
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutron, X-ray diffraction, DSC, Raman, Mössbauer and leaching studies of iron phosphate glasses and crystalline phases
Kajal Dadwala,
Margit Fábiánb,
Istvan Tolnaib,
Suruchi Sharmaa,
Rajinder Kaura,
Maria Gracheva bc,
Krisztina Kovácsc,
Zoltán Klencsárb and
Atul Khanna*a
bc,
Krisztina Kovácsc,
Zoltán Klencsárb and
Atul Khanna*a
aSensors and Glass Physics Laboratory, Department of Physics, Guru Nanak Dev University, Amritsar, 143005, Punjab, India. E-mail: atul.phy@gndu.ac.in
bHUN-REN Centre for Energy Research, Konkoly-Thege Miklós út 29-33, 1121 Budapest, Hungary
cInstitute of Chemistry, ELTE Eötvös Loránd University, 1117-Budapest, Pázmány P. s. 1/A, Hungary
First published on 17th February 2025
Abstract
xFe2O3–(100 − x)P2O5 glasses were synthesized by melt quenching and structure–property correlation studies were carried. Glasses containing 25 to 40 mol% Fe2O3 were prepared while the sample with 50 mol% Fe2O3 formed a crystalline sample containing Fe32+Fe43+[PO4]63− and Fe22+[P2O7]4− phases on melt-quenching. Glass density increases from 2.98 to 3.20 g cm−3, ionic packing fraction is in the range of 0.63–0.65 and the glass transition temperature decreases from 500 °C to 493 °C on increasing Fe2O3 concentration from 25 to 40 mol%. Pair distribution function analysis and Reverse Monte Carlo simulations of neutron diffraction datasets were used to calculate the atomic pair distributions, interatomic distances and co-ordination environments. The P–O co-ordination is essentially tetrahedral and is in the range: 3.9–3.7 (±0.1), the Fe–O co-ordination number decreases steadily from 4.8 to 4.2 (±0.1) with an increase in Fe2O3 concentration in the phosphate network, while O–O co-ordination is in the range: 6.6–6.3(±0.1), the decrease in these co-ordination numbers are due to an increase in oxygen deficiency in the glass network with an increase in Fe2O3 mol%. Fe–O and P–O pair distributions are asymmetrical indicating short-range disorder due to the existence of a wide range of bond-lengths with maxima at 1.79 Å and in the range: 1.45–1.51 Å respectively. Mössbauer studies carried out at room temperature and 80 K found that Fe exists in 2+ and 3+ valence states, and the glass and crystalline samples contained Fe2+ at least at three different sites. Raman studies found that the meta and pyrophosphate structural units are dominant species up to 35 mol% Fe2O3 concentration, while the orthophosphate units are in majority at 40 mol% of Fe2O3. The crystalline sample is a two phase material and contained both orthophosphate and pyrophosphate units with the former being the dominant species. Leaching studies on two iron phosphate glasses carried out in purified water at 90 °C found that dissolution of glasses decreases and the chemical durability increases drastically with an increase in Fe2O3 mol%.
1. Introduction
Nuclear waste management is an end stage of the nuclear fuel cycle and it is important to find matrices or materials that can be used to incorporate and immobilize high level radioactive waste.1,2 Borosilicate glasses have been widely used for high-level nuclear waste immobilization.3–6 However, the high-level waste containing halide and Mo ions, P and noble metal atoms are only sparingly soluble in borosilicate glasses, and this limits the waste loading, and increases the cost and time of vitrification.7 Iron phosphate and lead iron phosphate glasses are functional materials that are considered to be economical alternatives to borosilicate glasses for nuclear waste immobilization,8–12 because these are good solvents for heavy metal ions and possess excellent leaching resistance in acidic and neutral medium.13 Moreover, the addition of MoO3 enhances the thermal stability and water durability of iron phosphate glasses.14 The iron phosphate glasses are chemically more durable than alkali and alkaline-earth phosphate glasses due to the existence of Fe–O–P linkages, the Fe2+/Fe3+ in the phosphate glass network strengthens the cross-linking between the polyphosphate chains and enhances the chemical and thermal durability of glasses.15,16 The open structure and voids in iron phosphate glass network promotes the incorporation of large concentration of actinides and nuclear waste.17The basic building blocks of the glassy and crystalline phosphates are the P–O tetrahedral structural units that are connected by covalently bonded oxygens and that form various phosphate anions. The tetrahedra are categorized using the Qi terminology where i represents the number of bridging oxygens per tetrahedron. The three phosphate polyhedral units are: Q0, Q1 and Q2 respectively.18 The number of tetrahedral linkages through bridging oxygens between neighbouring P-tetrahedra can be classified by the oxygen to-phosphorus (O/P) ratio. Glass systems with O/P = 4, 3.5 and 3 are represented as Q0, Q1 and Q2 polyhedra and termed as orthophosphate, pyrophosphate and metaphosphate units respectively. The metaphosphates possess long chains of P-tetrahedra linked through two bridging oxygens, the pyrophosphates with single bridging oxygen terminates these chains and forms (P2O7)4− anions while the orthophosphates without any bridging oxygens are similar to isolated phosphate groups that exist in the crystalline phosphate compounds.18–20
The chemical durability of phosphate glasses can be enhanced considerably by adding Al2O3 and Fe2O3 in the glass network; the addition of these metal oxides replaces the easily hydrated –P–O–P– bonds by more hydrate resistant –M–O–P– bonds, where M are the metal cations.15,16 The glass formation range of xFe2O3–(100 − x)P2O5 system is reported to be 10 to 45 mol% of Fe2O3.21 The 40Fe2O3–60P2O5 glass is reported to exhibit the best chemical durability or lowest leaching rate among all the iron phosphate glasses,22–26 leaching is an important property of glasses/matrices that needs to be evaluated for their application in nuclear waste immobilization.
As the concentration of Fe2O3 increases in the phosphate network, the number of Fe–O–P linkages increases at the expense of P–O–P bonds and, the chain-like metaphosphate network breaks into dimeric pyrophosphate and finally into isolated orthophosphate groups.19,20 Iron phosphate glasses are reported to contain both Fe2+ and Fe3+ in the network, and the valence state of Fe is reported to depend on the use of either FeO or Fe2O3 as starting material in the synthesis of iron phosphate glasses.21 Molecular dynamics (MD) studies using Buckingham potential were carried out on both xFeO–(100 − x)P2O5 and xFe2O3–(100 − x)P2O5 glass systems, and Fe–O, P–O and O–O atomic pair correlations were determined.27 The experimental studies in which iron phosphate glasses were synthesized by melt-quenching have also reported large effects of melting temperature and melting time on relative concentration of Fe2+ and Fe3+ in the structure,19 however several of these experimental results can be erroneous as the investigators employed alumina or silica crucibles instead of more durable platinum crucibles for the synthesis of glasses.19,21,28,29 It is well established that alumina and silica crucibles can introduce significant concentration of Al3+ and Si4+ impurities in the oxide glass melt and give rise to the impurity Al–O and Si–O peaks in the total pair correlation functions and consequently give incorrect results on Fe–O and P–O co-ordination numbers and bond lengths since the Fe–O and P–O peaks exist at almost the same positions as Al–O and Si–O peaks respectively.
It is therefore important that iron phosphate glasses are prepared by melting the batch materials in more durable platinum crucible for the accurate determination of glass structure and properties. In the present study series of iron phosphate glasses were prepared by melting the starting materials in platinum crucibles and subsequently characterized by density measurements, X-ray diffraction, differential scanning calorimetry (DSC), Raman and Mössbauer spectroscopy, neutron diffraction, pair distribution function and Reverse Monte Carlo (RMC) simulations and leaching tests. The RMC analysis was used to calculate the partial atomic pair distributions, the cation–oxygen bond lengths and Fe–O and P–O co-ordination numbers; RMC simulations give accurate and reliable values of glass short-range structural properties since it does not suffer from the disadvantages of overlapping atomic pair correlation peaks in the total pair distribution function,30–34 that are difficult to deconvolute unambiguously when different atomic pairs exist at same or very similar distances. The chemical durability of two iron phosphate glasses were studied by measuring their leaching characteristics in MQ water at 90 °C for a period of 10 days.
2. Experimental
2.1 Glass synthesis
Disk shaped samples of the system: xFe2O3–(100 − x)P2O5 (x = 25, 30, 35, 40 and 50 mol%) were prepared by the melt-quenching (hereafter referred as 25FeP, 30FeP, 35FeP, 40FeP and 50FeP respectively). The starting materials for glass preparation were Fe2O3 (Aldrich India, 99%) and NH6PO4 (99%, Otto Kemi, India). The net weight of the batch mixture for all the samples was about 10 g. Appropriate amounts of materials were weighed and ground in an agate mortar-pestle for about 30 min. The batch mixture was then transferred into a platinum crucible. It was first heated in an electric furnace at 200 °C for 1 h and subsequently the temperature of the furnace was slowly increased to 550 °C to remove the residual gases (NH3 and H2O). The material was kept at this temperature for ∼1 h. Afterwards, the mixture was melted in another furnace and heated to 1300 °C, the melt was kept at this temperature for 1 h. The melt was then poured onto a brass block to get the final disk-shaped glass samples. All the glass samples were shining black in color. Samples upto 40 mol% Fe2O3 formed glasses, however the sample with 50 mol% Fe2O3 in the starting batch mixture did not produce a glass but solidified into a crystalline material on melt-quenching as confirmed by X-ray and neutron diffraction studies discussed below.2.2 Density measurements
The density, ρ of the disk-shaped glasses was measured by the Archimedes method using dibutylphatalate (DBP) as the immersion fluid on an electronic balance of sensitivity 10−4 g at laboratory temperature by using the following relation:
 | (1) |
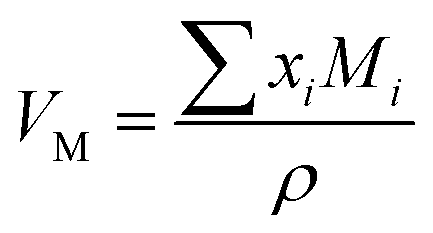 (where xi is the mol% of ith component and Mi is its molecular weight). The packing fraction, PF, of glasses is the ratio of ionic volume to molar volume and calculated by using the following formula:35
(where xi is the mol% of ith component and Mi is its molecular weight). The packing fraction, PF, of glasses is the ratio of ionic volume to molar volume and calculated by using the following formula:35
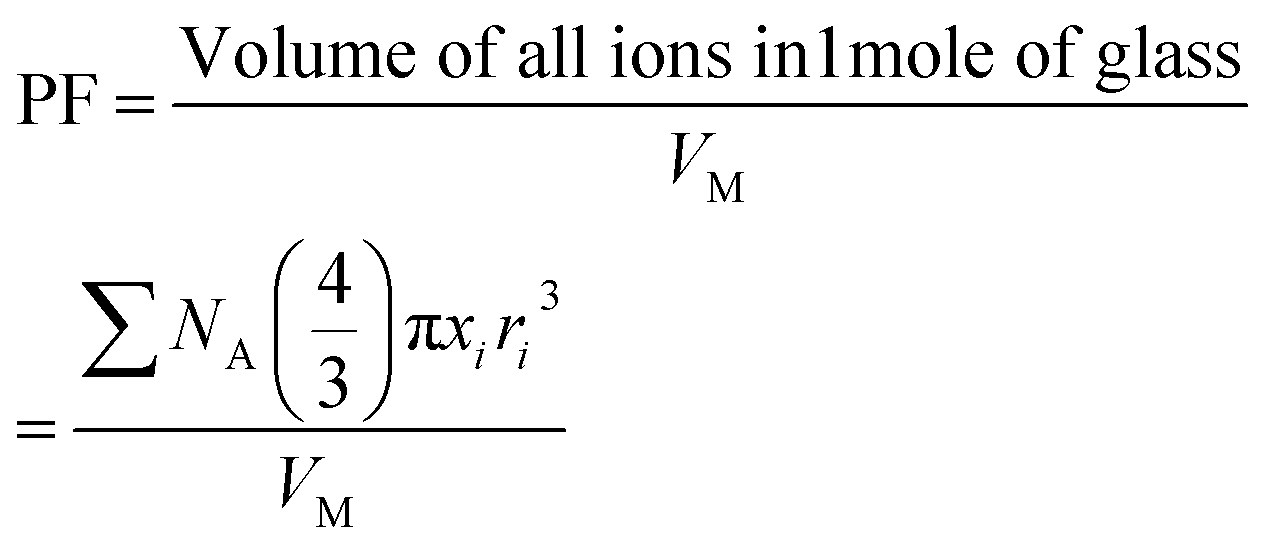 | (2) |
2.3 X-ray diffraction experiments
X-ray diffraction (XRD) studies were carried out on powdered samples on Bruker D8 Focus XRD system with Cu Kα1,2 radiation in the 2θ range of 10–90°. The X-ray tube was operated at 40 kV and 30 mA and the scattered X-ray intensity was measured with a scintillation detector using step size of 0.02° and scan rate of 0.5° per min.2.4 Differential scanning calorimetry
The thermal parameters such as glass transition temperature, Tg, crystallization temperature, Tc and the melting temperature, Tm were determined on SETARAM Labsys Evo DSC system. About 50 mg powder sample was taken in a platinum pan and the differential heat flow scan was recorded at a heating rate of 10 °C min−1 in the temperature range of 200–1200 °C under argon flow conditions (10 ml min−1). The glass transition temperature was taken as the mid-point value. The peak values of exothermic crystallization and endothermic melting peaks were taken as the crystallization and melting temperatures respectively. The maximum uncertainty in the measurement of Tg, Tc and Tm is ± 1 °C.2.5 Raman spectroscopy
Raman scattering studies were performed at room temperature on the iron phosphate samples on Renishaw In-Via Reflex micro-Raman spectrometer in the Raman shift range of 30–1500 cm−1 at a spectral resolution of 0.5 cm−1. The Stokes spectra were recorded by using a 514.5 nm argon-ion excitation laser, an edge filter, diffraction grating with 2400 lines per mm and a CCD detector.2.6 Neutron diffraction experiments
The neutron diffraction studies were carried out on powdered iron phosphate samples at the 2-axis Position Sensitive Detector (PSD) diffractometer, of Budapest Neutron Centre (BNC), Hungary, using monochromatic thermal neutrons of de Broglie wavelength, λ = 1.069 Å in the scattering vector or momentum transfer function (Q) range of 0.45 to 9.8 Å−1. The pulverized samples of mass 4 to 5 g were placed in thin-walled cylindrical vanadium can of diameter of 8 mm and the diffraction data was recorded for 24 h each. The raw neutron diffraction data of four glass samples was corrected for background, absorption and multiple scattering and normalized with vanadium rod standard. The structure factors, S(Q)'s were calculated from the raw diffraction data using the programme package available at the BNC facility.2.7 Pair distribution function (PDF) by Fourier transformation
The atomic pair distribution function, g(r) of each sample was calculated by the Fourier sine transformation of the neutron interference function amplified with Q i.e. F(Q)= Q(S(Q) − 1) by using the following formula:30–34
 | (3) |
and
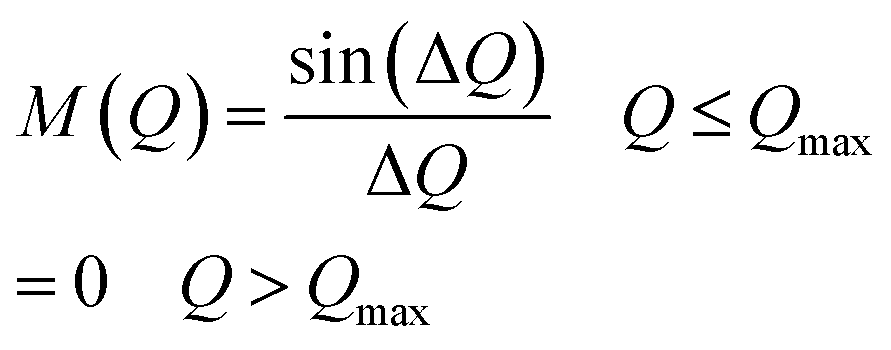
Qmax was selected around 9.8 Å−1 for calculating g(r) by (3).

2.8 Reverse Monte Carlo (RMC) simulations
The experimental neutron structure factors, S(Q) were simulated by the RMC technique using the RMC++ Version 1.5.1 software package.37–40 During the RMC simulation runs, the partial structure factors, Sij(Q) were calculated from the pair distribution functions gij(r) by Fourier transformation. If k is the total number of elements in the sample (k = 3 in the present glass system), then k(k + 1)/2 = 6 different atom pairs exist in its glass structure. The RMC technique minimizes the squared difference between the experimental S(Q) and the simulated one from a 3-dimensional atomic configuration by using the following equations
 | (4) |
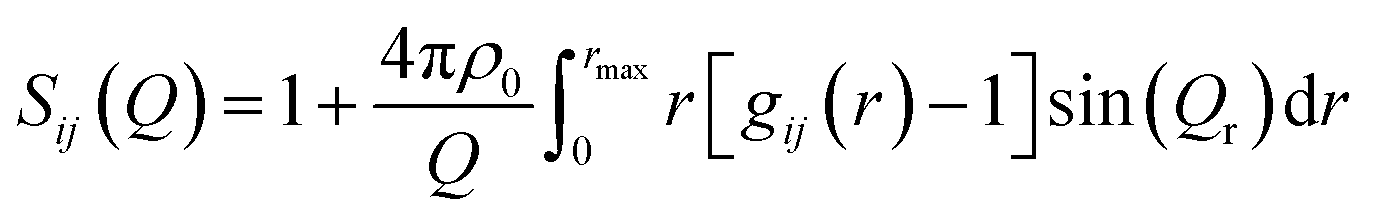 | (5) |
 | (6) |
The RMC simulations were started by building an initial random atomic configuration in a simulation box that was populated with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms of Fe, P and O. The atomic number densities, ρo were 0.0799, 0.0818, 0.0813 and 0.0802 atoms per Å−3 (corresponding to box edges of 26.79, 24.81, 24.86 and 24.97 Å) for the glass samples 25FeP, 30FeP, 35FeP and 40FeP respectively. The atoms inside the box were constrained to move apart by certain minimum interatomic distances (cut-off distances) that were provided in the RMC input program. No other constraints (such as Fe–O or P–O co-ordination constraints) were imposed on the atomic pairs during the RMC simulations. Repeated RMC runs were performed by varying the cut-off distances slightly, to produce reliable data with good stability and statistics. About 20 atomic configurations were obtained from the RMC calculations of each sample corresponding to more than 1100000 accepted atomic displacements inside the simulation box. After multiple runs, the experimental and the RMC simulated structure factor, S(Q) matched well and the atomic pair-correlation functions, gij(r), coordination number distributions, Nij, and bond angle distribution were calculated from the RMC simulated data.
000 atoms of Fe, P and O. The atomic number densities, ρo were 0.0799, 0.0818, 0.0813 and 0.0802 atoms per Å−3 (corresponding to box edges of 26.79, 24.81, 24.86 and 24.97 Å) for the glass samples 25FeP, 30FeP, 35FeP and 40FeP respectively. The atoms inside the box were constrained to move apart by certain minimum interatomic distances (cut-off distances) that were provided in the RMC input program. No other constraints (such as Fe–O or P–O co-ordination constraints) were imposed on the atomic pairs during the RMC simulations. Repeated RMC runs were performed by varying the cut-off distances slightly, to produce reliable data with good stability and statistics. About 20 atomic configurations were obtained from the RMC calculations of each sample corresponding to more than 1100000 accepted atomic displacements inside the simulation box. After multiple runs, the experimental and the RMC simulated structure factor, S(Q) matched well and the atomic pair-correlation functions, gij(r), coordination number distributions, Nij, and bond angle distribution were calculated from the RMC simulated data.
2.9 Mössbauer spectroscopy
57Fe Mössbauer spectra were recorded at room temperature (298–300 K) in transmission geometry by using a standard WissEl Mössbauer spectrometer setup along with a 57Co(Rh) radioactive source with an activity of ∼2.2 mCi. The source movement followed a sinusoidal velocity signal. Unfolded spectra were recorded in 2048 channels and were subsequently folded into 1024 channels for analysis. Isomer shift (δ) values are quoted with respect to that of α-Fe at room temperature. Circular absorbers were prepared with a diameter of 15.5 mm by using sample amounts that were expected to result in comparable (∼12 mg cm−2) Fe surface densities of the resulting samples based on the nominal sample stoichiometries. The powdered samples were in addition mixed in each case with 200 mg of cellulose in order to achieve a uniform absorber surface density. For the low temperature (T ≈ 80 K) measurements the samples were kept in a bath type cryostat (SVT-400-MOSS, Janis, Woburn, MA, USA) filled with liquid nitrogen, while the spectrometer operated with a triangular velocity waveform, in constant acceleration mode, recording spectra in 512 channels before folding. Unless explicitly noted otherwise, the resulting spectra were fitted to symmetrical quadrupole doublets by using version 4.0i of the MossWinn program.412.10 Leaching tests
The chemical durability of the two disk shaped bulk glass samples: 30FeP (weight = 1.772 g) and 40FeP (1.919 g) and the chemical evolution of the leaching solution in water purified with Millipore Milli-Q lab water system (Merck, USA). This purified water is referred to as MQ-water in the manuscript.MQ-water at 90 ± 1 °C were analyzed by calculating the normalized release and glass dissolution rate of P and Fe using the equations specified in the ASTM C1285-21 protocol.42 The procedure used for the leaching analysis is described elsewhere43 and leaching tests were carried over a total period of 10 days. After 1, 2, 3, 7, and 10 days, the containers were opened, and the leachates were filtered through a 0.45 μm syringe filter. Subsequently, the leachates were acidified with cc. HNO3 for inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements to measure the concentration of P and Fe concentration (in the units of mg l−1) that leached out in MQ-water using a PerkinElmer Avio 200 ICP-OES apparatus.43
3. Results and discussion
3.1 X-ray diffraction (XRD)
The XRD patterns of all iron phosphate samples are shown in Fig. 1 and it is found that samples containing 25, 30, 35 and 40 mol% of Fe2O3 show only broad humps in the 2θ range of 15° to 35°, which confirmed their amorphous nature. | ||
| Fig. 1 XRD patterns of iron phosphate samples. The sharp peaks in the sample 50FeP are due to the coexisting Fe7(PO4)6 (#) and Fe2P2O7 ($) crystalline phases. | ||
The sample: 50FeP prepared using 50 mol% Fe2O3 in the batch mixture, shows several sharp peaks; this sample is crystalline and its XRD peaks match with those of Fe(II)3Fe(III)4[PO4]6 and Fe(II)2[P2O7] phases both of which possess triclinic crystal structure (powder diffraction files # 72-2446 and 76-1762 respectively).
3.2 Density, molar volume and packing fraction
The density data of the iron phosphate samples prepared with 25, 30, 35, 40 and 50 mol% Fe2O3 is given in Table 1. Density increases from 2.98 g cm−3 to 3.59 ± 0.05 g cm−3 as the concentration of Fe2O3 is increased from 25 to 50 mol%, it may be noted that sample prepared with 50 mol% Fe2O3 in the starting batch mixture, is not a glass but is crystalline.| Sample code | Composition [mol%] | Density, ρ [g cm−3] | Molar mass (amu) | Molar volume VM [cm3 mol−1] | Packing fraction (PF) | Atomic number density ρo [Å−3] | |
|---|---|---|---|---|---|---|---|
| Fe2O3 | P2O5 | ||||||
| 25FeP | 25 | 75 | 2.98 | 146.38 | 49.1 | 0.64 | 0.0799 |
| 30FeP | 30 | 70 | 3.13 | 147.26 | 47.0 | 0.65 | 0.0818 |
| 35FeP | 35 | 65 | 3.18 | 148.15 | 46.6 | 0.64 | 0.0813 |
| 40FeP | 40 | 60 | 3.20 | 149.04 | 46.5 | 0.63 | 0.0802 |
| 50FeP | 50 | 50 | 3.59 | 150.82 | 42.0 | 0.67 | 0.0861 |
The molar masses of Fe2O3 and P2O5 are 159.6 u and 141.9 u respectively; therefore, the replacement of lighter P2O5 by the heavier Fe2O3 molecules in the glass network produces a steady enhancement in the density. Al-Hasni et al.44 reported density of 3.03 g cm−3 for the 40Fe2O3–60P2O5 glass which is lower than the density of 3.20 ± 0.05 g cm−3 of the glass of equal composition prepared and characterized in the present study.
The molar volume, VM defined as the volume occupied by 1 mole of glass molecules decreases from a value of 49.1 to 42.0 cm3, on increasing Fe2O3 concentration from 25 to 50 mol%. VM of iron phosphate glasses is found to be significantly higher than that of other oxide glasses such as borate and borosilicate glasses, for instance lead borate,45 lead borosilicate,45 lead aluminoborosilicate,46 bismuth borate47 and bismuth borosilicate48 have molar volume in the range of 24.2 to 42.7 cm3, the alkali and alkaline-earth silicate and aluminosilicate glasses have lower VM in the range of 16 to 30 cm3.49
The PF in iron phosphate glasses is in the range of 0.63–0.65 and it shows only a small variation with an increase in Fe2O3 concentration from 25 to 40 mol%, both VM and PF are important structural parameters of glasses that are used as matrices for radioactive waste immobilization. A larger VM and a smaller value of PF indicates greater concentration of voids that can incorporate other ions in the glass structure.
3.3 Thermal properties
DSC thermographs of the 4 iron phosphate glasses and one crystalline sample are displayed on Fig. 2. The glass transition temperature, Tg decreases steadily from 500 °C to 493 °C as Fe2O3 concentration is increased from 25 to 40 mol% (Table 2). This is because the weaker Fe–O (EFe–O = 409 kJ mol−1) bonds replace the significantly stronger P–O (EP–O = 597 kJ mol−1) bonds in the phosphate network,36 and this lowers the glass transition temperature. The value of Tg (493 ± 1 °C) in 40Fe2O3–60P2O5 glass is in good agreement with the value of Tg of 498 ± 5 °C reported for the same glass by Al-Hasni et al.44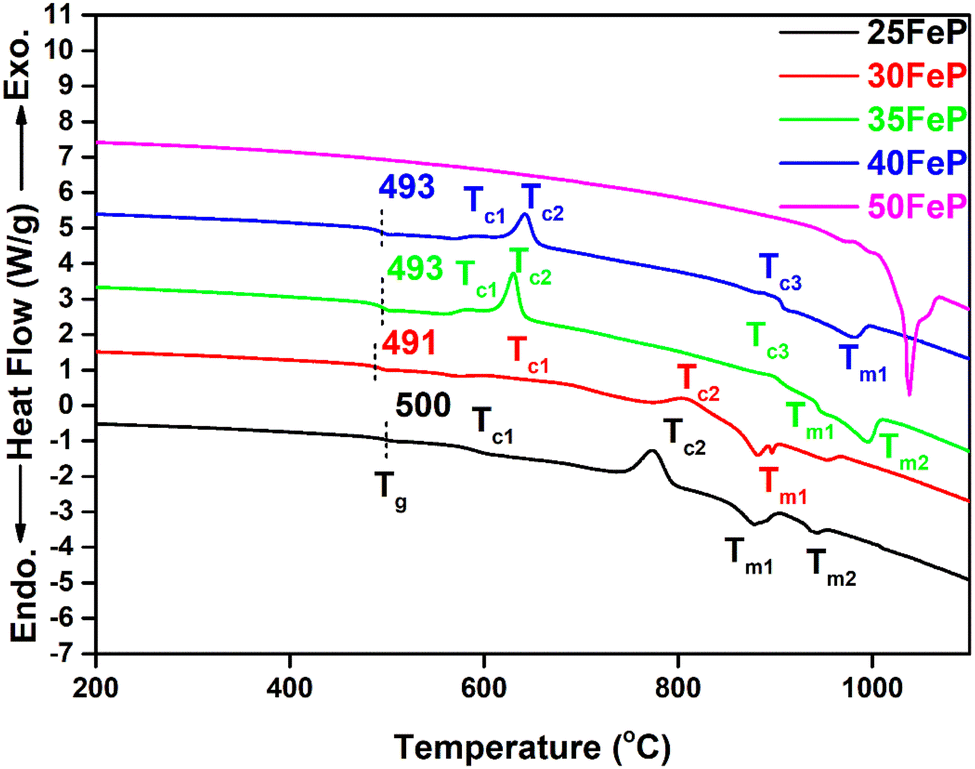 | ||
| Fig. 2 DSC curves of iron phosphate samples. The dotted line indicates the midpoint of glass transition. The sample 50FeP is crystalline and exhibits only endothermic liquidus peak. | ||
| Sample | Tg [oC] | Tc [oC] | Tm [oC] | EB [kJ mol−1] | |||
|---|---|---|---|---|---|---|---|
| Tc1 | Tc2 | Tc3 | Tm1 | Tm2 | |||
| 25FeP | 500 | 588 | 775 | — | 875 | 944 | 550 |
| 30FeP | 491 | 617 | 812 | — | 884 | 943 | 540 |
| 35FeP | 493 | 594 | 630 | 897 | 887 | 942 | 531 |
| 40FeP | 493 | 587 | 643 | 899 | 948 | 990 | 521 |
The average single bond enthalpy, EB in four glasses was calculated by using the following formula:50
 | (7) |
3.4 Vibrational spectra
The Raman spectra of iron phosphate samples are shown in Fig. 3. The sharp peak at low frequency of ∼65 cm−1 is the boson peak which is a universal feature of the disordered materials and glasses.51–55 This peak is not prominent in the 50FeP sample as it is crystalline and not glassy. Fig. 4 shows the Raman spectra of glassy samples; 25FeP, 30FeP, 35FeP and 40FeP deconvoluted using 11 independent Gaussian peaks in range 400–1500 cm−1 using peak positions at approximately same positions as reported in the earlier study by Stoch et al.56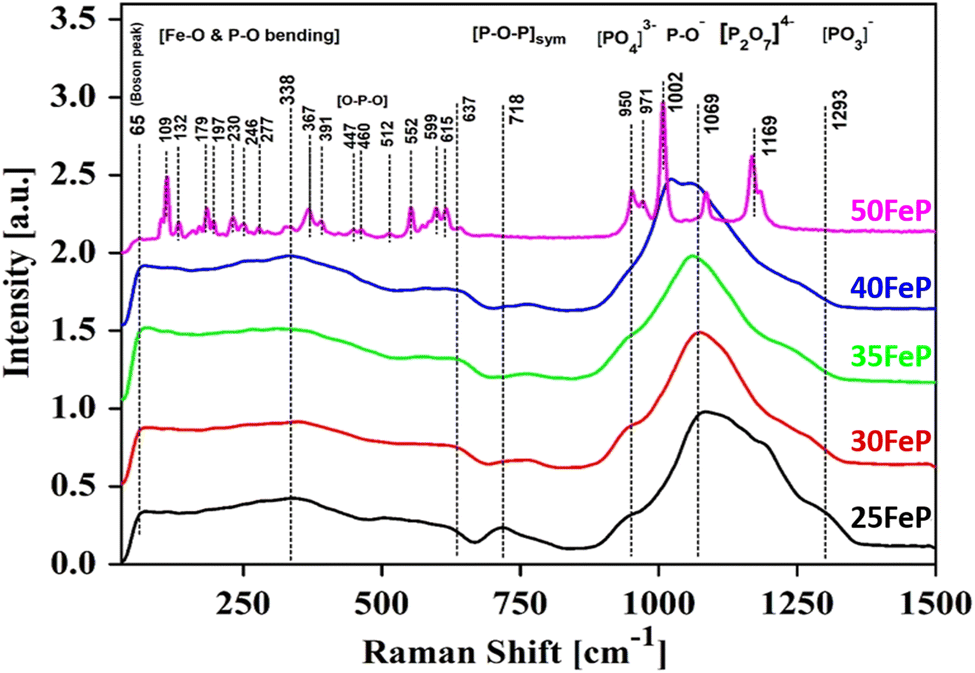 | ||
| Fig. 3 Raman spectra of four iron phosphate glasses and one crystalline sample. | ||
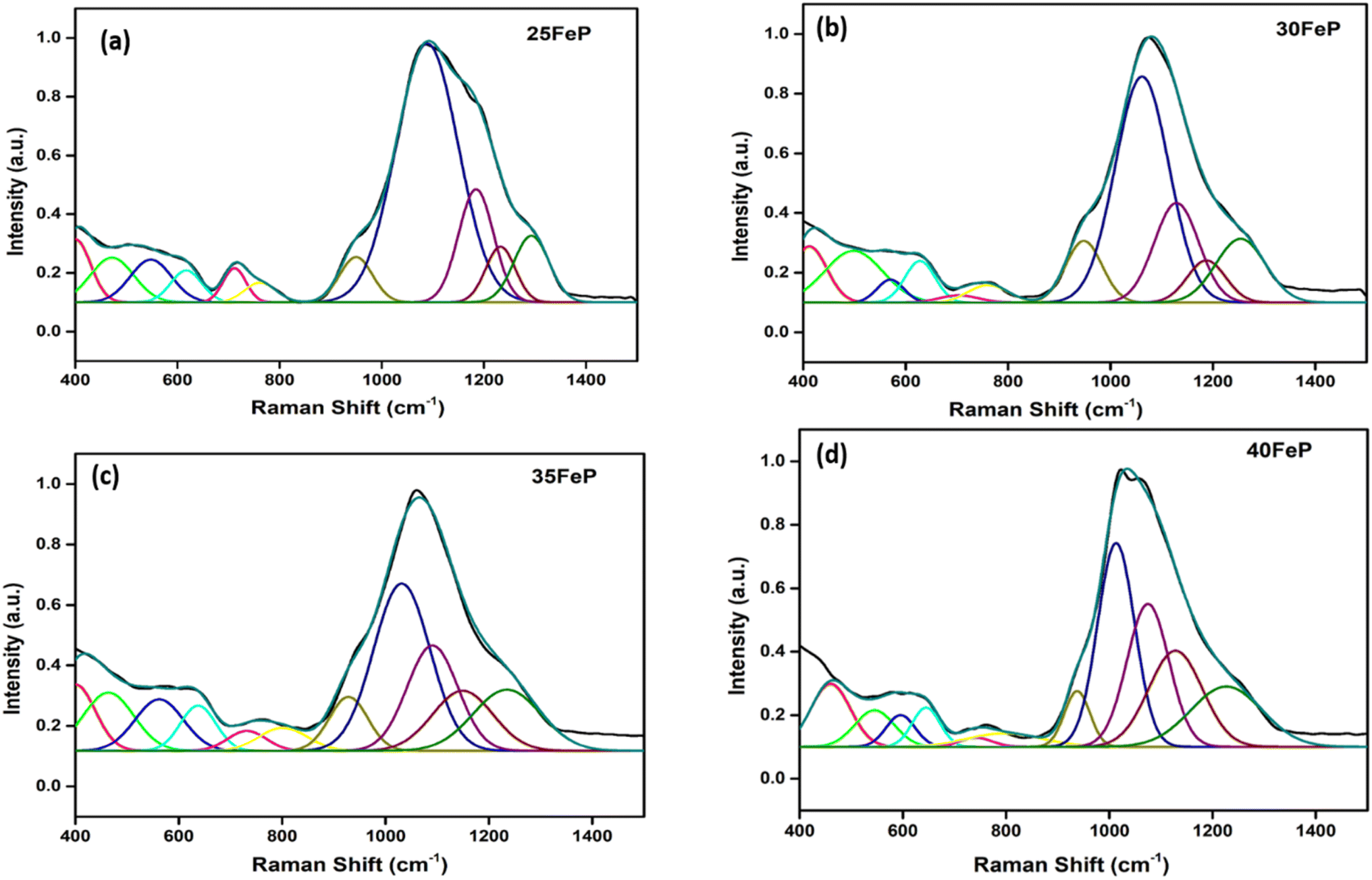 | ||
| Fig. 4 Decomposition of Raman spectrum for (a) 25FeP (b) 30FeP (c) 35FeP (d) 40FeP into Gaussian peaks. | ||
All the peaks in the mid-frequency region below 400 cm−1 are due to the bending vibration modes of Fe–O and P–O bonds.19,20 In this frequency range, glassy samples show a broad band centred at ∼340 cm−1 while the Raman spectra of 50FeP glass-ceramic sample shows several sharp peaks at 109, 132, 179, 197, 230, 246, 277, 336, 367 and 391 cm−1.20 In 50FeP glass ceramic sample, peaks were observed at 447 and 460 cm−1 which are due to O–P–O bending modes of Q0 units.19,20 The Raman band in glasses in the range of 664–823 cm−1 is due to the symmetric stretching vibrations of P–O–P bonds that contain bridging oxygens. Relative intensity of this band decreases with decrease in concentration of P2O5. The broad band in the Raman shift range: 869–1376 cm−1 in glasses is due to symmetric and asymmetric stretching vibration modes of bridging and non-bridging oxygens associated with different phosphate tetrahedra i.e., orthophosphate (Q0), pyrophosphate (Q1) and metaphosphate (Q2), respectively. The shoulder at ∼950 cm−1 is due to the symmetric stretching vibration of (PO4)3− anions or the orthophosphate groups. In case of 50FeP sample, two sharp peaks are observed at 950 and 971 cm−1 and these are due to vibrations of orthophosphate, (PO4)3− anions. The band at 1160 cm−1 is due to the asymmetric stretching vibrations of pyrophosphate, (P2O7)4− units of the Q1 groups.19,20
In the glass samples: 25FeP, 30FeP and 35FeP, the maximum intensity is of the Raman band at 1169 cm−1 and this indicates that pyrophosphate groups are the dominant short-range structural units in the glasses containing 25 to 35 mol% of Fe2O3, the pyrophosphate units are linked by Fe–O units.19,20 The Raman spectra of the glass sample 40FeP shows some differences, it has peak intensity at ∼1002 cm−1 instead at 1169 cm−1, the crystalline sample 50FeP also shows maximum intensity at ∼1002 cm−1. The peak at 1002 cm−1 is due to P–O− stretching vibrations of non-bridging oxygens in Q0 units in orthophosphate groups. Finally, the shoulder at 1293 cm−1 is due to the bond vibrations of metaphosphate (PO3)− units.19,20 The analysis of the area of the deconvoluted peaks of Raman spectra provides more information about the short-range structural transformation occurring in the iron phosphate glasses as a function of Fe2O3 concentration. The variation of area of the 11 peaks in the glass samples is shown in Fig. 5. It is found that the maximum area is of the peak at ∼1087 cm−1 and the area of this peak decreases significantly from 48 to 24% with an increase in Fe2O3 concentration from 25 to 40 mol%. This peak is due to symmetric stretching P–O vibrations in Q1-FeO6 units. Similarly, the areas of the peak at 472 cm−1 is due to bending vibrations of P–O bonds in Q1-FeO6 units, the area under this peak also decreases steadily. On the other hand, the area under the peaks at 1185 cm−1 (symmetric stretching P–O vibrations in Q1-FeO5 units), 1234 cm−1 (symmetric stretching P–O stretching vibrations in Q1-FeO4 units) and at 1293 cm−1 (asymmetric stretching P–O vibrations in Q1-FeO4) increases steadily (Fig. 5). These results reveal that the average Fe–O co-ordination number decreases with an increase in Fe2O3 content, a decrease in octahedral co-ordination of Fe with O indicates that the oxidation state of Fe must be decreasing simultaneously from +3 to +2, and this is indeed confirmed from the neutron diffraction and Mössbauer studies discussed below.
 | ||
| Fig. 5 Area (%) vs. concentration of Fe2O3 (mol%). | ||
Finally, on comparing the Raman spectra of glasses (samples 25FeP, 30FeP, 35FeP and 40FeP) with that of crystalline sample (50FeP), it is found that broad Raman bands in glasses occur at almost same positions as the sharp peaks in the crystalline sample, and hence it is concluded that the short-range structures of iron phosphate glass and crystalline samples are very similar, further the relative concentration of different short-range structural units i.e. meta, pyro and ortho units varies with an increase in Fe2O3 concentration.19,20
3.5 Short-range structure by neutron diffraction PDF and RMC analysis
The neutron scattering weight factors of all the six atomic pairs of glass samples are given in the Table 3. The neutron scattering weight factors of Fe–Fe, Fe–P and P–P atomic pairs are small while that of Fe–O, P–O and O–O atomic pair correlations have significantly higher values; therefore, the coordination number were calculated for the latter three atomic pair correlations.| Atomic pair | Weight factors, wij (%) | |||
|---|---|---|---|---|
| 25FeP | 30FeP | 35FeP | 40FeP | |
| Fe–Fe | 1.506 | 2.193 | 2.987 | 3.938 |
| Fe–P | 4.907 | 5.547 | 6.018 | 6.430 |
| Fe–O | 16.628 | 19.684 | 22.573 | 25.383 |
| P–P | 3.995 | 3.507 | 3.031 | 2.624 |
| P–O | 27.08 | 24.895 | 22.742 | 20.722 |
| O–O | 45.882 | 44.171 | 42.646 | 40.901 |
Fig. 6 shows the experimental neutron diffraction (S(Q) − 1) factors of 4 glass samples together with those calculated by the RMC technique. The experimental structure factors, (S(Q) − 1) of all the iron phosphate glass samples match well with the calculated structure factors. The neutron diffraction structure factor of the crystalline sample (50FeP) is shown in Fig. 7. The structure factors were used to calculate the pair distribution function, g(r) by Fourier transformation (eqn (3)) and these are displayed for all samples on Fig. 8a and b.
 | ||
| Fig. 6 Experimental and RMC calculated neutron structure factors (S(Q) − 1) of 4 iron phosphate glasses. Curves are displaced by 1 unit successively for clarity. | ||
 | ||
| Fig. 7 Neutron structure factor, S(Q) of crystalline iron phosphate sample. | ||
 | ||
| Fig. 8 Pair distribution function, g(r) of iron phosphate glasses (a) 25FeP to 40FeP and (b) crystalline sample (50FeP) calculated by Fourier transformation of S(Q) with Lorch modification function. | ||
The first peak in g(r) of the sample 25FeP is centered at 1.58 Å, this peak shifts to higher values of 1.62–1.65 Å in the samples 30FeP, 35FeP and 40FeP and it is centered at a significantly higher position of 1.84 Å in the crystalline sample 50FeP. This peak is due to superposition of P–O and Fe–O atomic pair correlations, and shifting of its position to longer distances with an increase in Fe2O3 concentration is due to a rise in the relative concentration of Fe–O atomic pairs in the glass network and also due to enhancement in the relative concentration of Fe2+ at the expense of Fe3+ (as found by Mössbauer studies discussed later). It is known that Fe2+–O bond lengths are longer than those of Fe3+–O linkages.21,28 The second peak in g(r) at ∼2.5 Å is due to O–O and P–P correlations and its intensity decreases with a decrease in the concentration of P2O5 in the samples. The third peak at ∼3.5 Å is due to Fe–Fe atomic pairs and its intensity grows significantly with an increase in Fe2O3 concentration. The pair distribution function of the sample 50FeP shows oscillations at distances, r > 6 Å due to long-range atomic order that exists in the crystalline sample (Fig. 8b). In the case of the crystalline sample 50FeP, the structure factor shows the sharp and intense peak at ∼2 Å−1 (Fig. 7), the First Sharp Diffraction Peak is due to medium range order of Fe–Fe and Fe–P atomic pairs. The higher medium range ordering is manifested in g(r) of the sample 50FeP (Fig. 8b) in which the intensity of the first peak at 1.84 Å is significantly less than that of the second peak at 3.4 Å. It is therefore concluded that Fe–Fe and Fe–P atomic pairs are dominant scattering species in the crystalline sample, whereas in case of glasses, the short range structural features of P–O and Fe–O atomic pairs are the dominant scattering species (Fig. 8a).
The P–O and Fe–O pair correlations strongly overlap in g(r) distributions, however the neutron data modelling by RMC technique provided the partial atomic pair distribution functions, gij(r) and the coordination numbers, Nij. The partial pair correlation functions of all the six atomic pairs i.e. gFe–O(r),gFe–P(r), gFe–O(r), gP–P(r),gP–O(r) and gO–O(r) are displayed in Fig. 9a–c and 10a–c.
 | ||
| Fig. 9 Partial atomic pair correlations of (a) Fe–O (gFe–O(r)), (b) P–O (gP–O(r)) and (c) O–O (gO–O(r)), in glass samples (curves are shifted successively along the y axis by 2.5 units for clarity). | ||
 | ||
| Fig. 10 Partial atomic pair correlations of (a) Fe–Fe (gFe–Fe(r)), (b) P–P (gP–P(r)) and (c) Fe–P (gFe–P(r)) in glass samples (curves are shifted successively along the y-axis by 2.5 units for clarity). | ||
The gFe–O(r) has a peak at 1.79 ± 0.05 Å in 25FeP glass sample (Fig. 9a). On increasing the Fe2O3 concentration from 25 to 40 mol%, the peak position in Fe–O pair correlation remains constant at 1.79 Å (Fig. 9a and Table 4). It is reported that the peaks in the range of 1.80–1.87 Å are due to Fe3+–O bonds,21,28 and further it is observed that the gFe–O is asymmetrical towards its tail end which indicates that there is significant peak due to Fe2+–O bonds, that are reported to show peaks at a longer distance of 2.12 Å in the iron phosphate glass network.21,28 Zhang et al. found from titration analysis that Fe2+/Fe3+ concentration ratio in glasses increases with melting time and temperature and with an increase in Fe2O3 concentration,19 these investigators used alumina instead of a more durable platinum crucible for glass melting, the alumina crucibles can introduce Al3+ impurities in the final glasses and Al3+ can reduce some of Fe3+ into Fe2+ and produce erroneous results on iron phosphate glass structure and properties. The reported results of increase in Fe2+ concentration by Zhang et al. in iron phosphate glasses with increase in melting time and temperature can also be due to steady increase in leaching of alumina crucible by iron phosphate melt that will introduce large concentration of Al3+ impurities and Al–O correlations in the phosphate network that have bond lengths very close to that of Fe–O linkages.19
| Sample code | Interatomic distances, gij(r) [Å] | |||||
|---|---|---|---|---|---|---|
| Fe–Fe | Fe–P | Fe–O | P–P | P–O | O–O | |
| 25FeP | 3.50 | 3.08 | 1.79 | 2.70 | 1.50 | 2.29, 2.75 |
| 30FeP | 3.41 | 3.05 | 1.79 | 2.76 | 1.50 | 2.25, 2.75 |
| 35FeP | 3.45 | 3.13 | 1.79 | 2.74 | 1.50 | 2.29, 2.75 |
| 40FeP | 3.45 | 3.10 | 1.79 | 2.70 | 1.48 | 2.29, 2.70 |
Hoppe et al. found from high energy X-ray diffraction studies that iron phosphate glasses prepared using FeO as a starting material (in the batch mixture) contained mostly Fe2+ while the glasses prepared from Fe2O3 contained mostly Fe3+ in the network, it is noteworthy that Hoppe et al. used alumina crucible for the synthesis of iron phosphate glasses.21 In the present study, Pt crucible, melting temperature of 1300 °C and melting time of ∼1 h has been used for glass synthesis, however large concentration of Fe2+ has been found in all samples (from Mössbauer studies discussed below) although Fe2O3 has been used as the starting material. It is therefore concluded that oxidation state of Fe in the final iron phosphate glass network doesn't seem to depend on the oxidation state of Fe in the starting materials.
The gP–O(r) and gO–O(r) distributions are shown in Fig. 9b and (c) respectively. The P–O partial pair correlation distributions are asymmetrical, and the most probable P–O bond length is in the range: 1.48–1.50 ± 0.05 Å in glasses. The O–O pair distribution is asymmetrical and there are at least two O–O distances of 2.27 ± 0.05 Å and 2.6 ± 0.05 Å in the phosphate network (Table 4). The Fe–Fe, Fe–P and P–P partial correlation functions are shown in Fig. 10a–c. The Fe–Fe partial pair distribution function, gFe–Fe(r), shows peak at 3.45 ± 0.05 Å (Fig. 10a). The P–P and Fe–P atomic pair correlation distributions shows the first peaks in the ranges: 2.70–2.90 ± 0.05 Å and 3.10–3.30 ± 0.05 Å, respectively. Within the sample series, the shape of the atomic pair distributions are quite similar to each other.
The cation-oxygen co-ordination numbers were calculated from the gij(r) plots and the corresponding rmin and rmax values that were used to calculate co-ordination number data are given in Table 5. The average co-ordination number of iron with oxygen (NFe–O) is 4.8 ± 0.1 in the 25FeP sample, it decreases steadily to a value of 4.2 in the glass sample 40FeP (Fig. 11a). Earlier Stoch et al.28 found from ab initio simulations studies on 40Fe2O3–60P2O5 glass that the short-range structure of this glass is very similar to that of crystalline FePO4 and basic building blocks are tetrahedral FeO4 and PO4 units.
| Sample code | Co-ordination number, Nij (±0.1) | ||
|---|---|---|---|
| Fe–O | P–O | O–O | |
| 25FeP | 4.8 (1.30–2.40) | 3.8 (1.30–2.35) | 6.6 (2.10–3.10) |
| 30FeP | 4.6 (1.40–2.35) | 3.9 (1.20–2.30) | 6.5 (2.00–3.10) |
| 35FeP | 4.3 (1.40–2.35) | 3.8 (1.30–2.35) | 6.4 (2.10–3.10) |
| 40FeP | 4.2 (1.40–2.35) | 3.7 (1.30–2.35) | 6.3 (2.00–3.10) |
 | ||
| Fig. 11 (a) Fe–O (b) P–O and (c) O–O co-ordination distributions in iron phosphate glasses. | ||
The P–O coordination (NP–O) is in the range of 3.8–3.7(±0.1) (Fig. 11b), therefore it is concluded that the P is essentially tetrahedrally co-ordinated with oxygens in the iron phosphate network. Finally, in case of O–O correlations, the average coordination number (Fig. 11c) is found to be in the range of 6.6–6.3 (±0.1). A steady decrease in Fe–O co-ordination number with an increase in Fe2O3 mol% is due to increasing oxygen deficiency in the glass network which also slightly lowers the P–O and O–O co-ordination numbers.
3.6 Fe oxidation states
The 57Fe Mössbauer spectra of the four glass samples (i.e. those with x = 25, 30, 35, 40) recorded at room temperature are displayed on Fig. 12. The spectra allowed us to distinguish one Fe3+ along with three different main Fe2+ iron microenvironments, each contributing to the spectra with a distinct quadrupole doublet component. The spectral shape indicates that the individual absorption peaks belonging to the doublet components are subject to non-Lorentzian broadening. This is due to a distribution in the isomer shift (δ) and/or in the quadrupole splitting (Δ) Mössbauer parameters, reflecting the presence of a high number of slightly different iron microenvironments associated with the different Fe2+ and Fe3+ species, which is indeed a typical trait of glasses. The spectra were fitted successfully with Voigt-based fitting57 by assuming a Gaussian distribution to be present only in the quadrupole splitting values of the doublets, except in the case of component Fe2+(C) (Fig. 12), for which the peaks' profile was better modelled by pure Lorentzian broadening. The Mössbauer parameters obtained from the fits are listed in Table 6.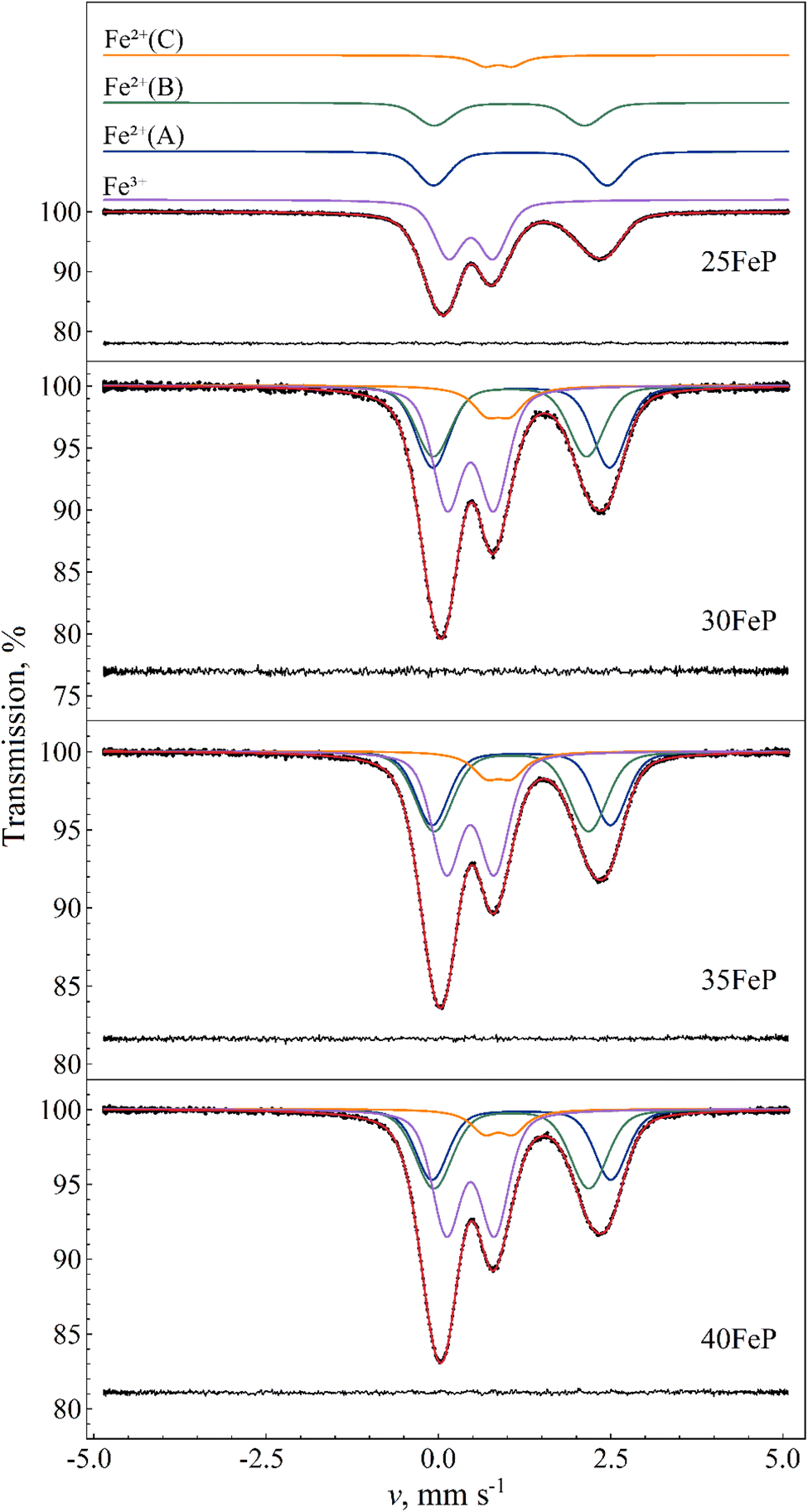 | ||
| Fig. 12 Room temperature 57Fe Mössbauer spectra (dots) of amorphous 25FeP, 30FeP, 35FeP and 40FeP samples, along with their fit envelope and decomposition of the resonant absorption peaks into subcomponents (solid line). For the sample 25FeP the subcomponents are shifted upwards for clarity. The fit residual is shown below the spectra. See Table 6 for the associated fit parameter values. | ||
| Sample code | 25FeP | 30FeP | 35FeP | 40FeP |
|---|---|---|---|---|
| x | 25 | 30 | 35 | 40 |
| χn2 | 1.20 | 1.07 | 1.04 | 1.10 |
|
||||
| Quadrupole doublet Fe2+(A) | ||||
| RA, % | 29.2(2.3) | 28.6(3.5) | 25.1(2.7) | 24.3(2.4) |
| δ, mm s−1 | 1.19(3) | 1.20(5) | 1.20(1) | 1.21(1) |
| 〈Δ〉, mm s−1 | 2.52(6) | 2.6(1) | 2.59(4) | 2.60(4) |
| σ(Δ), mm s−1 | 0.35(1) | 0.36(2) | 0.36(1) | 0.35(1) |
| W, mm s−1 | 0.256(6) | 0.255(9) | 0.245(6) | 0.249(5) |
|
||||
| Quadrupole doublet Fe2+(B) | ||||
| RA, % | 20.4(2.3) | 25.5(3.5) | 30.2(2.8) | 30.1(2.4) |
| δ, mm s−1 | 1.03(5) | 1.04(7) | 1.06(2) | 1.06(2) |
| 〈Δ〉, mm s−1 | 2.18(8) | 2.2(1) | 2.24(3) | 2.25(3) |
| σ(Δ), mm s−1 | 0.37(2) | 0.38(2) | 0.42(1) | 0.41(1) |
| W, mm s−1 | 0.256(6) | 0.255(9) | 0.245(6) | 0.249(5) |
|
||||
| Quadrupole doublet Fe2+(C) | ||||
| RA, % | 7.4(3) | 8.7(5) | 7.9(4) | 7.5(3) |
| δ, mm s−1 | 0.872(6) | 0.87(1) | 0.877(9) | 0.875(7) |
| 〈Δ〉, mm s−1 | 0.40(1) | 0.34(2) | 0.36(2) | 0.41(2) |
| σ(Δ), mm s−1 | 0(F) | 0(F) | 0(F) | 0(F) |
| W, mm s−1 | 0.41(1) | 0.49(3) | 0.49(2) | 0.47(2) |
|
||||
| Quadrupole doublet Fe3+ | ||||
| RA, % | 43.0(2) | 37.1(4) | 36.9(3) | 38.2(3) |
| δ, mm s−1 | 0.470(2) | 0.467(4) | 0.464(3) | 0.466(3) |
| 〈Δ〉, mm s−1 | 0.640(4) | 0.673(7) | 0.693(8) | 0.693(6) |
| σ(Δ), mm s−1 | 0.286(5) | 0.298(8) | 0.310(6) | 0.298(5) |
| W, mm s−1 | 0.256(6) | 0.255(9) | 0.245(6) | 0.249(5) |
Around 40% of the spectral area is associated with iron in the high-spin (HS) Fe3+ state in each case. With increasing Fe2O3 mol% the relative area fraction of the Fe3+ component drops by ca. 6% (from ∼43% to ∼37%) as the Fe2O3 concentration increases from 25% to 30% and above, which can refer to a simultaneously increased level of oxygen deficiency in the glass structure. The Fe3+ isomer shift and mean quadrupole splitting Mössbauer parameters, δ ≈ 0.47 mm s −1 and 〈Δ〉 ≈ 0.64–0.69 mm s −1 (Table 6), are consistent with octahedral oxygen coordination of high-spin Fe3+.
For the assignment of the obtained Fe3+ and Fe2+ doublet components it is informative to compare the corresponding Mössbauer parameters with those obtained for Fe7(PO4)6 in the crystalline sample 50FeP (Table 7). The 57Fe Mössbauer spectra of the latter sample measured at room temperature and at T = 80 K are shown in Fig. 13. Based on the associated X-ray diffractometry results (see Fig. 1) we assumed paramagnetic components of Fe7(PO4)6 and Fe2P2O7 to contribute to the spectra. The spectral decomposition was carried out accordingly, by taking into account the results of previous studies concerning Fe7(PO4)6 (ref. 58) and Fe2P2O7,59 and applying suitable constraints among others regarding the relative area fractions of the individual subcomponents. To fit these spectra properly in the thin absorber approximation limit, a minor asymmetry in the peak areas associated with the individual doublets had to be assumed (Table 7), which may be the consequence of thickness effects. The results indicate that in the sample 50FeP ca. 79% of iron is incorporated in the lattice of Fe7(PO4)6, and ca. 16% in Fe2P2O7 (Table 7), while a further ca. 5% contributes to a minor Fe2+ species (denoted with Fe2+(*) on Fig. 13) that remains unassigned. On the basis of the relatively low quadrupole splitting of the latter component, it may originate from Fe2+ species situated in a non-regular, distorted oxygen coordination environment. In the lattice of Fe7(PO4)6, Fe2+ occur with 6-fold (octahedral) and 5-fold (pyramidal) oxygen coordination60 with a relative occurrence of 2:1 in favor of the 5-fold coordination. On the basis of its higher relative area fraction and lower isomer shift value, among the Fe2+ components associated with Fe7(PO4)6 (i.e. Fe2+(a) and Fe2+(b) in Table 7) in the spectra of 50FeP it is the Fe2+(b) component that can be associated with the 5-fold coordinated Fe2+ species. In addition to the Fe2+ sites, the structure of Fe7(PO4)6 also includes two distinct Fe3+ sites.60 In both of these Fe3+ sites, the iron atoms exhibit a distorted octahedral oxygen coordination. The Mössbauer parameters we obtained for these sites are given under the names Fe7(PO4)6–Fe3+(a) and Fe3+(b) in Table 7.
| Sample code | 50FeP | |
|---|---|---|
| T, K | RT | 80 |
| χn2 | 1.15 | 1.08 |
| A+/A− | 1.08(1) | 1.10(1) |
|
||
| Fe7(PO4)6–Fe2+(a) | ||
| RA, % | 11.1(1) | 11.0(1) |
| δ, mm s−1 | 1.258(3) | 1.45(4) |
| 〈Δ〉, mm s−1 | 2.874(6) | 2.97(8) |
| σ(Δ), mm s−1 | 0(F) | 0(F) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe7(PO4)6–Fe2+(b) | ||
| RA, % | 23.0(2) | 22.8(2) |
| δ, mm s−1 | 1.151(3) | 1.298(3) |
| 〈Δ〉, mm s−1 | 2.529(4) | 2.939(5) |
| σ(Δ), mm s−1 | 0(F) | 0(F) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe7(PO4)6–Fe3+(a) | ||
| RA, % | 23.0(2) | 22.8(2) |
| δ, mm s−1 | 0.50(1) | 0.60(1) |
| 〈Δ〉, mm s−1 | 0.74(1) | 0.73(1) |
| σ(Δ), mm s−1 | 0.10(1) | 0.14(3) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe7(PO4)6–Fe3+(b) | ||
| RA, % | 22.2(2) | 22.0(2) |
| δ, mm s−1 | 0.37(1) | 0.42(3) |
| 〈Δ〉, mm s−1 | 0.71(1) | 0.80(7) |
| σ(Δ), mm s−1 | 0.09(1) | 0.14(3) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe2P2O7–Fe2+(1) | ||
| RA, % | 7.9(3) | 8.2(2) |
| δ, mm s−1 | 1.31(2) | 1.35(5) |
| 〈Δ〉, mm s−1 | 2.33(4) | 2.6(1) |
| σ(Δ), mm s−1 | 0.15(2) | 0(F) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe2P2O7–Fe2+(2) | ||
| RA, % | 7.9(3) | 8.2(2) |
| δ, mm s−1 | 1.24(2) | 1.28(5) |
| 〈Δ〉, mm s−1 | 2.47(4) | 2.7(2) |
| σ(Δ), mm s−1 | 0.15(2) | 0(F) |
| W, mm s−1 | 0.261(4) | 0.309(6) |
|
||
| Fe2+(*) | ||
| RA, % | 5(1) | 5(F) |
| δ, mm s−1 | 1.35(5) | 1.36(3) |
| 〈Δ〉, mm s−1 | 1.4(1) | 2.32(5) |
| σ(Δ), mm s−1 | 0(F) | 0(F) |
| W, mm s−1 | 0.7(1) | 0.28(5) |
 | ||
| Fig. 13 57Fe Mössbauer spectrum (dots) of 50Fe2O3·50P2O5 (50FeP) crystalline sample measured at 80 K (top) and at room temperature (bottom), along with the indication of the fit envelope and the subcomponents (solid line). For the measurement performed at 80 K, the fitted subcomponents are shifted upwards for clarity. Below the spectra the fit residual is displayed. See Table 7 for the associated fit parameter values. | ||
The Fe3+ isomer shift and mean quadrupole splitting Mössbauer parameters obtained for the glass samples are δ ≈ 0.47 mm s−1 and 〈Δ〉 ≈ 0.64–0.69 mm s−1 (Table 6). These values are consistent with octahedral oxygen coordination of high-spin Fe3+. They are also similar to the parameters of the Fe3+(a) and Fe3+(b) species of Fe7(PO4)6 (Table 7), with a closer match to the Fe3+(a) component. This corroborates that Fe3+ ions have distorted octahedral oxygen coordination in these glasses. The distortion of the oxygen octahedra appears to have a wide range of variability in the glass, as reflected by the considerable standard deviation of the distribution of quadrupole splitting values (Table 6). Note that some of the Gaussian broadening, which we modeled as a distribution in the quadrupole splitting values, may also be the result of a distribution in the isomer shift values.
Similarity between the δ and 〈Δ〉 values of Fe2+(b) in Fe7(PO4)6 and Fe2+(A) in the glass samples (Table 6) then suggests that Fe2+(A) may be associated mainly with 5-fold coordinated high-spin Fe2+ species in the glass. At the same time, contribution of Fe2+ species with 6-fold (octahedral) oxygen coordination to the Fe2+(A) component cannot be fully excluded, as such a species may also display similar δ and 〈Δ〉 Mössbauer parameter values; one such example is the Fe2+(2) species in Fe2P2O7 (Table 7) where iron has octahedral oxygen coordination.59
Considering the Fe–O coordination distribution function displayed in Fig. 11a, component Fe2+(B) in Table 6 needs then to be attributed mainly to 4-fold (e.g. tetrahedral) coordinated Fe2+ species in the glass. The latter attribution is further corroborated by the increasing tendency in the relative area fraction of Fe2+(B) with increasing Fe2O3 mol% (Table 6), which is in qualitative agreement with a similar tendency reflected by the Fe–O coordination distribution function (Fig. 11a).
The isomer shift (δ ≈ 0.87 mm s−1) and quadrupole splitting (Δ ≈ 0.4 mm s−1) of the Fe2+(C) component found in the spectra of the amorphous samples (Fig. 12) are considerably lower (Table 6) than respective values of Fe2+(A) and Fe2+(B), and may be attributed to e.g., 4-fold coordinated Fe2+ with square-planar oxygen coordination environment. This assignment is also supported by similar isomer shift and quadrupole splitting values observed for square-planar coordinated high-spin Fe2+ in the mineral gillespite (δ ≈ 0.75 mm s−1 wrt. α-Fe, Δ ≈ 0.51 mm s−1).61
Fig. 14 displays the sum of the relative area fractions of the Fe2+(B) and Fe2+(C) doublet components that were attributed to Fe2+ species with 4-fold oxygen coordination. Clearly, from 25FeP to 35FeP the relative weight of 4-fold coordinated Fe2+ environments increases. On the basis of Table 6, this increase occurs at the expense of both the 5- and 6-coordinated iron species. In line with the model of Hoppe,62 the gradual reduction of the coordination number of iron indicates that in the studied samples the concentration of iron already exceeded the limit where the available terminal oxygen ions of PO4 tetrahedra could ensure maximum (5–6) coordination number for all the iron ions. Based on the outstanding chemical durability of the 40FeP composition (see Table 9), the development of the glass structure leading to 40FeP must play a decisive role in shaping the advantageous material properties associated with this composition. Based on Fig. 14, from 35FeP to 40FeP the iron coordination numbers seem to be stationary. However, as calculated from atomic pair distribution functions, the concentration of iron atoms with 4-fold oxygen coordination increases further even in this range (Fig. 11a).
 | ||
| Fig. 14 Mössbauer-spectrum relative area fractions associated with 4-coordinated Fe species calculated based on the spectral decomposition of the room-temperature Mössbauer spectra of the 25FeP, 30FeP, 35FeP and 40FeP glass samples (Fig. 12). Among the identified components (Table 6), Fe2+ (B) and Fe2+ (C) were evaluated to be 4-coordinated species; the figure displays the sum of the relative area fractions of these components as a function of the Fe atomic concentration wrt. the total number of Fe and P atoms in the structure. See Table 6 for the associated Mössbauer parameters. Note that the depicted values were not corrected for possible differences in the recoilless fractions of Fe2+ and Fe3+ ions. | ||
Due to possible differences in the temperature dependence of the f(Fe2+) and f(Fe3+) recoilless fractions associated with Fe2+ and Fe3+ species respectively, the Fe2+/Fe3+ ratio in the glass may not be faithfully reflected by the corresponding relative spectral area ratios characteristic to the room temperature Mössbauer spectra of the glass samples (Fig. 12 and Table 6). In order to find a more reliable value of the Fe2+/Fe3+ ratio, the sample 30FeP was also measured at T = 80 K. The obtained spectrum and the associated fit parameters are shown in Fig. 15 and Table 8. On the basis of the isomer shift and mean quadrupole splitting (〈Δ〉) parameters (Table 8), in the latter spectrum (Fig. 15) one can clearly identify the components associated with the iron species Fe2+(A), Fe2+(B), Fe2+(C) and Fe3+. In contrast with the spectra of glasses recorded at room temperature (Fig. 12), which reflect an Fe2+/Fe3+ spectral area ratio of approximately 1.3–1.7 (Table 6), the relative area fractions of the Fe2+ and Fe3+ components derived on the basis of the spectrum in Fig. 15 indicate that the Fe2+/Fe3+ ratio is closer to 2 in these glasses, and hence Fe2+ species are clearly in majority concentration in the glass samples (Table 8). Concerning the sample 30FeP in particular, the Fe2+/(Fe2++Fe3+) ratio changes from 62.9(4)% (at room temperature) to 66.0(4)% (at 80 K), i.e. roughly by a factor of q ≈ 1.05. However, at 80 K the recoilless fraction of Fe2+ may still be smaller than that of Fe3+. Indeed, by assuming that at room temperature the f(Fe3+)/f(Fe2+) ratio of the recoilless fractions is ca. 1.3 as given, e.g., in 63, the associated factor turns out to be q ≈ 1.09, meaning that in the sample 30FeP the real Fe2+/(Fe2+ + Fe3+) ratio could reach 68–69%.
 | ||
| Fig. 15 57Fe Mössbauer spectrum (dots) of 30Fe2O3-70P2O5 (30FeP) glass measured at 80 K, along with the indication of the fit envelope and the subcomponents (solid line). The subcomponents are shifted upwards for clarity. Below the spectrum the fit residual is displayed. See Table 8 for the associated fit parameter values. | ||
K. Internal Lorentzian FWHM line width values (W) were constrained to be the same for all the subcomponents. See Table 6 for the corresponding room temperature data and further notations
| Sample code | 30FeP |
|---|---|
| χn2 | 1.43 |
|
|
| Quadrupole doublet Fe2+(A) | |
| RA, % | 31.4(3.9) |
| δ, mm s−1 | 1.34(2) |
| 〈Δ〉, mm s−1 | 2.82(5) |
| σ(Δ), mm s−1 | 0.38(2) |
| W, mm s−1 | 0.26(1) |
|
|
| Quadrupole doublet Fe2+(B) | |
| RA, % | 28.2(3.9) |
| δ, mm s−1 | 1.17(4) |
| 〈Δ〉, mm s−1 | 2.42(6) |
| σ(Δ), mm s−1 | 0.44(3) |
| W, mm s−1 | 0.26(1) |
|
|
| Quadrupole doublet Fe2+(C) | |
| RA, % | 6.4(3) |
| δ, mm s−1 | 1.01(2) |
| 〈Δ〉, mm s−1 | 0.41(3) |
| σ(Δ), mm s−1 | 0.23(2) |
| W, mm s−1 | 0.26(1) |
|
|
| Quadrupole doublet Fe3+ | |
| RA, % | 34.0(4) |
| δ, mm s−1 | 0.556(7) |
| 〈Δ〉, mm s−1 | 0.73(1) |
| σ(Δ), mm s−1 | 0.31(1) |
| W, mm s−1 | 0.26(1) |
The theoretical model we applied for the decomposition of the iron phosphate glass Mössbauer spectra was chosen on account of being the simplest model providing reasonable and consistent results along with excellent mathematical fits (Table 6 and Fig. 12). Nevertheless, the model we applied appears to be rather uncommon in related literature, making it pertinent to compare our results with those obtained using other approaches to the decomposition of Mössbauer spectra of iron phosphate glasses. One of the main difficulties in analyzing the spectra of these glasses is that the absorption peaks are non-Lorentzian, making traditional spectrum analysis techniques based on a few Lorentzian doublets inaccurate. Therefore, different solutions have been developed to address this problem and to extract information from the Mössbauer spectra of such glasses.
The most straightforward approach is to fit glass spectra, akin to those in Fig. 12, with one broadened Lorentzian doublet for Fe2+ and one for Fe3+,29,64–66 which will provide approximate average Mössbauer parameter values for the two different iron oxidation states. While not suitable to extract fine details from spectra of iron phosphate glass where Fe2+ or Fe3+ ions exist with multiple oxygen coordination environments, this method may be justified especially when the signal-to-noise ratio of the spectrum is not high enough to reveal deviations from the Lorentzian line profile. It also has the advantage of being easily reproducible in different laboratories, facilitating the comparison of associated results. However, even in this simple case two different solutions may be found reasonable regarding the Mössbauer parameters: one in which Fe3+ has a higher quadrupole splitting (QS), and therefore the left-most absorption peak belongs to Fe3+ in the fit model, and another in which Fe3+ has a lower QS, and the left-most Lorentzian of the model belongs to Fe2+ (similarly to the cases shown in Fig. 12). Usually, the former model provides the better fit, leading to room temperature Fe3+ quadrupole splitting values around 0.8–0.9 mm s−1,29,64,65 in contrast with the lower mean Fe3+ quadrupole splitting values of 0.64–0.70 mm s−1 we obtained (Table 6). However, when we apply the same model to the spectrum of 40FeP shown in Fig. 12 (which composition is also studied for example in ref. 22, 29, 63 and 66–68), we also find the larger QS solution to provide the better fit with main Mössbauer parameters of δ (Fe3+) ≈ 0.38 mm s−1, Δ(Fe3+) ≈ 0.89 mm s−1, δ(Fe2+) ≈ 1.23 mm s−1, Δ(Fe2+) ≈ 2.23 mm s−1. At the same time, the more accurate multi-component fits, displayed in Fig. 12, already favor the solution with the lower QS values of Fe3+ given in Table 6.
Another common method is the formal fit of the Fe2+ and Fe3+ spectrum parts with multiple (usually 3–4 for each oxidation state) broadened Lorentzian quadrupole doublets, without associating specific iron species with the individual components. The number of doublets is chosen to be high enough to achieve an acceptable fit of the spectra. The results are then reported as weighted averages of the isomer shift (IS) and quadrupole splitting values obtained for the individual oxidation states.22,63,67–71 An advantage of this method over the previous one is that due to the better fit of the spectra the obtained averages can reflect minor changes in the spectral shape more sensitively than by using only a single doublet for each oxidation state.
The main limitation of the two methods mentioned above is that they provide only a single average isomer shift and quadrupole splitting value for each oxidation state. This means that they cannot distinguish between different Fe2+ (or Fe3+) oxygen coordination environments that are simultaneously present in the sample. The average IS and QS values resulting from such analyses of Mössbauer spectra of iron phosphate glasses are typically found to refer mainly to 6-fold (octahedral or distorted octahedral) oxygen coordination for both Fe2+ and Fe3+ ions.22,29,63,66,68–71
A third method applied to decompose Mössbauer spectra of iron phosphate glasses utilizes the VBF (Voigt based fitting,72) or xVBF (extended Voigt based fitting57) method to fit quadrupole doublets by assuming correlated Gaussian distributions of the QS and IS Mössbauer parameter values of the individual doublets.28,73–75 This is the same method we also apply in the present work, with the significant difference that we have assumed the distributions of IS and QS values to be uncorrelated (or that of IS values being negligible). In the presence of IS-QS correlation the VBF and xVBF models can produce a wide variety of doublet peak profiles, where the amplitudes and the widths of the individual (Voigt-profile) peaks contributing to the doublet can differ from each other. By adjusting the parameter of correlation between distributions of IS and QS values of individual doublets of a fit model, iron phosphate glass spectra could be accounted for with high accuracy with only three main VBF or xVBF components and associated three main iron positions, two assumed for Fe3+ and one for Fe2+.28,73–75 By using this method, in different iron phosphate glass samples Fe3+ and Fe2+ ions were associated with positions having 6-, 5- and 4-fold and 6- and 5-fold oxygen coordination, respectively. As we see it, currently it is unclear whether correlation between distributions of IS and QS values of iron species should indeed be taken into account in the interpretation of iron phosphate glass Mössbauer spectra. Our results indicate (Fig. 12 and Table 6) that excellent fits can be achieved by assuming only four main iron species, even without considering the possibility of IS-QS distribution correlations. In comparison with previous reports, a novel component in our decomposition of the iron phosphate glass Mössbauer spectra (Fig. 12) is the minor doublet Fe2+(C) (Table 6) that displays Mössbauer parameters referring to Fe2+ in a square-planar oxygen coordination environment. The inclusion of this component into our model played a decisive role in the achievement of high-accuracy spectrum fits with a limited number of spectrum components.
3.7 Chemical durability of iron phosphate glasses
The chemical stability of two bulk glasses of composition 30Fe2O3–70P2O5 (sample code 30FeP) and 40Fe2O3–60P2O5 (sample code 40FeP) were analyzed over a period of 10 days in MQ-water kept at 90 °C and the concentration of dissolved Fe and P was measured after 1, 2, 3, 7 and 10 days and the experimental data are presented in Table 9. In case of glass sample 30FeP, the concentration of P in MQ-water increases from a value of 1.014 ± 0.100 to 15.855 ± 0.253 mg l−1 while that of Fe increases from a value of 0.092 ± 0.006 to 0.747 ± 0.049 mg l−1 over a time period of 10 days. In case of 40FeP glass, the concentration of P increases from a value of 0.337 ± 0.039 to 1.231 ± 0.195 mg l−1 while that of Fe remains nearly constant in the range: 0.078 ± 0.003 to 0.053 ± 0.013 mg l−1. It is found that the chemical stability of 40FeP glass is drastically higher than that of 30FeP glass and further the concentration of dissolved P is significantly higher than that of Fe. These findings are in agreement with the earlier reports that 40Fe2O3–60P2O5 glass has maximum chemical durability.22–26| Sample code & number of days | P (mg l−1) | Fe (mg l−1) |
|---|---|---|
| 30FeP | 1.014 ± 0.100 | 0.092 ± 0.006 |
| 1 Day | ||
| 40FeP | 0.337 ± 0.039 | 0.078 ± 0.003 |
| 1 Day | ||
| 30FeP | 6.699 ± 0.100 | 0.343 ± 0.008 |
| 2 Days | ||
| 40FeP | 0.552 ± 0.074 | 0.086 ± 0.015 |
| 2 Days | ||
| 30FeP | 11.755 ± 0.188 | 0.506 ± 0.019 |
| 3 Days | ||
| 40FeP | 0.958 ± 0.033 | 0.069±±0.010 |
| 3 Days | ||
| 30Fe | 12.807 ± 0.358 | 0.579 ± 0.0139 |
| 7 Days | ||
| 40FeP | 1.163 ± 0.060 | 0.069 ± 0.006 |
| 7 Days | ||
| 30FeP | 15.855 ± 0.253 | 0.747 ± 0.049 |
| 10 days | ||
| 40FeP | 1.231 ± 0.195 | 0.053 ± 0.013 |
| 10 Days |
4. Conclusions
Iron phosphate glasses with 25 to 40 mol% Fe2O3 were produced by melt quenching. The sample with a higher concentration of 50 mol% Fe2O3 has poor glass-forming ability and it produces crystalline sample containing Fe7[PO4]6 and Fe2P2O7 phases on melt-quenching. The glass transition, crystallization and liquidus temperatures were determined by DSC analysis. The ionic packing fraction show a small variation while the glass forming tendency decreases significantly with an increase in Fe2O3 concentration in the phosphate network. The RMC analysis of neutron diffraction datasets were used to calculate the partial atomic pair distributions and coordination environments in iron phosphate glasses. The most probable P–O and Fe–O bond distances in glasses are 1.50 Å and 1.79 ± 0.05 Å respectively. Mössbauer studies revealed that Fe exists in both 3+ and 2+ oxidation states in the glasses, with a Fe2+/Fe3+ occurrence ratio of ca. 2. Fe3+ ions were identified as having distorted octahedral oxygen coordination, while Fe2+ ions in the glass were found to have 4-fold and 5-fold oxygen coordination environments. The relative fraction of Fe2+ increases significantly with an increase in Fe2O3 concentration in the starting bath mixture, which also lowers the glass forming ability of the system. Iron coordination numbers from partial atomic pair distribution functions and Mössbauer results indicated that the occurrence of Fe2+ with 4-fold oxygen coordination increases with the concentration of Fe2O3. Neutron diffraction and Raman studies found that the short-range structure of iron phosphate glasses and crystalline phases are similar. Leaching experiments on iron phosphate glasses in purified water kept at 90 °C confirmed that chemical durability is enhanced considerably with an increase in iron concentration in the glass and that P dissolution is higher than that of Fe.Data availability
All data reported in the manuscript is available with the authors and will be provided on request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Atul Khanna, acknowledges UGC-DAE-CSR, Mumbai Centre, India for research grants (CRS-M-547) and the Central European Research Infrastructure (CERIC-20212041) for financial support to carry out the neutron diffraction experiments at Budapest Neutron Centre, Centre for Energy Research, Budapest, Hungary. Kajal Dadwal and Suruchi Sharma acknowledge UGC –DAE-CSR Mumbai Centre, India for PhD research fellowships. Maria Gracheva and Zoltán Klencsár express thanks to Gábor Benyács for the preparation of Mössbauer sample holders.References
- J. D. Vienna, Nuclear waste vitrification in the United States: recent developments and future options, Int. J. Appl. Glass Sci., 2010, 1, 309–321 CrossRef CAS.
- C. Corkhill and N. Hyatt, Nuclear Waste Management, IOP Publishing, Bristol, UK, 2018 Search PubMed.
- C. P. Kaushik, R. K. Mishra, P. Sengupta, A. Kumar, D. Das, G. B. Kale and K. Raj, barium borosilicate glass – a potential matrix for immobilization of sulfate bearing high-level radioactive liquid waste, J. Nucl. Mater., 2006, 358, 129–138 CrossRef CAS.
- M. J. Plodinec, Borosilicate Glasses for Nuclear Waste Imobilisation, Glass Technology, vol. 41, 2000, pp. 186–192 Search PubMed.
- M. Fábián, F. Pinakidou, I. Tolnai, O. Czompoly and J. Osan, Lanthanide (Ce, Nd, Eu) environments and leaching behavior in borosilicate glasses, Sci. Rep., 2021, 11, 13272–13286 CrossRef PubMed.
- M. Fábián, F. Gergely, J. Osan, T. Cendak, S. Kesari and R. Rao, Structural investigation of borosilicate glasses containing lanthanide ions, Sci. Rep., 2020, 10, 7835–7848 CrossRef PubMed.
- I. Pegg and I. Joseph, Hazardous and Radioactive Waste Treatment Technologies Handbook, ed. H. Chang, Boca Raton, CRC Press, 2001, ch. 4.2 Search PubMed.
- P. A. Bingham, S. D. Forder, R. J. Hand and A. Lavaysierre, Mössbauer studies of phosphate glasses for the immobilisation of toxic and nuclear wastes, Hyperfine Interact., 2005, 165, 135–140 Search PubMed.
- R. C. Ewing and L. Wang, Phosphates as nuclear waste forms, Rev. Mineral. Geochem., 2002, 48, 673–699 Search PubMed.
- D. Day, C. Ray and C. Kim, Final Report: Iron Phosphate Glasses: an Alternative for Vitrifying Certain Nuclear Wastes, Project No, DEFG07 e96ER45618, 2004 Search PubMed.
- B. C Sales and L. A. Boatner, Lead iron phosphate glass – a stable storage medium for high level nuclear waste, Science, 1984, 226, 45–48 CrossRef PubMed.
- B. C. Sales and L. A. Boatner, Physical and chemical characteristics of lead iron phosphate nuclear waste glasses, J. Non-Cryst. Solids, 1986, 79, 83–116 Search PubMed.
- L. Na Maa, Dissolution Behaviour of Phosphate Glasses, Doctoral Dissertation, Missouri University Rolla, 2014 Search PubMed.
- H. Takebe, M. Fujisawa, Y. Maeda and A. Saitoh, Effect of molybdenum oxide addition on the durability and structure of iron phosphate glasses, J. Ceram. Soc. Jpn., 2021, 129, 105–110 CrossRef CAS.
- S. T. Reis, A. Mogus-Milankovic, V. Licina, J. B. Yang, M. Karabulut, D. E. Day and R. K. Brow, Iron redox equilibrium, structure and properties of zinc iron phosphate glasses, J. Non-Cryst. Solids, 2007, 353, 151–158 CrossRef CAS.
- S. T. Reis, D. L. A. Faria, J. R. Martinelli, W. M. Pontuschka, D. E. Day and C. S. M. Partiti, Structural features of lead iron phosphate glasses, J. Non-Cryst. Solids, 2002, 304, 188–194 CrossRef CAS.
- X. Zhang, L. Zhang, H. Liu, B. Cao, L. Liu and W. Gong, Structure, morphology, size and application of iron phosphate, Rev. Adv. Mater. Sci., 2020, 59, 538–552 CAS.
- R. K. Brow, Review: structure of simple phosphate glasses, J. Non-Cryst. Solids, 2000, 263&264, 1–28 Search PubMed.
- L. Zhang and R. K. Brow, A Raman study of iron–phosphate crystalline compounds and glasses, J. Am. Ceram. Soc., 2011, 94, 3123–3130 Search PubMed.
- L. Zhang, R. K. Brow, M. E. Schlesinger, L. Ghussn and E. D. Zanotto, Glass formation from iron-rich phosphate melts, J. Non-Cryst. Solids, 2010, 356, 1252–1257 CrossRef CAS.
- U. Hoppe, M. Karabulut, E. Metwalli, R. K. Brow and P. Jóvári, The Fe–O coordination in iron phosphate glasses by x-ray diffraction with high energy photons, J. Phys.: Condens. Matter, 2003, 15, 6143–6153 CrossRef CAS.
- G. K. Marasinghe, M. Karabulut, C. S. Ray, D. E. Day, M. G. Shumsky, W. B. Yelon, C. H. Booth, P. G. Allen and D. K. Shuh, Structural features of iron phosphate glasses, J. Non-Cryst. Solids, 1997, 222, 144–152 CrossRef CAS.
- C. H. Booth, P. G. Allen and D. K. Shuh, Structural features of iron phosphate glasses, J. Non-Cryst. Solids, 1997, 222, 144–152 Search PubMed.
- M. G. Mesko and D. E. Day, Immobilization of spent nuclear fuel in iron phosphate glass, J. Nucl. Mater., 1997, 273, 27–36 CrossRef.
- M. Karabulut, G. K. Marasinghe, C. S. Ray, D. E. Day, O. Ozturk and G. D. Waddill, X-ray photoelectron and Mössbauer spectroscopic studies of iron phosphate glasses containing U, Cs and Bi, J. Non-Cryst. Solids, 1999, 249, 106–116 CrossRef CAS.
- B. Qian, X. Liang, S. Yang, S. He and L. Gao, Effects of lanthanum addition on the structure and properties of iron phosphate glasses, J. Mol. Struct., 2012, 1027, 31–35 CrossRef CAS.
- B. Al-Hasni and G. Mountjoy, Structural investigation of iron phosphate glasses using molecular dynamics simulation, J. Non-Cryst. Solids, 2011, 357, 2775–2779 CrossRef CAS.
- P. Stoch, W. Szczerba, W. Bodnar, M. Ciecinska, A. Stoch and E. Burkel, Structural properties of iron-phosphate glasses: spectroscopic studies and ab initio simulations, Phys. Chem. Chem. Phys., 2014, 16, 19917–19927 Search PubMed.
- S. T. Reis, W. M. Pontuschka, A. Moguš-Milanković and C. S. Partiti, Structural features of iron phosphate glass, J. Am. Ceram. Soc., 2017, 100, 1976–1981 CrossRef CAS.
- N. Kaur, A. Khanna and A. C. Hannon, Neutron diffraction investigation of copper tellurite glasses with high real-space resolution, J. Appl. Crystallogr., 2021, 54, 1647–1655 Search PubMed.
- A. Khanna, M. Fábián, Hirdesh, P. S. R. Krishna, C. J. Benmore, A. Kaur, A. Kaur, A. B. Shinde, P. Rajput and S. N. Jha, Structural analysis of WO3-TeO2 glasses by neutron, high energy X-ray diffraction, reverse Monte Carlo simulations and XANES, J. Non-Cryst. Solids, 2018, 495, 27–34 CrossRef CAS.
- A. Kaur, A. K. Hirdesh, M. Fábián, P. S. R. Krishna and A. B. Shinde, Structure of lead tellurite glasses and its relationship with stress-optic properties, Mater. Res. Bull., 2019, 110, 239–246 CrossRef CAS.
- A. Khanna, A. Kaur, M. Fábián, Hirdesh and A. Kaur, Structure of lead silicate glasses and its corelation with photoelastic properties, Indian J. Phys., 2021, 95, 2187–2199 CrossRef CAS.
- R. Kaur, A. Khanna and M. Fábián, Neutron diffraction investigation of strontium tellurite glass, anti-glass and crystalline phases, Phase Transitions, 2020, 93, 1016–1029 CrossRef CAS.
- S. Bista, A. O. Donovan-Zavada, M. Tyler, M. Franke, M. Affatigato and S. Feller, Packing in alkali and alkaline earth borosilicate glass systems, Eur. J. Glasses Sci. Technol. B Phys. Chem. Glasses, 2009, 50, 224–228 Search PubMed.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, 1992 Search PubMed.
- O. Gereben, P. Jóvári, L. Temleitner and L. Pusztai, A new version of the RMC++ Reverse Monte Carlo programme, aimed at investigating the structure of covalent glasses, J. Optoelectron. Adv. Mater., 2007, 9, 3021–3027 CAS.
- G. Evrard and L. Pusztai, Reverse Monte Carlo modelling of the structure of disordered materials with RMC++: a new implementation of the algorithm in C++, J. Phys.: Condens. Matter, 2005, 17, S1–S13 CrossRef CAS.
- M. Fábián, E. Svab and K. Krezhov, Network structure with mixed bond-angle linkages in MoO3–ZnO–B2O3 glasses: neutron diffraction and reverse Monte Carlo modelling, J. Non-Cryst. Solids, 2016, 433, 6–13 CrossRef.
- R. Kaur, A. Khanna, A.-C. Dippel, O. Gutowski, F. González and M. González-Barriuso, Structure of strontium tellurite glass, anti-glass and crystalline phases by high-energy X-ray diffraction, reverse Monte Carlo and Rietveld analysis, Acta Crystallographica Section B: Structural ScienceCrystal Engineering and Materials, 2020, 76, 108–121 CrossRef CAS PubMed.
- Z. Klencsár, E. Kuzmann and A. Vértes, User-friendly software for Mössbauer spectrum analysis, J. Radioanal. Nucl. Chem., 1996, 210(1), 105–118 CrossRef.
- Standard Test Methods for Determining Chemical Durability of Nuclear, Hazardous, and Mixed Waste Glasses and Multiphase Glass Ceramics: the Product Consistency Test (PCT), 2021. DOI:10.1520/C1285-21.
- I. Tolnai, J. Osan, F. Pinakidou, Zs. Kovacs and M. Fabian, Effects of UO3 on the structure, thermal and chemical stability of borosilicate glass matrix, J. Non-Cryst. Solids, 2024, 637, 123054 CrossRef CAS.
- B. M. Al-Hasni, G. Mountjoy and E. Barney, Atomic structure of iron phosphate glasses, Int. J. Appl. Glass Sci., 2021, 12, 245–258 CrossRef CAS.
- A. Saini, A. Khanna, V. K. Michaelis, S. Kroeker, F. González and D. Hernandez, Structure property correlations in lead borate and borosilicate glasses doped with aluminium oxide, J. Non-Cryst. Solids, 2009, 355, 2323–2332 CrossRef CAS.
- A. Khanna, A. Saini, B. Chen, F. González and B. Ortiz, Structural characterization of PbO–B2O3–SiO2 Glasses, J. Optoelectron. Adv. Mater., 2014, 55(2), 65–73 CAS.
- A. Bajaj, A. Khanna, B. Chen, J. G. Longstaffe, U. Werner Zwanziger, J. W. Zwanziger, Y. Gómez and F. González, Structural investigation of bismuth borate glasses and crystalline phases, J. Non-Cryst. Solids, 2009, 355, 45–53 CrossRef CAS.
- A. Khanna, A. Saini, B. Chen, F. González and C. Pesquera, Structural study of bismuth borosilicate, aluminoborate and aluminoborosilicate glasses by 11B and 27Al MAS NMR spectroscopy and thermal analysis, J. Non-Cryst. Solids, 2013, 373–374, 34–41 CrossRef CAS.
- D. R. Neuville and C. Le Losq, Link between Medium and Long-range Order and Macroscopic Properties of Silicate Glasses and Melts, Rev. Mineral. Geochem., 2022, 87, 105–162 CrossRef CAS.
- N. Kaur, A. Khanna, M. Gónzález-Barriuso, F. González and B. Chen, Effects of Al3+, W6+, Nb5+ and Pb2+ on the structure and properties of borotellurite glasses, J. Non-Cryst. Solids, 2015, 429, 151–163 CrossRef.
- G. D'Angelo, G. Carini, 1 C. Crupi, M. Koza, G. Tripodo and C. Vasi, Boson peak in alkaline borate glasses: Raman spectroscopy, neutron scattering, and specific-heat Measurements, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79, 014206 Search PubMed.
- C. Mclntosh, J. Toulouse and P. Tick, The Boson peak in alkali silicate glasses, J. Non-Cryst. Solids, 1997, 222, 335 CrossRef.
- A. I. Chumakov, G. Monaco, A. Monaco, W. A. Crichton, A. Bosak, R. Rüffer, A. Meyer, F. Kargl, L. Comez, D. Fioretto, H. Giefers, S. Roitsch, G. Wortmann, M. H. Manghnani, A. Hushur, Q. Williams, J. Balogh, K. Parlinski, P. Jochym and P. Piekarz, Equivalence of the Boson Peak in Glasses to the Transverse Acoustic van Hove Singularity in Crystals, Phys. Rev. Lett., 2011, 106, 225501 CrossRef CAS PubMed.
- E. Stavrou, C. Tsiantos, R. D. Tsopouridou, S. Kripotou, A. G. Kontos, C. Raptis, B. Capoen and M. Bouazaoui, S Turrell and S Khatir, Raman scattering boson peak and differential scanning calorimetry studies of the glass transition in tellurium–zinc oxide glasses, J. Phys.: Condens. Matter, 2010, 22, 195103 CrossRef CAS PubMed.
- M. González-Jiménez, T. Barnard, B. A. Russell, N. V. Tukachev, U. Javornik, L.-A. Hayes, A. J. Farrell, S. Guinane, H. M. Senn, A. J. Smith, M. Wilding, G. Mali, M. Nakano, Y. Miyazaki, McM. Paul, G. C. Sosso and K. Wynne, Understanding the emergence of the boson peak in molecular glasses, Nat. Commun., 2023, 14, 215 CrossRef PubMed.
- P. Stoch, A. Stoch, M. Ciecinska, I. Krakowiak and M. Sitarz, Structure of phosphate and iron-phosphate glasses by DFT calculations and FTIR/Raman spectroscopy, J. Non-Cryst. Solids, 2016, 450, 48–60 CrossRef CAS.
- K. Lagarec and D. G. Rancourt, Extended Voigt-based analytic lineshape method for determining N-dimensional correlated hyperfine parameter distributions in Mössbauer spectroscopy, Nucl. Instrum. Methods Phys. Res., B, 1997, 129, 266 CrossRef CAS.
- J. M. Millet, C. Virely, M. Forissier, P. Bussière and J. C. Vedrine, Mössbauer spectroscopic study of iron phosphate catalysts used in selective oxidation, Hyperfine Interact., 1989, 46, 619–628 CrossRef.
- T. Ericsson, A. G. Nord, M. M. O. Ahmed, A. Gismelseed and F. Khangi, Fe2P2O7 and Fe2P4O12 Studied Between 5 - 800 K, Hyperfine Interact., 1990, 57, 2179–2186 CrossRef CAS.
- E. Kozlyakova, I. Danilovich, A. Volkov, K. Zakharov, O. Dimitrova, E. Belokoneva, L. Shvanskaya, E. Zvereva, D. Chareev, O. Volkova and A. Vasiliev, Tuning of physical properties of Fe7(PO4)6 by sodium intercalation, J. Alloys Compd., 2018, 744, 600–605 CrossRef CAS.
- G. M. Bancroft, A. G. Maddock and R. G. Burns, Applications of the Mössbauer Effect to silicate mineralogy–I. Iron silicates of known crystal structure, Geochim. Cosmochim. Acta, 1967, 31, 2219–2246 CrossRef CAS.
- U. Hoppe, A structural model for phosphate glasses, J. Non-Cryst. Solids, 1996, 195, 138–147 CrossRef CAS.
- P. A. Bingham, R. J. Hand, S. D. Forder, A. Lavaysierre, S. H. Kilcoyne and I. Yasin, Preliminary studies of sulphate solubility and redox in 60P2O5-40Fe2O3 glasses, Mater. Lett., 2006, 60(2), 844–847 CrossRef CAS.
- H. Bińczycka and J. A. Sawicki, Conversion Electron Mossbauer Spectroscopy Study of Iron Oxidation in Phosphate Glasses, J. Phys. D: Appl. Phys., 1983, 16, 197–201 CrossRef.
- K. Joseph, R. Asuvathraman, R. V. Krishnan, T. R. Ravindran, R. Govindaraj, K. V. G. Kutty and P. R. V. Rao, Iron phosphate glass containing simulated fast reactor waste: Characterization and comparison with pristine iron phosphate glass, J. Nucl. Mater., 2014, 452(1–3), 273–280 CrossRef CAS.
- I. Khan, K. Sunakawa, R. Higashinaka, T. D. Matsuda, Y. Aoki, K. Nomura, E. Kuzmann, Z. Homonnay, K. Sinkó, T. Naka, T. Nakane, S. Krehula, S. Music and S. Kubuki, Structural characterization and magnetic properties of iron-phosphate glass prepared by sol-gel method, J. Non-Cryst. Solids, 2020, 543, 120158 CrossRef CAS.
- D. L. Griscom, C. I. Merzbacher, N. E. Bibler, H. Imagawa, S. Uchiyama, A. Namiki, G. K. Marasinghe, M. Mesko and M. Karabulut, On the structure and radiation chemistry of iron phosphate glasses: New insights from electron spin resonance, Mössbauer, and evolved-gas mass spectroscopy, Nucl. Instrum. Methods Phys. Res., Sect. B, 1998, 141(1–4), 600–615 CrossRef CAS.
- C. S. Ray, X. Fang, M. Karabulut, G. K. Marasinghe and D. E. Day, Effect of melting temperature and time on iron valence and crystallization of iron phosphate glasses, J. Non-Cryst. Solids, 1999, 249, 1–16 CrossRef CAS.
- X. Y. Yu, D. E. Day, G. J. Long and R. K. Brow, Properties and structure of sodium-iron phosphate glasses, J. Non-Cryst. Solids, 1997, 215(1), 21–31 CrossRef CAS.
- M. Karabulut, E. Metwalli, D. E. Day and R. K. Brow, Mössbauer and IR investigations of iron ultraphosphate glasses, J. Non-Cryst. Solids, 2003, 328(1–3), 199–206 CrossRef CAS.
- P. A. Bingham, R. J. Hand, S. D. Forder and A. Lavaysierre, Vitrified metal finishing wastes. II. Thermal and structural characterisation, J. Hazard. Mater., 2005, 122(1–2), 129–138 CrossRef CAS PubMed.
- D. G. Rancourt and J. Y. Ping, Voigt-based methods for arbitrary-shape static hyperfine parameter distributions in Mössbauer spectroscopy, Nucl. Instrum. Methods Phys. Res., B, 1991, 58, 85–97 CrossRef.
- M. Karabulut, G. K. Marasinghe, C. S. Ray, D. E. Day, G. D. Waddill, C. H. Booth, P. G. Allen, J. J. Bucher, D. L. Caulder and D. K. Shuh, An investigation of the local iron environment in iron phosphate glasses having different Fe(II) concentrations, J. Non-Cryst. Solids, 2002, 306(2), 182–192 CrossRef CAS.
- P. Stoch, M. Ciecińska, P. Zachariasz, J. Suwalski, L. Górski and T. Wójcik, Mössbauer spectroscopy study of 60P2O5-40Fe2O3 glass, Nukleonika, 2013, 58(1), 63–66 CAS.
- P. Stoch and A. Stoch, Mössbauer spectroscopy study of 60P2O5-40Fe2O3 glass crystallization, Nukleonika, 2015, 60(1), 133–136 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |