
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA silica-supported palladium oxide catalyst (PdO@MCM-41) selectively cleaves ether linkages in lignin model compounds and alkali lignin via intramolecular hydrogen transfer†
Noor Ul Hudaa,
Anwar Ul-Hamid b and
Muhammad Zaheer*a
b and
Muhammad Zaheer*a
aDepartment of Chemistry and Chemical Engineering, Syed Babar Ali School of Science and Engineering, Lahore University of Management Sciences (LUMS), Lahore 54792, Pakistan. E-mail: muhammad.zaheer@lums.edu.pk
bCore Research Facilities, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
First published on 24th February 2025
Abstract
Lignin is a potential renewable feedstock for the production of aromatic chemicals but due to the recalcitrant nature of its aryl ether bonds (C–O), and recondensation of depolymerized products, it is challenging to produce aromatic compounds with selectivity in high yield. Here we present that a heterogeneous catalyst containing highly dispersed palladium oxide (PdO) particles supported on mesoporous silica (MCM-41) catalyzes oxidant-free oxidation (dehydrogenation) of hydroxyl group at α-carbon of β-O-4 linkage in lignin model compounds and alkali lignin. The catalyst was synthesized via a molecular approach utilizing molecular designed dispersion of palladium diketonate complex followed by calcination. The oxidized lignin models provide high individual yields of monomeric products such as phenol (97%) at moderate temperature (120 °C) through intramolecular hydrogen transfer in green solvents (ethanol and water). The process, therefore, doesn't require any external oxidant, or reductant for cleaving the most abundant β-O-4 linkage of lignin model compounds and tolerates electron donating or withdrawing substitutes at the benzene ring. The approach was successfully extended to alkali lignin where 89% of lignin oil was produced from alkali lignin containing high yield (26 wt%) of monomeric products such as vanillin (2 wt%), benzaldehyde (12 wt%) and benzoic acid (12 wt%).
1 Introduction
The development of efficient strategies for the transformation of lignocellulosic biomass into valuable chemicals and biofuels is currently of enormous interest.1–4 Particularly, lignin is the largest renewable feedstock and aromatic biopolymer contributing to nearly 30% of the total non-fossil organic carbon. So, it is a potential renewable feedstock compared to continuously depleting fossil fuels for producing useful aromatic chemicals.5,6 Lignin, so far, has been underutilized as a low-quality fuel for the production of heat or considered a waste side product in pulp production.7 The selective depolymerization of lignin into well-defined aromatics could extract a higher value of lignin for the profitable biorefinery system.8,9Although there are numerous routes for the conversion of the carbohydrate portion of lignocellulose,10 <5% lignin is consumed to yield commercial aromatic products, due to its intricate polymeric structure, well-known chemical inactivity, and also condensation reactions during depolymerization.11 The multifaceted structure of lignin can be regarded as an amorphous polymer of phenylpropanoid units interlinked by ether (α-O-4, β-O-4, 4-O-5) and C–C (β–β, β–5, 5–5) bonds, among which β-O-4 glycerolaryl ether linkages represents the predominant connection and comprises around 60% of the innate lignin linkages.12,13 C–O bonds have lesser dissociation energy (209–348 kJ mol−1) than C–C bonds (226–494 kJ mol−1), hence represent the principle and facile target for depolymerization of lignin. The development of efficient degradation of the most abundant and relatively weaker β-O-4 linkages is a key to lignin valorization, as it could lead toward low molecular weight feedstock for bulk and fine chemical industries.14–17
To date, various approaches in both homogeneous and heterogeneous catalysis have been reported in the literature to cleave C–O bond in β-O-4 linkages in lignin. Among different approaches, oxidative18,19 and reductive20–22 catalytic systems have been extensively studied by several research groups, but a number of these approaches tend to need high temperatures and suffer from undesired hydrogenation of aromatic rings. In this regard, a strategy that can activate the dominant β-O-4 linkages, under milder and redox-neutral conditions, involving intramolecular hydrogen transfer has been considered an efficient approach to lignin depolymerization.23–26
The typical structure of β-O-4 linkages usually contains hydroxyl groups at Cα position which can be dehydrogenated to offer hydrogen for the cleavage of C–O bond in lignin, thus providing a suitable platform for a redox-neutral approach. Numerous homogeneous and heterogeneous catalytic systems27–33 operating under mild or redox neutral conditions have been reported for the redox neutral C–O bond cleavage of lignin and regardless of this progress, a heterogeneous catalyst for complete redox neutral cleavage of C–O bonds in β-O-4 linkages in lignin model compounds, commercial lignin (Kraft, alkali etc.) and especially in native lignin is still required. Our focus is on addressing the limitations of existing approaches, which often rely on stoichiometric amounts of various oxidants or reductants, thereby increasing reagent costs. Additionally, these methods typically require high temperatures for redox-neutral cleavage in lignin model compounds and lignin.8,34
Mesoporous silica with its high surface area, well-defined pore structures and acidity are potential candidates to decrease the repolymerization of lignin. These materials provide an optimal environment for converting long chain lignin polymers to monomeric phenolic compounds.35,36 In this study, we demonstrate that PdO nanoparticles supported on MCM-41 (PdO@MCM-41) can effectively cleave C–O bonds in lignin model compounds, and particularly in alkali lignin, to produce aromatic monomers under complete redox-neutral conditions at moderate temperature of 120 °C. Notably, our catalytic system efficiently converts alkali lignin, yielding over 89% bio-oil, with aromatic monomers constituting 26 wt%, including significant yields of benzaldehyde (12 wt%) and benzoic acid (12 wt%).
2 Experimental
Synthesis and characterization of catalyst is explained in our recent publication.372.1 Synthesis of β-O-4 lignin model compounds
Lignin model substrates were synthesized according to a modified approach reported in literature.38 In general, phenol (2a–e, 12 mmol) and K2CO3 (15 mmol) in acetone (50 mL) were added into 100 mL round bottom flask equipped with reflux condenser and magnetic stirrer. 2-Bromoacetophenone (1, 10 mmol, 1.99 g) was then added dropwise in the solution and suspension was refluxed with continuous stirring for 6 h. After complete consumption of 1 (TLC check), the reaction was filtered to remove K2CO3 and the filtrate was concentrated in vacuum. The resulting solid was recrystallized in diethyl ether to obtain 2-phenoxy-1-phenyl ethenones (3a–d) in excellent (∼90%) yields.

The synthesized phenoxyphenylethanone (3a–d, 10 mmol) was then dissolved in mixture of water/THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) and 20 mmol NaBH4. The reaction mixture was stirred at room temperature for 6 h. After that, aqueous NH4Cl (30 mL) was added in the reaction mixture to quench excess NaBH4. The mixture was filtered and the product was extracted with ethyl acetate (3 × 30 mL). Ethyl acetate layer was collected, washed with water and dried over MgSO4. Finally, the extract was concentrated in vacuum to obtain off-white solid product.
:5) and 20 mmol NaBH4. The reaction mixture was stirred at room temperature for 6 h. After that, aqueous NH4Cl (30 mL) was added in the reaction mixture to quench excess NaBH4. The mixture was filtered and the product was extracted with ethyl acetate (3 × 30 mL). Ethyl acetate layer was collected, washed with water and dried over MgSO4. Finally, the extract was concentrated in vacuum to obtain off-white solid product.
2.2 Catalytic studies
In general procedure for redox neutral C–O bond cleavage in lignin model compound; 2-phenoxy-1-phenyl ethanol (PP-ol), a reaction tube (Schlenk Flask) charged with a magnetic stirrer bar and catalyst (20 mg) was used. The reaction tube was evacuated and refilled with nitrogen gas (three times). Lignin model substrate (0.1 mmol, 21 mg) and ethanol/water (1:1, 3 mL) were added in the reaction tube under a continuous flow of nitrogen. The reaction mixture was refluxed at 120 °C in an oil bath for 6 h. After cooling the reaction mixture to room temperature, it was centrifuged to separate the catalyst. The products were extracted with ethyl acetate (30 mL). The extract was concentrated under reduced pressure and the products were qualified by GC-MS and quantified by GC-FID using mesitylene as internal standard (DB-wax column 30 m × 0.25 mm × 0.25 μm) and helium as a carrier gas.
For redox neutral C–O bond cleavage in alkali lignin, a reaction tube (Schlenk Flask) charged with a magnetic stirrer bar and catalyst (20 mg) was used. The reaction tube was evacuated and refilled with nitrogen gas (three times). Alkali lignin (50 mg) and ethanol/water (1:1, 3 mL) were added in the reaction tube under a continuous flow of nitrogen. The reaction mixture was refluxed at 100 °C in an oil bath for 12 h. After cooling, the reaction mixture was acidified with HCl to pH 1. This results in precipitation of unconverted lignin, which was then separated by filtration, dried and weighed. The unconverted lignin was retrieved from filter cake by washing with THF. The filtrate obtained was extracted with ethyl acetate (30 × 3 mL) to achieve formed monomeric products. After extraction, ethyl acetate was recovered by rotary evaporation, which results in brown oily liquid termed as ethyl acetate soluble bio-oil. The whole process of the product separation is shown in Scheme S1.† The product identification of bio-oil was then analyzed with GC-MS, GC-FID and FTIR.
3 Results and discussion
The catalyst (PdO@MCM-41) was synthesized following the procedure recently published by us.39 Briefly, MCM-41 material was first synthesized via a modified Stöber method, impregnated with a palladium diketone complex [Pd(acac)2] and calcined at 550 °C. As reported earlier, hydrogen bonding interaction between the complex and the support as well as the careful choice of the solvent ensures uniform distribution of the complex within the pores of the support.39 Compared to theoretical metal loading of 3 wt%, the catalyst retained 1.7 wt% of the metal, as shown by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Table S1†) which confirmed significant interaction between the complex and the support rather than a physical adsorption.EDX elemental mapping of PdO@MCM-41 was carried out to confirm the presence of Pd in the catalyst. We can observe the uniform distribution of Pd on silica, as shown in Fig. S1.† To get a deeper insight about the morphology and composition of the catalysts, transmission electron microscopy (TEM) was used. The micrographs presented in Fig. 1a show silica nanoparticles roughly around 65 nm in size and elliptical morphology. After the incorporation of metal, the morphology of MCM-41 slightly changed as well as their size (roughly to 50 nm). In high resolution-TEM image (inset in Fig. 1b) shows a nanoparticle with lattice fringe with d-spacing of 0.264 nm, which was indexed to (101) plane of PdO.40 The size of PdO nanoparticles was 2–5 nm.
 | ||
| Fig. 1 (a and b) TEM images of MCM-41 and PdO@MCM-41. The inset in (b) show lattice fringes with interplanar distance of 0.26 nm corresponding to 101 plane of tetragonal PdO, (c) XRD diffractrogram of MCM-41 and PdO@MCM-41, and (d) nitrogen physiosorption isotherms of MCM-41 and PdO@MCM-41. | ||
The nature of the metallic species was further confirmed by X-ray powder diffraction (PXRD) analysis. As the MCM-41 support was amorphous, it appeared as a broad band at 23° (2θ) in the diffractogram (Fig. 1c).41 The peak at 2θ value of 33.8° was assigned to 101 reflection of tetragonal PdO (ICDD# 04-002-4417)42 confirming the presence of metal oxide nanoparticles. The presence of PdO species was expected as the catalyst was not reduced after calcination in air. TEM and XRD results correlate well, confirming the successful synthesis of PdO@MCM-41.
Nitrogen physisorption was used to determine the surface area and pore size distribution of the MCM-41 material and the catalyst. The adsorption–desorption isotherms of MCM-41 and PdO@MCM-41 are depicted in Fig. 1d. Both materials showed isotherms close to type-IV with hysteresis typically shown by the mesoporous materials. We observed a reduction in the surface area of the MCM-41 from 255 m2 g−1 to 184 m2 g−1 upon metal loading. The details of nitrogen physisorption measurements are given in Table S2.†
3.1 Catalytic studies on lignin models
2-Phenoxy-1-phenylethanol (PP-ol) was used as a dimeric model compound of lignin to investigate the cleavage of internal β-O-4 units. Firstly, catalytic dehydrogenation (oxidation) of benzylic –OH in the model compound could provide product whose subsequent hydrogenolysis provide phenol and acetophenone. In other words, activation of the Cα–O bond of PP-ol, transfers hydrogen to the Cβ–O bond leading to the formation of phenol and acetophenone.26Firstly, the effect of temperature (80–120 °C) on product yield was investigated (Fig. 2). The yields of phenol (b) and acetophenone (c) gradually increased with rising temperature, reaching the maximum at 120 °C. The reaction could also take place on lower temperatures albeit with lower conversion and yields of the products (Fig. 2). The reaction performed at 120 °C for 6 h provided a complete conversion of PP-ol into phenol (97%) and acetophenone (54%), as shown in (Table 1, entry 1). The individual yields of both products were found by GC-FID (Fig. S2†). Instead of the expected 1:1 ratio, almost 1:2 ratio of acetophenone and phenol was achieved (entry 1). This anomaly can be attributed to further hydrogenation of acetophenone leading to the formation of 1-phenylethanol as side product whose presence was confirmed through GC-FID analysis of the standard compound (Fig. S2†).
 | ||
| Fig. 2 Effect of temperature on fragmentation of lignin model compound. Reaction conditions: 0.1 mmol substrate, 20 mg catalyst (1.6 mol% Pd), 120 °C, 6 h. Yields were determined by Gas Chromatography-Flame Ionization Detector (GC-FID) using mesitylene as an internal standard. | ||
|
||
|---|---|---|
| Entry | Catalyst | Yield (a, b, c)% |
| 1 | PdO@MCM-41 | <1, 97, 54 |
| 2 | No catalyst | >99, <1, <1 |
| 3 | Pd@MCM-41 | <1, 35, 11 |
| 4 | Pd@alumina | >99, <1, <1 |
| 5 | Pd@C | >99, <1, <1 |

To verify the indispensable role of the catalyst, the reaction was also allowed to be carried out under similar conditions without a catalyst, no desired product was obtained (Table 1, entry 2). The catalyst was also reduced under a hydrogen atmosphere to form Pd(0) nanoparticles on MCM-41 to compare its activity with PdO@MCM-41. Pd(0)@MCM-41 was then analyzed for catalytic activity and low yields of phenol (35%) and acetophenone (11%) were obtained, which showed that Pd(0) nanoparticles were found to be less active for the β-O-4 cleavage of PP-ol (Table 1, entry 3). For catalytic comparison, the reaction was also carried out by using commercial Pd@C and Pd@alumina, but PdO@MCM-41 showed the best performance as the commercial catalysts were found inactive for β-O-4 cleavage of PP-ol (Table 1, entry 4, 5).
Various other supported catalysts (Pd/C27, PdNi/FeMIL-100,33 ReO/AC,8 Ni-W/ZrP,34 Ni/ZSM-543) have reported a redox neutral C–O bond cleavage of β-O-4 model compounds and depolymerization of alkali lignin, which endows the heterogeneous catalyst as a research hotspot in the conversion of lignin model compounds and alkali lignin into aromatic products. However, these catalytic systems often rely on the addition of external hydrogen sources and require higher temperatures to achieve satisfactory yields of aromatic products.
PdO can catalyze dehydrogenation and oxidation reactions, which may be beneficial for cleaving specific lignin linkages without requiring external oxidants. This characteristic promotes a more environmentally sustainable process and can enhance selectivity for targeted reaction pathways.
To bring the best solvent mixture to light, the reaction was carried out with solvents having different polarity such as xylene and mixtures of ethanol/H2O, dioxane/H2O, and methanol/H2O (Table 2). Literature indicates that reaction temperatures for water-solvent mixtures are typically 473 K or lower, compared to those for pure solvents. This suggests that utilizing water–solvent mixtures can enhance the depolymerization process and ultimately improve product yields.44,45 Thus, mixtures of different solvents with H2O were used for the reaction. Solvents like methanol/H2O and dioxane/H2O (entry 2, 3) give better yields of products but an equimolar mixture of ethanol/H2O favored the reaction affording higher individual yields of phenol (97%) and acetophenone (54%). No conversion of the reactant was observed in acetonitrile (entry 4), xylene (entry 5), and 1,4-dioxane (entry 6). The reaction in pure H2O and pure ethanol gave lower yields of phenol and acetophenone, suggesting that H2O acts as a co-solvent only in the presence of another solvent and pure ethanol is not a good solvent for this reaction (entry 7, 8). Thus relying on the yields, a mixture of ethanol–H2O was used as a solvent for remaining catalytic studies.
| Entry | Solvent | Products yield (a, b, c) |
|---|---|---|
| a Reaction conditions: catalysts (PdO@MCM-41), substrate (0.1 mmol, 21.4 mg), catalyst amount (20 mg, 1.7 mol% Pd), ethanol/water (1:1), temperature (120 °C), reaction time (6 hours). Results were screened with GC-FID using mesitylene as an internal standard. |
||
| 1 | Ethanol/H2O | <1%, 97%, 54% |
| 2 | Methanol/H2O | <1%, 68%, 13% |
| 3 | Dioxane/H2O | <1%, 64%, 20% |
| 4 | Xylene | No reaction |
| 5 | Dioxane | No reaction |
| 6 | Acetonitrile | No reaction |
| 7 | H2O | <1%, 15%, <1% |
| 8 | Ethanol | <1%, 40%, 10% |
3.2 Exploring the reaction pathway
As no external hydrogen reagent was added, we believe that the hydroxyl group of PP-ol was first dehydrogenated by the catalyst to produce the ketonic intermediate (PP-on). C–O bond hydrogenolysis of the ketone then led to the formation of monomeric products such as phenol and acetophenone (Scheme 1). In order to further confirm the mechanistic steps, control reactions were carried out. As catalytic dehydrogenation of PPol should produce molecular hydrogen, we used a water burette to collect the gas produced during the reaction. Analysis of the collected gas with GC-TCD confirmed the presence of hydrogen (Fig. S3†) and operative dehydrogenative mechanism rather than oxidation of the alcohol (PP-ol). | ||
| Scheme 1 Control experiments used to determine the reaction pathway (a) reaction with PP-ol using ethanol/H2O solvent, (b) reaction with PP-on, (c) reaction with PP-ol using 1,4-dioxane solvent, (d) reaction with (1-methoxy-2-phenoxyethyl)bezene as a substrate. | ||
In our case, ketonic intermediate (PP-on) produced via catalytic dehydrogenation of starting alcohol (PP-ol), further converted to monomeric products (acetophenone and phenol) via C–O bond hydrogenolysis utilizing the hydrogen produced in the first step (Scheme 1a). In order to confirm this, starting the reaction from keto intermediate did not produce expected acetophenone and phenol products (Scheme 1b) as no intramolecular hydrogen was available to promote C–O bond scission. So, C–O bond cleavage might have occurred due to intramolecular hydrogen transfer mechanism.33 In summary, the activation of the Cα–O bond in PP-ol resulted in hydrogen transfer to Cβ–O bond of the keto intermediate leading to its hydrogenolysis to produce phenol and acetophenone.
However, it is equally likely that the hydrogen required for the hydrogenolysis might have been given by ethanol. Ethanol is a protic solvent that is known to donate hydrogen in the reaction conditions we employed. So, to decouple the contribution of hydrogen from ethanol, we performed the reaction using aprotic, non-hydrogen donor solvent 1,4-dioxane (Scheme 1c). Interestingly, 100% conversion was obtained in 1,4-dioxane. Moreover, the reaction using dehydrogenated product as a substrate with ethanol/H2O as solvent did not produce any products as an intramolecular hydrogen source is not present for this transformation (Scheme 1b). Therefore, ethanol alone as a hydrogen source is not sufficient for the C–O bond cleavage to yield products.
Lastly, we also performed the reaction by using methoxy group as a substituent instead of Cα–OH in starting substrate (Scheme 1d), the reaction did not take place indicating that the H atoms on CαO–H and Cα–H are necessary for the cleavage of lignin model compound indicating intramolecular hydrogen transfer mechanism.
Furthermore, the stability and durability of the catalyst were investigated. Upon completion of the reaction, the catalyst was separated via filtration, washed with ethanol and DI water, dried in a vacuum and then used in the next cycle under similar reaction conditions. The catalyst retained its activity up to three successive runs after which it started decreasing (Fig. S4†). After the third cycle onwards, a small amount of 2-phenoxy-1-phenyl ethenone (PP-on) was also produced indicating its incomplete conversion into phenol and acetophenone due to decreased catalytic activity. The drop in the activity was attributed to the loss of the metal during recycling as confirmed through ICP analysis of the reused catalyst (Table S1†).
To further investigate the potential leaching of Pd, a hot filtration experiment was conducted. After the reaction, the solid supported catalyst was separated from the reaction mixture by filtration, while the solution was still hot. The filtrate containing leached Pd was then tested for activity under optimized reaction conditions. The results showed that the filtrate did not exhibit any catalytic activity, indicating that the leached Pd in the solution did not contribute to the reaction (Fig. S5†). This suggests that the observed catalytic performance is primarily due to silica supported Pd catalyst, rather than any Pd that may have leached into the solution.
3.3 Substrate scope
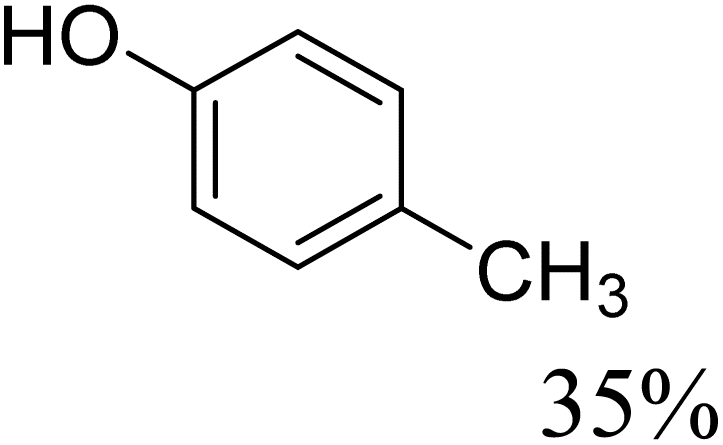
To further assess the catalytic activity of PdO@MCM-41, the catalyst was then tested for the redox neutral cleavage of C–O bond in β-O-4 linkages substituted with methoxy groups at different aryl positions. As presented in Table 3, the dominant products in all the tests were acetophenone and phenol derivates, indicating that selective cleavage of aryl C–O bonds had occurred. In all the reactions, conversion was >99.9% while the yields of the products varied. The substrate with no substitution group showed the best activity with 54% acetophenone and 97% phenol, indicating that the steric hindrance was the key factor for this reaction (entry 1). However, when methoxy groups were present at the R4 and R5 positions on the benzene ring, the yields of the phenolic products decreased. We observed lower yields of substituted phenols with electron-donating groups compared to acetophenone, which can be attributed to differences in reaction kinetics. The electron-donating effect of the methoxy group destabilizes the transition state, making C–O bond cleavage less favorable and consequently inhibiting the efficient formation of substituted phenols. Additionally, the bulkiness of the methoxy groups introduced steric hindrance, further obstructing the reaction site and contributing to the reduced yields. The combined effect of increased electron density and steric hindrance likely caused the observed decrease in product yields in the presence of these groups (entry 2–4).33| Entry | Substrate | Yield | Yield |
|---|---|---|---|
| a Reaction conditions: substrate (0.1 mmol, 21.4 mg), PdO@MCM-41 (20 mg, 1.7 mol% Pd), ethanol/water (1:1), T = 120 °C, t = 6 h. Results are screened with GC-FID, using mesitylene as internal standard. |
|||
| 1 |  |
 |
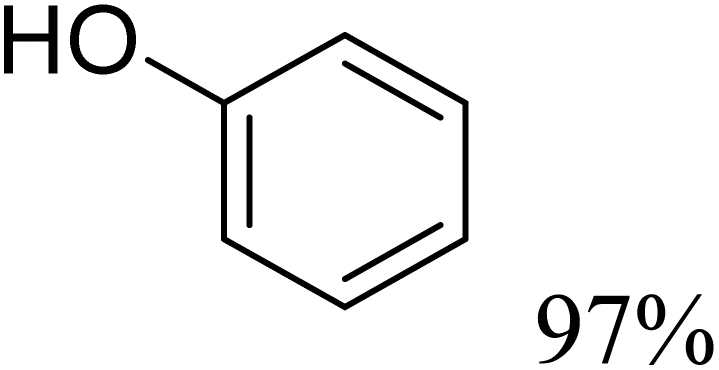 |
| 2 |  |
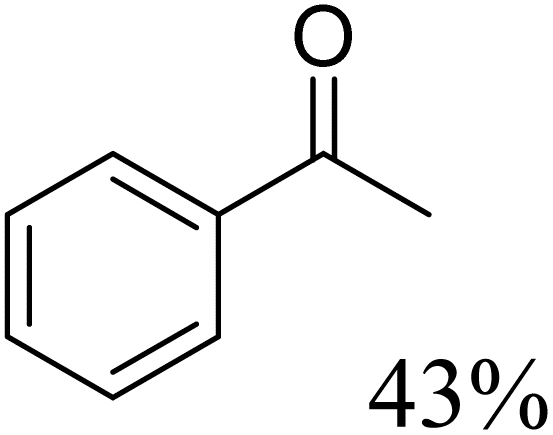 |
 |
| 3 |  |
 |
 |
| 4 | 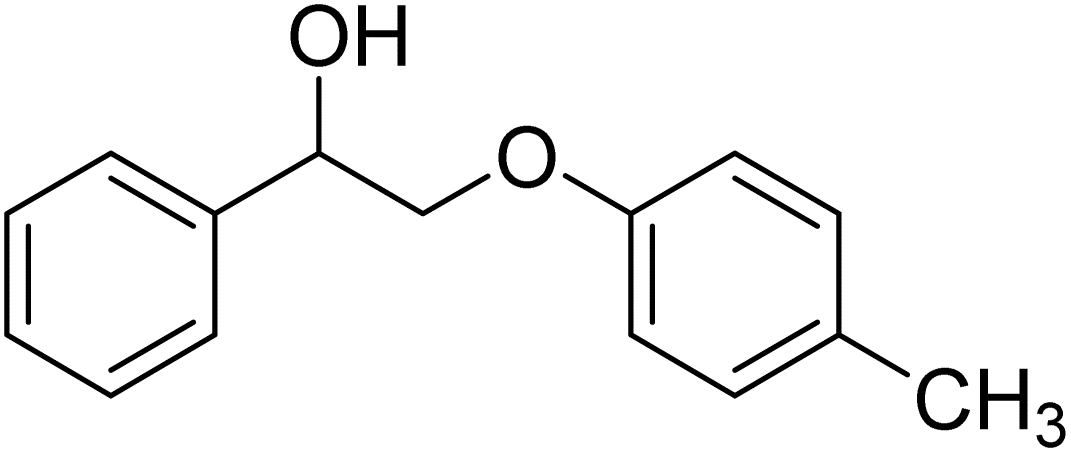 |
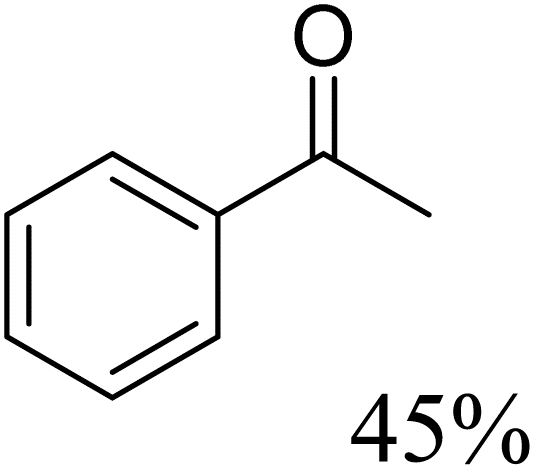 |
 |
3.4 Catalytic studies on alkali lignin
Encouraged by the results obtained from lignin model compounds, the methodology was extended to the conversion of alkali lignin to aromatic monomers (Fig. 3). On the treatment of alkali lignin over the designed catalyst in ethanol/water at 120 °C for 6 h, ethyl acetate soluble bio-oil was obtained in high yield (89 wt%). GC-MS analysis of the oil showed the presence of monomeric compounds including benzaldehyde, vanillin benzoic acid and ethyl esters. The esters detected might be produced by the esterification of ethanol with lignin-derived intermediates.46 Some unidentified peaks and peaks with <1% yield were not considered. | ||
| Fig. 3 Depolymerization of alkali lignin under redox-neutral reaction conditions. | ||
Next, we tried to quantify monomeric products in bio oil with GC-FID analysis using available standards (Fig. S6†). Aromatic compounds such as vanillin, benzaldehyde and benzoic acid were obtained in 2 wt%, 12 wt% and 12 wt% yield respectively corresponding to a total 26 wt% yield for the monomeric products.
Fourier transform infrared spectroscopy (FTIR) characterization was performed, which depicts the variation in the distribution of functional groups in alkali lignin and its depolymerization products (Fig. 4). In FTIR spectrum of alkali lignin, the presence of hydroxyl groups was confirmed by the presence of a peak at 3430 cm−1 corresponding to O–H stretching vibrations. The peaks at 2969 cm−1 and 2768 cm−1 were assigned to stretching vibrations of methyl (–CH3) and methylene (–CH2–) group. The two peaks at 1597 cm−1 and 1434 cm−1 were attributed to the aromatic rings47 while the one at 1730 cm−1 represented the stretching vibration of carbonyl group (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–).48
O–).48
 | ||
| Fig. 4 FTIR analysis of alkali lignin and bio-oil. | ||
The catalytic reaction enhanced the carbonyl peak significantly in bio-oil, which as in accordance with the dehydrogenation of hydroxyl groups to carbonyl groups. The bands corresponding to the vibrations of aromatic rings represented at 1597 cm−1 and 1434 cm−1 became stronger in bio-oil as compared to alkali lignin, which indicated the enrichment of aromatic compounds in bio-oil.
HSQC 2D NMR is one of the most widely used approaches for the elucidation of lignin structure49 and was used to find representative interunit linkages in the structure of alkali lignin and bio oil obtained from it (Fig. 5). The prominent interunit linkages (β-O-4, β-5) were observed between δ 2.7–4.5 ppm and δ 43–60 ppm in 1H and 13C regions as shown in Fig. 5a. Careful analysis of 2D NMR spectra provided us with information about structural changes before and after the reaction. In the side chain region, the primary linkages in the starting lignin structure were the cross signals of β-O-4 ether linkages (A). Signals at δ C/δ H 57/4.8 ppm (Aα), δ C/δ H 67/4.13 ppm (Aβ) and δ C/δ H 403.40 ppm (Aγ) corresponded to Cα–Hα, Cβ–Hβ and Cγ–Hγ correlations of the β-O-4′ ether substructures50 (Fig. 5a). In addition to β-O-4 rich linkages phenylcoumaran (C) was also detected in lignin structure with Cγ–Hγ correlations at δ C/δ H 56–57/3.35–3.56 ppm along with Cβ-5 linkages with Cβ–Hβ correlations at δ C/δ H 41–42/2.6–3.2 ppm.51 Since β-5 depicted a higher bond dissociation energy (110 kcal mol−1) than that of β-O-4 ether linkage (69.5 kcal mol−1), thus few β-5 signals were still present in treated lignin spectra (Fig. 5b). The signals of Aα, Aβ and Aγ corresponding to A linkage of β-O-4 moiety significantly narrowed in post-reacted lignin, clearly indicating the cleavage of major β-O-4 linkage.
 | ||
| Fig. 5 (a) Representative interunit linkages in alkali lignin determined by 2D HSQC NMR (b) HSQC 2D NMR spectra of alkali lignin and (c) HSQC 2D NMR spectra of depolymerized alkali lignin. | ||
Signals of different monomers could be clearly seen (δ C/δ H 127–134/7.4–7.9) in the aromatic region of the bio-oil (Fig. 5b). Moreover, the signals of CO arise, indicating the occurrence of carbonyl or carboxyl groups. The solid 13C NMR was then employed to confirm the presence of carboxyl groups for acids and esters. Fig. S7† confirmed the presence of carboxyl group at a chemical shift of 170 ppm in 13C NMR spectrum of bio-oil.52 These results confirm the cleavage of lignin interunit linkages during redox neutral depolymerization using our catalytic system.
The reusability of PdO@MCM-41 for the depolymerization of alkali lignin was also tested. The results in Fig. 6b show an obvious decrease in catalytic activity as bio-oil yield was gradually reduced after each run. With the increasing number of cycles, the catalytic activity decreased perhaps due to the adsorption of products at the active sites. After three cycles, the catalyst was characterized by SEM and ICP analysis. SEM image in Fig. 6a showed slight agglomeration of the catalyst particles which might decrease the catalytic activity. Moreover, the ICP analysis (Table S1†) showed a subtle decrease in Pd content of the used (recycled) catalyst which also contributed to the activity loss.
 | ||
| Fig. 6 (a) SEM image of the used catalyst after depolymerization of alkali lignin and (b) recyclability tests of PdO@MCM-41 catalyst. | ||
4 Conclusions
In conclusion, this study highlights the efficacy of palladium oxide (PdO) supported on MCM-41 as a catalyst for oxidant free oxidation (dehydrogenation) of hydroxyl groups at the α-carbon of β-O-4 linkages in lignin model compounds and dehydrogenation of alkali lignin into aromatic products. Phenol and acetophenone were produced with individual yields of 97% and 54% respectively for the conversion of 2-phenoxy-1-phenyl ethanol at 120 °C under an inert atmosphere for 6 h. The current catalyst also demonstrated good activity in the direct conversion of alkali lignin in ethanol/H2O under inert atmosphere, resulting in the production of benzaldehyde and benzoic acid in good yields. Our mechanistic studies suggested that PdO nanoparticles played a crucial role in dehydrogenation of C–αOH group in PP-ol to CO which led to the weakening of β-O-4 bond and intramolecular-hydrogen produced monomeric products. The advantages of operating under completely redox-neutral conditions, employing minimal catalyst loading, and obviating the need for external hydrogen or oxygen sources underscore the potential of this method for economically viable production of valuable platform chemicals from lignin.
Data availability
The data that support the findings of this study are available in the ESI† of this article. Additional data is available on request.Author contributions
N. H. contributed to the investigation, formal analysis and writing original draft. A. H contributed in formal analysis and methodology. M. Z. was involved in conceptulization, project supervision, funding and writing–reviewing the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. Z. is thankful to the Higher Education Commission (HEC) of Pakistan for the financial support under National Research Program for Universities (NRPU) grant number 20-15804.References
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. Im Kim and H. Choudhari, Green Chem., 2015, 17, 1492–1499 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak and C. L. Liotta, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- B. Zhang, Z. Qi, X. Li, J. Ji, W. Luo, C. Li, A. Wang and T. Zhang, ACS Sustain. Chem. Eng., 2018, 7, 208–215 CrossRef.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustain. Chem. Eng., 2019, 7, 12105–12116 CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, science, 2014, 344, 1246843 CrossRef PubMed.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- I. Klein, C. Marcum, H. Kenttämaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- C. Ruß and B. König, Green Chem., 2012, 14, 2969–2982 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- C. Scimmi, L. Sancineto, J. Drabowicz and C. Santi, Int. J. Mol. Sci., 2022, 23, 4378 CrossRef CAS PubMed.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- Y. Jing, L. Dong, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2020, 13, 4181–4198 CrossRef CAS PubMed.
- M. Zaheer, J. Hermannsdörfer, W. P. Kretschmer, G. Motz and R. Kempe, ChemCatChem, 2014, 6, 91–95 CrossRef CAS.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., 2015, 127, 260–264 CrossRef.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- R. Zhu, B. Wang, M. Cui, J. Deng, X. Li, Y. Ma and Y. Fu, Green Chem., 2016, 18, 2029–2036 RSC.
- M. Ahsan Usman, M. Naeem, M. Saeed and M. Zaheer, Inorg. Chim. Acta, 2021, 521, 120305 CrossRef CAS.
- M. V. Galkin, C. Dahlstrand and J. S. Samec, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS PubMed.
- M. V. Galkin and J. S. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef CAS PubMed.
- M. V. Galkin, A. T. Smit, E. Subbotina, K. A. Artemenko, J. Bergquist, W. J. Huijgen and J. S. Samec, ChemSusChem, 2016, 9, 3280–3287 CrossRef CAS PubMed.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef CAS PubMed.
- N. Luo, M. Wang, H. Li, J. Zhang, T. Hou, H. Chen, X. Zhang, J. Lu and F. Wang, ACS Catal., 2017, 7, 4571–4580 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- J.-w. Zhang, G.-p. Lu and C. Cai, Green Chem., 2017, 19, 4538–4543 RSC.
- D. Wang, X. Lu, H. Guo, P. Xiu, J. Chen, Y. Qin, H. M. Robin, C. Xu and X. Gu, Biomass Convers. Biorefin., 2023, 13, 5943–5955 CrossRef CAS.
- T. Klamrassamee, N. Laosiripojana, D. Cronin, L. Moghaddam, Z. Zhang and W. O. Doherty, Bioresour. Technol., 2015, 180, 222–229 CrossRef CAS PubMed.
- J.-Y. Kim, S. Y. Park, I.-G. Choi and J. W. Choi, Chem. Eng. J., 2018, 336, 640–648 CrossRef CAS.
- N. Ul Huda, A. Ul-Hamid, M. A. Khan, S. Shahida and M. Zaheer, ChemPlusChem, 2023, 88, e202300338 CrossRef CAS PubMed.
- Y. Liu, C. Li, W. Miao, W. Tang, D. Xue, C. Li, B. Zhang, J. Xiao, A. Wang and T. Zhang, ACS Catal., 2019, 9, 4441–4447 CrossRef CAS.
- N. Ul Huda, A. Ul-Hamid, M. A. Khan, S. Shahida and M. Zaheer, ChemPlusChem, 2023, 88, e202300338 CrossRef CAS PubMed.
- J. Lin, T. Mei, M. Lv, Z. Zhao and X. Wang, RSC Adv., 2014, 4, 29563–29570 RSC.
- R. K. Biswas, P. Khan, S. Mukherjee, A. K. Mukhopadhyay, J. Ghosh and K. Muraleedharan, J. Non-Cryst. Solids, 2018, 488, 1–9 CrossRef CAS.
- M.-Y. Kim, E. A. Kyriakidou, J.-S. Choi, T. J. Toops, A. J. Binder, C. Thomas, J. E. Parks II, V. Schwartz, J. Chen and D. K. Hensley, Appl. Catal., B, 2016, 187, 181–194 CrossRef CAS.
- M. Zhou, B. K. Sharma, P. Liu, J. Ye, J. Xu and J.-C. Jiang, ACS Sustain. Chem. Eng., 2018, 6, 6867–6875 CrossRef CAS.
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S.-F. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustain. Chem. Eng., 2016, 4, 6894–6904 CrossRef CAS.
- J. H. Jang, A. R. C. Morais, M. Browning, D. G. Brandner, J. K. Kenny, L. M. Stanley, R. M. Happs, A. S. Kovvali, J. I. Cutler and Y. Román-Leshkov, Green Chem., 2023, 25, 3660–3670 RSC.
- Z. Tang, Y. Zhang and Q. Guo, Ind. Eng. Chem. Res., 2010, 49, 2040–2046 CrossRef CAS.
- C. Fushimi, S. Katayama, K. Tasaka, M. Suzuki and A. Tsutsumi, AIChE J., 2009, 55, 529–537 CrossRef CAS.
- J. Long, B. Guo, J. Teng, Y. Yu, L. Wang and X. Li, Bioresour. Technol., 2011, 102, 10114–10123 CrossRef CAS PubMed.
- S. Constant, H. L. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. Van Es, R. J. Grisel, B. M. Weckhuysen, W. J. Huijgen and R. J. Gosselink, Green Chem., 2016, 18, 2651–2665 RSC.
- J. Dai, G. N. Styles, A. F. Patti and K. Saito, ACS Omega, 2018, 3, 10433–10441 CrossRef CAS PubMed.
- L. Shao, Q. Zhang, T. You, X. Zhang and F. Xu, Bioresour. Technol., 2018, 264, 238–243 CrossRef CAS PubMed.
- J. Mao, K. M. Holtman, J. T. Scott, J. F. Kadla and K. Schmidt-Rohr, J. Agric. Food Chem., 2006, 54, 9677–9686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08934k |
| This journal is © The Royal Society of Chemistry 2025 |