibility of the simulation.
3. Application examples
To evaluate the proposed procedure, simulations were performed using m-xylylenediamine (XLN) and trimesic acid (TMA) as well as 1,4-bis(3-aminopropyl)piperazine (DAPP) and TMA as crosslinking monomer inputs to the software. XLN is a versatile crosslinking agent with two reactive amino groups, enabling it to interact with a wide range of acidic or anhydride monomers to form highly crosslinked polymer networks. These crosslinked structures exhibit excellent mechanical strength, chemical resistance, and thermal stability. DAPP is a commonly used crosslinking agent with two amino groups that can react with a variety of acidic or anhydride monomers (such as TMA) to form highly crosslinked polymer membranes. These crosslinked membranes show great potential for application in water treatment, gas separation, and nanofiltration.26–28 TMA has also been widely studied as a building monomer for crosslinked polymers. Because it contains three carboxyl groups, it can form stable amide bonds with polyamine compounds to construct membrane structures with high mechanical strength and chemical stability.29,30 Based on its aromatic structure, TMA gives the membrane material high thermal stability and mechanical strength, which makes its application under high temperature and high pressure conditions possible.31,32 In the field of gas separation, the combination of DAPP and TMA can improve the selectivity for specific gases and optimize industrial separation processes.27In the application cases of XLN and TMA, we mainly selected the membrane structure with a 40% DC to deeply study the internal thin layer structure of the obtained membrane. In the application cases of DAPP and TMA, we obtained four membrane structures with different DC value, respectively, to verify the ability of the program to generate cross-linked polymer membranes with different DC value.
3.1 XLN and TMA
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of free carboxyl and amine groups. The process is automated and the user only needs to input the necessary parameters.
:1 ratio of free carboxyl and amine groups. The process is automated and the user only needs to input the necessary parameters.
 | ||
| Fig. 3 (a) Snapshot of the initial system. (b) Snapshot of the system with a density of 1.19 g cm−3 at 40% DC. (c) Visualization of pore size distribution. Red represents the part with a larger pore size, green represents the part with a medium pore size, and blue represents the part with a smaller pore size. The minimum pore size is 1.1002 Å and the maximum pore size is 6.5 Å. (d) Pore size distribution of cross-linked polymer membranes, the data in the figure are diameters. | ||
It can be seen from the Fig. 3(d), PSD from the simulation results exhibits a normal distribution. The main peak value of pore size ranges between 3 Å and 5 Å, corresponding to the green and yellow parts in Fig. 3(c). It is consistent with the PSD of the nanofiltration membrane from the experiment,37–40 verifying the accuracy of the membrane structure obtained by the program. In addition, secondary peaks are observed in both the small pore size area (<3 Å) and the larger pore size area (>5 Å). The distribution of small pore regions may be related to the close packing of polymer segments, while the distribution of large pore regions may be caused by insufficient local cross-linking.
In addition to this, the order parameter S(r) and radial density ρ(r) were evaluated to more accurately investigate the local structure within the cross-linked thin layer, which is mainly controlled by the interactions between the phenyl groups of the bonded and non-bonded monomers.41 Fig. 4 explains the calculation basis of these parameters. Here, r denotes the radial distance between the centers of mass of the phenyl rings, while θ(r) refers to the angle formed by the normal vectors of neighboring phenyl rings at a distance r. The order parameter S(r) is expressed mathematically as
 | (1) |
 | (2) |
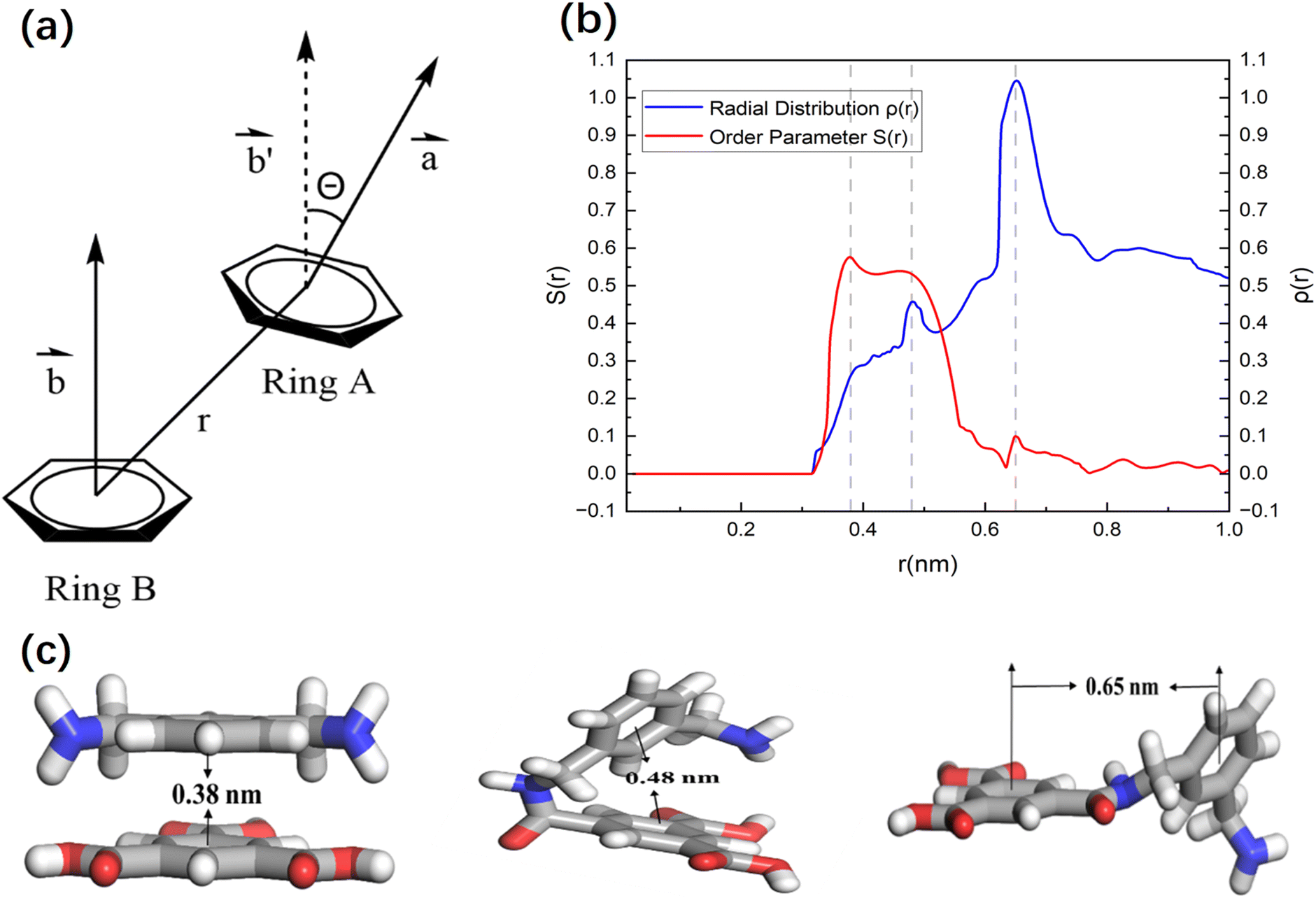 | ||
| Fig. 4 (a) Schematic representation of the order parameter calculation: r represents the radial distance between the centers of mass of benzene rings, while θ(r) denotes the angle formed by the normal vectors of adjacent benzene rings at a distance r. (b) The red dashed line is the order parameter S(r) of the adjacent monomers, and the blue solid line is the radial density ρ(r) curve. Based on the distribution of the peaks in the S(r) and ρ(r) spectra and their correlation, three representative structures were identified: π–π parallel stacking rings at r ≈ 0.38 nm, T-shaped rings at r ≈ 0.48 nm, and regular bonding rings at r ≈ 0.65 nm. (c) Schematic diagram of three benzene ring stacking methods and their corresponding radial distances. | ||
As shown in Fig. 4(b), the order parameter S(r) exhibits a sharp peak at r ≈ 0.38 nm, corresponding to the π–π stacking structure, while a smaller peak appears at r ≈ 0.65 nm, attributed to regularly bonded benzene rings. On the other hand, the radial density distribution ρ(r) reveals a significant peak at r ≈ 0.65 nm, which corresponds to the distribution of S(r). This prominent peak in ρ(r) suggests that the majority of local structures in the cross-linked polymer film thin layer are covalently bonded rings with slightly twisted orientations. Additionally, another peak observed at r ≈ 0.48 nm corresponds to T-shaped rings, indicating a higher occurrence of bent structures in the thin layer. These structural differences may result in larger pore sizes compared to the ordered π–π stacking structures. These results are basically consistent with the study of order function in other cross-linked polyamide membranes, and also verify the rationality of the membrane structure obtained by our procedure.
The position r ≈ 0.38 nm corresponds to the parallel π–π stacking between benzene rings. At this distance, the molecules tend to be arranged in a regular and orderly manner, and S(r) shows a significant peak. Although there are two geometric stacking modes at r ≈ 0.48 nm and r ≈ 0.65 nm, there may be a certain degree of rotational freedom between the benzene rings in these two stacking modes. Different from the π–π stacking, the molecules in these two models are in a relatively free or random orientation state, and it reduces the S(r) value.
3.2 DAPP and TMA
:1 ratio of free carboxyl and amine groups.
 | ||
| Fig. 5 (a) Structure formula of DAPP and TMA monomers and the crosslinked polyamide. (b) Snapshot of the initial system. (c) 3D pore distribution map. The box size is 6.0 × 6.0 × 6.0 nm3 and the density is 0.8 g cm−3. | ||
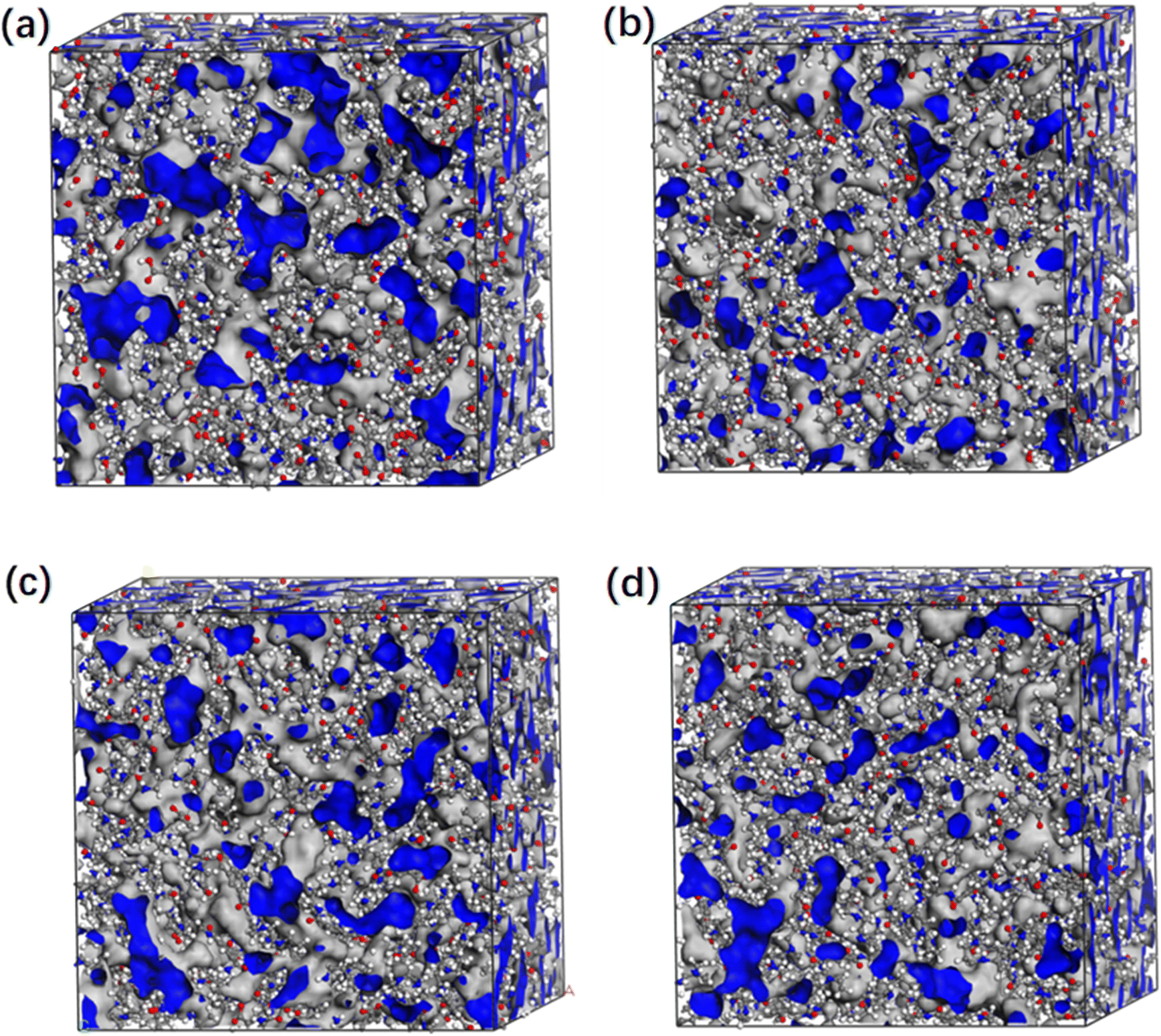 | ||
| Fig. 6 Pore distribution map of four polymer membranes with DC: (a) 20%; (b) 40%; (c) 60% and (d) 80%, respectively. The grey areas represent the occupied space and the blue areas represent the free volume. | ||
 | ||
| Fig. 7 The PSD data of the four polymer membranes were obtained using the 21-step equilibrium method with DC: (a) 20%; (b) 40%; (c) 60% and (d) 80%, respectively, with peak values correspond to pore sizes of 4.1 Å, 3.8 Å, 4.2 Å, and 4.2 Å, respectively. The data observed in the figure are all normally distributed. | ||
 | ||
| Fig. 8 PSD of the solvated membranes formed by DAPP and TMA with different DC. Aggregate pores are larger pores formed by loose packing or incompletely cross-linked regions between polymer chains. Network pores are small, uniform pores formed by a highly cross-linked network of polymer chains. | ||
Fig. 8 shows the PSD of the hydrated membranes. Membranes with low DC usually have more structural irregularities, resulting in larger holes or aggregate pores, which show a wider PSD. As the DC increases, more covalent bonds are formed, resulting in a denser membrane structure. This reduces the free volume within the membranes, effectively reducing the pore size. Therefore, the PSD becomes more concentrated and the pore size decreases. These findings are in strong agreement with the results reported in related study.42
All the PSD of different membranes exhibit a normal distribution. It is in consistent with the experimental results. The pore diameter of a general nanofiltration membrane is usually between 0.1 nm and 1 nm (ref. 37–40) from the experiment, which is in good agreement with the PSD of the nanofiltration membrane in our simulation. At the same time, in another simulation of a typical polyamide membrane, the researchers measured the pore diameter of the dry membrane made of MPD and TMA to be 0.4 nm,9 which is consistent with the pore size data measured in this case. It confirms that the PX-MDsim is suitable for building the nanofiltration membrane by amino-carboxyl condensation reaction.
4. Conclusions
In this study, PX-MDsim is proposed, an innovative MD simulation platform designed for the construction and simulation of PA membranes via automated cross-linking amino-carboxyl condensation reaction. PX-MDsim effectively extends the functionality of PXLink, providing greater flexibility in simulating a variety of monomers containing amino and carboxyl groups. This paper introduces two application examples in detail. The rationality and reliability of the membrane structure obtained by the software platform were verified by analyzing the order parameters and pore size distribution of the PA membrane obtained by crosslinking XLN and TMA. The flexibility and efficiency of the platform in constructing PA membranes with different DC values were highlighted by simulating the crosslinking process of DAPP and TMA and verifying the pore size distribution of the results.In future work, we plan to further optimize the algorithm of PX-MDsim to accelerate the computation and improve the accuracy of force field parameter generation. It is worth emphasizing that PX-MDsim not only provides an efficient simulation method for current polymer research and membrane material design, but more importantly, it can easily generate a large amount of simulation data. These large-scale data create conditions for the application of machine learning and artificial intelligence in the design of new materials. By leveraging the diverse datasets generated by PX-MDsim, we hope to lay the foundation for AI-driven materials science research in the future, thereby promoting the process of automated and intelligent materials development. This prospect will enable PX-MDsim to play an even more important role in the modern technology of new material design and optimization.
This software is free and open to everyone. For specific usage methods, please refer to the software documentation in the repository.
Data availability
PX-MDsim is a free software and open to everyone. Installation instructions, source code, latest documentation, and example usage can be accessed on the GitHub repository (https://github.com/AI4SCI-HDU).Author contributions
Yiran Peng: investigation, data curation, writing – original draft. Chi Zhang: investigation, data curation. Guangle Bu: data curation. Kai Fan: conceptualization, data curation. Xingren Chen: methodology. Ming Wu: investigation, data curation. Lin Zhang: conceptualization, methodology, writing– review & editing. Lijun Liang: conceptualization, methodology, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (22276045, 22138010); Joint R&D of the Yangtze River Delta Science and Technology Innovation Community (2023CSJGG1000); Key R&D project of Anhui Province (No. 202304a05020022). We would like to thank Xiang Huang from Shaoxing Testing Institute of Quality and Technical Supervision, Shaoxing 312000, China for assisting with processing data during manuscript revisions.References
- P. Sarkar, C. Wu, Z. Yang and C. Y. Tang, Chem. Soc. Rev., 2024, 53, 4374–4399 RSC
.
- J. Usman, S. I. Abba, F. J. Abdu, L. T. Yogarathinam, A. G. Usman, D. Lawal, B. Salhi and I. H. Aljundi, RSC Adv., 2024, 14, 31259–31273 RSC
- M. A. Ahmed, S. A. Mahmoud and A. A. Mohamed, RSC Adv., 2024, 14, 18879–18906 RSC
- T. R. Nickerson, E. N. Antonio, D. P. McNally, M. F. Toney, C. Ban and A. P. Straub, Chem. Sci., 2023, 14, 751–770 RSC
- W. Liu, J. L. Livingston, L. Wang, Z. Wang, M. del Cerro, S. A. Younssi, R. Epsztein, M. Elimelech and S. Lin, Nat. Rev. Methods Primers, 2024, 4, 10 CrossRef CAS
- H. Takabatake, M. Taniguchi and M. Kurihara, Membranes, 2021, 11, 138 CrossRef CAS PubMed
- M. Peydayesh, Membranes, 2022, 12, 518 CrossRef CAS PubMed
- M. Q. Seah, W. J. Lau, P. S. Goh, H.-H. Tseng, R. A. Wahab and A. F. Ismail, Polymers, 2020, 12, 2817 CrossRef CAS PubMed
- C. Zhang, G. Bu, M. S. J. Sajib, L. Meng, S. Xu, S. Zheng, L. Zhang and T. Wei, Comput. Phys. Commun., 2023, 291, 108840 CrossRef CAS
- K. Li, S. Li, L. Liu, W. Huang, Y. Wang, C. Yu and Y. Zhou, Phys. Chem. Chem. Phys., 2019, 21, 19995–20002 RSC
- Y. Li, M. Wang, L. Wang, T. Chen, W. Feng, T. Wang and L. Zhao, Phys. Chem. Chem. Phys., 2023, 25, 25309–25321 RSC
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier and R. Shan, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS
- S. Duangdangchote, D. S. Seferos and O. Voznyy, Digital Discovery, 2024, 3, 2177–2182 RSC
- A. V. Karatrantos, O. Couture, C. Hesse and D. F. Schmidt, Polymers, 2024, 16, 1373 CrossRef CAS PubMed
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell Jr, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed
- K. Vanommeslaeghe, E. P. Raman and A. D. Mackerell Jr, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed
- A. D. Mackerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. Mackerell Jr and R. W. Pastor, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed
- J. Cheng, H. Wang, Y. Zhang, X. Wang and G. Liu, Org. Biomol. Chem., 2024, 22, 7549–7559 RSC
- J. Kang, C. Wang, D. Li, G. He and H. Tan, Phys. Chem. Chem. Phys., 2015, 17, 16519–16524 RSC
- G. S. Larsen, P. Lin, K. E. Hart and C. M. Colina, Macromolecules, 2011, 44, 6944–6951 CrossRef CAS
- K. A. Veetil, S. Kannan, E. K. Sun, M. H. Kabir, O. Choi, I. Hossain and T.-H. Kim, Sep. Purif. Technol., 2025, 359, 130755 CrossRef
- Y. Oya, M. Nakazawa, K. Shirasu, Y. Hino, K. Inuyama, G. Kikugawa, J. Li, R. Kuwahara, N. Kishimoto, H. Waizumi, M. Nishikawa, A. Waas, N. Odagiri, A. Koyanagi, M. Salviato and T. Okabe, Chem. Phys. Lett., 2021, 762, 138104 CrossRef CAS
- M. S. Ragab, M. R. Shehata, M. M. Shoukry, M. Haukka and M. A. Ragheb, RSC Adv., 2022, 12, 1871–1884 RSC
- J. Smith, R. Wang and T. Liu, Polym. Chem., 2023, 14, 1456–1473 RSC
- X. Li, Y. Zhao and L. Chen, J. Polym. Res., 2023, 30, 45001 Search PubMed
- J. A. Reglero Ruiz, M. Trigo-López, F. C. García and J. M. García, Polymers, 2017, 9, 414 CrossRef PubMed
- D. Rana and T. Matsuura, Environ. Sci.:Water Res. Technol., 2015, 1, 431–459 Search PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS
- M. Pinheiro, R. L. Martin, C. H. Rycroft, A. Jones, E. Iglesia and M. Haranczyk, J. Mol. Graphics Modell., 2013, 44, 208–219 CrossRef CAS PubMed
- D. Ongari, P. G. Boyd, S. Barthel, M. Witman, M. Haranczyk and B. Smit, Langmuir, 2017, 33(51), 14529–14538 CrossRef CAS PubMed
- C. D. Roth, S. C. Poh and D. X. Vuong, Nanotechnology Applications for Clean Water, 2nd edn, 2014, pp. 201–207 Search PubMed
- Y. Chen, C. Shen and F. Fang, Desalination, 2020, 494, 114640 Search PubMed
- Y. H. Teow, J. Y. Sum, K. C. Ho and A. W. Mohammad, Osmosis Eng., 2021, 53–95 Search PubMed
- D. J. Johnson and N. Hilal, Desalination, 2022, 524, 115480 CrossRef CAS
- T. Wei, L. Zhang, H. Zhao, H. Ma, M. S. J. Sajib, H. Jiang and S. Murad, J. Phys. Chem. B, 2016, 120, 10311–10318 CrossRef CAS PubMed
- J. He, J. Yang, J. R. McCutcheon and Y. Li, J. Membr. Sci., 2022, 658, 120731 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |