
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyne dichotomy and hydrogen migration in binuclear cyclopentadienylmetal alkyne complexes†
Huidong Li*ab,
Jinfeng Luoa,
Haoyu Chena,
Ruilin Lua,
Yucheng Hua,
Huijie Wanga,
Yanshu Wanga,
Qunchao Fan*a,
R. Bruce King *b and
Henry F. Schaefer IIIb
*b and
Henry F. Schaefer IIIb
aSchool of Science, Key Laboratory of High Performance Scientific Computation, Xihua University, Chengdu 610039, China
bCenter for Computational Quantum Chemistry, University of Georgia, Athens, Georgia 30602, USA. E-mail: rbking@chem.uga.edu; huidongli@mail.xhu.edu.cn
First published on 24th February 2025
Abstract
The structures and energetics of the binuclear cyclopentadienylmetal alkyne systems Cp2M2C2R2 (M = Ni, Co, Fe; R = Me and NMe2) have been investigated using density functional theory. For the Cp2M2C2(NMe2)2 (M = Ni, Co, Fe) systems the relative energies of isomeric tetrahedrane Cp2M2(alkyne) structures having intact alkyne ligands and alkyne dichotomy structures Cp2M2(CNMe2)2 in which the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond of the alkyne has broken completely to give separate Me2NC units depending on the central metal atoms. For the nickel system Cp2Ni2C2(NMe2)2 as well as the related nickel systems Cp2Ni2(MeC2NMe2) and Cp2Ni2C2Me2 the tetrahedrane structures are clearly preferred energetically consistent with the experimental syntheses of several stable Cp2Ni2(alkyne) complexes. The tetrahedrane and alkyne dichotomy structures have similar energies for the Cp2Co2C2(NMe2)2 system whereas the alkyne dichotomy structures are significantly energetically preferred for the Cp2Fe2C2(NMe2)2 system. The potential energy surfaces for the Cp2M2(MeC2NMe2) and Cp2M2C2Me2 systems (M = Co, Fe) are complicated by low-energy structures in which hydrogen migration occurs from the alkyne methyl groups to one or both alkyne carbon atoms to give Cp2M2(C3H3NMe2) and Cp2M2(C3H3Me) derivatives with bridging metalallylic ligands, Cp2M2(CH2
C triple bond of the alkyne has broken completely to give separate Me2NC units depending on the central metal atoms. For the nickel system Cp2Ni2C2(NMe2)2 as well as the related nickel systems Cp2Ni2(MeC2NMe2) and Cp2Ni2C2Me2 the tetrahedrane structures are clearly preferred energetically consistent with the experimental syntheses of several stable Cp2Ni2(alkyne) complexes. The tetrahedrane and alkyne dichotomy structures have similar energies for the Cp2Co2C2(NMe2)2 system whereas the alkyne dichotomy structures are significantly energetically preferred for the Cp2Fe2C2(NMe2)2 system. The potential energy surfaces for the Cp2M2(MeC2NMe2) and Cp2M2C2Me2 systems (M = Co, Fe) are complicated by low-energy structures in which hydrogen migration occurs from the alkyne methyl groups to one or both alkyne carbon atoms to give Cp2M2(C3H3NMe2) and Cp2M2(C3H3Me) derivatives with bridging metalallylic ligands, Cp2M2(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCHNMe2) and Cp2M2(CH2CCHMe) with bridging allene ligands, as well as Cp2M2(CH2CH–CNMe2) and Cp2M2(CH2CH–CHMe) with bridging vinylcarbene ligands. For the Cp2M2C2Me2 (M = Co, Fe) systems migration of a hydrogen atom from each methyl group to an alkyne carbon atom can give relatively low-energy Cp2M2(CH2CH–CHCH2) structures with a bridging butadiene ligand. Five transition states have been identified in a proposed mechanism for the conversion of the Cp2Co2/MeCCNMe2 system to the cobaltallylic complex Cp2Co2(C3H3NMe2) with intermediates having agostic C–H–Co interactions and an activation energy barrier sequence of 13.1, 17.0, 15.2, and 12.0 kcal mol−1.
CCHNMe2) and Cp2M2(CH2CCHMe) with bridging allene ligands, as well as Cp2M2(CH2CH–CNMe2) and Cp2M2(CH2CH–CHMe) with bridging vinylcarbene ligands. For the Cp2M2C2Me2 (M = Co, Fe) systems migration of a hydrogen atom from each methyl group to an alkyne carbon atom can give relatively low-energy Cp2M2(CH2CH–CHCH2) structures with a bridging butadiene ligand. Five transition states have been identified in a proposed mechanism for the conversion of the Cp2Co2/MeCCNMe2 system to the cobaltallylic complex Cp2Co2(C3H3NMe2) with intermediates having agostic C–H–Co interactions and an activation energy barrier sequence of 13.1, 17.0, 15.2, and 12.0 kcal mol−1.
1. Introduction
The chemistry of transition metal complexes with intact alkyne ligands originated with the 1956 discovery of cobalt carbonyl derivatives of the type (alkyne)Co2(CO)6, readily synthesized in good yield from reactions of alkynes with Co2(CO)8 under mild conditions1–3 The structures of such (alkyne)Co2(CO)6 derivatives are characterized by a central Co2C2 tetrahedron having formal single bonds along each of the six edges of the central tetrahedron. In these structures the alkyne ligand donates four electrons to the central singly bonded Co2 unit thereby giving each cobalt atom the favored 18-electron configuration (Fig. 1). The facile synthesis of (alkyne)Co2(CO)6 derivatives has led to extensive applications in synthetic organic chemistry4,5 and catalysis.6–9 | ||
| Fig. 1 Structures of some experimentally known alkyne metal complexes. | ||
Reactions of iron carbonyls with alkynes typically require more vigorous conditions and lead to more complicated product mixtures. Dominant in such reaction mixtures are iron carbonyl complexes of ligands formed by the cyclization of two or three alkyne units including cyclobutadiene, cyclopentadienone, tropone, and ferracyclopentadiene (ferrole) ligands.10 However, such alkyne oligomerizations are inhibited by bulky substituents. Thus among the products obtained from iron carbonyls and di-tert-butylacetylene is t-Bu2C2Fe2(CO)6, shown by X-ray crystallography to have a central Fe2C2 tetrahedron similar to that in the (alkyne)Co2(CO)6 derivatives (Fig. 1).11 The FeFe distance of 2.316 Å in this Fe2C2 tetrahedron is ∼0.13 Å shorter than the Co–Co single bond distance of 2.462 Å in the analogous tBu2C2Co2(CO)6. This suggests the formal FeFe double bond in t-Bu2C2Fe2(CO)6 required to give each iron atom the favored 18-electron configuration.
Alkyne oligomerization to give cyclic ligands in the iron carbonyl system can also be inhibited by dialkylamino substituents. Thus the reaction of Et2NCCNEt2 with iron carbonyls12 was found to give a product of stoichiometry (Et2NC)2Fe2(CO)6 shown by X-ray crystallography to contain two separate Et2NC diethylaminocarbyne units resulting from complete breaking of the CC triple bond in the original alkyne in a process known as dichotomy (Fig. 1).13 Each Et2NC unit contributes three electrons to the central Fe2 unit so an Fe–Fe single bond in (Et2NC)2Fe2(CO)6 is sufficient to give each iron atom the favored 18-electron configuration. A density functional theory study has shown how dialkylamino substituents can favor alkyne dichotomy as compared with alkyl substituents without any donor atoms such as nitrogen.14
The robustness of the metal-cyclopentadienyl linkage makes CpM (Cp = η5-C5H5) units good building blocks for transition metal complexes of diverse types that are likely to be more stable than their metal carbonyl counterparts. In this connection the CpNi unit is isoelectronic and isolobal with the Co(CO)3 unit. Thus it is not surprising that several stable Cp2Ni2(alkyne) complexes have been synthesized by reactions of Cp2Ni2(CO)2 with alkynes.15–17 Similarly, the chemistry of Cp2Co2(alkyne) derivatives might be expected to parallel that of (alkyne)Fe2(CO)6 derivatives. However, Cp2Co2(alkyne) derivatives have not yet been synthesized since reactions of CpCo(CO)2 with alkynes typically lead to alkyne cyclodimerization to give very stable CpCo(cyclobutadiene) derivatives or to alkyne cyclotrimerization to give benzene derivatives.18
Extending the isolobal analogy further suggests analogy between the chemistry of (alkyne)Mn2(CO)6 and Cp2Fe2(alkyne) derivatives. However, the alkyne chemistry of manganese carbonyl, which is of more recent origin than that of cobalt and iron carbonyls, follows a different pattern involving carbonyl richer systems. This reflects the need for each manganese atom to acquire two more ligand electrons than a cobalt atom to attain the favored 18-electron configuration. Thus the experimentally known diallkylaminoacetylene derivatives (Et2NC2NEt2)Mn2(CO)8, (Et2NC2Me)Mn2(CO)8, and (Me2NC2Ph)Mn2(CO)8 are all examples of (alkyne)Mn2(CO)8 derivatives with central Mn2C2 tetrahedrane units analogous to the (alkyne)Co2(CO)6 derivatives but with one additional carbonyl group on each manganese atom.12 This original work on alkyne manganese carbonyl chemistry focused on alkynes with dialkylamino substituents. Later the manganese carbonyl chemistry of alkynes with methyl substituents was found to be complicated by hydrogen migration from the methyl group to one or both alkyne carbon atoms to give species with bridging allene or manganallyl units (Fig. 2).19
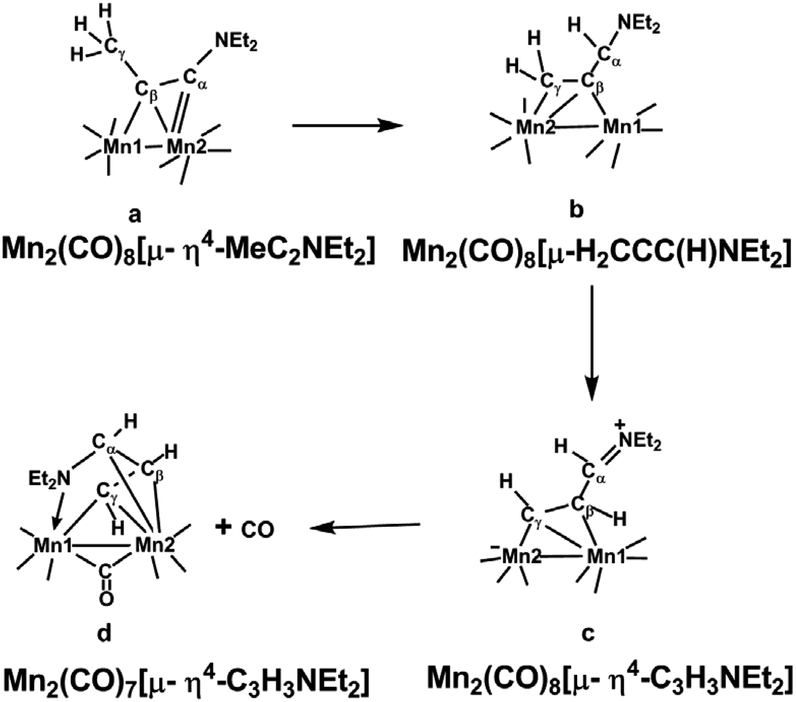 | ||
| Fig. 2 Products formed by hydrogen migration from Mn2(CO)8(MeC2NEt2) structurally characterized by X-ray crystallography.19 | ||
We report here comprehensive density functional theory studies on Cp2M2(alkyne) (M = Ni, Co, Fe) systems with the three alkynes Me2NCCNMe2, MeCCNMe2, and MeCCMe having the objective of guiding future experimental work in this area. In addition, our theoretical studies provide insight into hydrogen migration processes in alkyne metal carbonyl chemistry by suggesting a new mechanism for hydrogen migration in the Cp2Co2(MeC2NMe2) system. The systems having dimethylamino substituents on the alkyne carbon atoms were chosen to avoid the complication of possible hydrogen migration from methyl groups directly bonded to an alkyne carbon as observed experimentally for the manganese carbonyl derivatives.19 The nickel derivatives Cp2Ni2(alkyne) are of interest both by being known experimentally15–17 as well as by being analogues of the extensive series of (alkyne)Co2(CO)6 derivatives.1–3 Analogy between the experimentally still unknown Cp2Co2(alkyne) systems and the (alkyne)Fe2(CO)6 systems suggest the possibility of alkyne dichotomy in low-energy Cp2Co2(alkyne) structures, particularly those with dialkylamino substituents.
2. Theoretical methods
The hybrid meta-GGA DFT methods M06-L20,21 and ωB97xD22 including empirical dispersion correction implemented in the Gaussian16 program,23 were chosen for the calculations in order to consider possible agostic hydrogen and dispersion interactions. The B3PW91-D3 method24 with Grimme's D3 dispersion25 scheme was also used. These methods have been reported to give better overall performance for organometallic compounds than the first-generation functionals.21,26 Double-ζ plus polarization (DZP) basis sets were used in conjunction with the M06-L method for the DFT optimizations. For carbon one set of pure spherical harmonic d functions with orbital exponent αd(C) = 0.75 was added to the standard Huzinaga–Dunning contracted DZ sets. This basis set is designated as (9s5p1d/4s2p1d).27,28 For hydrogen, a set of p polarization functions αp(H) = 0.75 was added to the Huzinaga-Dunning DZ sets. For the first row transition metals the Wachters' primitive sets were used in our loosely contracted DZP basis sets, but augmented by two sets of p functions and one set of d functions, and contracted following Hood et al., and designated as (14s11p6d/10s8p3d).29,30 For testing the basis set effects on the geometry optimizations and electronic structures, the M06-L method with the def2-TZVP basis sets31,32 was used to optimize fully the geometries based on the initial geometries predicted at the M06-L/DZP level. The results predicted at the higher level M06-L/def2-TZVP method are reported in the text. Other results based on the DZP basis sets are given in the ESI.†The geometries of all structures were edited by hand starting from an XRD template and fully optimized by using the above DFT method with the (120, 974) integration grid. Small imaginary frequencies for the optimized structures were assumed to originate from numerical integration errors. Each structure is designated as mN–M–nX, where M is the symbol of the transition metal atom, m is the number of dimethylamino groups, n is the order of the structure in a sequence of increasing relative energies predicted by the M06-L/DZP method, and X designates the spin states, using S and T for singlets and triplets, respectively. The root-mean-square deviation (RMSD) value suggesting the structural difference of some singlet and triplet structural pairs were calculated by the RMSD code.33 Only the energies predicted by the M06-L/DZP method and ωB97xD/DZP method are reported in the text. The distances (in Ångstroms) displayed in each figure are predicted by the M06-L/DZP method.
3. Results and discussion
3.1 Structures
C triple bond donating an electron pair to one of the nickel atoms so that the alkyne ligand is a net four-electron donor to the central Ni2 system. The predicted Ni–Ni distance of 2.303 in 2N–Ni–1S can be interpreted as the Ni–Ni single bond required to give each nickel atom the favored 18-electron configuration. This Ni–Ni distance in 2N–Ni–1S is very close to the experimental Ni–Ni distance of 2.33 Å in the diphenylacetylene complex Cp2Ni2C2Ph2 as determined by X-ray crystallography.34 Structure 2N–Ni–1S is the analogue of the well-known (alkyne)Co2(CO)6 derivatives1–3 in which each Co(CO)3 unit has been replaced with an isoelectronic CpNi unit.
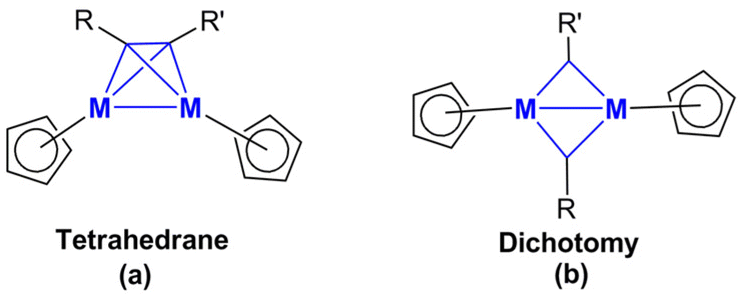 | ||
| Fig. 3 The tetrahedrane and alkyne dichotomy structures for the Cp2M2C2RR′ derivatives. | ||
 | ||
| Fig. 4 The optimized low-energy Cp2Ni2C2(NMe2)2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative emergies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The next higher energy Cp2Ni2C2(NMe2)2 structure 2N–Ni–2T, lying 13.6 kcal mol−1 above 2N–Ni–1S, appears to be a triplet excited state of 2N–Ni–1S (Fig. 4). The increase in the Ni–Ni distance in going from 2.303 Å in the singlet 2N–Ni–1S to 2.353 Å in the corresponding triplet 2N–Ni–2T is surprisingly small. The root-mean-square deviation (RMSD) was predicted to be 0.254 Å between the singlet 2N–Ni–1S and the triplet 2N–Ni–2T.
The Cp2Ni2C2(NMe2)2 structure 2N–Ni–3S, lying 24.2 kcal mol−1 above the lowest energy structure 2N–Ni–1S, has a different coordination environment. In 2N–Ni–3S one nickel atom is bonded to both alkyne carbon atoms with nickel-carbon distances of 1.899 Å and 1.779 Å corresponding to formal Ni–C single and Ni=C double bonds, respectively. The other nickel atom is connected to only one alkyne carbon atom with a Ni–C distance of 1.857 Å corresponding to a formal single bond but also to a nitrogen atom through a dative N→Ni bond of length 1.923 Å. The long Ni⋯Ni distance of 3.491 Å in 2N–Ni–3S indicates the lack of a nickel–nickel bond. However, since each nickel atom receives three electrons from the alkyne ligand, each nickel atom has the favored 18-electron configuration.
The Cp2Ni2C2(NMe2)2 structure 2N–Ni–4S is a different type of dichotomy structure, with two separate three-electron donor Me2NC dimethylaminocarbyne units bridging the nickel atoms with the carbyne carbon atom of such unit forming a Ni–C bond to each nickel atom (Fig. 4). Thus 2N–Ni–4S has similar stereochemistry of the central Ni2C2 unit to that of the central Fe2C2 unit in the experimental (Et2NC)2Fe2(CO)6 dichotomy structure (Fig. 1)13 except for a relatively long Ni⋯Ni distance of 2.911 Å suggesting the absence of a direct nickel–nickel bond. This is consistent with the 18-electron configuration of each nickel atom in a dimethylaminocarbyne structure Cp2Ni2(CNMe2)2 without a direct nickel–nickel bond.
For the cobalt system Cp2Co2C2(NMe2)2 the tetrahedrane and dichotomy structures are spaced very close in energy with the dichotomy structure 2N–Co–1S lying only 2.6 kcal mol−1 and 1.9 kcal mol−1 below the singlet and triplet tetrahedrane structures 2N–Co–3S and 2N–Co–2T, respectively (Fig. 5). In the dichotomy structure 2N–Co–1S each of the separate bridging Me2NC carbyne units donates three electrons to the central Co2 unit. The Co–Co distance in 2N–Co–1S of 2.351 Å suggests the formal single bond required to give each cobalt atom the favored 18-electron configuration. Structure 2N–Co–1S is thus the analogue of the experimentally known and structurally characterized (Et2NC)2Fe2(CO)6 (Fig. 1)11 in which each Fe(CO)3 unit has been replaced by an isoelectronic CpCo unit. The CoCo distance of 2.233 Å in the singlet tetrahedrane structure 2N–Co–3S is ∼0.1 Å shorter than the Co–Co distance in the dichotomy structure 2N–Co–1S as well as the Ni–Ni single bond distance in the tetrahedrane structure 2N–Ni–1S. Interpretation of the CoCo distance in 2N–Co–3S as a formal double bond gives each cobalt atom the favored 18-electron configuration. Thus 2N–Co–3S is thus an analogue of the experimentally known iron tetrahedrane complex11 (tBu2C2)Fe2(CO)6 in which each Fe(CO)3 unit has been replaced by an isoelectronic CpCo unit. The CoCo distance of 2.301 Å in the triplet tetrahedrane structure 2N–Co–2T is essentially identical to that in the singlet analogue 2N–Co–3S and thus can likewise be interpreted as a double bond thereby giving each cobalt the favored 18-electron configuration as in 2N–Co–3S. The root-mean-square deviation (RMSD) was predicted to be 0.235 Å between the singlet 2N–Co–3S and the triplet 2N–Co–2T. The CoCo double bond in the triplet 2N–Co–2T is of the σ + 2/2 π type with two orthogonal one-electron π-components similar to the FeFe double bond in the triplet state organometallic35 Cp2Fe2(μ-CO)3 or normal dioxygen. The two singlet isomers 2N–Co–1s and 2N–Co–3S can interchange mutually by overcoming a very low activation energy barrier of ∼6.0 kcal mol−1 (Fig. 6).
 | ||
| Fig. 5 The optimized low-energy Cp2Co2C2(NMe2)2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
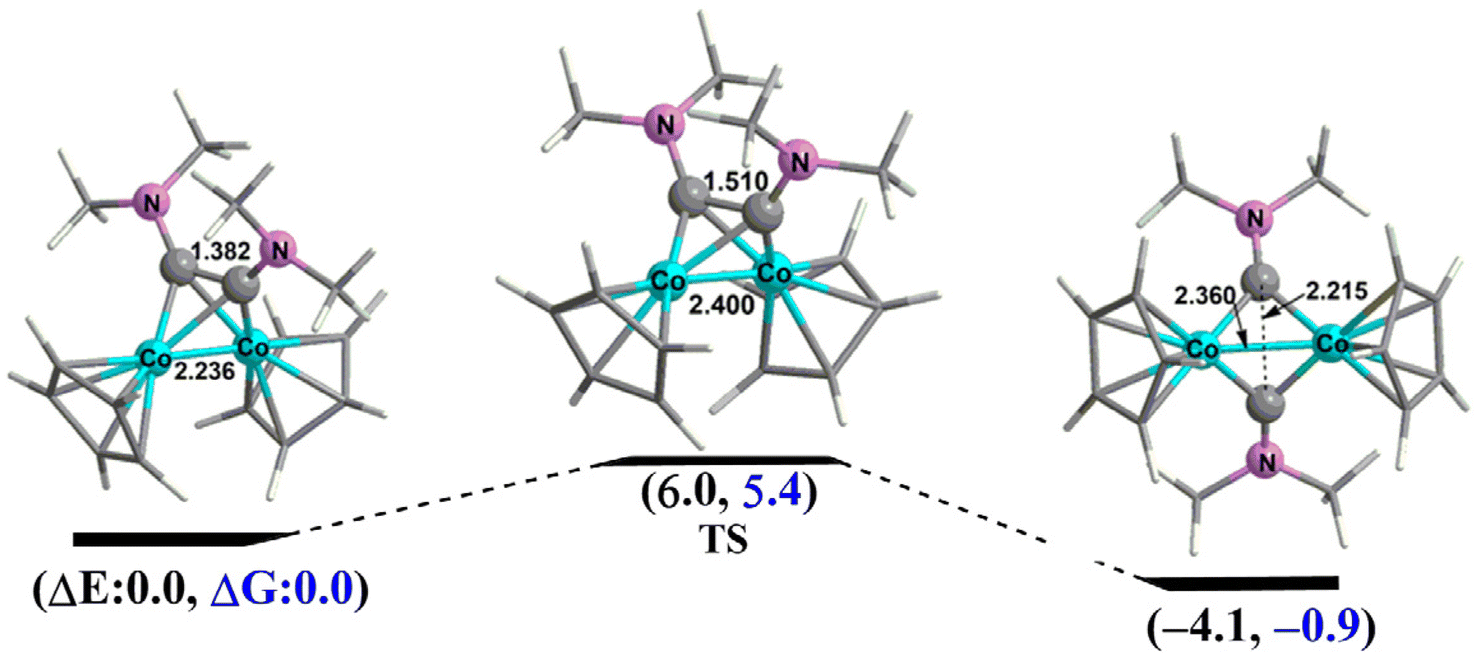 | ||
| Fig. 6 The reaction pathway between the singlet tetrahedrane and dichotomy Cp2Co2C2(NMe2)2 isomers 2N–Co–3S and 2N–Co–1s, respectively, showing the low energy barrier of ∼6.0 kcal mol−1. The numbers in parentheses are the relative energies predicted by M06-L/def2-TZVP method. | ||
Two higher energy Cp2Co2C2(NMe2)2 structures are of interest (Fig. 5). The singlet structure 2N–Co–4S, lying 18.2 kcal mol−1 in energy above 2N–Co–1S, has an intact Me2NCCNMe2 ligand functioning as a six-electron donor by bonding to the central Co2 unit through both π-components of its CC triple bond as well as through an N→Co dative bond of length 1.979 Å from one of the amino nitrogen atoms. The Co–Co distance of 2.631 Å in 2N–Co–4S is 0.3 to 0.4 Å longer than those in three lower energy Cp2Co2C2(NMe2)2 isomers in Fig. 5 and thus can be interpreted as a formal single bond. Such a Co–Co single bond is sufficient to give each cobalt atom in 2N–Co–4S the favored 18-electron configuration with a six-electron rather than four-electron donor bridging alkyne ligand. The other higher energy Cp2Co2C2(NMe2)2 structure 2N–Co–5T, lying 24.6 kcal mol−1 in energy above 2N–Co–1s, is a triplet dichotomy structure related to the singlet dichotomy structure 2N–Co–1s but with a significantly longer Co–Co distance of 2.542 Å. The root-mean-square deviation (RMSD) was predicted to be 0.651 Å between the singlet 2N–Co–1s and the triplet 2N–Co–5T. The triplet spin state of 2N–Co–5T suggests that the two cobalt atoms are squeezed to a normally bonding distance by the geometry of the two bridging dimethylaminocarbyne ligands but are not able to form a bond instead retaining an unpaired electron on each cobalt atom.
The iron system Cp2Fe2C2(NMe2)2 is the reverse of the nickel system since both the singlet and triplet dichotomy structures, 2N–Fe–1S and 2N–Fe–2T, respectively, are the lowest energy structures by substantial margins greater than 11 kcal mol−1 (Fig. 7). The root-mean-square deviation (RMSD) was predicted to be 0.255 Å between 2N–Fe–1S and 2N–Fe–2T. The FeFe distance of 2.335 Å in 2N–Fe–1S and 2.350 Å in 2N–Fe–2T both can be considered as the formal double bonds required to give each iron atom the favored 18-electron configuration. In the singlet 2N–Fe–1S the FeFe double bond is of the normal σ + π type similar to that in ethylene. However, in the triplet 2N–Fe–2T the FeFe double bond is of the σ + 2/2π type similar to that in the organometallic35 Cp2Fe2(μ-CO)3 or dioxygen.
 | ||
| Fig. 7 The optimized low-energy Cp2Fe2C2(NMe2)2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The higher energy Cp2Fe2C2(NMe2)2 structure 2N–Fe–3S, lying 13.3 kcal mol−1 above 2N–Fe–1S, has a tetrahedrane-like geometry but with a relatively long C–C distance of 1.691 Å suggesting a very weak interaction and a relatively short iron–iron distance of 2.268 Å approaching a formal triple bond. These C–C and FeFe distances suggest a resonance hybrid between a tetrahedrane and a dichotomy structure with both canonical forms having the favored 18-electron configuration for each iron atom.
The remaining two high-energy Cp2Fe2C2(NMe2)2 structures, namely 2N–Fe–4T and 2N–Fe–5S lying 15.2 and 17.6 kcal mol−1 above 2N–Fe–1S, respectively, each have a six-electron donor alkyne ligand using both π-components of the CC bond as well as a nitrogen lone pair through a dative N→Fe bond similar to the alkyne ligand in 2N–Co–4S (Fig. 7). Interpreting the 2.560 Å Fe–Fe distance in the triplet structure 2N–Fe–4T as a formal single bond gives each iron atom a 17-electron configuration consistent with a binuclear triplet. The 2.478 Å of FeFe distance in the singlet 2N–Fe–5S is ∼0.1 Å shorter than that in the otherwise similar 2N–Fe–4T and thus can be interpreted as the formal double bond required to give each iron atom the favored 18-electron configuration.
CNMe2 alkyne ligands or Me2NC carbyne ligands formed by complete rupture of the alkyne CC triple bond. The potential surfaces become considerably more complicated when one or both of the dimethylamino substituents on the alkyne are replaced by methyl substituents. Hydrogen migration from such methyl substituents to one or both alkyne carbon atoms can lead to structures with bridging allene, metalallylic, or vinylcarbene ligands (Fig. 8). The first two structure types have been observed experimentally by Adams and coworkers in binuclear manganese carbonyl complexes of the MeCCNEt2 ligand.19
 | ||
| Fig. 8 Structure types formed by hydrogen migration from methyl groups in methylalkynes (a to d); an observed sandwich-type structure (e). | ||
Consider first binuclear cyclopentadienylmetal complexes of the type Cp2M2(MeC2NMe2) (M = Ni, Co, Fe) having one methyl substituent on the alkyne ligand to provide a source of hydrogen atoms for migration to one or both alkyne carbon atoms (Fig. 8). Such hydrogen migration reactions can lead to complexes having bridging dimethylamino four-electron donor vinylcarbene (CH2CH–CNMe2) ligands, four-electron donor metalallylic ligands (C3H3NMe2), and four-electron donor allene ligands (CH2C=CHNMe2). For the nickel system Cp2Ni2(MeC2NMe2) the tetrahedrane structure 1N–Ni–1S having an intact alkyne ligand with no hydrogen migration remains the lowest energy structure with an Ni–Ni distance of 2.293 Å (Fig. 9) that is close to the Ni–Ni distance of 2.303 Å in the Cp2Ni2C2(NMe2)2 structure 2N–Ni–1S discussed above (Fig. 4). However, 1N–Ni–2S with a bridging four-electron donor vinylcarbene ligand formed by a single hydrogen migration and a Ni–Ni single bond distance of 2.382 Å lies only 9.2 kcal mol−1 in energy above 1N–Ni–1S. A triplet version of the vinylcarbene complex, namely 1N–Ni–3T with a longer Ni–Ni distance of 2.536 Å, lies 12.5 kcal mol−1 above 1N–Ni–1S. The root-mean-square deviation (RMSD) was predicted to be 0.348 Å between the singlet 1N–Ni–2S and the triplet 1N–Ni–3T.
 | ||
| Fig. 9 The optimized low-energy Cp2Ni2(MeC2NMe2) structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The singlet structure 1N–Ni–4S, lying 14.0 kcal mol−1 in energy above 1N–Ni–1S, has a six-electron donor bridging nickelallyllic ligand formed by double hydrogen migration bonded to the central Ni2 unit similar to the manganallyllic ligand found in the experimentally known (MeC2NEt2)Mn2(CO)7 complex.19 In 1N–Ni–4S the nickelallyllic ligand is bonded to one CpNi unit as a trihapto ligand forming three Ni–C bonds and to the other CpNi unit by forming one dative N→Ni bond and one Ni–C bond. This configuration of bonds gives each nickel atom the favored 18-electron configuration without the need for a Ni–Ni bond. This is consistent with the relatively long Ni⋯Ni non-bonding distance of 2.888 Å in 1N–Ni–4S suggesting the lack of a direct nickel–nickel bond.
Structures with hydrogen migration in the cobalt system Cp2Co2(MeC2NMe2) are predicted to have significantly lower energies than either the tetrahedrane or dichotomy isomers. Thus the lowest energy Cp2Co2(MeC2NMe2) structure 1N–Co–1S has a six-electron donor bridging cobaltallylic ligand bonded to the central Co2 unit similar to the nickelallylic ligand in 1N–Ni–4S or the manganallylic ligand in the experimentally known19 (MeC2NEt2)Mn2(CO)7 complex (Fig. 10). The Co–Co distance of 2.437 Å in 1N–Co–1S is clearly a bonding distance corresponding to the formal single bond that is required to give each cobalt atom the favored 18-electron configuration. This contrasts with 1N–Ni–4S having the same type of metalallyllic ligand but with the much longer Ni⋯Ni distance of 2.888 Å where no nickel–nickel bond is needed to give each nickel atom the favored 18-electron configuration.
 | ||
| Fig. 10 The optimized low-energy Cp2Co2(MeC2NMe2) structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The next lower energy Cp2Co2(MeC2NMe2) structure is the triplet 1N–Co–2T, lying 11.5 kcal mol−1 above 1N–Co–1S (Fig. 10). Structure 1N–Co–2T has a bridging vinylcarbene ligand similar to that in 1N–Ni–2S with a similar metal–metal bonding distance of 2.357 Å. However, since cobalt has one less electron than nickel, each cobalt atom in 1N–Co–2T has only a 17-electron configuration consistent with its triplet spin state. The next Cp2Co2(MeC2NMe2) structure 1N–Co–3T, lying 12.0 kcal mol−1 in energy above 1N–Co–1S, is a triplet version of 1N–Co–1S. The Co–Co distance of 2.717 Å in 1N–Co–3T is ∼0.3 Å longer than that of 2.437 Å in 1N–Co–1S implying the lack of a significant Co⋯Co interaction similar to the situation in 1N–Ni–4S. The root-mean-square deviation (RMSD) was predicted to be 0.254 Å between the singlet 1N–Co–1S and the triplet 1N–Co–3T.
The lowest energy Cp2Co2(MeC2NMe2) structure with an intact alkyne ligand without hydrogen migration and thus with a central Co2C2 tetrahedron, namely 1N–Co–4T at 12.6 kcal mol−1 above 1N–Co–1S, is also a triplet state structure. The Co–Co distance of 2.284 Å in 1N–Co–4T is essentially identical to the Ni–Ni distance of 2.293 Å in 1N–Ni–1S. However, the WBI value of 0.84 for 1N–Co–4T is larger than of that of 0.72 for 1N–Ni–1S, suggesting a σ + 2/2π type double bond. This gives each cobalt atom in 1N–Co–4T an 18-electron configuration consistent with its triplet spin state.
The lowest energy Cp2Fe2(MeC2NMe2) structure is a triplet structure 1N–Fe–1T with a six-electron donor bridging ferrallylic ligand similar to 1N–Co–3T (Fig. 10) and 1N–Ni–4S (Fig. 9). The WBI value for the Fe–Fe distance of 2.372 Å in 1N–Fe–1T is 0.92, which is much larger than that of 0.62 in 1N–Co–1S, suggesting a σ + 2/2π type double bond. This gives each iron atom in 1N–Fe–1T an 18-electron configuration consistent with its triplet spin state.
Hydrogen migration does not play a role in the next three Cp2Fe2(MeC2NMe2) structures in terms of relative energies (Fig. 11). Structures 1N–Fe–2S and 1N–Fe–4T, lying 3.5 kcal mol−1 and 11.0 kcal mol−1 in energy, respectively, above 1N–Fe–1T, are alkyne dichotomy structures completely analogous to the lowest energy Cp2Fe2C2(NMe2)2 structures 2N–Fe–1S and 2N–Fe–2T with very similar FeFe double bond distances. The root-mean-square deviation (RMSD) was predicted to be 0.104 Å between the singlet 1N–Fe–2S and the triplet 1N–Fe–4T. Structure 1N–Fe–3T, lying 5.2 kcal mol−1 above 1N–Fe–1T in energy, is a triplet tetrahedrane structure. Interpreting the FeFe distance of 2.332 Å as a formal double bond gives each iron atom the 17-electron configuration for a binuclear triplet.
 | ||
| Fig. 11 The optimized low-energy Cp2Fe2(MeC2NMe2) structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
CH–CHCH2) in which the methyl carbon atoms in the MeCCMe ligand become terminal carbon atoms and the alkyne carbon atoms become internal carbon atoms (d in Fig. 8). Such a bridging butadiene ligand consists of a metallacyclopentadiene (metallole) ring forming a tetrahapto bond to the other metal atom. In addition, one of the terminal hydrogens of the butadiene unit has an agostic C–H–M interaction with the ring metal atom. The overall structures of the Cp2M2(CH2CH–CHCH2) bridging butadiene complexes arising from hydrogen migration from each methyl group of dimethylacetylene resemble that of the experimentally known36 cobaltacyclopentadiene complex Cp2Co2(η4,η2-C4H4) with the added feature of the agostic C–H–M interaction. In addition to the bridging butadiene complexes hydrogen migrations from one of the methyl groups in the MeCCMe ligand can lead to the same types of bridging allene, metalallylic, and vinylcarbene ligands found in the Cp2M2(MeC2NMe2) complexes discussed above.The nickel system is the only one of the three Cp2M2C2Me2 (M = Ni, Co, Fe) systems that has a reasonably simple potential energy surface (Fig. 12). The lowest energy Cp2Ni2C2Me2 structure by more than 13 kcal mol−1 (M06-L) is the alkyne complex 0N–Ni–1S with a central Ni2C2 tetrahedrane unit similar to numerous experimentally known Cp2Ni2(alkyne) complexes.15–17 The next Cp2Ni2C2Me2 isomer in terms of relative energy, namely 0N–Ni–2T, can be regarded as the triplet excited state of 0N–Ni–1S with similar geometry. The next three structures in terms of energy, namely the singlet structures 0N–Ni–3S, 0N–Ni–4S, and 0N–Ni–5S lying 15.8, 15.8, and 18.7 kcal mol−1, respectively, above 0N–Ni–1S have bridging vinylcarbene, methylallene, and butadiene units, respectively. The butadiene derivative 0N–Ni–5S does not have an agostic C–H–Ni interaction since a four-electron donor butadiene ligand without any such agostic interactions is sufficient to give each nickel atom the favored 18-electron configuration in a structure with an Ni–Ni single bond. Structure 0N–Ni–6T, lying 17.5 kcal mol−1 above 0N–Ni–1S in energy, is a triplet version of the singlet structure 0N–Ni–3S with fundamentally the same geometry. The Ni–Ni distance of 2.547 Å in the triplet 0N–Ni–6T is significantly longer than the Ni–Ni distance of 2.402 Å in the singlet 0N–Ni–3S.
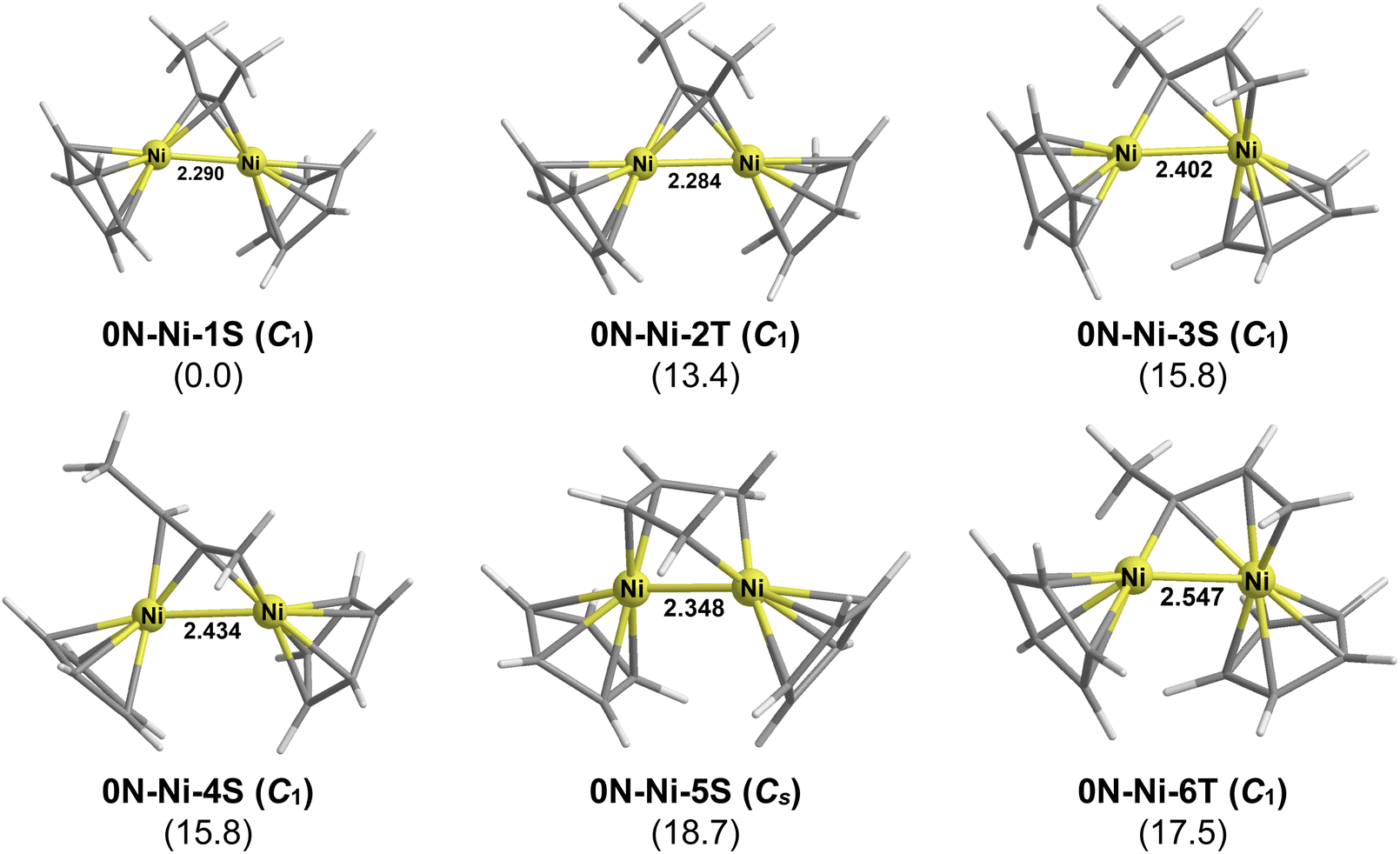 | ||
| Fig. 12 The optimized low-energy Cp2Ni2C2Me2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The potential energy surface becomes much more complicated for the cobalt system Cp2Co2C2Me2 with nine structures lying within 10 kcal mol−1 of the lowest energy structure (Fig. 13). The unchanged dimethylacetylene ligand without any hydrogen migration is found in the triplet structure 0N–Co–2T of relatively high C2v symmetry lying only 1.6 kcal mol−1 below the global minimum as well in the higher energy singlet structure 0N–Co–6S. Structures having central cobaltallylic units in various geometries and spin states are most prevalent being found in the lowest energy Cp2Co2C2Me2 structure 0N–Co–1T as well as the triplet structure 0N–Co–3T and the singlet structures 0N–Co–7S and 0N–Co–9S. The singlet structure 0N–Co–4S and the triplet structure 0N–Co–5T both have bridging butadiene ligands with one agostic C–H–Co interaction. A bridging methylallene ligand is found in structure 0N–Co–8T.
 | ||
| Fig. 13 The optimized low-energy Cp2Co2C2Me2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. Four of the other seven low-energy Cp2Fe2C2Me2 structures, namely 0N–Fe–3T, 0N–Fe–4T, 0N–Fe–7S, and 0N–Fe–8S, have bridging ferrallylic ligands with the three higher energy structures clearly having agostic hydrogen C–H–Fe interactions. A bridging butadiene unit with one agostic C–H–Fe interaction is found in 0N–Fe–6T and a bridging methylallene ligand is found in 0N–Fe–10T. | ||
The potential energy surface for the iron system Cp2Fe2C2Me2 is nearly as complicated as that of the corresponding cobalt system with ten structures lying within 22 kcal mol−1 of the lowest energy structure (Fig. 14). The lowest energy Cp2Fe2C2Me2 structure is the singlet structure 0N–Fe–1S with two bridging three-electron donor methylcarbyne groups arising from dichotomy of the CC triple bond in dimethylacetylene. Structure 0N–Fe–1S appears to be a particularly favorable structure since it lies more than 9 kcal mol−1 in energy below the next lowest energy structure. Interpreting the FeFe distance of 2.318 Å in 0N–Fe–1S as a formal double bond gives each iron atom the favored 18-electron configuration. The corresponding triplet alkyne dichotomy structure 0N–Fe–5T with essentially the same FeFe distance of 2.353 Å lies 12.8 kcal mol−1 above 0N–Fe–1S. In 0N–Fe–5T the triplet spin state arises from an FeFe double bond of the σ + 2/2π type similar to that in dioxygen as well as the triplet spin state organometallic derivative (η5-Me5C5)2Fe2(μ-CO)3, that has been structurally characterized by X-ray crystallography.35 The root-mean-square deviation (RMSD) was predicted to be 0.079 Å between the singlet 0N–Fe–1S and the triplet 0N–Fe–5T.
 | ||
| Fig. 14 The optimized low-energy Cp2Fe2C2Me2 structures. The symmetry of the structure is indicated in parentheses in the first line. The numbers in parentheses in the second line are the relative energies (ΔE in kcal mol−1) predicted by M06-L/def2-TZVP method. | ||
The second lowest energy Cp2Fe2C2Me2 structure, namely the triplet 0N–Fe–2T lying 10.6 kcal mol−1 above 0N–Fe–1S, has an intact dimethylacetylene ligand. Interpreting the FeFe distance (WBI = 1.24 in Table 1) of 2.150 Å as a triple σ + π + δ type bond gives each iron atom a 18-electron configuration for a binuclear triplet.
| Structure | Ni–Ni (Å) | WBI | MBI | Bond order |
|---|---|---|---|---|
| 2N–Ni–1S | 2.310 | 0.71 | 0.68 | 1 |
| 2N–Ni–2T | 2.359 | 0.62 | 0.55 | 1 |
| 2N–Ni–3S | 3.496 | 0.22 | 0.16 | 0 |
| 2N–Ni–4S | 2.790 | 0.32 | 0.19 | 0 |
| 2N–Co–1S | 2.360 | 0.71 | 0.54 | 1 |
| 2N–Co–2T | 2.314 | 0.82 | 0.81 | 2 |
| 2N–Co–3S | 2.236 | 0.85 | 0.87 | 2 |
| 2N–Co–4S | 2.624 | 0.54 | 0.47 | 1 |
| 2N–Co–5T | 2.556 | 0.63 | 0.42 | 1 |
| 2N–Fe–1S | 2.344 | 0.85 | 0.77 | 2 |
| 2N–Fe–2T | 2.360 | 0.86 | 0.72 | 2 |
| 2N–Fe–3S | 2.278 | 1.14 | 1.21 | 3 |
| 2N–Fe–4T | 2.555 | 0.61 | 0.54 | 1 |
| 2N–Fe–5S | 2.468 | 0.71 | 0.70 | 2 |
| 1N–Ni–1S | 2.300 | 0.72 | 0.71 | 1 |
| 1N–Ni–2S | 2.379 | 0.56 | 0.52 | 1 |
| 1N–Ni–3T | 2.531 | 0.45 | 0.38 | 1 |
| 1N–Ni–4S | 2.903 | 0.26 | 0.15 | 0 |
| 1N–Co–1S | 2.436 | 0.62 | 0.52 | 1 |
| 1N–Co–2T | 2.356 | 0.64 | 0.61 | 1 |
| 1N–Co–3T | 2.714 | 0.37 | 0.26 | 1 |
| 1N–Co–4T | 2.302 | 0.82 | 0.79 | 1 |
| 1N–Fe–1T | 2.370 | 0.92 | 0.86 | 2 |
| 1N–Fe–2S | 2.345 | 0.81 | 0.68 | 2 |
| 1N–Fe–3T | 2.345 | 0.87 | 0.83 | 2 |
| 1N–Fe–4T | 2.381 | 0.79 | 0.61 | 2 |
| 0N–Ni–1S | 2.294 | 0.72 | 0.73 | 1 |
| 0N–Ni–2T | 2.285 | 0.71 | 0.67 | 1 |
| 0N–Ni–3S | 2.400 | 0.53 | 0.49 | 1 |
| 0N–Ni–4S | 2.432 | 0.53 | 0.55 | 1 |
| 0N–Ni–5S | 2.342 | 0.58 | 0.55 | 1 |
| 0N–Ni–6T | 2.545 | 0.44 | 0.38 | 1 |
| 0N–Co–1T | 2.417 | 0.63 | 0.59 | 1 |
| 0N–Co–2T | 2.312 | 0.84 | 0.84 | 2 |
| 0N–Co–3T | 2.404 | 0.61 | 0.59 | 1 |
| 0N–Co–4S | 2.416 | 0.65 | 0.60 | 1 |
| 0N–Co–5T | 2.524 | 0.51 | 0.37 | 1 |
| 0N–Co–6S | 2.159 | 1.05 | 1.12 | 2 |
| 0N–Co–7S | 2.432 | 0.61 | 0.57 | 1 |
| 0N–Co–8T | 2.454 | 0.60 | 0.59 | 1 |
| 0N–Co–9S | 2.291 | 0.85 | 0.83 | 2 |
| 0N–Fe–1S | 2.326 | 0.79 | 0.56 | 2 |
| 0N–Fe–2T | 2.150 | 1.24 | 1.38 | 3 |
| 0N–Fe–3T | 2.542 | 0.64 | 0.60 | 2 |
| 0N–Fe–4T | 2.450 | 0.72 | 0.65 | 2 |
| 0N–Fe–5T | 2.338 | 0.80 | 0.56 | 2 |
| 0N–Fe–6T | 2.330 | 0.87 | 0.81 | 2 |
| 0N–Fe–7S | 2.280 | 0.87 | 0.80 | 2 |
| 0N–Fe–8S | 2.437 | 0.71 | 0.68 | 2 |
| 0N–Fe–9T | 2.398 | 0.57 | 0.58 | 1 |
| 0N–Fe–10T | 2.490 | 0.66 | 0.63 | 1 |
The remaining low-energy Cp2Fe2C2Me2 structure, namely the triplet 0N–Fe–9T lying 16.6 kcal mol−1 above 0N–Fe–1S, is the only Cp2M2(alkyne) isomer found in this work in which a cyclopentadienyl ring has migrated from one iron atom to the other (Fig. 14). This Cp migration results in one iron atom approaching a ferrocene-like environment by being bonded to a quasiterminal pentahapto η5-Cp ligand and to four carbon atoms of a bridging η4,η1-Cp ligand. This sandwich-type ligand arrangement coupled with an FeFe double bond of length 2.399 Å gives this central iron atom the favored 18-electron configuration provided that it bears a formal positive charge. The other iron atom in 0N–Fe–9T, which is bonded to one carbon atom of the bridging η4,η1-Cp ligand, has a C–H–Fe agostic interaction with a C–H unit of the quasiterminal Cp ring, and bears an intact terminal MeCCMe ligand functioning as a four-electron donor through both π systems. This set of ligands combined with the FeFe double bond gives this iron atom a 16-electron configuration provided that it bears a formal negative charge to balance the formal positive charge on the other iron atom. The 16-electron configuration of the iron bearing the alkyne ligand can account for the triplet spin state of 0N–Fe–9T.
3.2 Wiberg bond indices
The Wiberg Bond Indices and Mayer Bond Indices of the binuclear cyclopentadienylmetal alkyne systems Cp2M2C2R2 (M = Ni, Co, Fe; R = Me and NMe2) have been investigated by using the MultiWFN software (Table 1).37 They are comparable to each other; only the Wiberg Bond Indices (WBIs) are reported in the text. The Wiberg Bond Indices (WBIs) for metal–metal bonds involving transition metal atoms do not match the conventional chemical bond order assumptions, but instead are much smaller than such conventional formal bond orders. However, the relative WBI values for transition metal M–M bonds provide insight regarding the corresponding formal metal–metal bond orders. For the present systems, the WBIs range from 0.37 to 0.84 for M–M single bonds, from 0.71 to 0.92 for MM double bonds, and from 1.14 to 1.24 for MM triple bonds.
Taking the singlet Cp2Fe2C2(NMe2)2 structures as examples, the Bond Critical Points (BCPs), Ring Critical Points (RCPs), and Cage Critical Points (CCPs) have been plotted by using the MultiWFN software (Fig. 15). However, no obvious BCPs between the two iron atoms or between the nitrogen atom and the iron atom were observed. This suggests that there is no pure metal–metal bonding or nitrogen–metal bonding. Instead, multicenter bond characters (RCP and CCP) are displayed in these structures. However, use of the formal metal–metal bond orders suggested by the WBI data (Table 1) leads to reasonable electron bookkeeping for these molecules.
 | ||
| Fig. 15 QTAIM analysis of the singlet Cp2Fe2C2(NMe2)2 structures. | ||
3.3 A hydrogen migration mechanism
A possible double hydrogen migration scheme starting with the Cp2Co2(MeC2NMe2) complex to give the cobaltallylic complex Cp2Co2(C3H3NMe2) is depicted in Fig. 16. Five transition states have been identified. The initial complex 1 with two agostic C–H–Co interactions can be formed when the free MeCCNMe2 ligand reacts with a suitable CpCo source. Transfer of the hydrogen atom H2 in 1 from the γ-C to the β-C through transition state TS-1 can then occur with an activation energy barrier of ΔG = 12.8 kcal mol−1 to give 2. The other hydrogen atom H1 can then migrate from the γ-C to the α-C through transition states TS-2 and TS-3 with a activation energy barrier of ΔG = 18.2 kcal mol−1 to form 4 that retains the agostic C–H2–Co interaction. In 4, the C3H3 unit is bonded to one cobalt atom as a trihapto ligand using all three carbon atoms whereas only the β-C and γ-C atoms are bonded to the other cobalt atom. The Me2N unit then attacks one cobalt atom in 4 through transition state TS-4 with an activation energy barrier ΔG of 15.6 kcal mol−1 to displace the agostic hydrogen atom thereby giving 5 with no changes in the bonding of the hydrocarbon part of the Me2NC3H3 ligand. Finally, migration of a carbon atom from the cobalt atom bonded to three carbon atoms in 5 to the other cobalt atom through transition state TS-5 with an activation energy barrier ΔG of 8.2 kcal mol−1 gives the cobaltallylic derivative 6 corresponding to the lowest energy Cp2Co2(MeC2NMe2) isomer 1N–Co–1S in Fig. 10. The coordination mode of the bridging Me2NC3H3 cobaltallylic ligand in 6 is similar to that of the bridging Et2NC3H3 manganallylic ligand in the experimentally known and structurally characterized complex Mn2(CO)7(C3H3NMe2).19
 | ||
| Fig. 16 Proposed hydrogen migration path for the Cp2Co2(MeC2NMe2) system predicted by the M06-L/def2-TZVP method. | ||
4. Conclusion
The Cp2M2C2(NMe2)2 (M = Ni, Co, Fe) systems have the simplest potential energy surfaces of the Cp2M2(alkyne) systems studied in this work. Thus the low-energy structures in these systems are limited to tetrahedrane complexes with an intact alkyne ligand and a central M2C2 tetrahedron and the alkyne dichotomy products Cp2M2(CNMe2)2 in which the CC triple bond of the original alkyne is completely broken to give two separate bridging dimethylaminocarbyne ligands. The relative energies of these two structure types depend on the electron requirements of the central metal atoms. Thus for the nickel system Cp2Ni2C2(NMe2)2 the tetrahedrane complex, corresponding to extensive series of known Cp2Ni2(alkyne) complexes15–17 and having favorable 18-electron nickel configurations, is favored energetically by a substantial margin greater than 23 kcal mol−1. For the cobalt system Cp2Co2C2(NMe2)2 the singlet and triplet tetrahedrane structures and a singlet dichotomy structure have similar energies within ∼3 kcal mol−1. For the iron system Cp2Fe2C2(NMe2)2 both singlet and triplet dichotomy structures lie more than 12 kcal mol−1 in energy below the lowest energy structures with intact alkyne ligands. Furthermore, the intact alkyne ligands in the complexes Cp2Fe2(Me2NC2NMe2) are six-electron donors to the central Fe2 unit by forming an N→Fe dative bond from one of the Me2N substituents in addition to the usual π-bonds to iron atoms from both of the π-components in the alkyne CC triple bond.
The Cp2M2(alkyne) systems based on the alkynes MeCCNMe2 and MeCCMe, have methyl groups directly linked to alkyne carbon atoms. This makes their hydrogen atoms more accessible for migration to alkyne carbon atoms than the more remotely located methyl hydrogen atoms on the amino nitrogen atoms in the Cp2M2(Me2NCCNMe2) systems. Therefore, the potential energy surfaces of the MeCCNMe2 and MeCCMe systems are complicated. However, for the nickel systems Cp2Ni2(MeC2NMe2) and Cp2Ni2C2Me2 with enough d electrons from the nickel atoms, it is not necessary to completely cleave the CC bond for the nickel atoms to have the favored 18-electron configuration. Therefore the tetrahedrane complexes with intact alkyne ligands remain the lowest energy structures again corresponding to several experimentally known Cp2Ni2(alkyne) complexes.15,16 However, for the cobalt and iron systems derived from the MeCCNMe2 ligand, the singlet and triplet metalallylic structures Cp2M2(CHCHCHNMe2) arising from migration of two methyl hydrogen atoms, one to each alkyne carbon atom, are the lowest energy structures. This situation is similar to that in the experimentally known manganese carbonyl complexes (Et2NC2Me)Mn2(CO)8 of alkynes with methyl substituents where hydrogen migration from the methyl group to one or both alkyne carbon atoms is observed to give species with bridging allene or manganallyl units (Fig. 2).19 For the cobalt system derived from dimethylacetylene, the Cp2Co2(CH2CHCMe) vinylcarbene structure with migration of one methyl hydrogen atom to the adjacent alkyne carbon atom is the lowest energy structure. For the iron system derived from dimethylacetylene, the alkyne dichotomy Cp2Fe2(CMe)2 structure is the lowest energy structure.
The sets of low-energy Cp2M2(MeC2NMe2) and Cp2M2C2Me2 isomers formed by hydrogen migration processes also include structures with bridging allene ligands and with bridging vinylcarbene ligands formed by migration of a single methyl hydrogen to the remote alkyne carbon atom (the γ-carbon atom) and to the adjacent alkyne carbon atom (the β-carbon atom), respectively. In addition, the set of low-energy structures for the Cp2M2C2Me2 (M = Co, Fe) systems derived from dimethylacetylene includes derivatives with bridging butadiene ligands formed by migration of one hydrogen atom from each methyl group to an alkyne carbon atom.
Five transition states have been identified in a possible mechanistic sequence for double hydrogen migration in the Cp2Co2/MeCCNMe2 system to give the cobaltallylic complex Cp2Co2(C3H3NMe2) found to be the lowest energy structure of Cp2Co2(MeC2NMe2) stoichiometry. This mechanistic sequence has intermediates with C–H–Co interactions through agostic hydrogen atoms and successive activation energy barriers of 13.1, 17.0, 15.2, and 12.0 kcal mol−1.
Data availability
Data are presented in the ESI† and are available from Prof. Huidong Li.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The research in Chengdu was supported by the Sichuan Provincial Foundation for Distinguished Young Leaders of Disciplines in Science and Technology of China (GrantNos.2019JDJQ0051 and 2019JDJQ0050), the Chunhui Program of the Ministry of Education of China (Grant Z2017091), and the Open Research Subject of the Key Laboratory of Advanced Computation in Xihua University (Grant No. szjj2017-011 and szjj2017-012). The research at the University of Georgia was supported by the U. S. Department of Energy, Basic Energy Sciences, Division of Chemistry, Computational and Theoretical Chemistry (CTC) Program under Contract DE-SC0018412.References
- H. Greenfield, H. W. Sternberg, R. A. Friedel, J. H. Wotiz, R. Markby and I. Wender, Acetylenic dicobalt hexacarbonyls—organometallic compounds derived from alkynes and dicobalt octacarbonyl, J. Am. Chem. Soc., 1956, 78, 120–124 CrossRef CAS.
- R. S. Dickson and P. J. Fraser, Compounds derived from alkynes and carbonyl complexes of cobalt, Adv. Organomet. Chem., 1974, 12, 323–377 CrossRef CAS.
- M. J. Went, Synthesis and reactions of polynuclear cobalt alkyne complexes, Adv. Organomet. Chem., 1997, 41, 69–125 CrossRef CAS.
- S. E. Gibson and N. Mainolfi, The intermolecular Pauson–Khand reaction, Angew. Chem., Int. Ed., 2005, 44, 3022–3037 CrossRef CAS PubMed.
- K. M. Nicholas, Chemistry and synthetic utility of cobalt-complexed propargyl cations, Acc. Chem. Res., 1987, 20, 207–214 CrossRef CAS.
- N. Iwasawa, I. Ooi and K. Inaba, [4+2] Cycloaddition reaction of cyclic alkyne-{Co2(CO)6} complexes with dienes, Angew. Chem., Int. Ed., 2010, 49(41), 7534–7537 CrossRef CAS PubMed.
- I. Ooi, T. Sakurai and J. Takaya, Protonation-triggered generation of acylcobalt species from alkyne-[Co2(CO)6] complexes and their reaction with alkenes, Chem. Eur J., 2012, 18, 14618–14621 CrossRef CAS PubMed.
- N. A. Danilkina, A. G. Lyapunova and A. F. Khlebnikov, Ring-closing metathesis of Co2(CO)6-alkyne complexes for the synthesis of 11-membered dienediynes: overcoming thermodynamic barriers, J. Org. Chem., 2015, 80, 5546–5555 CrossRef CAS PubMed.
- B. Alcaide, C. Polanco and M. A. Sierra, Alkyne-Co2(CO)6 complexes in the synthesis of fused tricyclic beta-lactam and azetidine systems, J. Org. Chem., 1998, 63, 6786–6796 CrossRef CAS PubMed.
- W. Hübel, in Organic Syntheses via Metal Carbonyls, ed. I. Wender and P. Pino, Interscience—Wiley, New York, vol. 1, 1968, pp. 273–342 Search PubMed.
- F. A. Cotton, J. D. Jamerson and B. R. Stults, Metal-metal multiple bonds in organometallic compounds. 1. (Di-tert-butylacetylene)hexacarbonyldiiron and hexacarbonyldicobalt, J. Am. Chem. Soc., 1976, 98, 1774–1779 CrossRef CAS.
- R. B. King and C. A. Harmon, Novel products from reactions of aminoalkynes with metal carbonyls, Inorg. Chem., 1976, 15, 879–885 CrossRef CAS.
- G. G. Cash, R. C. Pettersen and R. B. King, X-ray crystal and molecular structure of [Et2NCFe(CO)3]2—example of division of an alkyne into 2 separate units by rupture of the C≡C bond, Chem. Commun., 1977, 30–31 RSC.
- H. Li, H. Feng, W. Sun, Y. Xie, R. B. King and H. F. Schaefer, Alkyne dichotomy: splitting of bis(dialkylamino)acetylenes, dimethoxyacetylene, bis(methylthio)acetylene, and their heavier congeners to give carbyne ligands in iron carbonyl derivatives, Organometallics, 2013, 32, 88–94 CrossRef.
- J. F. Tilney-Bassett and O. Mills, Cyclopentadienylnickel-acetylene complexes, J. Am. Chem. Soc., 1959, 81, 4757–4758 CrossRef CAS.
- J. F. Tilney-Bassett, Cyclopentadienylnickel-acetylene complexes, J. Chem. Soc., 1961, 577–581 RSC.
- E. J. Forbes and N. Iranpoor, Dicyclopentadienylnickel alkyne complexes: photochemical formation and mass spectra, J. Organomet. Chem., 1982, 236, 403–407 CrossRef CAS.
- D. Kumar, M. Deb, J. Singh, N. Singh, K. Keshav and A. J. Elias, Chemistry of the highly stable hindered cobalt sandwich compound (η5-Cp)Co(η4-C4Ph4) and its derivatives, Coord. Chem. Rev., 2016, 306, 115–170 CrossRef CAS.
- R. D. Adams, G. Chen and Y. Chi, Coordination and transformations of the ynamine ligand in a dimanganese complex, Organometallics, 1992, 11, 1473–1479 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 12, 194101–194107 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- (a) J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron gas correlation energy, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed; (b) J. P. Perdew, K. Burke and Y. Wang, Generalized gradient approximation for the exchange-correlation hole of a many-electron system, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- (a) S. Grimme, Density functional theory with London dispersion corrections. Wires Comput, Mol. Sci., 2011, 1, 211–228 CrossRef CAS; (b) M. Dierksen and S. Grimme, An efficient approach for the calculation of Franck-Condon integrals of large molecules, J. Chem. Phys., 2005, 122, 244101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- T. H. Dunning Jr, Gaussian basis functions for use in molecular calculations. I. Contraction of (9s5p) atomic basis sets for the first-row atoms, J. Chem. Phys., 1970, 53, 2823–2833 CrossRef.
- S. Huzinaga, Gaussian-type functions for polyatomic systems. I, J. Chem. Phys., 1965, 42, 1293–1302 CrossRef.
- A. J. H. Wachters, Gaussian basis set for molecular wavefunctions containing third-row atoms, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS.
- D. M. Hood, R. M. Pitzer and H. F. Schaefer, Electronic structure of homoleptic transition metal hydrides: TiH4, VH4, CrH4, MnH4, FeH4, CoH4, and NiH4, J. Chem. Phys., 1979, 71, 705–712 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) Calculate Root-mean-square deviation (RMSD) of Two Molecules Using Rotation, GitHub, http://github.com/charnley/rmsd Search PubMed; (b) W. Kabsch, A solution for the best rotation to relate two sets of vectors, Acta Crystallogr., 1976, A32, 922–923 CrossRef; (c) M. W. Walker, L. Shao and R. A. Volz, Estimating 3-D location parameters using dual number quaternions, CVGIP: Image Understanding, 1991, vol. 54, pp. 358–367 Search PubMed.
- O. S. Mills and B. W. Shaw, The structure of (diphenylacetylene)bis(cyclopentadienylnickel), J. Organomet. Chem., 1968, 3, 595–600 CrossRef.
- J. P. Blaha, B. E. Bursten, J. C. Dewan, R. B. Frankel, C. L. Randolph, B. A. Wilson and M. S. Wrighton, Dinuclear, 18-electron species having a triplet ground state—isolation, characterization, and crystal structure of photogenerated (η5-C5Me5)2Fe2(μ-CO)3, J. Am. Chem. Soc., 1985, 107, 4561–4562 CrossRef CAS.
- M. Rosenblum, B. North, D. Wells and W. P. Giering, Synthesis and chemistry of η4-cyclobutadiene(η5-cyclopentadienyl)cobalt, J. Am. Chem. Soc., 1972, 94, 1239–1245 CrossRef CAS.
- (a) T. Lu and F. M. Chen, A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu, A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 to S11: Atomic coordinates of the optimized structures for the cmp-n (n = 1 to 6) and TS-n (n = 1 to 5) complexes in Fig. 15; Tables S12 to S22: harmonic vibrational frequencies (in cm−1) and infrared intensities (in parentheses in km mol−1) for the cmp-n (n = 1–6) and TS-n (n = 1–5) complexes in Fig. 15; Tables S23: the optimized structures of the cmp-n (n = 1–6) and TS-n (n = 1–5) complexes in Fig. 15; Tables S24 to S26: the optimized structures of the Cp2Ni2C2(NMe2)2, Cp2Co2C2(NMe2)2 and Cp2Fe2C2(NMe2)2 complexes; Tables S27 to S29: the optimized structures of the Cp2Ni2(MeC2NMe2), Cp2Co2(MeC2NMe2) and Cp2Fe2(MeC2NMe2) complexes; Tables S30 to S32: The optimized structures of the Cp2Ni2C2Me2, Cp2Co2C2Me2 and Cp2Fe2C2Me2 complexes. Separate .xyz file of the optimized structures. See DOI: https://doi.org/10.1039/d4ra01410c |
| This journal is © The Royal Society of Chemistry 2025 |