DOI:
10.1039/D4RA08639B
(Paper)
RSC Adv., 2025,
15, 5277-5285
Unveiling the efficacy and mechanism of chlortetracycline degradation by MnFeCu-LDH/GO activating of peroxymonosulfate†
Received
8th December 2024
, Accepted 2nd February 2025
First published on 17th February 2025
Abstract
Antibiotics are frequently detected in aquatic environments, posing potential risks to ecological systems and human health. Among the various treatment strategies, advanced oxidation processes (AOPs) utilizing peroxymonosulfate (PMS) activation stand out due to their ease of activation and potent oxidative capabilities. This study synthesized manganese–iron–copper layered double hydroxides coupled with graphene oxide (MnFeCu-LDH/GO) using eco-friendly transition metals. Optimal conditions of pH 7, 298 K, and 0.2 g L−1 dosage of both catalyst and PMS achieved 98% CTC removal in 30 minutes. The composite was employed for the degradation of chlortetracycline (CTC) through PMS activation. Reactive oxygen species (ROS), including 1O2, ˙OH, and SO4˙−, were identified and quantified through trapping and quenching experiments, and 1O2 was identified, to be the most significant contributor to degradation. The catalyst demonstrated excellent reusability, maintaining high degradation efficiency after five cycles. These findings suggest that MnFeCu-LDH/GO-activated PMS is a promising method for mitigating antibiotic contamination in water.
1. Introduction
Antibiotics are essential in treating infectious diseases, promoting human health, and supporting animal growth. However, a significant portion, ranging from 30% to 90%, is excreted in urine and feces, either unchanged or as metabolites, and ultimately ends up in water systems.1 While concentrations of antibiotics in aquatic environments can range from nanograms per liter to milligrams per liter, even low levels pose potential risks to ecosystems and human health.2,3 The presence of antibiotics in the environment is a key factor in the development and spread of antibiotic-resistant genes (ARGs), a growing global health concern.4 Despite efforts to manage wastewater, current treatment plants are ineffective at fully removing antibiotics, as evidenced by the ongoing detection of a wide range of antibiotics in both effluent and sludge. Furthermore, untreated or inadequately treated wastewater from rural households, livestock farms, and aquaculture facilities in many developing countries often enters water bodies.5,6 The lack of a universally effective treatment method for antibiotic-contaminated wastewater underscores the urgent need for more efficient treatment technologies to address this issue.
Advanced Oxidation Processes (AOPs) have attracted significant attention due to their ability to generate highly reactive species (e.g., hydroxyl radicals (˙OH), etc.), which are effective in degrading trace, high-risk antibiotics.7,8 AOPs utilizing persulfates, such as peroxymonosulfate (PMS) and peroxydisulfate (PDS), produce reactive species like, ˙OH, sulfate radicals (SO4˙−), and singlet oxygen (1O2).9 These processes offer several advantages, including the removal of a wide range of organic pollutants, applicability across different pH levels, ease of peroxysulfate storage, and straightforward activation methods, making them promising for environmental remediation.10,11 Among various activation methods, heterogeneous metal catalysts are often studied for persulfate activation due to their cost-effectiveness, ease of use, and high efficiency.12,13 Consequently, there is an urgent need to develop environmentally friendly transition metals for catalyst development, ensuring both the effectiveness of AOPs based on persulfate activation and their environmental sustainability.
Layered double hydroxides (LDHs), also known as anionic clays or hydrotalcite-like materials, are a class of two-dimensional inorganic compounds with the ability to intercalate anionic species within their interlayer spaces.14,15 The versatility of LDHs arises from their tunable composition, which includes the ratio of lamellar metal elements and interlayer anions, as well as the homogeneous distribution of metal cations. These characteristics make LDHs suitable for various applications, especially in multiphase catalysis. LDHs are well-known for their ability to activate O–O bonds in hydrogen peroxide (H2O2) to generate hydroxyl radicals (˙OH),16,17 which suggests their potential to activate O–O bonds in persulfate salts (PMS) as well. Despite this potential, the use of LDHs for PMS activation remains relatively underexplored. Yang et al. have investigated Co-based LDHs and their ability to activate PMS for organic dye degradation,18 demonstrating effective catalytic activity. However, the leaching of metal ions, particularly Co2+, from these catalysts during activation poses significant risks to human health and the environment, as Co2+ ions are toxic and potentially carcinogenic even in small amounts.19,20
Transition metals such as iron and manganese are expected to replace Co to achieve green activation of PMS. For example, studies have found that FeMn-LDH activated PMS to achieve the degradation of sulfamethoxazole.21 Compared with Co catalyst, the activation performance still needs to be further improved. Here, we have noticed that some scholars have introduced copper into the catalyst, which can effectively improve the electron transfer efficiency.22 Therefore, the introduction of copper into ferromanganese hydrotalcite to prepare ternary hydrotalcite may also effectively improve the efficiency of catalyst activation PMS. In addition, Hybridization with graphene oxide (GO) can also improve catalytic activity and stability.23 However, at present, the performance of ternary LDH/GO activated PMS and the electron transfer path in the activation process are still unclear.
In the present study, we proposed to prepare MnFeCu-LDH/GO composite material by hybridising MnFeCu-LDH with GO, to achieve stable, efficient and green activation of PMS. The impact of various factors, including catalyst amount, initial pH, and temperature, on CTC degradation efficiency was investigated. The generation of ROS during the degradation process was analyzed using electron paramagnetic resonance (EPR) spectroscopy, and their contribution to CTC degradation were further assessed. The physicochemical properties of the catalysts, both before and after PMS activation, were compared, and their cyclic stability was evaluated through catalyst recycling and reuse, alongside monitoring CTC degradation efficiency. This study aims to introduce a novel approach for removing antibiotics from aquatic environments.
2. Material and methods
2.1 Chemicals reagents
Chlortetracycline, manganese nitrate tetrahydrate, copper nitrate trihydrate, ferric nitrate nonahydrate, p-benzoquinone, and furfuryl alcohol were purchased from Shanghai Aladdin Company. Hydrochloric acid, sodium hydroxide, sodium carbonate, tert-butanol, and anhydrous ethanol were procured from the Shanghai Macklin Company.
2.2 Preparation of catalysts
MnFeCu-LDHs were synthesized using a hydrothermal method. First, 1 mmol of iron nitrate and manganese nitrate, along with 2 mmol of copper nitrate, were dissolved in 100 mL of deionized water and sonicated for 30 minutes. This solution was then added dropwise to a sodium hydroxide and sodium carbonate solution under vigorous stirring, maintaining a pH of 10. The mixture was transferred to a hydrothermal reactor and heated at 140 °C for 16 hours. Afterward, the product was centrifuged, washed with ethanol and water until neutral, and dried in a vacuum oven at 55 °C for 24 hours. To prepare the MnFeCu-LDH/GO composite, 0.05 g of graphene oxide (GO) was ultrasonically dispersed in water for 8 hours. The GO solution was added at a rate of 3 mL min−1 during the metal solution addition. The final composite product was then subjected to centrifugation, washing, and drying.
2.3 Characterization of catalyst
The surface morphology, crystal structure, and shape of the synthesized ternary hydrotalcites were characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray diffraction (XRD). Furthermore, X-ray photoelectron spectroscopy (XPS) was employed to analyze the ternary hydrotalcites both before and after the reaction, enabling the comparison of elemental valence state transformations during the reaction process.
2.4 Catalytic degradation experiments of CTC
The experiments were conducted under consistent conditions: 40 mg of the catalyst was added to 200 mL of CTC solution (10 mg L−1) at 25 °C and an initial pH of 7, yielding a catalyst concentration of 0.2 g L−1. The degradation experiments in this study were conducted under dark conditions. The catalyst was stirred for 30 minutes using a magnetic stirrer in the absence of light to achieve adsorption–desorption equilibrium. A solution of 40 mg of PMS was then added, resulting in a concentration of 0.2 g L−1. Samples were taken at 2.5, 5, 10, 15, 20, and 30 minutes, passed through 0.22 μm microporous membrane within 30 seconds. The concentrations of CTC were measured by liquid chromatography equipped with UV/visible and photodiode array detection (HPLC-DAD; Agilent 1260). HPLC grade water, methanol and acetonitrile (with 0.025 M KH2PO4) were used as the mobile phase. The analytes were separated in an Agilent C18 column (4.6 × 75 mm, 5 μm).
2.5 Data analysis
Statistical analysis was performed using univariate and one-way ANOVA with SPSS 26.0 software (IBM Corporation, Armonk, NY). A value of p < 0.01 was considered statistically significant.
3. Results and discussion
3.1 Characterization of MnFeCu-LDH/GO
The surface morphology and crystal type of the synthesized ternary hydrotalcites were characterized using scanning electron microscopy (SEM), and X-ray diffraction (XRD). Additionally, the ternary hydrotalcites were characterized using X-ray photoelectron spectroscopy (XPS) before and after the reaction, for comparison of elemental valence states before and after the reaction. The results of the characterization of MnFeCu-LDH/GO are presented in Fig. 1. Fig. 1(a) and (b) present the SEM micrographs of MnFeCu-LDH/GO, which reveal that MnFeCu-LDH/GO exhibits a lamellar structure. The single particles display relatively flat hydroxide layer surfaces and discernible stacking structures.24 Moreover, TEM images (Fig. S1 in ESI†) clearly depict the layered structure of the MnFeCu-LDH/GO composite, confirming the intercalation of graphene oxide (GO) within the MnFeCu-LDH structure. Furthermore, the petal-like LDH is observed as being attached to graphene with a large surface area, thus providing additional active sites for the catalytic reaction.
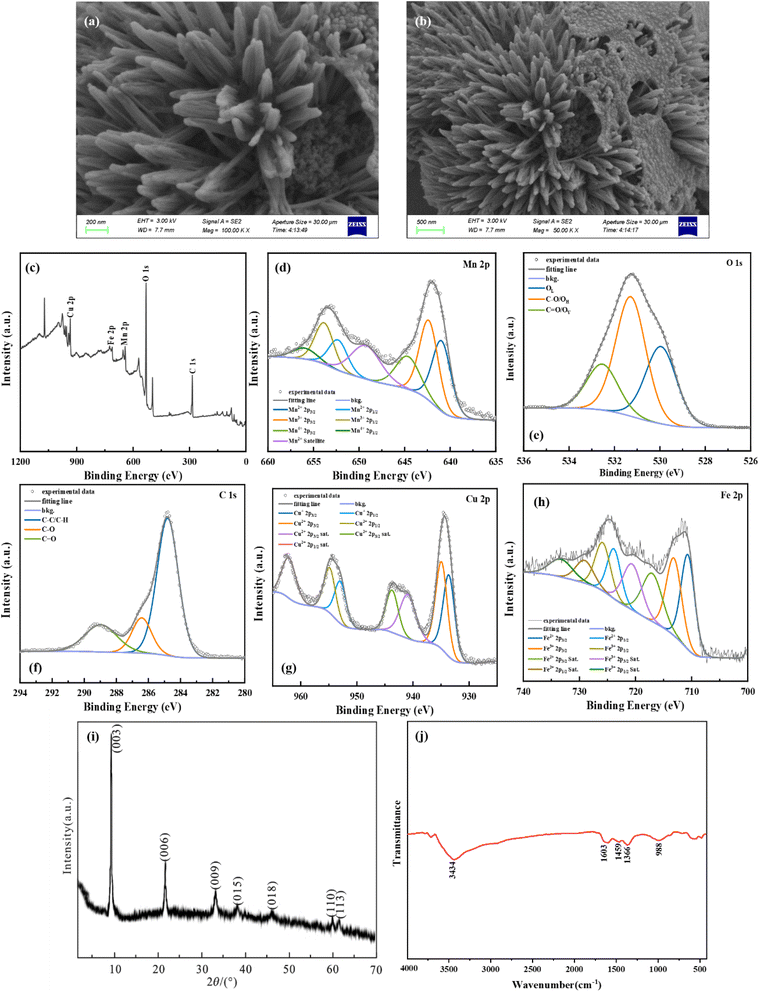 |
| | Fig. 1 The characterization of MnFeCu-LDH/GO: (a) and (b) present the SEM micrographs of MnFeCu-LDH/GO, (c) to (h) present the full XPS spectrum, the Mn 2p fine spectrum, the O 1s fine spectrum, the C 1s fine spectrum, the Cu 2p fine spectrum, and the Fe 2p fine spectrum of MnFeCu-LDH/GO, (i) present XRD pattern of MnFeCu-LDH/GO, (j) present Fourier infrared spectra. | |
Fig. 1(c)–(h) present the full XPS spectrum, the Mn 2p fine spectrum, the O 1s fine spectrum, the C 1s fine spectrum, the Cu 2p fine spectrum, and the Fe 2p fine spectrum of MnFeCu-LDH/GO, respectively. The carbon content is markedly elevated following the doping of graphene oxide. The peak splitting results indicate that the contents of Mn2+, Mn3+, and Mn4+ were 36.49%, 41.46%, and 22.05%, respectively. Similarly, the contents of Fe2+ and Fe3+ were 52.18% and 47.82%, respectively, while the contents of Cu+ and Cu2+ were 48.35% and 51.65%, respectively. Furthermore, there is a notable reduction in the lattice oxygen content from 45.20% to 32.24% in MnFeCu-LDH, while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond content increases from 6.64% to 19.45%. Fig. 1(i) depicts the XRD pattern of MnFeCu-LDH/GO, which exhibits a peak shape analogous to that of MnFeCu-LDH, accompanied by pronounced characteristic diffraction peaks of hydrotalcite. Fig. 1(j) depicts the Fourier infrared spectra of MnFeCu-LDH/GO. Following the addition of graphene oxide, the peak observed at 3434 cm−1 in MnFeCu-LDH/GO is attributed to the hydroxyl group stretching vibration, while the peak at 988 cm−1 is ascribed to the C–O bonding stretching vibration. Additionally, the characteristic metal–oxygen bonding peaks are diminished near 473 cm−1, indicating a reduction in the metal–oxygen bonding content following the doping of graphene oxide.25 This finding aligns with the observed decline in the oxygen content of the lattice, as evidenced by the XPS analysis.
O bond content increases from 6.64% to 19.45%. Fig. 1(i) depicts the XRD pattern of MnFeCu-LDH/GO, which exhibits a peak shape analogous to that of MnFeCu-LDH, accompanied by pronounced characteristic diffraction peaks of hydrotalcite. Fig. 1(j) depicts the Fourier infrared spectra of MnFeCu-LDH/GO. Following the addition of graphene oxide, the peak observed at 3434 cm−1 in MnFeCu-LDH/GO is attributed to the hydroxyl group stretching vibration, while the peak at 988 cm−1 is ascribed to the C–O bonding stretching vibration. Additionally, the characteristic metal–oxygen bonding peaks are diminished near 473 cm−1, indicating a reduction in the metal–oxygen bonding content following the doping of graphene oxide.25 This finding aligns with the observed decline in the oxygen content of the lattice, as evidenced by the XPS analysis.
3.2 Degradation efficiency of pollutants
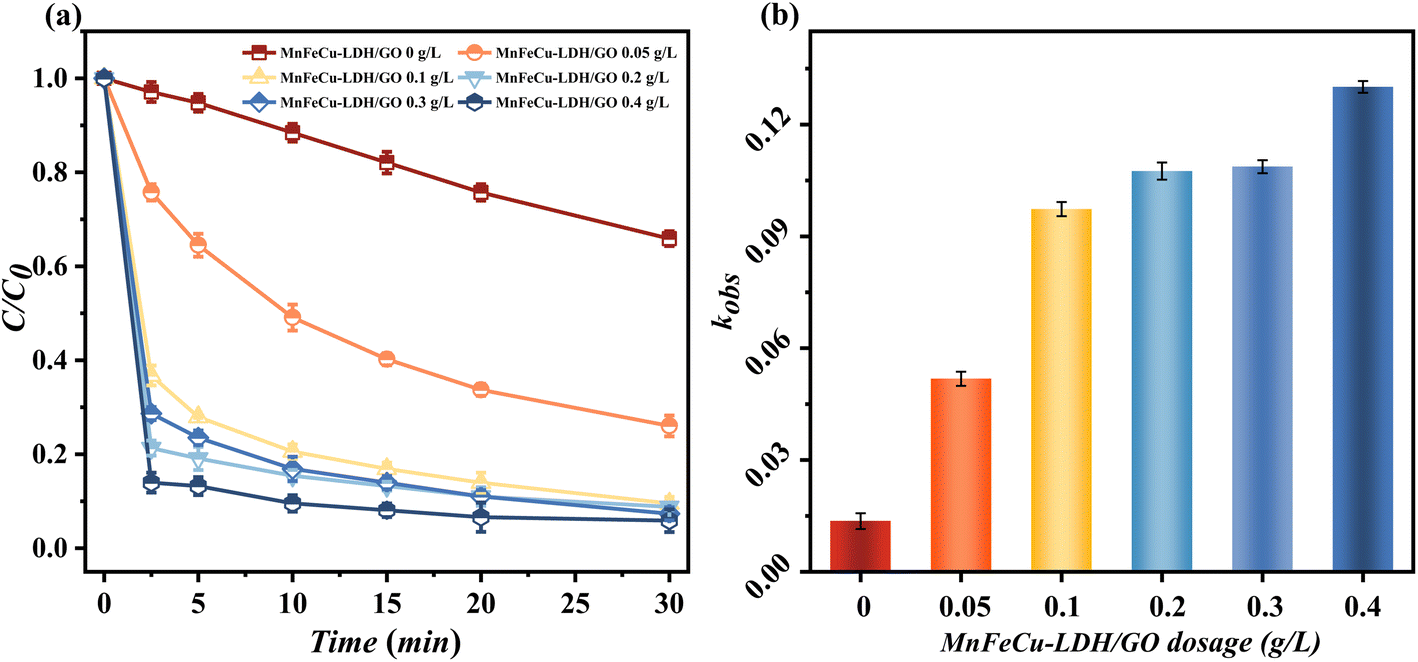
The effect of varying CTC concentrations on the degradation efficiency was firstly investigated (Fig. S2†). The degradation of chlortetracycline (CTC) at various initial concentrations, with 10 mg L−1 being the highest concentration tested. As the initial concentration of CTC increases, the degradation efficiency decreases, with lower degradation observed at higher concentrations. Therefore, we chose 10 mg L−1 as the initial concentration in the subsequent experiment. Fig. 2 illustrates the degradation effect of MnFeCu-LDH/GO-activated PMS for CTC degradation at varying catalyst dosages. The experimental results show that PMS alone has a limited ability to degrade CTC, achieving only 24.1% degradation within 30 minutes. This is likely due to PMS's inherent oxidation capacity, which produces a small amount of ˙OH free radicals in water.26 However, when MnFeCu-LDHs were added as a catalyst (0.2 g L−1), the concentration of CTC decreased rapidly within 2.5 minutes, with a removal rate of 78.68%, which further increased to 91.18% after 30 minutes. These results indicate that MnFeCu-LDHs effectively activate PMS, producing a variety of active species that facilitate the efficient degradation of CTC. Further experiments with varying catalyst concentrations (0.05, 0.1, 0.3, and 0.4 g L−1), while maintaining the PMS dosage at 0.2 g L−1, showed that increasing the catalyst concentration from 0.05 g L−1 to 0.2 g L−1 significantly improved the degradation efficiency. The degradation kinetic constants were fitted using a pseudo-first-order kinetic model, as shown in Fig. 2(b), the degradation rate constant increased with catalyst concentration (from 0.048 min−1 to 1.31 min−1). This is because a higher catalyst concentration provides more active sites and free radicals for PMS activation, thus enhancing CTC degradation. However, when the catalyst concentration increased further to 0.4 g L−1, the degradation curve leveled off, and the improvement in CTC degradation became minimal. This suggests that PMS activation reached saturation at this concentration, with PMS concentration becoming the rate-limiting factor for CTC catalytic degradation.
 |
| | Fig. 2 The degradation efficiency of CTC at different dosage MnFeCu-LDH/GO, (a) present removal rate, (b) present obvious rate constant of degradation kinetics (kobs) (catalyst dosage: 0.05–0.4 g L−1, PMS dosage: 0.2 g L−1, reaction temperature = 25 °C). | |
The effect of pH on MnFeCu-LDH/GO activating PMS to degrade CTC was studied as shown in Fig. 3(a). At an initial pH of 4, the catalytic degradation efficiency of MnFeCu-LDH/GO was 68.38%, 79.41%, and 87.50% at 5, 15, and 30 minutes, respectively. At an initial pH of 6, the catalytic degradation efficiencies at 5, 15, and 30 minutes were 68.44%, 77.21%, and 85.29%. While, the 0.5 hour degradation efficiencies of CTC exhibited a slight increase at pH values of 7, 8, and 10, with respective values of 91.18%, 93.38%, and 97.06%. Overall, MnFeCu-LDH/GO demonstrates greater property with pH increasing.
 |
| | Fig. 3 The degradation efficiency at different initial pH and temperature of MnFeCu-LDH/GO, (a) present degradation efficiency at different initial pH, (b) present degradation efficiency at different initial temperature (reaction conditions: initial CTC concentration = 10 mg L−1, catalyst dosage = 0.2 g L−1, PMS dosage = 0.2 g L−1 and reaction temperature = 25 °C). | |
This was done The CTC degradation ability of MnFeCu-LDH/GO-activated PMS at different temperatures was investigated. The results of this investigation are presented in Fig. 3(b). The CTC degradation capacity of MnFeCu-LDH/GO-activated PMS was markedly enhanced at all temperatures. At 298 K, the catalytic degradation efficiency reached 73.96% after 30 minutes. At 308 K, the efficiency increased significantly to 88.17% within the same duration. A rapid degradation was observed at 318 K, where 98.82% of the target compound was degraded. At 328 K, the degradation efficiency achieved 99.40%, TOF of MnFeCu-LDH/GO catalyst was prepared to be 0.77 h−1, indicating the reaction reached near-complete degradation in a shorter time at elevated temperatures. When compared to other advanced oxidation processes (AOPs) and photocatalytic systems, our MnFeCu-LDH/GO catalyst shows promising results. For instance, similar composite catalysts, such as Mn-based layered double hydroxides (LDHs) activated by PMS, have been shown to efficiently degrade organic pollutants, but typically, the degradation efficiency reaches about 80–90% under similar experimental conditions. Overall, the reaction rate constants of MnFeCu-LDH/GO at various temperatures were markedly enhanced.
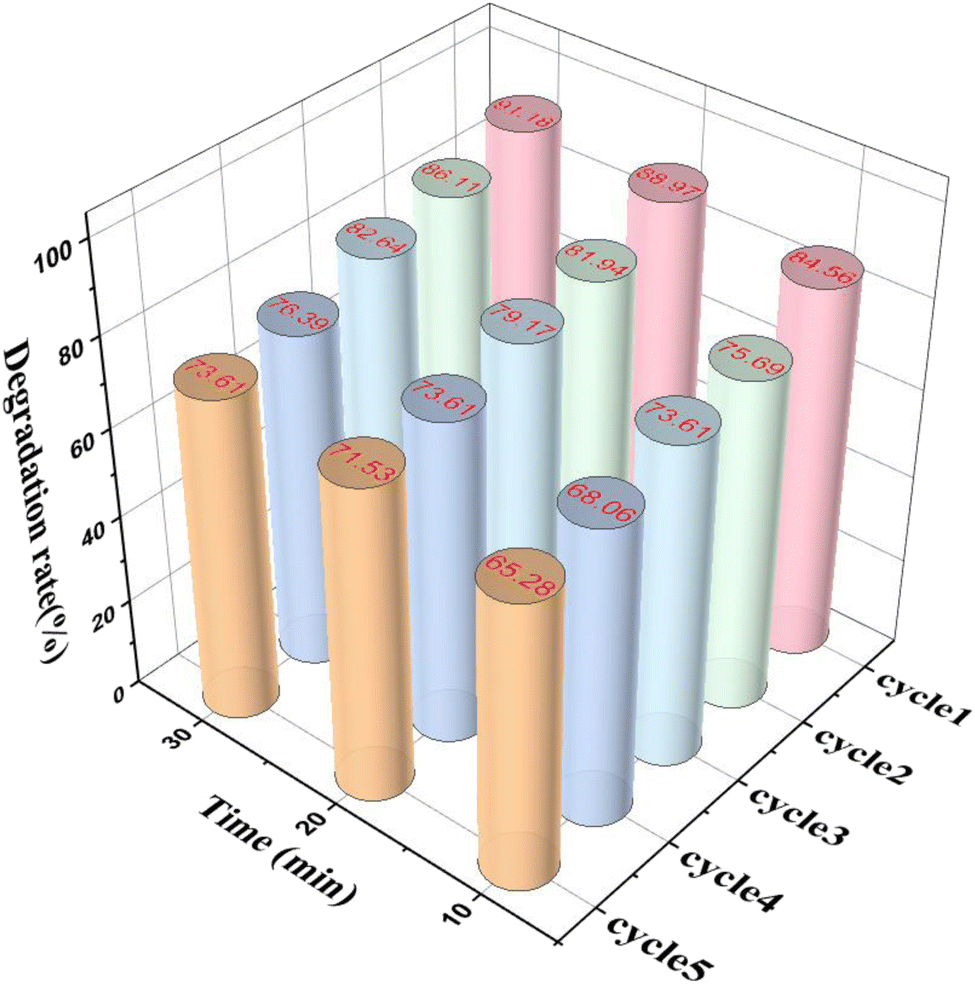
The MnFeCu-LDH/GO catalyst was recovered post-reaction by centrifugation, followed by drying, milling, and subsequent reuse for the catalytic degradation of CTC. The cyclic performance results are shown in Fig. 4, after three consecutive cycles, the catalytic degradation efficiencies for MnFeCu-LDH/GO were 73.61% at 10 minutes and 82.64% at 30 minutes, respectively. These findings underscore the superior reusability and sustained catalytic performance of MnFeCu-LDH/GO.
 |
| | Fig. 4 The reusability of MnFeCu-LDH/GO (reaction conditions: initial CTC concentration = 10 mg L−1, catalyst dosage = 0.2 g L−1, PMS dosage = 0.2 g L−1, reaction temperature = 25 °C and initial pH = 7). | |
The post-reaction solutions with initial pH values of 4, 6, 7, 8, and 10 were subsequently collected and filtered through a 0.22 μm microporous membrane to assess the concentration of dissolved metal ions from MnFeCu-LDH/GO. The results, as shown in Fig. 5, indicate that the doping of graphene oxide significantly reduced the dissolution of metal ions from MnFeCu-LDH/GO compared to the undoped MnFeCu-LDH. The total quantities of dissolved metal ions from MnFeCu-LDH/GO at pH 4, 6, 7, 8, and 10 were 4145, 3010, 2335, 1995, and 1970 ppb, respectively, markedly lower than those observed for MnFeCu-LDH, which were 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 565, 7825, 8005, 5910, and 2720 ppb, respectively. This corresponds to a reduction in dissolved metal ions of 67.01%, 61.53%, 70.83%, 66.24%, and 27.57% at the respective pH values. These findings suggest that the graphene oxide-modified catalysts not only exhibit enhanced environmental stability but also demonstrate more sustainable performance in terms of metal ion dissolution.
565, 7825, 8005, 5910, and 2720 ppb, respectively. This corresponds to a reduction in dissolved metal ions of 67.01%, 61.53%, 70.83%, 66.24%, and 27.57% at the respective pH values. These findings suggest that the graphene oxide-modified catalysts not only exhibit enhanced environmental stability but also demonstrate more sustainable performance in terms of metal ion dissolution.
 |
| | Fig. 5 Leaching of ions after the reactions at different initial pH of MnFeCu-LDH/GO, (a) for MnFeCu-LDH, (b) for MnFeCu-LDH/GO (reaction conditions: initial CTC concentration = 10 mg L−1, catalyst dosage = 0.2 g L−1, PMS dosage = 0.2 g L−1 and reaction temperature = 25 °C). | |
3.3 Mechanism of degradation of CTC by MnFeCu-LHDs/PMS
To investigate the generation of reactive oxygen species (ROS) in the reaction system, DMPO was used to capture ˙OH and SO4˙− radicals, while TEMP was employed to capture O2˙− and 1O2 species. The characteristic signal peaks for each ROS were analyzed using electron paramagnetic resonance (EPR) (Fig. 6). The results confirmed the presence of distinct signal peaks corresponding to ˙OH, SO4˙−, O2˙−, and 1O2 within the reaction system. Notably, the most pronounced signal peaks for 1O2, ˙OH, and O2˙− appeared at 1 minute and 10 minutes after the addition of PMS. In contrast, the signal peaks for SO4˙− were strongest at 1 minute but significantly diminished by 10 minutes. This reduction in intensity at the 10 minute mark suggests that SO4˙− is less stable and plays a less significant role in the later stages of the reaction.
 |
| | Fig. 6 Effect of radicals quenching on degradation of CTC and EPR spectra in MnFeCu-LDH/GO activating PMS system, (a) present ˙OH and SO4˙−, (b) and (c) present 1O2 and O2˙−, respectively, (d) present degradation efficiency of CTC under different quencher. | |
To further elucidate the contribution of individual reactive species to the degradation of CTC, various quenching agents were introduced into the reaction system. Anhydrous ethanol (EtOH) was used to quench both ˙OH and SO4˙– radicals, tertiary butyl alcohol (TBA) was applied to quench ˙OH, p-benzoquinone (p-BQ) to quench O2˙–, and furfuryl alcohol (FFA) was used to quench 1O2.27,28 The results, presented in Fig. 6, demonstrate a reduction in CTC degradation efficiency following the addition of these quenchers. Specifically, the 0.5 hour removal efficiency decreased from 91.18% (in the absence of quenchers) to 76.47% upon the addition of ethanol. Similarly, the introduction of TBA resulted in a 0.5 hour removal efficiency of 85.29%, indicating the involvement of both sulfate and hydroxyl radicals in the system. Upon the addition of FFA, the removal efficiency decreased significantly to 38.24%, suggesting a much stronger inhibitory effect compared to EtOH and TBA. This result underscores the critical role of 1O2 in the reaction. Furthermore, the reduction in O2˙– after p-BQ addition supports the conclusion that 1O2 generation is a key factor. Collectively, these findings confirm that non-radical processes, specifically those involving 1O2, represent the primary mechanism governing CTC degradation in this system.
The XPS analysis (Fig. 7) reveals the presence of divalent and trivalent manganese and iron ions, along with monovalent and divalent copper ions in the MnFeCu-LDH/PMS system, which are capable of activating PMS to generate potent oxidizing species through redox reactions. Notably, the proportions of these transition metals' valence states underwent significant changes before and after the reaction. Specifically, the proportion of Mn2+ decreased from 39.67% to 35.84%, while Mn3+ increased from 44.9% to 49.28%. Similarly, the proportion of Fe3+ decreased slightly from 36.05% to 35.37%, while Fe2+ increased from 63.95% to 64.63%. For copper, the proportion of Cu+ increased from 40.51% to 44.36%, whereas Cu2+ decreased from 59.49% to 55.64%. Furthermore, a 2.93% reduction in lattice oxygen content observed in the O 1s spectrum suggests the involvement of oxygen-containing functional groups on the catalyst surface in the PMS activation process.
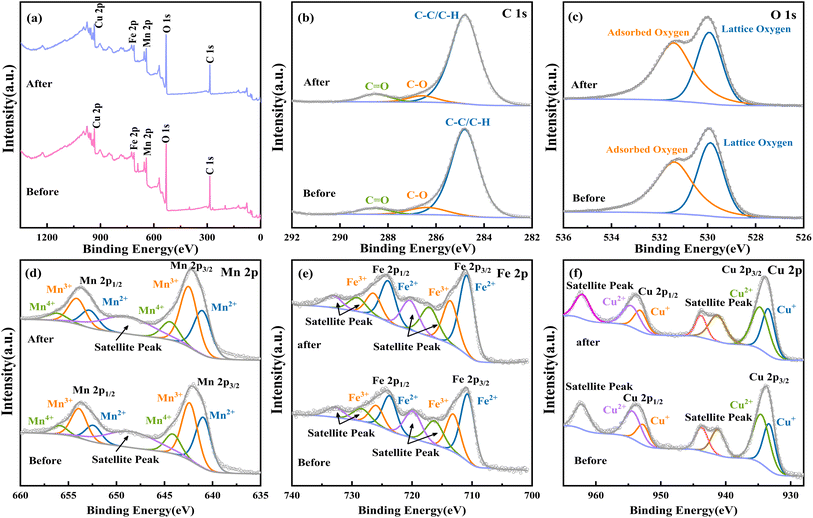 |
| | Fig. 7 XPS spectrum of Mn–Fe–Cu LDH before and after reaction, (a) present total spectra, (b) to (f) present C spectra, O spectra, Mn spectra, Fe spectra, and Cu spectra, respectively. | |
Based on these observations, the following activation mechanism for PMS in the MnFeCu-LDH system is proposed: the Mn2+, Fe3+, and Cu2+ ions in MnFeCu-LDH catalyze the activation of PMS (HSO5−) via electron transfer processes (eqn (1)–(3)), producing sulfate radicals (SO4˙−). These SO4˙− radicals can then react with H2O or OH− to form hydroxyl radicals (˙OH) (eqn (4)).29,30 The high-valence Mn3+, Fe3+, and Cu2+ ions are reduced to their lower valence states by HSO5− (eqn (5)), while oxidation of other metal ions occurs, leading to continuous redox cycling. The redox potentials for Mn3+/Mn2+ (1.51 V), Fe3+/Fe2+ (0.77 V), Cu2+/Cu+ (0.34 V), and Mn4+/Mn3+ (0.15 V) facilitate synergistic interactions, thereby enhancing the overall catalytic activity of the multi-metal system31,32 (eqn (6)–(10)). Moreover, Mn2+, Fe2+, and Cu+ ions bind to H2O and OH− on the catalyst surface, contributing further to PMS activation (eqn (11) and (12)). In addition, singlet oxygen (1O2) is produced through PMS decomposition and the reaction between O2˙− and H2O (eqn (13) and (14)). Consequently, the degradation of CTC is driven by the synergistic action of multiple reactive oxygen species, including SO4˙−, ˙OH, 1O2, and O2˙−.
| | |
Mn2+/Mn3+ + HSO5− → Mn3+/Mn4+ + SO4˙− + OH−
| (1) |
| | |
Fe2+ + HSO5− → Fe3+ + SO4˙− + OH−
| (2) |
| | |
Cu+ + HSO5− → Cu2+ + SO4˙− + OH−
| (3) |
| | |
SO4˙− + OH− → ˙OH + SO42−
| (4) |
| | |
Fe3+/Mn3+/Cu2+ + HSO5− → Fe2+/Mn2+/Cu+ + SO5˙− + H+
| (5) |
| | |
Mn3+ + Fe2+ → Mn2+ + Fe3+
| (6) |
| | |
Mn3+ + Cu+ → Mn2+ + Cu2+
| (7) |
| | |
Fe3+ + Cu+ → Fe2+ + Cu2+
| (8) |
| | |
Cu2+ + Mn3+ → Cu+ + Mn4+
| (9) |
| | |
Fe3+ + Mn3+ → Fe2+ + Mn4+
| (10) |
| | |
Fe2+/Mn2+/Cu+–OH− + HSO5− → Fe2+/Mn2+/Cu+–OHOSO3− + OH−
| (11) |
| | |
Fe2+/Mn2+/Cu+–OHOSO3− → Fe3+/Mn3+/Cu2+ + SO4˙−
| (12) |
| | |
HSO5− + SO52− → HSO5− + SO4− + 1O2
| (13) |
| | |
2O2˙− + H2O → HOO− + OH− + 1O2
| (14) |
Overall, the addition of GO improves the surface area and provides additional active sites for the catalytic reaction. Furthermore, GO helps in stabilizing the MnFeCu-LDH particles, preventing their aggregation and leaching during the reaction. GO also facilitates the electronic transfer, which enhances the activation of PMS and, consequently, the degradation efficiency of CTC. This explanation has been incorporated into the revised discussion section to clarify how GO contributes to the observed improvements.
4. Conclusion
The synthesis of MnFeCu-LDH/GO was successfully achieved and their application to activated PMS for CTC degradation was investigated. The findings revealed that the MnFeCu-LDH/GO exhibited excellent catalytic activity and were capable of efficiently removing CTC from aqueous environments.
(1) At an initial pH of 7 and a catalyst/PMS dosage of 0.2 g L−1, the CTC removal rate reached 91.18% within 30 minutes.
(2) Increasing both the catalyst dosage and reaction temperature further enhances the efficiency of CTC degradation. However, when the catalyst dosage exceeds 0.2 g L−1, the enhancement effect becomes negligible. Elevated PMS concentrations can lead to the formation of SO5˙− through reactions with SO4˙− and ˙OH, which ultimately reduces the overall oxidizing potential.
(3) The involvement of various reactive species in the CTC degradation process was confirmed through quenching experiments and EPR detection. Among these, 1O2 contributed the most to degradation, followed by ˙OH and SO4˙−.
(4) The presence of different valence states of metal ions on the catalyst surface promotes redox cycling due to the differences in their redox potentials, thereby generating a synergistic effect that enhances catalytic activity. After five cycles of use, the catalyst maintained substantial catalytic degradation efficiency for CTC, demonstrating excellent reusability.
Data availability
All relevant data are within the paper.
Author contributions
Dongfeng Shi: investigation, writing – review & editing, resources. Xiyin Mao: investigation, writing – original draft. Min Fei: validation, formal analysis. Cong Liang: data curation, writing – review & editing. Yuyang Luo: validation, data curation. Zhidan Xu: software, project administration. Liyong Hu: methodology, conceptualization, supervision.
Conflicts of interest
There are no conflicts to declare.
References
- Y. J. Tian, L. Feng, R. N. Li, J. Li, Z. W. Du, L. Q. Zhang and Y. Z. Liu, Sci. Total Environ., 2021, 798, 149001 CrossRef CAS PubMed.
- W. W. Ben, B. Zhu, X. J. Yuan, Y. Zhang, M. Yang and Z. M. Qiang, Water Res., 2018, 130, 38–46 CrossRef CAS PubMed.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- X. H. Liu, S. Y. Lu, W. Guo, B. D. Xi and W. L. Wang, Sci. Total Environ., 2018, 627, 1195–1208 CrossRef CAS PubMed.
- T. T. H. Pham, P. Rossi, H. D. K. Dinh, N. T. A. Pham, P. A. Tran, T. Ho, Q. T. Dinh and L. F. De Alencastro, J. Environ. Manage., 2018, 214, 149–156 CrossRef CAS PubMed.
- Y. Xu, J. Ai and H. Zhang, J. Hazard. Mater., 2016, 309, 87–96 CrossRef CAS PubMed.
- J. Liu, Q. Yang, D. B. Wang, X. M. Li, Y. Zhong, X. Li, Y. C. Deng, L. Q. Wang, K. X. Yi and G. M. Zeng, Bioresour. Technol., 2016, 206, 134–140 CrossRef CAS PubMed.
- G. Boczkaj and A. Fernandes, Chem. Eng. J., 2017, 320, 608–633 CrossRef CAS.
- X. T. Pang, Y. Guo, Y. T. Zhang, B. B. Xu and F. Qi, Chem. Eng. J., 2016, 304, 897–907 CrossRef CAS.
- X. B. Dong, B. X. Ren, Z. M. Sun, C. Q. Li, X. W. Zhang, M. H. Kong, S. L. Zheng and D. D. Dionysiou, Appl. Catal., B, 2019, 253, 206–217 CrossRef CAS.
- X. F. Zhao, C. G. Niu, L. Zhang, H. Guo, X. J. Wen, C. Liang and G. M. Zeng, Chemosphere, 2018, 204, 11–21 CrossRef CAS PubMed.
- P. J. Duan, T. F. Ma, Y. Yue, Y. W. Li, X. Zhang, Y. A. Shang, B. Y. Gao, Q. Z. Zhang, Q. Y. Yue and X. Xu, Environ. Sci.:Nano, 2019, 6, 1799–1811 RSC.
- W. L. Guo, S. N. Su, C. L. Yi and Z. M. Ma, Environ. Prog. Sustainable Energy, 2013, 32, 193–197 CrossRef CAS.
- Y. Yuan, S. Garg, J. Ma and T. D. Waite, Environ. Sci. Technol., 2021, 55, 13274–13285 CrossRef CAS PubMed.
- Y. Feng, D. L. Wu, Y. Zhou and K. M. Shih, Chem. Eng. J., 2017, 330, 906–913 CrossRef CAS.
- X. L. Fan, Q. Q. Cao, F. Y. Meng, B. Song, Z. Q. Bai, Y. Zhao, D. D. Chen, Y. Zhou and M. Song, Chemosphere, 2021, 266, 128992 CrossRef PubMed.
- Y. X. Wang, Z. M. Ao, H. Q. Sun, X. G. Duan and S. B. Wang, Appl. Catal., B, 2016, 198, 295–302 CrossRef CAS.
- S. S. Yang, P. X. Wu, J. Q. Liu, M. Q. Chen, Z. Ahmed and N. W. Zhu, Chem. Eng. J., 2018, 350, 484–495 CrossRef CAS.
- R. N. Guo, L. C. Nengzi, Y. Chen, Y. H. Li, X. Y. Zhang and X. W. Cheng, Chem. Eng. J., 2020, 398, 125676 CrossRef CAS.
- R. N. Guo, Y. L. Zhu, X. W. Cheng, J. J. Li and J. C. Crittenden, J. Hazard. Mater., 2020, 399, 122966 CrossRef CAS PubMed.
- L. H. Hou, X. M. Li, Q. Yang, F. Chen, S. N. Wang, Y. H. Ma, Y. Wu, X. F. Zhu, X. D. Huang and D. B. Wang, Sci. Total Environ., 2019, 663, 453–464 CrossRef CAS PubMed.
- L. Wang, N. Jiang, H. Xu, Y. Luo and T. Zhang, Environ. Sci. Technol., 2023, 57, 12523–12533 CrossRef CAS PubMed.
- G. X. Huang, C. Y. Wang, C. W. Yang, P. C. Guo and H. Q. Yu, Environ. Sci. Technol., 2017, 51, 12611–12618 CrossRef CAS PubMed.
- T. Doughty, A. Zingl, M. Wunschek, C. M. Pichler, M. B. Watkins and S. Roy, ACS Appl. Mater. Interfaces, 2024, 16, 40814–40824 CrossRef CAS PubMed.
- C. Gong, F. Chen, Q. Yang, K. Luo, F. B. Yao, S. N. Wang, X. L. Wang, J. W. Wu, X. M. Li, D. B. Wang and G. M. Zeng, Chem. Eng. J., 2017, 321, 222–232 CrossRef CAS.
- Z. D. Li, Y. M. Sun, W. L. Huang, C. Xue, Y. Zhu, Q. W. Wang and D. F. Liu, J. Environ. Sci., 2020, 88, 46–58 CrossRef CAS PubMed.
- X. H. Liu, X. C. Guo, Y. Liu, S. Y. Lu, B. D. Xi, J. Zhang, Z. Wang and B. Bi, Environ. Pollut., 2019, 254, 112996 CrossRef CAS PubMed.
- G. Wang, D. Z. Li, S. Y. Wang, Z. B. Zhao, S. H. Lv and J. S. Qiu, Sep. Purif. Technol., 2021, 254, 117667 CrossRef CAS.
- P. Wang, X. Q. Zhang, B. Zhou, F. P. Meng, Y. S. Wang and G. W. Wen, J. Environ. Chem. Eng., 2023, 11, 111191 CrossRef CAS.
- R. X. Yuan, L. Hu, P. Yu, H. Y. Wang, Z. H. Wang and J. Y. Fang, Chemosphere, 2018, 198, 204–215 CrossRef CAS PubMed.
- H. X. Zeng, W. Q. Zhang, L. Deng, J. M. Luo, S. Q. Zhou, X. Liu, Y. Pei, Z. Shi and J. Crittenden, J. Colloid Interface Sci., 2018, 515, 92–100 CrossRef CAS PubMed.
- L. J. Zhou, J. Li, Y. D. Zhang, L. Y. Kong, M. Jin, X. D. Yang and Q. L. L. Wu, Ecotoxicol. Environ. Saf., 2019, 183, 109511 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *cd
*cd