DOI:
10.1039/D4RA08731C
(Paper)
RSC Adv., 2025,
15, 5316-5326
Photocatalytic hydrocarbon production from aqueous acetic acid using TiO2 with simultaneous photodeposition of Cu†
Received
12th December 2024
, Accepted 18th January 2025
First published on 17th February 2025
Abstract
Photocatalytic techniques are considered clean, sustainable and cost-effective in energy conversion and environmental restoration. The large band gap, light harvesting limitation and rapid electron–hole pair recombination can suppress the photocatalytic efficiency in photocatalytic applications. Metal deposition has become one of the most important technical means to improve photocatalytic efficiency. This study has dealt with photocatalytic hydrocarbon and hydrogen production from the acetic acid solution with simultaneous in situ Cu deposition on TiO2 photocatalyst surface. Due to having favorable redox potential and work function values, the photodeposition and Schottky junction formation of Cu occurred smoothly on the TiO2 surface, which further contributed to accelerating the interfacial charge transfer and photocatalytic activity. The reaction conditions (Cu2+ loading, reaction pH and initial concentration of acetic acid) were optimized to enhance photocatalytic methane production. Under the optimum condition, the Cu/TiO2 photocatalytic hydrocarbon production was maximum (4136 μmol g−1), approximately 9 times better than those obtained with pure TiO2 (450 μmol g−1). The surface morphological and optical properties of photodeposited Cu/TiO2 samples were characterized before and after the photocatalytic reaction with utmost precision and thoroughness using a TEM, XPS, DRS, PL, N2 adsorption–desorption isotherm and BET surface area analysis. The DRS and PL study confirm that in situ Cu-deposition on TiO2 reduced the energy bandgap and improved the light-harvesting area, photogenerated electron–hole pair separation and migration efficiency, respectively. Cycle experiments disclose that the simultaneous Cu-deposited photocatalyst has excellent stability and reusability. A reaction mechanism was proposed for the photocatalytic hydrocarbon formation from the acetic acid by Cu/TiO2 photocatalytic reaction.
1. Introduction
In 2015, the “Paris Agreement” attempted to limit global warming by reducing greenhouse gas emissions, especially carbon dioxide (CO2) emissions from the burning of fossil fuels (coal, oil and natural gas), the major contributor to climate change.1 Clean energy sources (solar, wind, hydropower, geothermal, biomass and nuclear energy) can help to establish decarbonization attempts.2 The utilization of multiple energy resources can release various hazardous and poisonous compounds into the environment.3 Meanwhile, with the depletion of natural non-renewable energy resources, demand increases for the development of renewable and eco-friendly energy resources.4 In the past few decades, numerous researches were conducted for the effective ways to the energy demands5 and environmental remedies.6 Although there are several ways to produce methane, including natural gas extraction, steam reforming, pyrolysis, anaerobic digestion and Sabatier, the photocatalytic transformation of organic substances is an alluring strategy for the production of hydrocarbons considering the environmental issues.7 Recently, photocatalytic techniques have been effectively employed in organic synthesis, detoxification, water splitting, nitrogen fixation, CO2 reduction and energy production due to their readily availability, photostability and less toxicity.8 They can boost the chemical reaction in the visible, infrared and ultraviolet light ranges.9 The catalyst's crystal size, surface area, electron–hole pair recombination rate, and applied light wavelength affect its photocatalytic activity.10,11 A positive hole (h+) forms as a result of an electron (e−) moving from the valence band (VB) to the conduction band (CB) when the energy of the irradiation light is equal to or higher than the energy band gap (Eg) of the photocatalyst. Finally, the reduction and oxidation of the target structure are performed by the photogenerated electrons and positive holes.12
TiO2 has been one of the many semiconductor-based photocatalysts due to its advantageous electrical and optical characteristics, thermodynamic and chemical stability, crystallinity, non-toxicity, resilience to corrosive conditions and comparatively low cost.13,14 Nevertheless, because of the quick recombination of photogenerated holes and electrons (h+/e−) and the high band gap (Eg = 3.2 eV), TiO2 itself works better in the ultraviolet (UV) region only.15 Metal deposition appears to be a viable strategy to overcome these wavelength limitations, whereby the specific interaction between the metal and TiO2 determines whether or not the catalysts' characteristics may be improved. The redox potential of Cu is situated between the VB and CB of TiO2, which allows Cu for smooth deposition on the TiO2 surface. Similarly, the larger work value of Cu than the TiO2 favored the Schottky-junction formation and enhanced the photocatalytic efficiency by hindering electron–hole recombination. 1978, Kraeutler and Bard first reported methane production from acetic acid solution using the Pt-loaded TiO2 photocatalyst.16 Previously, the insertion of metals (Ag, Au, Pd, Pt, Co, Cu, Mg, Ni, Sn, Fe, Mn, Au, La, Ru and Rh) into the TiO2 lattice also resulted in a reduction in the energy band gap and a slower rate of (h+/e−) recombination.17–24 Mozia et al. reported the physicochemical,25 suspended26 and Fe-modified7,27 effects of TiO2 photocatalytic decomposition of acetic acid for hydrocarbon evolution under UV light (365 nm) irradiation at room temperature. Heciak et al.28 and Amorós-Pérez et al.29 also reported the Cu-modified TiO2 catalytic production of biogas (mainly CH4) and hydrogen under UV light irradiation at room temperature. Asal et al. also looked at the synthesis of methane from acetic acid over La3+-modified TiO2 and found that the output was marginally enhanced under the same reaction conditions, the output was marginally enhanced.23 Betts et al. reported the P25 TiO2 photocatalytic alkane production from acetic acid and propionic acid.30 Recently, Cao et al. reported significantly enhanced methane production from acetic acid using the platinized TiO2 under argon and CO2 conditions.31 However, combining TiO2 with inexpensive, readily available metal oxides (Fe2O3, SnO2, Al2O3, ZnO, Ag2O, NiO, Cu2O and CuO) also significantly enhanced the photocatalytic efficiencies in various magnitudes.32–41
The present work mainly focused on photocatalytic hydrocarbon (primarily methane) production from aqueous acetic acid solution employing TiO2 with in situ simultaneous photodeposition of copper under atmospheric conditions. Compared to other reports, the Cu/TiO2 photocatalyst demonstrated more efficient methane synthesis and the simultaneous Cu deposition on TiO2 was a relatively straightforward process. Finally, the photocatalytic reaction smoothly proceeded under ultraviolet (365 nm) and visible light (405 nm) irradiation without any noble expensive metals (Ag, Au and Pt).
2. Experimental section
2.1. Materials
All the reagents were analytical grade and used as received without further purification. CH3COOH (99.7%) and CuCl2 (95.0%) were purchased from FUJIFILM Wako Pure Chemical Corporation, Cu(CH3COO)2·H2O (99.0%) was purchased from Kanto Chemical Co., Ltd., TiO2 (P-25) photocatalyst was purchased from Degussa Co., Ltd., Germany (anatase 75%, rutile 25%, surface area 53 m2 g−1, particle size 25 nm) and CuO (surface area 29 m2 g−1, particle size 33 nm) was purchased from Sigma-Aldrich Chemicals Pvt. Ltd.
2.2. Characterization
The surface morphology was analyzed by transmission electron microscope on JEM-2100 TEM (JEOL, Japan) and the surface structure characteristic of the products was recorded on X-ray photoelectron spectroscopy PHI Quantera SXM photoelectron spectrometer with Al Kα radiation. Optical properties were inspected by the UV-vis diffuse reflectance sorption (DRS) spectroscopy on the JASCO V-750 UV-vis instrument equipped with an integrating sphere adaptor in the wavelength range of 200–700 nm. Photoluminescence (PL) spectroscopic measurement was carried out by Shimadzu fluorescence spectrometer (RF-5300PC, Japan). The photocatalysts' BET surface area, total pore volume and average pore size were assessed from the N2 adsorption–desorption isotherm utilizing a BELSORP-miniII (MicrotracBEL) apparatus.
2.3. Photocatalytic methane evolution
All the photocatalytic hydrocarbon and hydrogen production experiments were carried out in a heat resistance Pyrex glass vessel (inner volume: 123 mL). The photocatalyst (50 mg of TiO2) and co-catalyst (5.6 ppm of CuO, 0.45 wt% of Cu2+ relative to the weight of TiO2) were dispersed in 40 mL of aqueous acetic acid (1.0 M) solution, heated to 50 °C with constant stirring, then illuminated by UV-vis (365 nm, ∼12.0 mW cm−2) and visible light (405 nm) for 3 h, respectively. The produced hydrocarbon was examined by gas chromatography (GC-353B, GL Science, Japan) using a flame ionization detector. The column was packed with Porapak Q (mash, 80–100) and the carrier gas was N2 (99.9%, 29.5 mL min−1). Similarly, the evolved hydrogen and consumed oxygen were analyzed using a gas chromatographer (GL Sciences, GC-3200) equipped with a thermal conductivity detector, with the injection part, column and detector maintained at 50 °C.
3. Results and discussion
3.1. Effect of metallic source on photocatalytic methane production under UV/vis irradiation
The effect of metallic source on photocatalytic methane production from the aqueous solution of acetic acid using TiO2 with in situ simultaneous deposition of copper was studied under UV light (365 nm) irradiation (Fig. 1a). The photocatalytic methane production with pure TiO2 was 450 μmol g−1. The larger band gap (3.2 eV) and rapid electron hole-pair recombination are responsible for the poor catalytic activity. Due to the simultaneous deposition of Cu2+ ions from CuCl2, methane production increased 4.5-fold. Further, the individual deposition of Cu(CH3COO)2 and CuO as Cu2+ ion sources elevated methane production by 7.7 and 7.9 folds, respectively. 9.2 times enhanced methane was achieved from the physical mixing (PM) of CuO and TiO2 compared to sole TiO2. The redox potential of Cu2+ is 0.342 V vs. standard hydrogen electrode (SHE). It could be reduced by the photocatalyst when the CB of the semiconductor is more negative than the reduction potential of the Cu2+.42 The CB and VB edges for the TiO2 photocatalyst are –0.46 V and + 2.7 V vs. SHE, respectively.43 As a result, the photodeposition of Cu2+ on the TiO2 surface generally occurred during the photocatalytic reaction and its metal reduction is also thermodynamically feasible.44
 |
| | Fig. 1 (a) Effect of Cu sources on the photocatalytic methane production from acetic acid solution, and (b) comparison of hydrocarbon production rates under UV light (365 nm) irradiation. | |
3.2. Comparison of hydrocarbon production rate under UV light irradiation
The comparison of hydrocarbon production rate using TiO2 with simultaneous photodeposition of copper ions under UV/vis light irradiation with the extension of irradiation time was studied, as presented in Fig. 1b. The methane production rate gradually increased with the increase of irradiation time. All Cu2+ ion sources showed almost similar efficiency with or without physical mixing conditions. The physical mixing of CuO/TiO2 showed little improvement in ethane production due to the favorable cyclic conversion of CuO → Cu2+ → Cu1+ → Cu0. The metallic copper (Cu0) further reacts with dissolved oxygen and regenerates the CuO, reaching a cyclic redox reaction (eqn (1) and (2)). Since it was observed that the simultaneous photodeposition of copper on the TiO2 enhanced the photocatalytic methane production, optimal conditions such as Cu2+ ion concentration, acetic acid concentration and reaction pH were optimized.
3.3. Photocatalytic performance conditions
Numerous operational factors, including catalyst and co-catalyst loading, substrate initial concentration and reaction pH, can affect the photocatalytic redox reaction. The following factors were optimized to achieve the maximum methane production under 12 h of UV/vis light illumination at 50 °C, whereas Cu(OAc)2 was used as a Cu2+ ion source throughout the optimization reactions.
3.3.1. Effect of Cu2+ ion concentration. The effect of the co-catalyst (Cu2+) concentration on the photocatalytic hydrocarbon production activity using TiO2 with simultaneous deposition of Cu under UV/vis light irradiation was investigated (Fig. S1a†). Various amounts of Cu2+ were simultaneously introduced into the acetic acid solution along with the TiO2 to determine the impact of co-catalyst loading on the photocatalytic hydrocarbon production rate with constant stirring conditions. The insertion of Cu2+ on the TiO2 disclosed improved photocatalytic performance. The work function value of Cu (4.65 eV) is larger than the work function of TiO2 (4.50 eV). A larger work function of metal ions is favorable for forming the Schottky junction effect at the metal-TiO2 interface. As a result, metal with appropriate work functions can facilitate electron transfer, assist in overcoming the Schottky energy barrier and enhance photocatalytic efficiency.44–46 Experimental results indicated that methane production gradually increased with the increase of Cu2+ concentration, reaching a maximum of 5.6 mg L−1 (0.45 wt% of Cu2+ relative to the weight of TiO2), achieving the highest methane production (4136 μmol g−1). The further increase of Cu2+ loading decreased the methane production due to the coagulation of Cu2+ and the incident light blockage on the photocatalyst surface.47
3.3.2. Effect of CH3COOH concentration. In the photodegradation of organic compounds, the degradation rate increases to a certain point due to the rise of reactant concentration. The effect of acetic acid concentration on Cu/TiO2 photocatalytic hydrocarbon production was studied under UV/vis light irradiation (Fig. S1b†). The amount of methane production sharply increased with the increase in acetic acid concentration from 0 to 1.0 mol L−1. However, the highest amount (4730 μmol g−1) of methane was evolved at 2.0 mol L−1 concentration of acetic acid, further increase of the reactant concentration, the production became steady due to the excess adsorption of acetic acid on the Cu/TiO2 photocatalyst and reached saturation level.48 With an increase of the initial concentration of acetic acid from 1.0 mol L−1 to 2.0 mol L−1, the methane production rate was not so high compared to the concentration, which resulted in 1.0 mol L−1 being considered the optimum concentration and all the further experiments were carried out accordingly.
3.3.3. Effect of reaction pH. The pH of the reaction medium and the charge on the catalyst's surface can affect the adsorption and dissociation of the reactant. The pH effect on Cu/TiO2 photocatalytic hydrocarbon production was assessed by varying the pH from 1.5 to 3.5 (Fig. S1c†). 0.1 M HCl and 0.1 M NaOH reagents were used for the pH adjustment. The point zero charge (PZC) of TiO2 P25 was approximately pH = 5.2 and the photocatalyst surface became chargeless at PZC and had no interaction with polar substances like acetic acid.49 The photocatalytic degradation process is influenced by the PZC of TiO2 and the dissociation constant (pKa) of acetic acid (pKa = 4.76). The surface of TiO2 becomes positively and negatively charged when the pH is less and more than pHPZC, respectively. At lower pH (pH < pKa), acetic acid remains unchanged and at relatively higher pH (pH > pKa), CH3COO− ions were produced. When pH > pHPZC, the TiO2 surface is negatively charged and the adsorption of CH3COO− ions on TiO2 surface is difficult because of static repulsion.47,50 Herein, due to the increase of reaction pH from 1.5 to 2.5, the methane production sharply increased from 36 μmol g−1 to 4136 μmol g−1. With a further increase in pH from 3.5, the methane production rapidly decreased from 4136 μmol g−1 to 1886 μmol g−1. Numerous investigations have demonstrated the crucial effect of reaction pH and charge on the photocatalyst surface, which can significantly affect the degradation of different carboxylic acids.47
3.4. Effect of irradiation time
The effect of irradiation time on Cu/TiO2 photocatalytic hydrocarbon and hydrogen production from aqueous acetic acid solution was studied under UV/vis light irradiation (Fig. 2a). The amount of hydrocarbon production increased with the increase in irradiation time. In particular, CH4 and C2H6 production sharply increased up to 30 h and 28 h (Fig. 2a, and S2a†), respectively. Further, the methane production remains steady till 96 h of irradiation. At the beginning of the reaction, in the presence of the optimal amount of dissolved oxygen, the number of electrons decreased, which was utilized for proton reduction, induced hydrocarbon production and reduced hydrogen generation. As a result, hydrogen production increased sharply after 32 h of continuous irradiation (Fig. S2b†). Meanwhile, oxygen in the system decreased sharply. The metal Cu0 had been photo-precipitated on the TiO2 surface due to the shortage of oxygen (Fig. S3†). As a result, methane and ethane production continued to increase until about 30 h but leveled off after the oxygen disappeared. The role of O2 consumption during the Cu/TiO2 photocatalytic reaction favored hydrocarbon production and reduced hydrogen generation (Fig. S2c†). Herein, CuO dissolves in the acetic acid aqueous solution and exists as Cu2+ ions. The Cu2+ ions were first reduced to metallic Cu0 on the TiO2 surface by reacting with the photogenerated electrons (eqn (1)). However, no discoloration was observed after 24 h of UV light irradiation. After 48 h of irradiation, the discoloration occurred due to the complete photo-precipitation of metallic Cu on the TiO2 surface, which suggests no existence of dissolved oxygen in the reaction system (Fig. S3b†). The Fermi level can be induced toward greater negative potentials using Cu nanoparticles, which can store photogenerated electrons.51 Therefore, it was assumed that the photo-precipitated metallic Cu was also dissolved in the acetic acid aqueous solution. To observe this, a considerable amount of metallic Cu powder was dissolved in the acetic acid solution and continuously stirred for 8 h at 50 °C, where a prolonged dissolution rate was observed (Fig. S3a†).52 Therefore, in this system, the proportion of Cu metal directly dissolved in the aqueous acetic acid solution was tiny and it was considered that the Cu metal reacted with oxygen in the system and was oxidized to CuO (eqn (2)) before being dissolved in the aqueous acetic acid solution.53 Further, the color change of the solution was compared before and after the reaction. In addition, the discolored solution returned to its initial color by introducing atmospheric oxygen into the reaction system for 3 h at 50 °C at continuous stirring conditions (Fig. S3b†).| | |
Cu2+ + e− → Cu+ + e− → Cu0
| (1) |
 |
| | Fig. 2 (a) Effect of irradiation time on Cu/TiO2 photocatalytic CH4 production, and (b) stability test results of CH4 production. | |
Therefore, it was considered that the metal Cu was oxidized to CuO again and dissolved in the aqueous acetic acid solution. It re-exists as Cu2+ ions; this conversion was hampered due to the absence of oxygen (Fig. S3c†). From the GC-TCD analysis, the oxygen in the system completely disappeared in about 30 h of illumination (Fig. S2c†). The solution began to change color almost at the same time. Therefore, oxygen plays a vital role in this system. Methane and ethane production continued to increase until about 30 h and finally leveled off after the complete vanishing of oxygen.
3.5. Reusability and stability test
To assess the stability and reusability of the Cu/TiO2 photocatalysts, the hydrocarbon and hydrogen production from the acetic acid solution was carried out for eleven consecutive cycles (Fig. 2b and S4†). The reactor was fully evacuated before starting a new cycle. The photocatalytic methane production slightly increased till the fourth cycle due to the proper catalytic activation. Further, the production remained almost unchanged till the tenth cycle compared to the first cycle (Fig. 2b). For the ethane production (Fig. S4a†), a slight decrease was observed till the fifth cycle and a further slight elevation was found and became almost the same as the first cycle, indicating the excellent catalytic durability and reusability of Cu/TiO2.54 Compared to the first cycle, hydrogen production improved catalytic performance until the last cycle (Fig. S4b†). As mentioned earlier; oxygen favored the oxidation of acetic acid to produce methane. Every cycle consumed almost the same amount of oxygen and became near zero in 24 h of Cu/TiO2 photocatalytic reaction (Fig. S4c†) for hydrocarbon production.
3.6. Visible light-driven methane production with Cu/TiO2
The photocatalytic methane production from aqueous acetic acid solution utilizing TiO2 with simultaneous photodeposition of Cu from different sources (CuO, CuCl2 and Cu(CH3COO)2) was studied under visible light irradiation (405 nm) to explore the visible light response. The DRS result (Fig. 6a) proved that the simultaneous deposition of Cu on TiO2 has increased the light absorption ability of Cu/TiO2 than the pure TiO2. No methane was detected from the photolysis of acetic acid. Similarly, no methane was tranced from the photocatalytic reaction of CuO due to its smaller band gap (1.2 eV) and faster electron–hole pair recombination rate.55 Due to the large bandgap (3.2 eV), pure TiO2 works better under ultraviolet light region only. Herein, a small amount of methane (0.4 μmol g−1) was achieved from the photocatalytic reaction of pure TiO2 P25 due to the less activation under visible light irradiation and fast undesirable electron–hole pair recombination. The deposition of specific metals on the TiO2 surface significantly improved its photocatalytic efficiency.56,57 The same amount of methane (0.4 μmol g−1) was obtained from the catalytic reaction of CuO/TiO2. Furthermore, using CuCl2 as a Cu2+ ion source increased the photocatalytic methane production by 3.5-fold compared to pure TiO2. The prior physical mixing of CuO/TiO2 does not influence methane production due to the physical condition of CuO. However, the formation of Cu2+ ion and further reduction to metallic Cu0 occurs inside the aqueous solution of acetic acid. The highest amount of methane (29 times compared to TiO2 P25) was obtained using Cu(OAc)2 as a copper ion supplier in simultaneous Cu deposition on TiO2. The physical state and dissociation rate of Cu(OAc)2 responded better than that of CuO and CuCl2 under visible light irradiation.
3.7. Comparison of hydrocarbon production rate under visible light irradiation
The photocatalytic hydrocarbon production rate from the acetic acid solution under visible light irradiation using TiO2 with simultaneous deposition of Cu2+ was also investigated. The Cu2+/TiO2 (CuO) photocatalytic methane production (39 μmol g−1) was relatively higher than that of the physical mixing (32 μmol g−1) condition (Fig. 3b). Moreover, the highest production (2.4 times of CuO/TiO2) was obtained using Cu2+/TiO2 (Cu(OAc)2) photocatalysts under the same conditions. The co-catalytic performance of CuO was relatively lower than Cu(OAc)2 due to the lower solubility and dissociation. It's considered to produce more hydroxyl ions when dissolved in an aqueous acetic acid solution and the photogenerated holes are occupied by hydroxyl radicals. On the other hand, copper acetate behaves the opposite way and works smoothly under visible light irradiation due to its relatively faster dissociation.58 Meanwhile, a trace amount of ethane was also detected at the mentioned conditions.
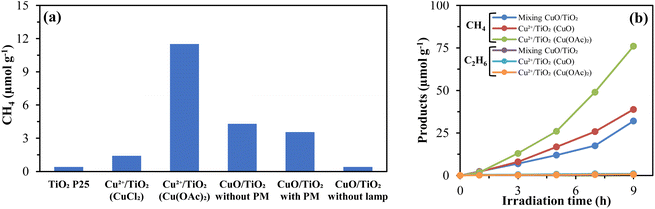 |
| | Fig. 3 (a) Effect of Cu sources on the photocatalytic methane production from acetic acid solution, and (b) comparison of hydrocarbon production rates under visible light (405 nm). | |
3.8. Characterization
3.8.1. XPS analysis. XPS studies were performed to investigate the surface chemical composition and electronic state of Cu/TiO2. The survey and high-resolution spectra of Cu 2p of the studied catalyst are presented in Fig. 4a–d and the binding energy is depicted in Table S2.† From the experimental results (Fig. 4c), two characteristic peaks for Cu 2p3/2 and Cu 2p1/2 were detected at 933.8 eV and 953 eV, respectively, corresponding to Cu2+ (Fig. 4, Table S2†). A satellite peak was observed at 942 eV, indicating the presence of divalent Cu, which was seen in the pure CuO but considerably smaller in the fresh CuO/TiO2 due to the low content of Cu.59 After 3 h of irradiation by UV (365 nm) and visible (405 nm) light, no Cu compound was detected. However, the Cu 2p3/2 peak was re-existed at 932.7 eV after 48 h of UV irradiation. During the simultaneous photodeposition of CuO into the aqueous acetic acid solution, Cu2+ ions first form following the dissolution process. It further reduced to metallic Cu0 on the TiO2 surface by reacting with the photogenerated electrons (eqn (1)). Further, Cu0 is oxidized to CuO by reacting with the dissolved oxygen and dissolved in the aqueous acetic acid solution and re-exists as Cu2+ ions (Fig. S3c†), due to this cyclic conversion, at a certain point Cu ion was not detected. In addition, although CuO/TiO2 was simply kneaded with CuO and TiO2 in an agate mortar, the peak position of CuO shifted slightly to the lower energy side.29,60 This is considered to be caused by interaction with TiO2 due to kneading.
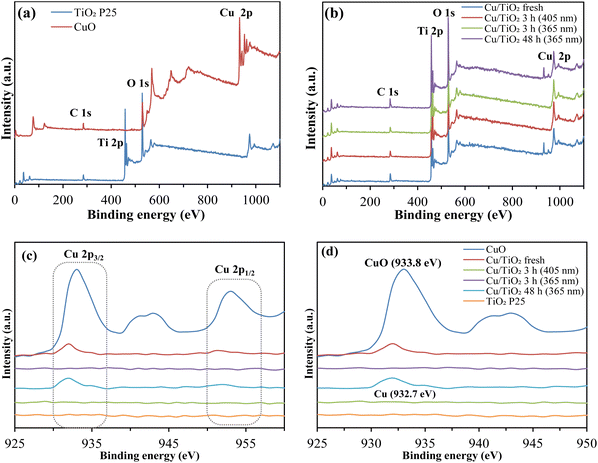 |
| | Fig. 4 XPS (a and b) survey spectra and high-resolution spectra of the studied catalysts (c) in the region of Cu 2p and (d) binding energy at varied conditions. | |
3.8.2. Morphological investigation. The morphological change of Cu/TiO2 before and after the irradiation was characterized by TEM analysis. Fig. 5a depicts the formation of a nanocluster of loaded Cu2+ on the surface of TiO2.61 In contrast, after 3 h of UV (365 nm) light radiation, the nanostructure of CuO notably decreased. A small amount of fragmented Cu2+ nanoparticles were detected around the surface edge of TiO2 (Fig. 5b).62,63
 |
| | Fig. 5 TEM images of Cu/TiO2 (a) before and (b) after 3 hours of irradiation. | |
3.8.3. BET analysis. To check the specific surface area and pore structure, the N2 adsorption–desorption isotherms of the catalysts were measured at 77 K. Fig. S5(a–d)† depicted the N2 adsorption–desorption isotherms of TiO2 and Cu/TiO2 before and after the irradiation. The effect of load Cu2+ on the surface area of TiO2 was investigated. All the catalysts were classified as type IV in the IUPAC adsorption–desorption isotherm classification and there are H3-type hysteresis loops were found between P/P0 = 0.6 and P/P0 = 1. It is proven that the tested catalysts are mesoporous materials.64,65 Table S1† illustrates the surface area and pore structure parameters of tested catalysts.66 The irradiation decreased the BET surface area, total pore volume and average pore diameter. Though the pore volume and diameter gradually reduced with the increase in irradiation time, the BET surface area remained unchanged after 3 and 48 hours of irradiation (Table S1†).
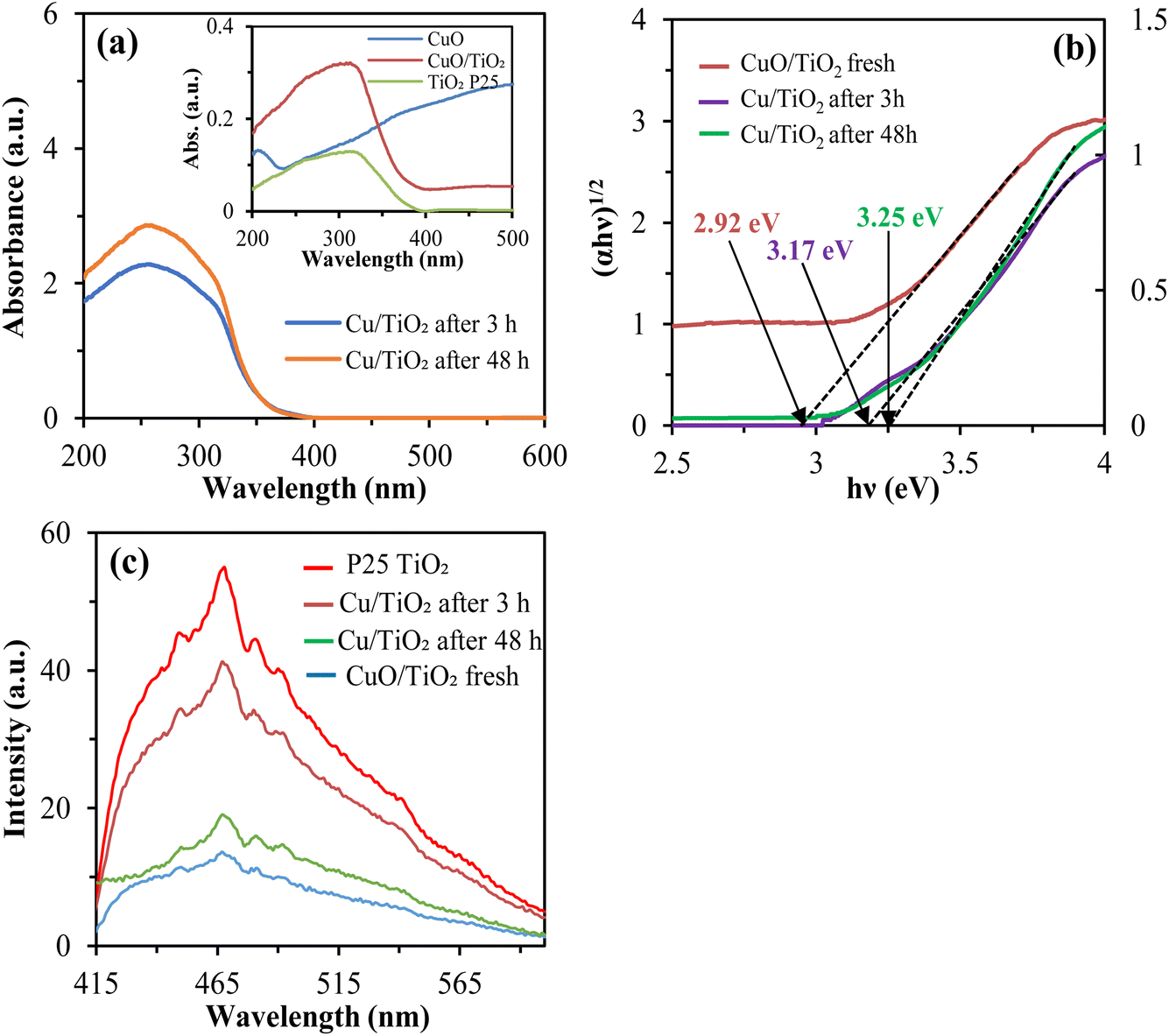
3.8.4. DRS analysis. The light-harvesting capability of the following catalysts was characterized by ultraviolet-visible diffuse reflectance spectrum (DRS) analysis. From the “Kubelka–Munk (K–M)” transformation (Fig. 6a), CuO/TiO2 showed absorption in the visible region before the irradiation due to the presence of CuO (inset).67 CuO can absorb light of broad wavelengths but has no significant photocatalytic activity due to a smaller bandgap. After the irradiation (3–48 h), the catalyst had almost no absorption in the visible region. Still, the absorption in the ultraviolet region was significantly higher than that of the before irradiation.
 |
| | Fig. 6 (a) DRS spectra, (b) Tauc-plot and (c) PL spectra of the studied catalysts. | |
Further, the Tauc equation was implemented to calculate the energy bandgap as follows;
| | |
(αhν)1/n = A(hν − Eg)
| (4) |
where
α denotes the absorption coefficient,
h is the Planck constant,
ν is the light frequency,
A is the characteristic constant and
Eg is the bandgap energy, respectively. Electronic inter-band excitation helps to determine the transition steps; when
n = 1/2, it depicts the direct transition and
n = 2 stands for the indirect transition, respectively. For anatase-type titanium oxide,
n = 2 is set because the inter-band excitation is an indirect transition.
68 From
eqn (4), the bandgap energy can be obtained by plotting (
αhν)
1/n on the vertical axis and
hν on the horizontal axis and extrapolating a straight line (
Fig. 6b). Where the energy band gap of fresh CuO/TiO
2 was found 2.92 eV, which indicating the Cu deposition on the TiO
2 surface significantly reduced the bandgap. Further, the energy band gap of Cu/TiO
2 was increased to 3.17 eV and 3.25 eV after 3 h and 48 h of light irradiation, which indicates the state change of Cu due to the photocatalytic reaction.
3.8.5. PL analysis. The photoluminescence (PL) spectral technique was utilized to examine the catalysts' electron hole-pair separation and migration efficiency. The smaller peak of the photoluminescence spectrum indicates the suppression of the recombination of photogenerated electron–hole pairs. From the experimental results (Fig. 6c), the recombination of electrons and holes is considerably capable of suppressing CuO/TiO2 before irradiation compared to P25 TiO2. Comparing the catalyst after 3 h and 48 h irradiation, the peak was smaller in the sample after 48 h irradiation, suggesting that more metallic Cu2+ was photo-deposited on the TiO2 surface and delayed the recombination rate. The possible reasons for the improved photocatalytic activity of Cu/TiO2 are either increasing light absorption or effectively capturing the photoinduced electrons due to the proper amount of oxygen vacancies and Cu ions on the TiO2 surface. Where Cu ions can work as electron traps to reduce the recombination rate of h+/e− pairs (eqn (1)). Therefore, both Cu2+ and Cu+ ions are able of accepting the photogenerated electrons.69
3.9. Photocatalysis mechanism
To better understand the simultaneous Cu-deposition on the TiO2 in photocatalytic hydrocarbon and hydrogen production from acetic acid solution, a possible reaction mechanism has been proposed combining the mentioned theories and experimental consequences (Fig. 7). Electron transfer to the conduction band of CuO is feasible due to its location just below the conduction band of TiO2 under the UV light irradiation. For the same reason, the hole transfer from TiO2's valence band to CuO's valence band is permitted and the combined effect of Cu/TiO2 increases hydrocarbon and hydrogen production.36 When light energy larger than the TiO2 bandgap is absorbed by the TiO2 semiconductor, the photogenerated electrons move toward the conduction band, resulting in photogenerated holes becoming available in the valence band (eqn (5)). The photogenerated electrons are used to reduce Cu2+ ions into metals in the aqueous acetic acid solution and metallic Cu is deposited on the TiO2 surface. Since electrons are consumed one after another, it is considered that the oxidation reaction by holes is accelerated. The photo-precipitated Cu metal is oxidized by dissolved oxygen in the system to become CuO (eqn (2) and Fig. S3c†), further dissolved in an acetic acid aqueous solution and re-existed as Cu2+ ions. As long as oxygen exists in the system, this cycle always causes the precipitation of fresh metal Cu so that the photocatalytic activity can be maintained semi-permanently.
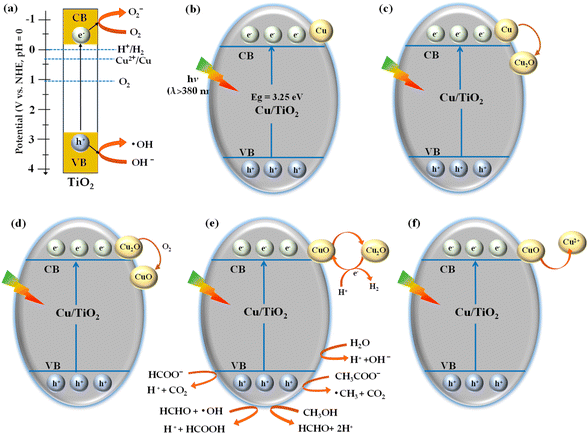 |
| | Fig. 7 Mechanism for (a) oxygen reduction, and hydroxide ion oxidation and (b–f) photocatalytic hydrocarbon and H2 production. | |
Next, the oxidation reaction mechanism of acetic acid considered in this experiment is shown below (Scheme 1 and Fig. 7b–f). Where the photogenerated holes and ˙OH radicals are influenced by the rate of water-splitting (eqn (6)). Further, the adsorbed CH3COOH dissociates to CH3COO− and comes into contact with the photogenerated holes to produce ˙CH3 and CO2, followed by the Photo-Kolbe reaction (eqn (7) and (8)). At this stage, ˙CH3 radicles react with water to release CH4 and with another ˙CH3 to produce C2H6 (eqn (9) and (10)). Methanol (CH3OH), formaldehyde (HCHO) and formic acid (HCOOH) were detected in the liquid phase during the photocatalytic decomposition of acetic acid.27 The photogenerated hole reacts with the methanol to produce formaldehyde (eqn (11) and (12)) and further formic acid was detected as an intermediate due to the oxidation reaction in the presence of ˙OH and hole (eqn (13)). Finally, the formate ion comes in contact with photogenerated holes and produces H+, which is further reduced by the photogenerated electron to evolve H2 (eqn (15)).
 |
| | Scheme 1 Reaction mechanism for the hydrocarbon production from acetic acid.47,70,71 | |
Next, the effects of dissolved oxygen and hydroxide ions on the generation of hydrogen and hydrocarbons were considered. Since the reduction potential of oxygen is on the nobler side than the potential of the conduction band of TiO2, dissolved oxygen is thought to be reduced using electrons photogenerated on titanium oxide to form superoxide anions. Therefore, when the amount of dissolved oxygen increases, the number of electrons used for proton reduction decreases, which is thought to harm hydrogen generation. In addition, it is believed that when the pH increases and the number of hydroxide ions increases, the methane produced decreases because the photogenerated holes are consumed and hydroxyl radicals are generated. The conduction band potential of TiO2 is −0.2 V (Fig. 7b), which is more negative than the oxidation–reduction potential of Cu2+/Cu of +0.34 V, so in this stage, Cu ions can be reduced to metallic Cu. A comparative TiO2 photocatalytic methane production efficiency from acetic acid result is presented in Table S3.†
4. Conclusions
In summary, the photocatalytic activity of in situ Cu-deposited TiO2 was studied for the hydrocarbon and hydrogen production from aqueous acetic acid solution under UV light irradiation. Factors that affect the photocatalytic reaction, such as substrate initial concentration, irradiation time, catalyst and co-catalyst dosage and reaction pH were optimized. The structural and optical properties of Cu/TiO2 were characterized by XPS, TEM, BET surface area, DRS and PL analysis. The appropriate work function of Cu contributed to Schottky junction formation, which facilitates electron transfer, assists in overcoming the Schottky energy barrier and leads to enhanced photocatalytic efficiency. The energy band gap and light absorption capability of Cu/TiO2 were significantly improved due to the Cu deposition. PL results confirm that the photogenerated charge separation and migration capability also improved for Cu/TiO2. The optimized Cu/TiO2 exhibited the highest methane production (4136 μmol g−1), 9.2-fold more than pure TiO2 without using any noble metal. The most straightforward technique reduced the recombination rate and energy band gap, resulting in the simultaneous production of hydrocarbon and hydrogen, which also smoothly proceeded under visible light (405 nm) irradiation and atmospheric conditions. Additionally, the excellent stability of Cu/TiO2 was proven through cycling experiments. Finally, the Cu/TiO2 can be considered a potential photocatalyst for efficiently converting solar power to chemical energy.
Data availabilty
The data that support this study's findings are available from the corresponding author upon reasonable request. Additionally, any ESI,† including figures and datasets generated during the study, will be provided in the ESI† section of the manuscript.
Author contributions
Monir Uzzaman: writing – original draft, writing – review & editing, investigation, formal analysis, methodology, visualization, data curation. Mai Furukawa: writing – review & editing. Ikki Tateishi: writing – review & editing. Hideyuki Katsumata: writing – review & editing, validation. Mst. Farhana Afrin: writing – review & editing, methodology. Satoshi Kaneco: conceptualization, supervision, methodology, validation, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was partially funded by Grant-in-Aid for Scientific Research (B) 21H03642 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- R. Falkner, Int. Aff., 2016, 92, 1107–1125 CrossRef.
- I. Dincer and C. Acar, Int. J. Energy Res., 2015, 39, 585–606 CrossRef.
- G. Yadav and M. Ahmaruzzaman, Inorg. Chem. Commun., 2022, 138, 109288 CrossRef CAS.
- Y. Yang, C. Zhou, W. Wang, W. Xiong, G. Zeng, D. Huang, C. Zhang, B. Song, W. Xue and X. Li, Chem. Eng. J., 2021, 405, 126547 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- M. S. S. Danish, L. L. Estrella, I. M. A. Alemaida, A. Lisin, N. Moiseev, M. Ahmadi, M. Nazari, M. Wali, H. Zaheb and T. Senjyu, Metals, 2021, 11, 80 CrossRef CAS.
- S. Mozia, A. Heciak and A. W. Morawski, J. Photochem. Photobiol. A Chem., 2010, 216, 275–282 CrossRef CAS.
- I. Paramasivam, H. Jha, N. Liu and P. Schmuki, Small, 2012, 8, 3073–3103 CrossRef CAS PubMed.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol. C Photochem. Rev., 2013, 15, 1–20 CrossRef CAS.
- Z. Zhang, C.-C. Wang, R. Zakaria and J. Y. Ying, J. Phys. Chem. B, 1998, 102, 10871–10878 CrossRef CAS.
- H. D. Jang, S.-K. Kim and S.-J. Kim, J. Nano. Res., 2001, 3, 141–147 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol. C Photochem. Rev., 2008, 9, 1–12 CrossRef CAS.
- N. Yahya, F. Aziz, N. A. Jamaludin, M. A. Mutalib, A. F. Ismail, W. N. W. Salleh, J. Jaafar, N. Yusof and N. A. Ludin, J. Environ. Chem. Eng., 2018, 6, 7411–7425 CrossRef CAS.
- K. Nishijima, B. Ohtani, X. Yan, T. Kamai, T. Chiyoya, T. Tsubota, N. Murakami and T. Ohno, Chem. Phys., 2007, 339, 64–72 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505–516 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 2239–2240 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- C. He, D. Shu, M. Su, D. Xia, M. Abou Asi, L. Lin and Y. Xiong, Desalination, 2010, 253, 88–93 CrossRef CAS.
- G. Colón, M. Maicu, M. C. s Hidalgo and J. A. Navío, Appl. Catal., B, 2006, 67, 41–51 CrossRef.
- X.-J. Zheng, L.-F. Wei, Z.-H. Zhang, Q.-J. Jiang, Y.-J. Wei, B. Xie and M.-B. Wei, Int. J. Hydrogen Energy, 2009, 34, 9033–9041 CrossRef CAS.
- M. A. Barakat, H. Schaeffer, G. Hayes and S. Ismat-Shah, Appl. Catal., B, 2005, 57, 23–30 Search PubMed.
- J. Serafin, M. Ouzzine, J. Sreńscek-Nazzal and J. Llorca, J. Photochem. Photobiol. A Chem., 2022, 425, 113726 CrossRef CAS.
- S. Asal, M. Saif, H. Hafez, S. Mozia, A. Heciak, D. Moszyński and M. S. A. Abdel-Mottaleb, Int. J. Hydrogen Energy, 2011, 36, 6529–6537 CrossRef CAS.
- Z. H. N. Al-Azri, W.-T. Chen, A. Chan, V. Jovic, T. Ina, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 329, 355–367 CrossRef CAS.
- S. Mozia, A. Heciak and A. W. Morawski, Appl. Catal., B, 2011, 104, 21–29 CrossRef CAS.
- S. Mozia, A. Heciak, D. Darowna and A. W. Morawski, J Photochem. Photobiol. A Chem., 2012, 236, 48–53 CrossRef CAS.
- S. Mozia, A. Heciak and A. W. Morawski, Catal. Today, 2011, 161, 189–195 CrossRef CAS.
- A. Heciak, A. W. Morawski, B. Grzmil and S. Mozia, Appl. Catal., B, 2013, 140, 108–114 CrossRef.
- A. Amorós-Pérez, L. Cano-Casanova, M. Á. Lillo-Ródenas and M. C. Román-Martínez, Catal. Today, 2017, 287, 78–84 Search PubMed.
- L. M. Betts, F. Dappozze and C. Guillard, Appl Catal, A, 2018, 554, 35–43 CrossRef CAS.
- Y. Cao, S. N. Lou and T. Ohno, Mater. Lett., 2023, 347, 134552 Search PubMed.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol. C Photochem. Rev., 2012, 13, 169–189 CrossRef CAS.
- A. Madhumitha, V. Preethi and S. Kanmani, Int. J. Hydrogen Energy, 2018, 43, 3946–3956 CrossRef CAS.
- D. Guerrero-Araque, P. Acevedo-Peña, D. Ramírez-Ortega, L. Lartundo-Rojas and R. Gómez, J. Chem. Technol. Biotechnol., 2017, 92, 1531–1539 Search PubMed.
- S. Chen, Q. Shi, Z. Xue, X. Sun and W. Xiang, Int. J. Hydrogen Energy, 2011, 36, 8915–8926 Search PubMed.
- T. Miwa, S. Kaneco, H. Katsumata, T. Suzuki, K. Ohta, S. Chand Verma and K. Sugihara, Int. J. Hydrogen Energy, 2010, 35, 6554–6560 CrossRef CAS.
- A. Pérez-Larios, R. Lopez, A. Hernández-Gordillo, F. Tzompantzi, R. Gómez and L. M. Torres-Guerra, Fuel, 2012, 100, 139–143 CrossRef.
- C. Wang, X. Cai, Y. Chen, Z. Cheng, X. Luo, S. Mo, L. Jia, P. Lin and Z. Yang, Chem. Eng. J., 2017, 317, 522–532 Search PubMed.
- Md. T. Uddin, Y. Nicolas, C. Olivier, W. Jaegermann, N. Rockstroh, H. Junge and T. Toupance, Phys. Chem. Chem. Phys., 2017, 19, 19279–19288 RSC.
- M. Muscetta, R. Andreozzi, L. Clarizia, I. Di Somma and R. Marotta, Int. J. Hydrogen Energy, 2020, 45, 28531–28552 CrossRef CAS.
- G. Li, J. Huang, J. Chen, Z. Deng, Q. Huang, Z. Liu, W. Guo and R. Cao, ACS Omega, 2019, 4, 3392–3397 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 2004, vol. 85 Search PubMed.
- T. Aarthi and G. Madras, Catal. Commun., 2008, 9, 630–634 CrossRef CAS.
- P. Gomathisankar, D. Yamamoto, H. Katsumata, T. Suzuki and S. Kaneco, Int. J. Hydrogen Energy, 2013, 38, 5517–5524 CrossRef CAS.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 Search PubMed.
- T. Ano, F. Kishimoto, S. Tsubaki, Y.-H. Lu, J. N. Hohman, M. M. Maitani, M. Salmeron and Y. Wada, J. Phys. Chem. C, 2021, 125, 13984–13989 CrossRef CAS.
- M. Uzzaman, M. F. Afrin, M. Furukawa, I. Tateishi, H. Katsumata and S. Kaneco, ChemEngineering, 2024, 8, 75 CrossRef CAS.
- M. H. Suhag, I. Tateishi, M. Furukawa, H. Katsumata, A. Khatun and S. Kaneco, ChemEngineering, 2022, 6, 43 CrossRef CAS.
- M. Zeng, Bull. Korean Chem. Soc., 2013, 34, 953–956 Search PubMed.
- E. N. Bakatula, D. Richard, C. M. Neculita and G. J. Zagury, Environ. Sci. Pollut. Res., 2018, 25, 7823–7833 Search PubMed.
- P. Gomathisankar, K. Hachisuka, H. Katsumata, T. Suzuki, K. Funasaka and S. Kaneco, Int. J. Hydrogen Energy, 2013, 38, 11840–11846 CrossRef CAS.
- G. Li, N. M. Dimitrijevic, L. Chen, T. Rajh and K. A. Gray, J. Phys. Chem. C, 2008, 112, 19040–19044 CrossRef CAS.
- X. J. Zheng, Y. J. Wei, L. F. Wei, B. Xie and M. B. Wei, Int. J. Hydrogen Energy, 2010, 35, 11709–11718 CrossRef CAS.
- K. Rajendran, M. Sharma, A. Jaison, M. Ankitha, A. D. Tiwari, C. P. Vinod and D. Jagadeesan, Catal. Sci. Technol., 2023, 13, 2330–2339 Search PubMed.
- P. C. Okoye, S. O. Azi, T. F. Qahtan, T. O. Owolabi and T. A. Saleh, Mater. Today Chem., 2023, 30, 101513 CrossRef CAS.
- L. Xu, G. Zheng, S. Pei and J. Wang, Optik, 2018, 158, 382–390 Search PubMed.
- K. Nagaveni, M. S. Hegde, N. Ravishankar, G. N. Subbanna and G. Madras, Langmuir, 2004, 20, 2900–2907 CrossRef CAS PubMed.
- T. Aguilar, J. Navas, R. Alcántara, C. Fernández-Lorenzo, J. J. Gallardo, G. Blanco and J. Martín-Calleja, Chem. Phys. Lett., 2013, 571, 49–53 CrossRef CAS.
- B. Srinivas, B. Shubhamangala, K. Lalitha, P. Anil Kumar Reddy, V. Durga Kumari, M. Subrahmanyam and B. R. De, Photochem. Photobiol., 2011, 87, 995–1001 Search PubMed.
- B. Xin, P. Wang, D. Ding, J. Liu, Z. Ren and H. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS.
- A. Moghanian, F. Sharifianjazi, P. Abachi, E. Sadeghi, H. Jafarikhorami and A. Sedghi, J. Alloys Compd., 2017, 698, 518–524 Search PubMed.
- Y. Gao and S. A. Elder, Mater. Lett., 2000, 44, 228–232 Search PubMed.
- S. Xu and D. D. Sun, Int. J. Hydrogen Energy, 2009, 34, 6096–6104 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- M. Uzzaman, M. H. Suhag, H. Katsumata, I. Tateishi, M. Furukawa and S. Kaneco, Catal. Sci. Technol., 2024, 14, 267–278 RSC.
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134–135, 349–358 CrossRef CAS.
- W. E. Vargas and G. A. Niklasson, Appl. Opt., 1997, 36, 5580 CrossRef CAS PubMed.
- A. Dolgonos, T. O. Mason and K. R. Poeppelmeier, J. Solid State Chem., 2016, 240, 43–48 CrossRef CAS.
- B. Xin, P. Wang, D. Ding, J. Liu, Z. Ren and H. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS.
- X.-J. Zheng, L.-F. Wei, Z.-H. Zhang, Q.-J. Jiang, Y.-J. Wei, B. Xie and M.-B. Wei, Int. J. Hydrogen Energy, 2009, 34, 9033–9041 CrossRef CAS.
- M. F. Afrin, M. Furukawa, I. Tateishi, H. Katsumata, M. Uzzaman and S. Kaneco, J. Compos. Sci., 2025, 9(2), 68 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mai Furukawaa,
Ikki Tateishib,
Hideyuki Katsumata
*a,
Mai Furukawaa,
Ikki Tateishib,
Hideyuki Katsumata