
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihydropyrimidine-2-thione derivatives as SARS-CoV-2 main protease inhibitors: synthesis, SAR and in vitro profiling†
Anees Saeed a,
Ayesha Tahira,
Muhammad Shaha,
Fahad Hussaina,
Abdul Sadiqb and
Umer Rashid*a
a,
Ayesha Tahira,
Muhammad Shaha,
Fahad Hussaina,
Abdul Sadiqb and
Umer Rashid*a
aDepartment of Chemistry, COMSATS University Islamabad, Abbottabad Campus-22060, Pakistan. E-mail: umerrashid@cuiatd.edu.pk
bDepartment of Pharmacy, Faculty of Biological Sciences, University of Malakand, Chakdara, 18000 Dir (L), KP, Pakistan
First published on 26th February 2025
Abstract
Despite the passage of approximately five years since the outbreak, an efficacious remedy for SARS-CoV-2 remains elusive, highlighting the urgent imperative for developing SARS-CoV-2 potent inhibitors. In our current study, we have unmasked the hitherto unrealized potential of dihydropyrimidine-2-thiones against the Main Protease (Mpro) of SARS-CoV-2. Employing a predictive docking tool, we identified promising lead compounds and optimized them via comprehensive Structural Activity Relationship (SAR) studies. Key design elements included proton donor/acceptor groups, six-membered rings, and fluorinated moieties to enhance interactions. These leads underwent in vitro inhibition assays to enhance their interaction with key Mpro amino acid residues. Our findings indicated that all synthesized compounds exhibited significant inhibition of the Mpro. Compounds 12j (IC50 = 0.063 μM), and 12l (IC50 = 0.054 μM) displayed exceptional in vitro binding affinities. In addition to their string inhibitory activity, CC50 values were assessed, confirming acceptable cytotoxicity profiles for potent compounds. Molecular dynamic simulation substantiated the binding mechanism revealing that compound 12l maintains robust stability with the target protein. Furthermore, compounds predicted to have minimal oral toxicity and high intestinal absorption make them promising candidates for drug development. These findings paved the way for the potent clinical application of these dihydropyrimidine-2-thiones as efficient SARS-CoV-2 therapeutics.
Introduction
COVID-19 is caused by a deadly Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) that targets the human respiratory system. Coronaviruses such as NL63, OC43, 229E, and HKU1, have generally caused mild respiratory infections like the common cold in humans.1–5 However, highly pathogenic strains like SARS-CoV-1 (2003), MERS-CoV (2012), and SARS-CoV-2 (2019) have emerged in the past 20 years, posing severe threats to public health. The first SARS-CoV-2 outbreak, reported in Wuhan, China, in 2019, signified the beginning of a global pandemic. SARS-CoV-2, a positive-sense, single-stranded RNA virus within the mammalian beta-coronavirus genus causes respiratory and lung damage leading to potentially fatal illnesses.6 On March 11, 2020, the World Health Organization (WHO) declared COVID-19, a global pandemic, due to the rapid spread of the virus and the rising mortality rate.7,8 As of November 25, 2024, WHO reported 776.41 million confirmed cases, 7.075 million deaths, and over 13.64 billion vaccinations globally. Notably, SARS-CoV-2 is among the most infectious virus in the coronavirus family impacting both human and animal populations.8,9SARS-CoV-2 encodes 16 non-structural proteins (NSPs), key structural proteins, membrane (M), nucleocapsid (N), and spike (S) and a Main Protease (Mpro) or 3C-like protease (3CLpro), Papain Like Protease (PLpro), and RNA-dependent RNA polymerase (RdRp).10–13 The viral genome, approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nucleotides long, contains a 5′-cap structure, a 3′-poly (A) tail, and multiple open reading frames (ORFs). NSPs possess PLpro and Mpro which play a crucial role in viral replication. Mpro, comprised of three domains (I–III) and a unique Cys–His dyad active site, is highly conserved across coronaviruses, highlighting its potential as a target for antivirals.10,14–18 With S1, S1′, S2, and S3 forming the active binding pocket for substrate engagement, Mpro facilitates viral protein maturation through peptide bond cleavage, a process validated by mixed quantum mechanics/molecular mechanics (QM/MM) simulations.19,20 Due to its essential function in the viral life cycle and lack of homologs in humans, Mpro is an ideal candidate for therapeutic intervention and drug design.21–23 Fig. 1 illustrates the Mpro's role in viral replication, making protease inhibition a prime target for antiviral drug development.
000 nucleotides long, contains a 5′-cap structure, a 3′-poly (A) tail, and multiple open reading frames (ORFs). NSPs possess PLpro and Mpro which play a crucial role in viral replication. Mpro, comprised of three domains (I–III) and a unique Cys–His dyad active site, is highly conserved across coronaviruses, highlighting its potential as a target for antivirals.10,14–18 With S1, S1′, S2, and S3 forming the active binding pocket for substrate engagement, Mpro facilitates viral protein maturation through peptide bond cleavage, a process validated by mixed quantum mechanics/molecular mechanics (QM/MM) simulations.19,20 Due to its essential function in the viral life cycle and lack of homologs in humans, Mpro is an ideal candidate for therapeutic intervention and drug design.21–23 Fig. 1 illustrates the Mpro's role in viral replication, making protease inhibition a prime target for antiviral drug development.
 | ||
| Fig. 1 SARS-CoV-2 Mpro inhibition and mechanism of action. | ||
Currently, FDA-approved COVID-19 inhibitors (Fig. 2) are limited. However, PF-07321332 (nirmatrelvir, Ki = 0.003 μM) (1a), demonstrates safety, selectivity, and high antiviral activity (EC50 of 0.074 μM).4,24 To counteract its rapid metabolism by CYP3A, the HIV protease inhibitor ritonavir (1b) was added, forming Paxlovid, which received FDA approval in 2021 for treating mild to moderate COVID-19. Past studies have identified effective Mpro inhibitors, such as PF-00835231 (1c) during the SARS-CoV-1 outbreak in 2003.25 In 2021, Pfizer developed an oral SARS-CoV-2 Mpro inhibitor. Japan approved ensitrelvir (S-217622) (1d) in 2022, which showed potent Mpro inhibition with IC50 = 0.013 μM and EC50 = 0.37 μM.26,27 Several other Mpro inhibitors (Fig. 3), including lufotrelvir (2a), ebselen (2b), and masitinib (2c) are in clinical trials while repurposed drugs like boceprevir (IC50 = 4.13 μM) also exhibit notable inhibitory effects.28–32
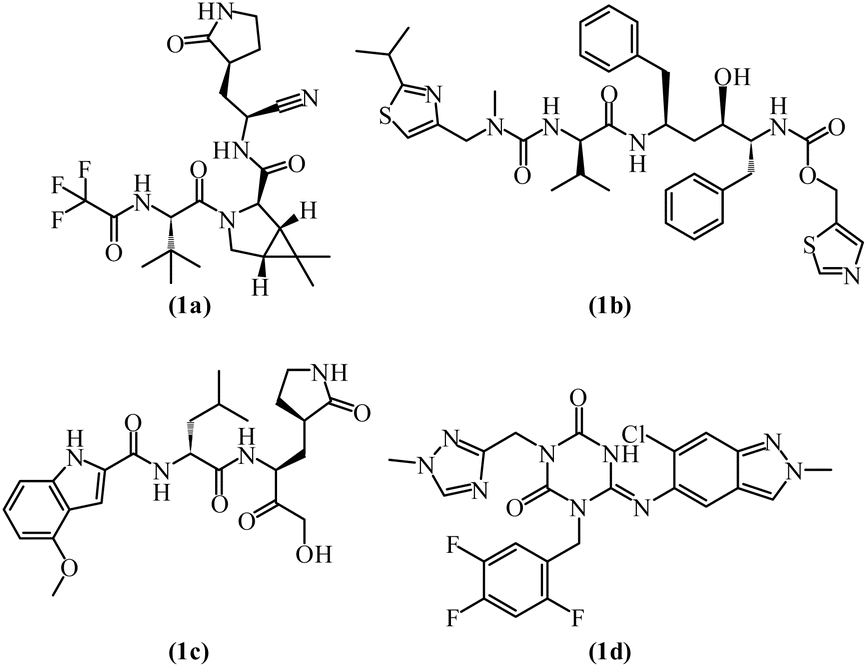 | ||
| Fig. 2 FDA-approved medication for the treatment of SARS-CoV-2 (1a–d). | ||
 | ||
| Fig. 3 Medication in clinical trials for the treatment of SARS-CoV-2 (2a–c). | ||
This work seeks to identify novel dihydropyrimidine-2-thione-based inhibitors with favourable pharmacokinetic properties to combat SARS-CoV-2 effectively by focusing on the design of dihydropyrimidine-2-thione-based derivatives targeting SARS-CoV-2 Mpro, aiming to maximize scaffold inhibitory potency. Structure-Based Drug Design (SBDD) techniques were used to optimize and evaluate these scaffolds.33 The sulphur-rich dihydropyrimidine-2-thione has shown antiviral, antibacterial, and potential anticancer activities. It disrupts viral replication and hosts immune responses.34–36 Notably, dihydropyrimidine derivatives such as batzelladine A & B exhibit antiviral properties, while monastrol is known for anticancer activity.37,38 The antihypertensive drug terazosin exemplified its diverse biological activities.39
Materials and methods
General
Solvents and reagents used for current work were purchased from commercial sources and were used without purification. Compounds include substituted aromatic aldehydes (6a–c), thiourea, ethyl 4,4,4-trifluoro-3-oxobutanoate, diverse primary amines (L-glutamic acid (7a), L-aspartic acid (7b), sulfanilic acid (7c), L-histidine (7d), L-tyrosine (7e)), bromoacetyl bromide and diverse secondary amines (11a–c) were purchased from Sigma-Aldrich. 400 MHz/100 MHz Bruker NMR spectrometer was used to record 1H and 13C NMR spectra respectively in the DMSO solvent. The solvent was used as an internal reference in NMR analysis. Chemical shifts in NMR analysis are recorded in δ scale part per million (ppm). Thin Layer Chromatography (TLC) was used to monitor the progress of all reactions for current research work on 2 × 5 cm precoated aluminum sheets with silica gel (60-F254), the coating layer thickness specification was 0.25 mm (Merck). Liquid chromatography-mass spectrometry (LC-MS) was performed using Agilent Technologies 1200 series high-performance liquid chromatography system with C18 reversed-phase column (particle size: 3.5 μm, length: 100 mm, internal diameter: 4.6 mm, vendor: Agilent Technologies). Elemental analyses were conducted using an Elemental Vario EI III CHN analyzer. Elemental analysis (±0.4% of the calculated values) was performed for all the tested compounds. Final products were checked for their purity on a HPLC system using a C18 RP column (particle size: 5 μm, length: 150 mm, internal diameter: 4.6 mm, vendor: Shimadzu) and an isocratic solvent system (mentioned in the experimental part) at room temperature. Biologically screened compounds are >95% pure as determined by HPLC.General method for the synthesis of compounds (6a–c)
Dihydropyrimidine-2-thiones (6a–c) core was synthesized by 10 mmol aldehydes (4-(trifluoromethyl)benzaldehyde (3a), 4-nitrobenzaldehyde (3b) and methyl 4-formyl benzoate (3c)) reaction with thiourea (4) (10 mmol) and ethyl 4,4,4-trifluoro-3-oxobutanoate (5) (12 mmol) through classical multicomponent Biginelli approach using acetonitrile solvent under reflux for 6 h's in the presence of SnCl2·2H2O. Reaction was monitored by TLC, after completion of the reaction the reaction mixture was poured into ice-cold distilled water and allowed to stir for 5 minutes. Precipitates formed, filtered off, dried, washed with cold ethanol, and recrystallized with ethanol to afford pure dihydropyrimidine-2-thiones (6a–c).General method for the synthesis of compounds (8a–h)
Compounds (8a–h) are synthesized by the reaction of the substituted dihydropyrimidine-2-thiones (20 mmol, 6a–c) with diverse primary amines (25 mmol, 7a–e) in the presence of K2CO3 base (1.2 equivalent) in a solvent dimethylformamide (DMF) on heating. The reaction was monitored by TLC, after completion, the reaction mixture was poured into ice water. The precipitates were filtered and washed with cold water and purified with the help of column chromatography (hexane:ethyl acetate 9:1 system) to afford pure compounds (8a–h). Characterization data of intermediate compounds is presented in the ESI† file.
General method for the synthesis of compounds (10a–h)
To the stirred solution of (5 mmol) synthesized dihydropyrimidine-thione amide derivatives (8a–h) in 10 mL acetone solvent, at room temperature, and K2CO3 as base and bromoacetyl bromide (9) (7.5 mmol) was added dropwise with continuous stirring. The reaction was monitored by TLC and stirring continued for 4 hours. At the end of the reaction precipitated solid was obtained which was further recrystallized by dioxane:ethanol (1:1) to furnish S-acetylated product/compounds (10a–h). Characterization data of intermediate compounds is presented in the ESI† file.
General method for the synthesis of compounds (12a–l)
Diverse secondary amines (5 mmol) (11a–c) along with K2CO3 base (1.5 eq.) were taken in DMF (10 mL). To this stirred solution previously synthesized S-bromoacylated products (10a–h) (7.5 mmol) were added. The reaction was monitored by TLC. Stirring continued till the completion of the reaction. Water was added after completion of the reaction to afford precipitation of the products (12a–l). In some products, precipitation was not observed. If precipitates were not formed product was not extracted in the organic layer. Later on, all products were purified through column chromatography.(6-(Trifluoromethyl)-2-((2-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]-pyrazin-7(8H)-yl)acetyl)thio)-4-(4 (trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-glutamic acid (12a)
Light yellow solid, yield = 48%, m.p. 199–201 °C; Rf = 0.47; (n-hexane/EA; 3:1); HPLC purity = 97.5% (C18 RP, acetonitrile/H2O-80:20), TR = 12.1 min. 1H NMR (400 MHz, DMSO-d6) δ 12.39 (s, 1H, COOH), 11.75 (s, 1H, COOH), 9.38 (s, 1H, py-NH), 8.41 (d, J = 4.72 Hz, –NH), 7.81 (d, J = 8.52 Hz, –2H, ArH), 7.27 (d, J = 8.52 Hz, –2H, ArH), 5.46 (s, 1H, –CH), 4.49–4.45 (m, 1H, glu), 4.26 (t, J = 5.92 Hz, 2H, pip-CH2), 3.83 (s, 2H, CH2), 3.28 (s, 2H, pip-CH2), 3.11 (t, J = 5.92 Hz, 2H, pip-CH2), 2.27–2.19 (m, 2H, glu), 2.03–1.93 (m, 2H, glu). 13C NMR (100 MHz, DMSO-d6) δ 190.5, 174.1, 173.6, 162.6, 147.6, 139.7 (q, J = 36.0 Hz, –C–CF3), 133.8 (q, J = 32.49 Hz, –C–CF3), 131.7 (q, J = 32.12 Hz, –C–CF3), 128.1 (2C), 126.9, 125.9, 124.2, 123.4, 122.4, 122.2 (q, J = 269 Hz, –CF3), 119.9, 116.2, 110.7, 64.8, 59.6, 57.7, 48.4, 41.5, 32.2, 23.4. Analysis calculated for C26H22F9N7O6S; C, 42.69; H, 3.03; F, 23.37; N, 13.40; O, 13.12; S, 4.38; found C, 42.56; H, 3.04; N, 13.42; LCMS: m/z = 732.1 [M + H]+.
(6-(Trifluoromethyl)-2-((2-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]-pyrazin-7(8H)-yl)acetyl)thio)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-aspartic acid (12b)
White solid, yield = 65%, m.p. 204–206 °C; Rf = 0.48; (n-hexane/EA; 3:1); HPLC purity = 98.6% (C18 RP, acetonitrile/H2O-80:20), TR = 11.9 min. 1H NMR (400 MHz, DMSO-d6) δ 12.46 (s, 1H, COOH), 11.81 (s, 1H, COOH), 9.36 (s, 1H, py-NH), 8.40 (d, J = 6.08 Hz, –NH), 7.81 (d, J = 8.36 Hz, –2H, ArH), 7.27 (d, J = 8.36 Hz, –2H, ArH), 5.46 (s, 1H, –CH), 4.44–4.39 (m, 1H, glu), 4.22 (t, J = 5.8 Hz, 2H, pip-CH2), 3.83 (s, 2H, CH2), 3.53–3.45 (m, 2H, pip-CH2), 3.28 (s, 2H, pip-CH2), 3.10 (t, J = 5.8 Hz, 2H, glu). 13C NMR (100 MHz, DMSO-d6) δ 188.7, 173.5, 172.9, 164.1, 148.8, 144.9, 142.4, 140.5, 140.2, 132.7, 132.9 (q, J = 36.7 Hz, –C–CF3), 132.8 (q, J = 32 Hz, –C–CF3), 131.6 (q, J = 36.5 Hz, –C–CF3), 127.5, 125.7, 125.2, 124.3, 123.6, 122.4 (q, J = 274 Hz, –CF3), 117.7, 115.6, 90.7, 61.3, 57.2, 52.8, 50.5, 49.8, 42.7, 36.2. Analysis calculated for C25H20F9N7O6S; C, 41.85; H, 2.81; F, 23.83; N, 13.66; O, 13.38; S, 4.47; found C, 41.70; H, 2.82; N, 13.72; LCMS: m/z = 718.1 [M + H]+.
4-(6-(Trifluoromethyl)-2-((2-(3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]-pyrazin-7(8H)-yl)acetyl)thio)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carboxamide)benzenesulfonic acid (12c)
Off white solid, yield = 52%, m.p. 182–184 °C; Rf = 0.51; (n-hexane/EA; 3:1); HPLC purity = 97.3% (C18 RP, acetonitrile/H2O-80:20), TR = 14.4 min. 1H NMR (400 MHz, DMSO-d6) δ 10.38 (s, 1H, OH, sulpha), 9.36 (s, 1H, py-NH), 8.69 (s, –NH, sulpha), 7.82 (d, J = 8.32 Hz, –2H, ArH), 7.71 (d, J = 8.56 Hz, –2H, ArH), 7.41 (d, J = 8.56 Hz, –2H, ArH, sulpha), 7.27 (d, J = 8.36 Hz, –2H, ArH), 5.46 (s, 1H, –CH), 4.26 (t, J = 5.84 Hz, 2H, pip-CH2), 3.83 (s, 2H, CH2), 3.28 (s, 2H, pip-CH2), 3.10 (t, J = 5.80 Hz, 2H, pip-CH2). 13C NMR (100 MHz, DMSO-d6) δ 188.7, 166.1, 147.5, 144.9, 142.4, 140.7, 139.4, 139.1, 136.3, 133.1, 132.8 (q, J = 36.3 Hz, –C–CF3), 132.6 (q, J = 32.4 Hz, –C–CF3), 131.3 (q, J = 36.6 Hz, –C–CF3), 128.0, 127.5, 125.7, 125.2, 124.7, 123.2, 122.1 (q, J = 271 Hz, –CF3), 121.3, 119.3, 117.2, 100.1, 61.6, 57.2, 51.2, 50.2, 42.8. Analysis calculated for C27H20F9N7O5S2; C, 42.81; H, 2.66; F, 22.57; N, 12.94; O, 10.56; S, 8.46; found C, 42.94; H, 2.67; F, 22.57; N, 13.00; O, 10.56; S, 8.46; LCMS: m/z = 758.0 [M + H]+.
(6-(Trifluoromethyl)-2-((2-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)acetyl)thio)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)histidine (12d)
Off white solid, yield = 51%, m.p. 226–228 °C; Rf = 0.45; (n-hexane/EA; 3:1); HPLC purity = 97.3% (C18 RP, acetonitrile/H2O-80:20), TR = 13.7 min. 1H NMR (400 MHz, DMSO-d6) δ 12.16 (brs, 1H, COOH, His), 9.36 (brs, 1H, py-NH), 8.86 (s, 1H, imidazole, NH), 8.49 (d, J = 6.6 Hz, 1H, CONH, His), 8.03 (s, 1H, imidazole, ArH), 7.82 (d, J = 8.4 Hz, –2H, ArH), 7.26 (d, J = 8.4 Hz, –2H, ArH), 7.07 (s, 1H, imidazole, ArH), 5.46 (s, 1H, –CH), 4.36–4.31 (m, 1H, CHCH2, His), 4.25 (t, J = 576 Hz, 2H, pip-CH2), 3.83 (s, 2H, CH2), 3.53–3.45 (m, 2H, CHCH2, His), 3.28 (s, 2H, pip-CH2), 3.10 (t, J = 5.72 Hz, 2H, pip-CH2). 13C NMR (100 MHz, DMSO-d6) δ 188.7, 175.3, 164.5, 148.8, 144.9, 142.4, 140.5, 140.2, 138.8, 135.2, 133.1 (q, J = 36.3 Hz, –C–CF3), 132.7 (q, J = 36.7 Hz, –C–CF3), 131.4 (q, J = 36.4 Hz, –C–CF3), 130.4, 127.5, 125.7, 125.2, 124.9, 123.0, 122.1 (q, J = 272.4 Hz, –CF3) 118.8, 117.7, 115.6, 90.7, 61.3, 57.2, 54.0, 52.8, 49.8, 42.7, 29.6. Analysis calculated for C27H22F9N9O4S; C, 43.85; H, 3.00; F, 23.12; N, 17.05; O, 8.65; S, 4.33; found C, 43.73; H, 3.04; F, 23.12; N, 17.23; O, 8.65; S, 4.33; LCMS: m/z = 740.5 [M + H]+.
(6-(Trifluoromethyl)-2-((2-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]-pyrazin-7(8H)-yl)acetyl)thio)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-tyrosine (12e)
Cream yellow solid, yield = 54%, m.p. 191–193 °C; Rf = 0.47; (n-hexane/EA; 3:1); HPLC purity = 97.6% (C18 RP, acetonitrile/H2O-80:20), TR = 14.3 min. 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H, COOH, Tyr), 9.37 (s, 1H, py-NH), 9.08 (s, 1H, Tyr–OH), 8.53 (d, J = 5.28 Hz, 1H, CO–NH), 7.82 (d, J = 8.2 Hz, –2H, ArH), 7.26 (d, J = 8.16 Hz, –2H, ArH), 7.03 (d, J = 7.52 Hz, 2H, Tyr–ArH), 6.76 (d, J = 7.56 Hz, 2H), 5.46 (s, 1H, –CH), 4.98–4.88 (m, 1H, CHCH2 of Tyr), 4.26 (t, J = 5.8 Hz, 2H, pip-CH2), 3.83 (s, 2H, CH2), 3.71 (dd, J = 12.44 Hz, J = 9.68 Hz, 1H, CH2 of Tyr), 3.49 (dd, J = 12.16 Hz, J = 5.16 Hz, 1H, CH2 of Tyr), 3.28 (s, 2H, pip-CH2), 3.10 (t, J = 5.8 Hz, 2H, pip-CH2). 13C NMR (100 MHz, DMSO-d6) δ 188.4, 177.9, 166.4, 164.5, 157.8, 148.3, 143.8, 141.7, 138.3, 132.3, 131.4, 130.4 (2C), 128.5, 127.6, 125.7 (2C), 125.5 (2C), 124.5, 122.1, 117.3 (2C), 91.5, 61.5, 55.1, 49.3, 48.6, 41.9, 34.1. Analysis calculated for C30H24F9N7O5S; C, 47.06; H, 3.16; F, 22.33; N, 12.81; O, 10.45; S, 4.19; found C, 46.88; H, 3.17; F, 22.33; N, 12.85; O, 10.45; S, 4.19; LCMS: m/z = 766.1 [M + H]+.
(2-((2-(3-Aminopiperidin-1-yl)acetyl)thio)-6-(trifluoromethyl)-4-(4-(trifluoromethyl)-phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-aspartic acid (12f)
Light yellow solid, yield = 61%, m.p. 188–190 °C; Rf = 0.45; (DCM/MeOH; 5:1); HPLC purity = 97.8% (C18 RP, acetonitrile/H2O-80:20), TR = 7.9 min. 1H NMR (400 MHz, CDCl3) δ 12.43 (s, COOH, 1H), 11.80 (s, COOH, 1H), 9.36 (s, py-NH, 1H), 8.40 (d, J = 6.16 Hz, –NH), 7.81 (d, J = 8.4 Hz, 2H, ArH), 7.28 (d, J = 8.4 Hz, 2H, ArH), 5.46 (s, 1H, –CH), 4.44–4.38 (m, 1H, glu), 3.50–3.45 (m, 4H, –CH), 3.31 (s, 2H, CH2), 3.16–3.12 (m, 1H, CH), 2.67 (d, J = 3.92 Hz, 2H, NH2), 2.48 (dd, J = 17.28 Hz, J = 5.16 Hz, 6.2 Hz, 1H, CH), 1.60–1.49 (m, 2H, glu), 1.41–1.38 (m, 2H, –CH2). 13C NMR (100 MHz, CDCl3) δ 188.6, 173.6, 172.4, 166.2, 144.9, 143.1, 132.3, 130.6, 127.5 (2C), 125.3 (2C), 124.3, 123.8, 91.5, 63.1, 59.6, 57.5, 53.6, 49.8, 47.6, 35.8, 34.1, 21.7. Analysis calculated for C24H25F6N5O6S; C, 46.08; H, 4.03; F, 18.22; N, 11.20; O, 15.35; S, 5.13; found C, 45.89; H, 4.05; F, 18.22; N, 11.23; O, 15.35; S, 5.13; LCMS: m/z = 626.1 [M + H]+.
(2-((2-(3-Aminopiperidin-1-yl)acetyl)thio)-6-(trifluoromethyl)-4-(4-(trifluoromethyl)-phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-tyrosine (12g)
Cream yellow solid, yield = 51%, m.p. 199–201 °C; Rf = 0.48; (n-hexane/EA; 3:1); HPLC purity = 97.6% (C18 RP, acetonitrile/H2O-80:20), TR = 13.5 min. 1H NMR (400 MHz, CDCl3) δ 12.36 (s, 1H, COOH, Tyr), 9.36 (s, 1H, py-NH), 9.09 (s, 1H, Tyr–OH), 8.54 (d, J = 5.36 Hz, 1H, CO–NH), 7.81 (d, J = 8.2 Hz, 2H, ArH), 7.28 (d, J = 8.2 Hz, 2H, ArH), 7.03 (d, J = 7.56 Hz, 2H, Tyr–ArH), 6.75 (d, J = 7.56 Hz, 2H, Tyr–ArH), 5.45 (s, 1H, –CH), 4.44–4.38 (m, 1H, CHCH2 of Tyr), 3.30 (s, 2H, CH2 of Tyr), 3.18–3.14 (m, 3H, –CH), 3.08 (t, J = 5.28 Hz, 2H, CH2), 2.67 (d, J = 4.4 Hz, 2H, NH2), 2.47 (dd, J = 17.12 Hz, J = 5.08 Hz, 1H, CH), 1.65–1.50 (m, 2H, –CH2), 1.44–1.38 (m, 2H, –CH2). 13C NMR (100 MHz, CDCl3) δ 188.7, 177.7, 166.0, 157.7, 145.9, 143.0, 134.2 (q, J = 32.2 Hz), 132.2, (q, J = 32.4 Hz), 130.9, 129.7 (2C), 126.9 (2C), 125.8 (q, J = 280 Hz, CF3), 125.4, (2C) 121.6, 116.2 (2C), 91.9, 63.9, 61.7, 59.4, 56.6, 53.6, 47.7, 38.9, 34.4, 21.6. Analysis calculated for C29H29F6N5O5S; C, 51.71; H, 4.34; F, 16.92; N, 10.40; O, 11.88; S, 4.76; found C, 51.91; H, 4.32; F, 16.92; N, 10.37; O, 11.88; S, 4.76; LCMS: m/z = 674.1 [M + H]+.
(2-((2-(2-(Methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)acetyl)thio)-6-(trifluoromethyl)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)histidine (12h)
Light brown solid, yield = 48%, m.p. 231–233 °C; Rf = 0.46; (n-hexane/EA; 3:1); HPLC purity = 97.6% (C18 RP, acetonitrile/H2O-80:20), TR = 13.3 min. 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H, COOH, His), 9.46 (s, 1H, py-NH), 8.68 (s, 1H, imidazole, NH), 8.53 (s, 1H, CONH, His), 7.94 (s, 1H, pyrazol–ArH), 7.79 (s, 1H, imidazole, ArH), 7.75 (d, J = 8.16 Hz, –2H, ArH), 7.24 (d, J = 8.16 Hz, –2H, ArH), 7.10 (s, 1H, imidazole, ArH), 5.28 (s, 1H, –CH), 4.34–4.29 (m, 1H, CHCH2, His), 4.19–4.14 (m, 4H, 2× CH2), 3.48–3.42 (m, 2H, CHCH2, His), 3.10 (s, 3H, SO2–CH3), 3.01 (s, 2H, CH2). 13C NMR (100 MHz, CDCl3) δ 189.8, 175.3, 164.5, 150.8, 144.9, 142.4, 138.8, 135.2, 133.5 (q, J = 36.6 Hz, –C–CF3), 131.2 (q, J = 36.1 Hz, –C–CF3), 130.6, 130.4, 127.5, 126.1, 125.7, 125.2 (q, J = 272.8 Hz, –CF3), 124.9, 124.4, 123.0, 122.8, 118.8, 90.7, 63.8, 57.2, 55.4, 54.6, 54.0, 41.0, 29.6. Analysis calculated for C27H24F6N8O6S2; C, 44.14; H, 3.29; F, 15.52; N, 15.25; O, 13.07; S, 8.73; found C, 44.11; H, 3.31; F, 15.52; N, 15.22; O, 13.07; S, 8.73; LCMS: m/z = 735.12 [M + H]+.
(2-((2-(2-(Methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)acetyl)thio)-6-(trifluoromethyl)-4-(4-(trifluoromethyl)phenyl)-1,4-dihydropyrimidine-5-carbonyl)-L-tyrosine (12i)
Off white solid, yield = 49%, m.p. 221–223 °C; Rf = 0.47; (n-hexane/EA; 3:1); HPLC purity = 97.7% (C18 RP, acetonitrile/H2O-80:20), TR = 13.4 min. 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H, COOH, Tyr), 9.45 (s, 1H, py-NH), 9.24 (s, 1H, Tyr–OH), 8.50 (d, J = 2.64 Hz, 1H, CO–NH), 7.83 (s, 1H, pyrazol–ArH), 7.76 (d, J = 7.88 Hz, –2H, ArH), 7.24 (d, J = 8.72 Hz, –2H, ArH), 7.04 (d, J = 7.6 Hz, 2H, Tyr–ArH), 6.75 (d, J = 7.48 Hz, 2H), 5.29 (s, 1H, –CH), 4.94–4.89 (m, 1H, CHCH2 of Tyr), 4.20–4.13 (m, 4H, 2× CH2), 3.66 (dd, 1H, J = 12.12 Hz, J = 9.64 Hz, CH2 of Tyr), 3.48 (dd, 1H, J = 11.92 Hz, J = 5.04 Hz, CH2 of Tyr), 3.12 (s, 3H, CH2), 2.98 (s, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 189.8, 174.9, 164.5, 156.4, 150.8, 144.9, 142.4, 132.3, 132.0, 132.4 (q, J = 36.9 Hz, –C–CF3), 131.8 (q, J = 36.3 Hz, –C–CF3), 129.6, 127.5, 126.1, 125.7, 125.2, 124.9, 124.4, 123.6, 122.5 (q, J = 273 Hz, –CF3), 115.8, 90.7, 63.8, 57.2, 55.4, 55.0, 54.6, 41.0, 37.2. Analysis calculated for C30H26F6N6O7S2; C, 47.37; H, 3.45; F, 14.99; N, 11.05; O, 14.72; S, 8.43; found C, 47.19; H, 3.46; F, 14.99; N, 11.02; O, 14.72; S, 8.43; LCMS: m/z = 761.1 [M + H]+.
(2-((2-(2-(Methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)acetyl)thio)-4-(4-nitrophenyl)-6-(trifluoromethyl)-1,4-dihydro pyrimidine-5-carbonyl)-L-glutamic acid (12j)
Yellow solid, yield = 56%, m.p. 234–236 °C; Rf = 0.41; (DCM/MeOH; 5:1); HPLC purity = 97.8% (C18 RP, acetonitrile/H2O-80:20), TR = 9.1 min. 1H NMR (400 MHz, DMSO-d6) δ 12.36 (s, 1H, COOH), 11.70 (s, 1H, COOH), 9.36 (s, 1H, py-NH), 8.44 (d, J = 4.4 Hz, –NH), 8.26 (d, J = 8.7 Hz, 2H, ArH), 7.95 (d, J = 8.7 Hz, 1H, pyrazol–ArH), 7.76 (s, 2H, ArH), 5.47 (s, 1H, –CH), 4.52–4.42 (m, 1H, glu), 4.24–4.22 (m, 4H, 2× CH2), 3.12 (s, 3H, SO2–CH3), 2.97 (s, 2H, CH2), 2.31 (t, J = 7.24 Hz, 2H, glu), 2.29–1.92 (m, 2H, glu). 13C NMR (100 MHz, DMSO-d6) δ 188.3, 177.0, 176.5, 151.7, 146.4, 145.7, 131.8 (q, J = 32.5 Hz, C–CF3), 129.0, 126.4 (q, J = 273.1 Hz, CF3), 124.5, 124.0, 123.7, 121.2, 91.7, 64.9, 56.0, 55.1, 48.9, 41.9, 32.6, 23.8. Analysis calculated for C25H24F3N7O10S2; C, 42.68; H, 3.44; F, 8.10; N, 13.93; O, 22.74; S, 9.11; found C, 42.53; H, 3.45; F, 8.10; N, 13.97; O, 22.74; S, 9.11; LCMS: m/z = 704.1 [M + H]+.
4-(2-((2-(2-(Methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl)acetyl)thio)-4-(4-nitrophenyl)-6-(trifluoromethyl)-1,4-dihydropyrimidine-5-carboxamido)benzene-sulfonic acid (12k)
Brownish solid, yield = 64%, m.p. 254–256 °C; Rf = 0.43; (n-hexane/EA; 3:1); HPLC purity = 97.6% (C18 RP, acetonitrile/H2O-80:20), TR = 12.5 min. 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1H, SO2–OH), 9.42 (s, 1H, py-NH), 8.49 (s, 1H, CO–NH), 8.32 (d, J = 8.9 Hz, 2H, ArH), 8.23 (d, J = 8.4 Hz, 2H, ArH), 7.89 (d, J = 8.1 Hz, 2H, ArH), 7.87 (s, 1H, pyrazol–ArH), 7.86 (d, J = 8.4 Hz, 2H, ArH), 5.41 (s, 1H, –CH), 4.17–4.17 (m, 4H, 2× CH2), 3.11 (s, 2H, CH2), 3.09 (s, 3H, SO2–CH3). 13C NMR (100 MHz, CDCl3) δ 189.8, 166.1, 150.8, 147.3, 144.9, 140.7, 136.3, 133.1, 132.8 (q, J = 32.8 Hz, –C–CF3), 129.6, 128.0, 124.7, 123.1, 122.5, 121.3, 119.8, 100.1, 64.2, 57.4, 56.0, 55.8, 41.0. Analysis calculated for C26H22F3N7O9S3; C, 42.80; H, 3.04; F, 7.81; N, 13.44; O, 19.73; S, 13.18; found C, 42.93; H, 3.05; F, 7.81; N, 13.48; O, 19.73; S, 13.18, LCMS: m/z = 730.0 [M + H]+.
(4-(4-(Methoxycarbonyl)phenyl)-6-(trifluoromethyl)-2-((2-(3-(tri fluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl) acetyl)thio)-1,4-dihydropyrimidine-5-carbonyl)-L-histidine (12l)
White solid, yield = 57%, m.p. 195–197 °C; Rf = 0.44; (DCM/MeOH; 5:1); HPLC purity = 98.6% (C18 RP, acetonitrile/H2O-80:20), TR = 8.8 min. 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H, COOH, His), 9.53 (s, 1H, py-NH), 8.69 (s, 1H, imidazole, NH), 8.53 (s, 1H, CONH, His), 7.94 (s, 1H, imidazole, ArH), 7.92 (d, J = 8.6 Hz, 2H, ArH), 7.44 (d, J = 8.7 Hz, 2H, ArH), 7.10 (s, 1H, imidazole, ArH), 5.28 (s, 1H, –CH), 4.35–4.30 (m, 1H, CHCH2, His), 4.35 (t, 2H, pip-CH2), 3.92 (s, 3H, –OCH3), 3.79 (s, 2H, CH2), 3.41–3.48 (m, 2H, CHCH2, His), 3.31 (s, 2H, pip-CH2), 3.26 (t, 2H, pip-CH2), 2.33 (t, 3H, CH3). 13C NMR (100 MHz, DMSO-d6) δ 188.7, 175.3, 168.0, 164.5, 148.8, 145.4, 144.9, 140.5, 140.2, 138.8, 135.2, 132.8, 132.6 (q, J = 32.4 Hz, –C–CF3), 132.5 (q, J = 36.0 Hz, –C–CF3), 129.6, 124.9, 123.6, 122.8, 118.8, 117.7, 115.6, 90.7, 61.3, 56.9, 54.0, 52.8, 52.1, 49.8, 42.7, 29.6. Analysis calculated for C28H25F6N9O6S; C, 46.09; H, 3.45; F, 15.62; N, 17.28; O, 13.16; S, 4.39; found C, 45.91; H, 3.46; F, 15.62; N, 17.33; O, 13.16; S, 4.39; LCMS: m/z = 730.1 [M + H]+.
Docking studies
Docking studies were conducted by AutoDock4 version (v4.2.6). The crystal structure of SARS-CoV-2 Mpro, along with its native ligand, was retrieved from the Protein Data Bank (PDB) with accession code 6XHM, which offers the target's highest resolution. The docking protocol was adapted from established procedures. The chemical structures of the designed intermediates and inhibitors were sketched using ChemDraw software, followed by geometry optimization, and energy minimization with the Avogadro tool using the MMFF94 force field. Optimized structures were saved in PDB format and converted to PDBQT format using AutoDock Tools (ADTs) with appropriate torsion settings and flexible bonds. Upon ligand preparation, the protein was downloaded from PDB and underwent pre-docking optimization, which included the removal of water molecules, addition of hydrogen atoms, and assigning of Kollman charges. The protein was first saved in PDB format followed by conversion to PDBQT format via ADTs. The active site of protein (6XHM) was identified based on coordinates (X: 10.541898, Y: 14.409504, Z: 27.098183), obtained using Discovery Studio Visualizer v24.1.0.23298. A grid box with the blind dimensions was set to encompass the entire ligand, ensuring sufficient coverage for binding conformations sampling. The file is saved in .gpf format. Docking parameters were defined using genetic algorithm configurations with 10 runs and 2500000 energy evaluations per run. AutoDock4 version (v4.2.6) was employed for docking simulations, which predicted binding energies. The resulting .dpf file was generated and subsequently used to produce .glg and .dlg files.40,41 A more favorable pose is associated with a lower score and the scoring function used kcal per mole as the unit. After docking, protein–ligand interactions, including H-bonding, π–π stacking, π–sigma stacking, etc., were analyzed using 2D and 3D visualization through Discovery Studio Visualizer v24.1.0.23298.42
Methodology of SARS-CoV-2 Mpro assay
The in vitro enzymatic inhibition assay was conducted using the SARS-CoV-2 Mpro Inhibitor Screening Assay Kit supplied by BPS Biosciences. This assay provides a reliable and user-friendly platform for evaluating potential inhibitors of Mpro. The assay followed the manufacturer's recommended protocols and was performed using a 96-well plate format. Inhibitor solutions were prepared by diluting the synthesized compound into the Mpro assay buffer. The assay utilized a fluorogenic substrate containing a cleavage site between nsp4 and nsp5, which, under normal conditions, is cleaved by Mpro. The cleavage released the EDANS fluorophores for the Dabcyl quencher, producing a measurable fluorescent signal. Fluorescence intensity was measured using a PerkinElmer 2030 Victor X Multilabel Plate Reader with excitation and emission wavelengths set to 355 nm and 535 nm, respectively. GC376, a known Mpro inhibitor, was included as a standard reference drug. In the 1st step, 5 ng of MBP-tagged Mpro enzyme was combined with 30 μL of assay buffer containing 1 mM dithiothreitol (DTT). Synthesized compounds dissolved in DMSO (10 μL) were added to the mixture and pre-incubated for 1 h at rt. Subsequently, 10 μL of the fluorescent substrate was added to initiate the enzymatic reaction, bringing the final reaction volume to 50 μL. The final concentration of the substrate and inhibitor in the reaction mixture was maintained at 50 μL. The incubation was carried out at rt for 12–17 hours. The assay tested a range of inhibitor concentrations (0.005–50 μL) to determine IC50 values. Positive control of well-containing enzymes, 1% DMSO, and substrate showed no inhibition of enzymatic activity. As reference control GC376 inhibitor was tested at 100, 10 and 0.1 μM. The IC50 values of the synthesized compounds were calculated using the nonlinear regression (curve fit) function in the GraphPad Prism 8.0 software.43Mpro cytotoxic assay
The cytotoxic activity of the synthesized compounds was assessed in Vero-E6 cells using the MTT assay with minor modifications. Stock solutions of the compounds were initially dissolved in 10% aqueous DMSO solution, followed by further dilution in DMEM to prepare working solutions. Vero-E6 cells were seeded in 96-well plates at a density of 1 × 104 cells per well and incubated for 24 hours at 37 °C in the five percent CO2 atmosphere. Various concentrations of the synthesized compound were then applied to the cells, and they were incubated for 48 hours under the same conditions. Cytotoxicity was determined by adding 10 μL of MTT reagent (5 mg mL−1 in PBS) to each well, followed by additional incubation for 4 hours to allow formazan crystal formation. The resulting crystals were dissolved in 100 μL of DMSO, and absorbance was measured at 570 nm using a microplate reader.44 The percentage of cell viability was plotted against the compound concentration, and the 50% cytotoxic concentration (CC50) was subsequently calculated using the non-linear regression function in GraphPad Prism 8.0.43,45,46Molecular dynamic simulation
After docking studies, the best and most potent COVID-19 inhibitor and conformation were selected for the molecular dynamic simulation (MDS) to evaluate their interaction at the molecular level and in real body conditions. GROMACS software was used to perform molecular dynamic simulation. Charmm GUI server was used to generate input files for MD simulation. The protein–ligand complex was enclosed in a cubic box and solved using the TIP3P water model. The system was equilibrated with a human body temperature at 37 °C and 1 atm pressure. The system was minimized using 50000 steps before performing MD simulation.
After MD simulation, root means square deviation (RMSD) analysis of protein and complex backbone has been performed to evaluate the structural changes of protein before and after ligand inhibition. RMSD gives a clear picture of overall protein dynamics (such as folding and unfolding) and conformational changes during the MD simulation. On the other hand, the Root Mean Square Fluctuation (RMSF) calculation analyzes the movement/fluctuation of protein regions and residue during the simulation. Another key parameter to understand the ligand inhibition potential and protein–ligand complex at the molecular level is the number of hydrogen bonding interactions throughout simulations.
Results and discussion
Design rational
We have conducted a comprehensive literature review on the surface topology of the Mpro active site and existing Mpro inhibitors (repurposed and rationalized). DHPM-based inhibitors have demonstrated anti-HIV,47,48 anti-SARS-CoV-1,49–51 and anti-SARS-CoV-2 (ref. 49, 50 and 52–54) properties. Due to the notable pharmacological relevance, we focused on DHPM's derivatives featuring EWGs and EDGs to design Mpro inhibitors. Our laboratory has previously explored the DHPM scaffold and reported its diverse activity profiles targeting various biological assays.55–61 We designed and docked DHPM derivatives against the best resolution X-ray crystallographic structure of the Mpro (PDB id: 6XHM), identified three promising DHPM-2-thiones (6a–c) with the lowest binding energies as a hit for guiding our lead identification process. To refine these hits, SAR analysis was employed to delineate critical structural elements that enhance biological activity. Subsequently, lead compounds (8a–h) were docked, and their IC50 values were determined (Fig. 5). This systematic approach enables precise lead optimization. The Mpro comprises four pockets S1, S1′, S2, and S3 pockets. Notable inhibitors such as N3, 13b, 11a, 11b, X77, boceprevir, and dipyridamole include five- and six-membered rings, showcasing the S1 pocket's capacity to accommodate bulky substituents.62,63 The S1′ pocket is the hydrated region within the active site that forms a water bridge with Thr26, and requires proton donors and acceptors, essential to maintain the water bridge in S1′ and H-bond within S1, inhibitors lacking these groups often exhibit reduced activity. The NGSC motif (Asn142–Gly143–Ser144–Cys145) in S1′ is crucial for forming H-bonds with inhibitors.64–66 The S2 pocket of Mpro is deeply embedded in nature, favoring six-membered ring and halogenated moieties enhancing bindings affinities e.g. nirmatrelvir, 13b and 11b.63 The hydrophobic S3 pocket supports cyclic structures and branched alkyl chains (e.g. X77, 11a, PF-00835231, boceprevir, and 13b),63 with full occupancy observed for six-membered ring.67 In line with these findings, we synthesized DHPM derivatives with proton donor/acceptor groups, aromatic bicyclic moieties, aliphatic moieties, and six-membered rings to optimize interactions across Mpro. Additionally, we strategically incorporated fluorine into our inhibitors, a modification known to increase the inhibitory potency of various medications such as anti-HIV (e.g. efavirenz) and antivirals (e.g. raltegravir, emtricitabine, ledipasvir, and tenofovir). Its presence is particularly relevant to the S1, S2, and S3 pockets where its electronegativity enhances the H-bonding and hydrophobic interactions, improving binding affinity.68 The synthesis of these potent compounds was carried out. To validate in silico findings, in vitro analysis was conducted confirming biological activity safety & efficacy. These results provided quantitative data for further optimization in drug development (Fig. 4). | ||
| Fig. 4 Design strategy for SARS-CoV-2 Mpro inhibitors. | ||
Chemistry
In the present study, we have synthesized a series of DHPM-based SARS-CoV-2 Mpro inhibitors. Initially, a range of aromatic aldehydes, thiourea and ethyl 4,4,4-trifluoro-3-oxobutanoate underwent a multi-component, one-pot classical Biginelli reaction in the presence of tin(II) chloride catalyst, leading to the formation of a variety of substituted DHPM-thiones (6a–c) (Scheme 1). | ||
| Scheme 1 DHPM-thiones (6a–c) synthesis through Biginelli reaction. | ||
Subsequently, DHPM-thione derivatives reacted with various primary amines via a substitution reaction, resulting in the synthesis of amide derivatives of DHPM-thiones (8a–h) (Scheme 2).
 | ||
| Scheme 2 Synthesis of DHPM-thiones amide derivatives (8a–h) by reaction of DHPM's & 1° amines. | ||
In the next step, bromoacetylated DHPM-thione (10a–h) (Scheme 3) were prepared through the reaction of amide derivatives of DHPM-thione with bromoacetyl bromide.
 | ||
| Scheme 3 Bromoacetylated DHPM-thiones (10a–h) synthesis by reaction of (8a–h) with (9). | ||
Finally, potent SARS-CoV-2 inhibitors (12a–l) based on DHPM-thiones scaffold were synthesized by reacting the bromoacetylated DHPM-thiones with diverse secondary amines (Scheme 4).
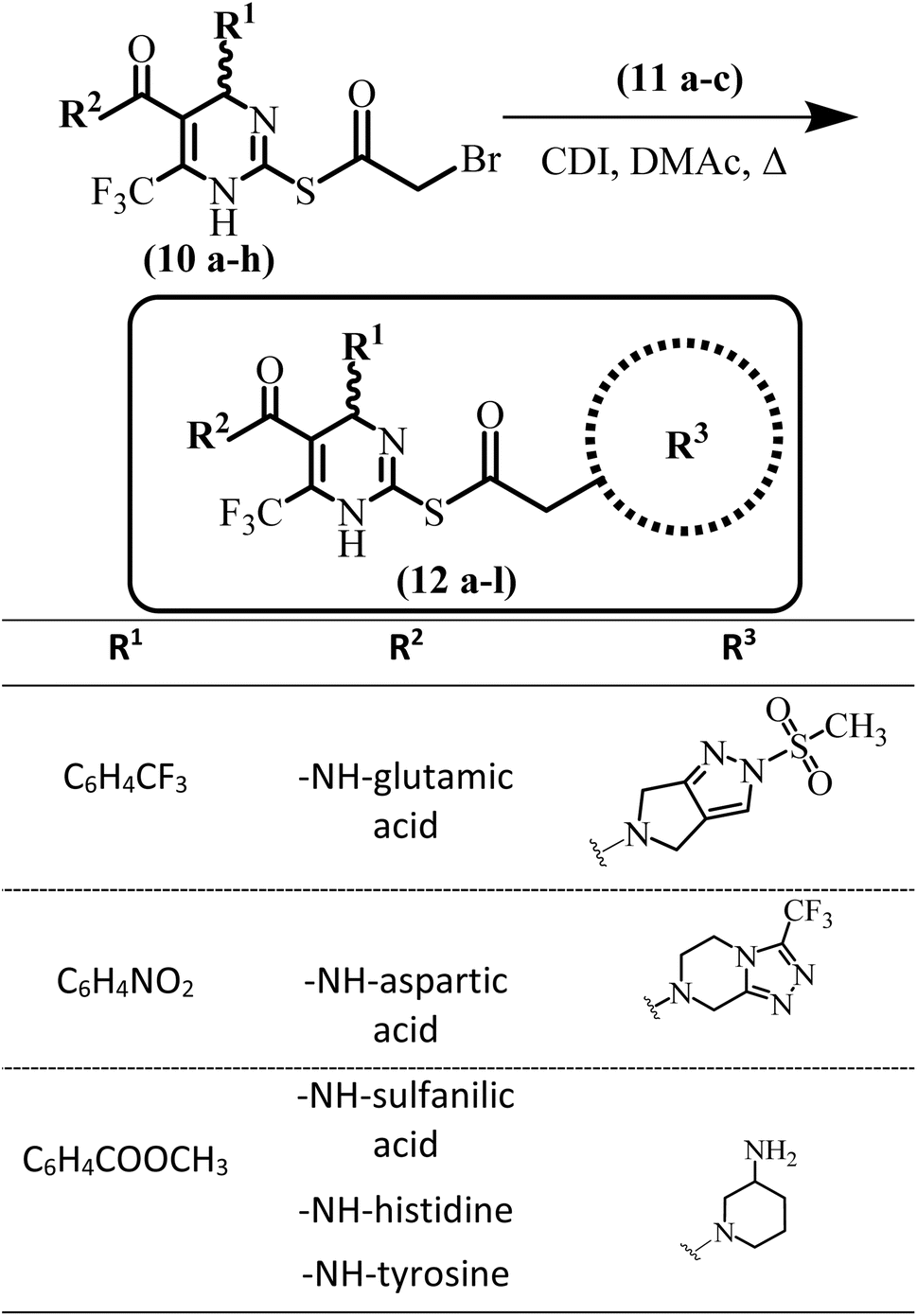 | ||
| Scheme 4 Reaction of bromoacetylated products (10a–h) with diverse 2° amines. | ||
In vitro pharmacology [inhibitory concentrations (IC50) and cytotoxicity concentration (CC50)]
The Mpro of SARS-CoV-2 plays a critical role in the virus replication by cleaving viral polyproteins, making it a pivotal target for the therapeutic intervention. Numerous covalent and non-covalent inhibitors such as carmofur, PX-12, and GC376 have been developed to combat SARS-CoV-2. These inhibitors feature N-heterocycles, that mimic the glutamine amino acid at P1 active site. Covalent inhibitors target the catalytic Cys145 residue leading to potent antiviral activity through both cellular and enzymatic inhibition. Noteworthy, ML188.47, a non-covalent Mpro inhibitor, has demonstrated significant efficacy against Mpro.69–71In this study we identified lead compounds through comprehensive Structure–Activity Relationship (SAR) analysis, followed by their optimization. Various amino acids were conjugated on the western side of the DHPM-thione scaffold using different synthetic strategies. The half-minimal inhibitory concentration (IC50) values of these intermediates were determined using Mpro assay kit. The in vitro inhibition results of the intermediates are summarized in Fig. 5. SAR analysis revealed that intermediate 8a containing a trifluoromethyl benzene moiety on the northern side and glutamic acid at the western side of scaffold DHPM-thione, exhibited potent inhibitory activity with an IC50 of 23.2 μM ± 0.92. Substituting glutamic acid with aspartic acid in 8b reduced inhibitory activity (IC50 = 44.7 ± 1.36 μM), indicating that the carbon chain length influences inhibition. Conjugation with sulfanilic acid in 8c enhanced inhibitory potency (IC50 = 28.2 μM ± 1.71). Based on these findings, we hypothesized that cyclic moieties could enhance activity. Replacing sulfanilic acid with histidine in 8d resulted in a marginally altered IC50 of 30.7 μM ± 1.43 while substituting with tyrosine in 8e yielded a significant inhibitory effect (IC50 = 25.3 μM ± 1.02). Modifying the northern side with nitrobenzene, while retaining glutamic acid in 8f and sulfanilic acid in 8g on the western side, resulted in reduced potency, with IC50 values of 36.8 μM ± 1.36 and 55.5 μM ± 1.43 respectively. Further SAR exploration incorporating histidine (8h) at the western side, paired with methyl benzoate at the northern side resulted in IC50 values of 34.9 μM ± 1.042 μM. The histidine derivative 8h demonstrated superior inhibitory activity. In conclusion, the SAR analysis highlights the critical role of cyclic moieties on the western side of the DHPM-thione scaffold, along with strategic substitution on the northern side, in influencing the inhibitory efficacy against the SARS-CoV-2 Mpro. This SAR investigation underscores the potential of strategic substitution to enhance the potency of Mpro inhibitors (Fig. 5).
 | ||
| Fig. 5 Intermediates (8a–h) synthesized during the SARS-CoV-Mpro inhibitor development and standard drug GC376. | ||
In our study, we synthesized a series of dihydropyrimidine-2-thione-based compounds (12a–l) based on SAR insights and evaluated in vitro inhibitory potential against SARS-CoV-2 Mpro. GC376 (a well-known SARS-CoV-2 inhibitor) was used as a benchmark to compare the in vitro results. Several exhibited notable inhibitory activities at the sub-micromolar range against SARS-CoV-2 Mpro, ranging from moderate to excellent.
Notably, compound 12l demonstrated remarkable potency, with an IC50 value of 0.054 μM ± 0.001, significantly surpassing the benchmark inhibitor GC376 (IC50 = 0.175 μM ± 0.004). The incorporation of histidine on the western side of 12l provided a key structure–activity relationship (SAR) insight essential for Mpro inhibition. However, contrary to expectations, (12d) and (12h), which also features histidine, exhibited lower inhibitory activity (IC50 = 14.38 μM ± 1.05 and 16.27 μM ± 0.81, respectively), suggesting that additional structural elements critically modulate efficacy.
The methyl benzoate moiety at the northern side of 12l further contributed to its enhanced activity, underscoring its importance in Mpro inhibition. Additionally, halogenated substituents, particularly fluorine, on the northern side influenced inhibitory potential across (12a–i). While most fluorinated compounds, including (12b), (12c), (12d), (12f), and (12h), displayed limited activity, compounds (12a), (12e), (12g), and (12h) exhibited improved inhibition (IC50 = 0.15 μM ± 0.01, 0.73 ± 0.04, 0.21 ± 0.01, and 16.27 μM ± 0.81, respectively) relative to GC376. The IC50 values for (12a–l) are summarized in Fig. 6.
 | ||
| Fig. 6 In vitro Mpro inhibition results of synthesized compounds 12a–l. | ||
Furthermore, (12a) and (12e) display superior efficacy when combined with additional functional groups, such as glutamic acid and tyrosine, on the western side. Further lead optimization revealed that 12j (IC50 = 0.063 μM ± 0.001) and 12k (IC50 = 0.146 μM ± 0.001) exhibited excellent inhibitory activity. These derivatives leveraged glutamic and sulfanilic acid moieties on the western side and nitrogen- and sulfur-containing groups on the northern and eastern sides, respectively.
The exceptional potency of compound 12l was attributed to the fluorinated triazolo-pyrazine group on its eastern side, a feature also observed in the potent 12a and 12e. Moreover, the piperidinyl amine group in (12g) and the sulfonyl pyrrolo-pyrazole moiety in 12i, 12j, and 12k further enhanced inhibition. These findings underscore the critical role of diverse substituents including histidine, halogens (fluorine), and functional groups containing nitrogen, oxygen, and sulfur in optimizing the efficacy of Mpro inhibitors.
Notably, compounds 12a, 12j, 12k, and 12l outperformed GC376 in Mpro inhibition, while others displayed moderate to good activity. Among the most potent inhibitors, 12j and 12l each achieved over 50% inhibition against Mpro. Dose–response experiments for 12j, 12k, and 12l confirmed their IC50 values, as depicted in Fig. 7. Compound 12l emerged as a highly promising candidate, demonstrating exceptional inhibitory potency against SARS-CoV-2 Mpro, making it a viable candidate for further drug development.
 | ||
| Fig. 7 Dose–response curves for IC50 values of compounds 12a, 12j, 12k, 12l, and standard drug GC376 ± SEM; n = 3. | ||
The cytotoxic potential of the synthesized DHPM-2-thione derivatives were evaluated to determine their half-maximal cytotoxic concentration (CC50) against SARS-CoV-2. The MTT assay revealed a wide spectrum of cytotoxic responses among the compounds, ranging from low to high. By employing non-linear regression analysis, the data were converted into the percent cell viability, enabling the precise calculation of CC50 values. Furthermore, these values were utilized to compute the selectivity index (SI) as the ratio of CC50 to IC50, providing insight into the therapeutic safety margins of the compounds.
The IC50 and CC50 graphs for the compounds are summarized in Fig. 7 and 8 respectively. Among these compounds, (12c) exhibited the highest CC50 of 247 μM, (lowest cytotoxicity) followed by 12k (CC50 = 239 μM), 12i (CC50 = 243 μM), 12j (CC50 = 211 μM) and 12l (CC50 = 198 μM). Particularly, structural optimization contributed significantly to improving CC50 values for selected derivatives, highlighting the critical role of the chemical modifications in enhancing their safety profiles. The CC50 value for these compounds, as observed on Vero E6 cells, offers a detailed comparison of their cytotoxic profile.
 | ||
| Fig. 8 Half-maximal cytotoxicity concentration (CC50) of final compounds 12c, 12i, 12j, 12k and 12l on Vero E6 cell ± SEM; n = 3. | ||
Docking studies
Docking studies were conducted to thoroughly analyze the spatial orientation and binding configurations of ligand–enzyme complexes. The crystal structure of the Mpro, co-crystallized with native ligands N-[(2S)-1-({(2S,3S)-3,4-dihydroxy-1-[(3S)-2-oxopyrrolidin-3-yl]butan-2-yl}amino)-4-methyl-1-oxopentan-2-yl]-4-methoxy-1H-indole-2-carboxamide and EDO (1,2-ethanediol), was retrieved from the Protein Data Bank (PDB) under accession code 6XHM, featuring the best resolution. To confirm the reliability of the docking protocol, a re-docking of the native ligand with the Mpro was performed. The observed root-mean-square deviation (RMSD) values were <2.0 Å, indicating a high degree of consistency between the experimental and modelled configurations.Subsequently, synthesized DHPM-thione derivatives (12a–l) were subjected to docking analysis against the binding sites of SARS-CoV-2 Mpro, in alignment with SAR study guidelines. Extensive literature reports that Mpro/3CLpro comprises four binding pockets: a bulky S1 pocket that accommodates six-member rings and facilitates H-bond formation, a hydrated S1′ pocket, a narrow and hydrophobic S2 pocket and the S3 pocket capable of binding both aliphatic and aromatic moieties. Key amino acid residues involved in the drug design for each pocket include: Phel40, Leul41, Asnl42, Hisl63, Glul66, Hisl72 (S1 pocket); Thr24, Thr25–26, Leu27, His41, Met49, Asnl42, Glyl43, Serl44, Cysl45 (S1′ pocket); His41, Met49, Tyr54, Glnl89, His164, Asp187, Arg188 (S2 pocket); and Metl65, Leul67, Prol68, Glnl89, Thrl90, Alal91, Glnl92, Leu167, Gly170 (S3 pocket).64,72–80
To optimize the lead compounds, a detailed SAR analysis was performed against the binding pockets of the Mpro using an advanced computational docking approach to assess binding interactions and to identify potent intermediate for further inhibitor development. Intermediates (8a–h) were docked into Mpro binding pockets, with four intermediates (8a, 8c, 8e, 8h) showing promising potential. To further enhance the binding affinity of the intermediates, the introduction of additional functional groups is required, which could improve the overall potency of inhibitors.
The 2D interaction plot of intermediate 8a (Fig. 9(a)) revealed six conventional H-bonds, three halogens (fluorine) interactions, two π–alkyl interactions, and a π–sulfur interaction. Specifically, intermediate 8a interacted with all four Mpro pockets: H-bonds formed with His163, halogen (fluorine) interactions with Phe140 and Leu141, and a π–alkyl interaction with His172 in the S1 pocket. The S1′ pocket exhibited H-bonds and a π–sulfur interaction Ser144 and Cys145 respectively. The S2 pocket showed halogen (fluorine) interactions and H-bonds interactions with His164 and His41, while the S3 pocket displayed H-bond interactions with Gln189 and Thr190 along with a π–alkyl interaction with Met165. The estimated free binding energy value of 8a in the binding pocket of Mpro was −6.9 kcal mol−1, indicating moderate binding affinity and promising potential for further development as an inhibitor.
 | ||
| Fig. 9 Two-dimensional (2D) interaction plot of intermediate (a) 8a (b) 8c. | ||
Intermediate 8c (Fig. 9(b)) exhibited a similar interaction profile with six conventional H-bonds, three halogens (fluorine) interactions, a π–sulfur and a π–alkyl interaction. H-bond formed with Asn142 and Glu166 in the S1 pocket and with Gly143 in the S1′ pocket. In S2 pocket halogen (fluorine) and π–alkyl interactions were observed with His41, while halogen (fluorine) and π–sulphur interactions with Asp187 and Met49.
The S3 pocket demonstrated H-bond interactions with Glnl89, Thrl90, and Glnl92, along with π–sulphur and π–alkyl interactions with Met49 and Met165 respectively. The estimated free binding energy of the intermediate 8c was −7.4 kcal mol−1, indicating moderate to strong binding affinity, suggesting its potential for optimization and further investigation as an inhibitor.
The 2D interaction plot for intermediate 8e (Fig. 10(a)) demonstrated six H-bonds, three halogens (fluorine) interactions, and two π–sulfur interactions. The H-bonds were formed with His163, Asn142, and Glu166 in the S1 pocket, while halogen (fluorine) interactions were observed with Leu141. The S1′ pocket H-bonds were formed with Cys145 and Ser144 accompanied by a π–sulfur interaction with Met49. The S2 pocket exhibited halogen (fluorine) interactions with His164 and His41 along with a π–sulfur interaction. The S3 pocket showed halogen (fluorine) interactions with Met165 and an H-bond with Gln189. The estimated free binding energy of the 8e was −7.3 kcal mol−1 indicating moderate to strong binding affinity, suggesting its potential for optimization as an inhibitor.
 | ||
| Fig. 10 Two-dimensional (2D) interaction plot of intermediate (a) 8e (b) 8h. | ||
The 2D interaction plot of intermediate 8h (Fig. 10(b)) indicated diverse interactions, including four H-bonds, a halogen (fluorine) interaction, one π–sulfur interaction, one sulphur–X interaction, a π–π T shaped interaction, and one π–alkyl interaction. H-bonds were observed with Glu166, HIS163, and LEU141 in S1 pocket. In the S1′ a π–sulfur interaction was noted with Cys145; however, this pocket is not addressed properly as the NGSC motif is not fully engaged. Additionally, the S3 pocket is not occupied comprehensively, suggesting further modifications and indicating the need for further optimization to enhance binding interactions in the pocket. The S2 pocket exhibited a π–π T-shaped interaction with His42, a π–alkyl interaction with Met49 and H-bond with Asp187. The S3 pocket demonstrated halogen (fluorine) interaction with Gln189 and a π–alkyl interaction with Met165. The estimated free binding energy of 8h was −7.7 kcal mol−1 demonstrating highly favorable binding interaction and promising potential for further development as an inhibitor.
The SAR analysis of intermediates 8a, 8c, 8e, and 8h confirmed their potential as effective Mpro inhibitors. These intermediate display robust binding interactions across all four defined binding pockets of SARS-CoV-2 Mpro, including H-bonds, halogen (fluorine), π–S, S–X, π–π T-shaped, and π–alkyl, contributing to their high binding affinities. The estimated free binding energies of intermediates ranged from −6.9 to −7.7 kcal mol−1 with RMSD values suggesting stable docked conformation. These findings underscore the promising therapeutic potential of these intermediates, warranting further optimization for Mpro inhibitors.
The two-dimensional (2D) interaction plot for compound (12a), presented in Fig. 11(a) revealed strong binding interactions with Mpro. Compound (12a) demonstrates versatile interactions with all four defined pockets of Mpro, including four halogen–fluorine interactions, six hydrogen bonds (H-bonds), and three π–alkyl interactions. The compound's proton donor and acceptor functionalities enable it to engage key residues across Mpro's pocket. Specifically, in the S1 pocket, (12a) forms halogen (F) interactions & π–alkyl interactions with Leu141 and halogen (F) interactions Phe140, H-bonds interaction with Glu166 & His163. In the S1′ pocket, H-bonds were observed with NGSC motif Ser144, and H-bond & π–alkyl interactions were observed with Cys145. In the S2 hydrophobic pocket, halogen (F) interactions & H-bond observed with His164. Furthermore, in the large S3 pocket, which accommodates both cyclic and aliphatic motifs, π–alkyl interactions with Pro168, H-bond observed with Gln189, and halogen (F) interactions were observed with Met165 & Thr190 and the chloro thiophene group. The estimated free binding energy of compound 12a was calculated to be −10.2 kcal mol−1, indicating a potent inhibitor against Mpro. In contrast, compound 12j, as shown in Fig. 11(b), demonstrated an even more extensive set of interactions with the Mpro. This compound formed seven H-bonds, three halogens (F) interactions, and a π–alkyl interaction. The proton donor and acceptor moieties of compound 12j facilitated its engagement with key residues across all Mpro pockets. In the S1 pocket, 12j formed H-bonds with Asn142 & Glu166, halogen (fluorine) interaction with Leu141 & Phe140, and H-bonds and π–alkyl interaction with His163.
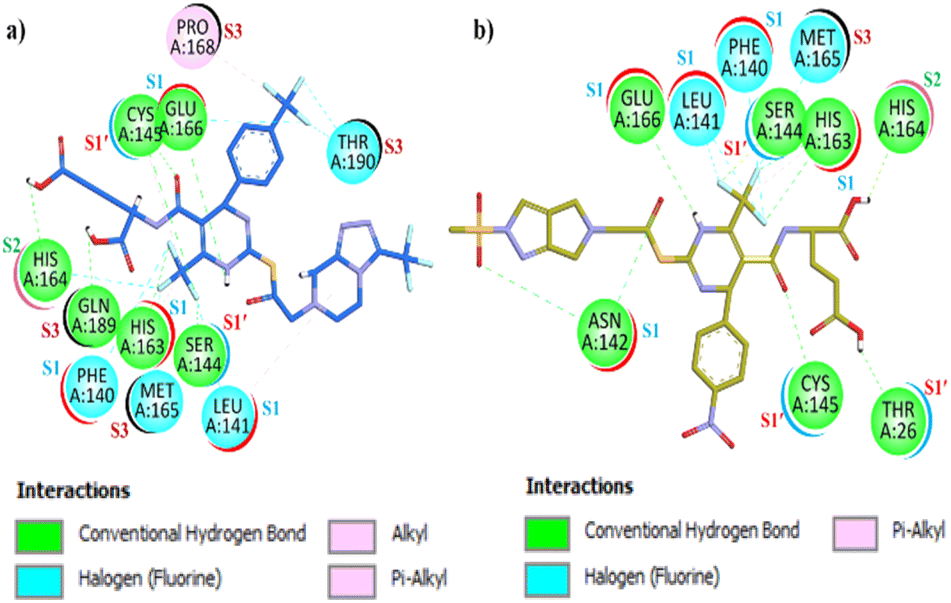 | ||
| Fig. 11 Two-dimensional (2D) interaction plot of synthesized compound (a) (12a) (b) (12j). | ||
In the S1′ pocket, H-bonds were observed with NGSC residues Ser144, Cys145 & Thr26 involving both carbonyl and fluorine groups. In the S2 pocket H bonding is observed with His164, and in the S3 pocket, Met165 halogen (fluorine) interaction was observed with (trifluoromethyl)benzene. The estimated free binding energy value of 12j was −11.5 kcal mol−1, reflecting its exceptional binding affinity as an Mpro inhibitor. Visual inspection of the docking results revealed that while all synthesized compounds exhibited significant interactions with Mpro, compound 12l displayed the most favorable interactions. It effectively engaged all the key pocket residues and demonstrated superior binding affinity compared to the standard drug against Mpro. The synthesized compound (12l) demonstrated excellent interactions with the SARS-CoV-2 Mpro. As shown in Fig. 12(a), a two-dimensional (2D) interaction plot reveals that compound 12l forms eight conventional H-bonds, six halogen (F) bonds, one π–sulphur interactions, two π–alkyl bonds, and a π–lone pair interaction. The compound comprehensively engages all key residues across all four pockets of Mpro. The presence of proton-withdrawing and proton-donating groups in 12l facilitates H-bonding interactions via the carbonyl group and (trifluoromethyl)benzene with key amino acid residues Asnl42, Hisl63. Additionally, halogen (F) bond interactions were observed with residues Glu166 and Leu141 of the S1 pocket, via (trifluoromethyl)-triazole and (trifluoromethyl)benzene moieties. In the S1′ pocket, Cys145 forms π–alkyl and conventional H-bond interactions with the carbonyl group and trifluoro moiety. The important residue Thr26 engages via H-bonding via the imidazole moiety. Furthermore, a π–sulphur interaction with Met49 and H-bonding with Glyl43 and Serl44, facilitated by carbonyl and trifluoro moieties, were also observed. All the NGSC motifs of the S1′ pocket of Mpro were effectively addressed by the compound 12l. In the hydrophobic S2 pocket, π–alkyl interactions with His41, and Met49 were facilitated by the benzene ring, while a π–lone pair interaction with Gln189 was noted. Halogen (F) bond interaction was observed with His164 & Arg188 via the trifluoromethyl-triazole moiety. In the S3 pocket Metl65, and Leul67 exhibit halogen (F) interaction through fluorine moiety of DHPM and triazole. Additionally, π–alkyl interaction with Pro168, halogen (F) interaction with Thr190, and H-bonding interaction with Gln192 was observed via the trifluoromethyl-triazole moiety in the S3 pocket of the Mpro target. The estimated free binding energy for 12l in the binding pocket of SARS-CoV-2 Mpro was −11.9 kcal mol−1, demonstrating a superior binding affinity against Mpro making it a highly promising inhibitor. For comparison GC376 (the standard drug), as shown in Fig. 12(b), exhibited diverse but fewer interactions with Mpro. The interactions include five H-hydrogen bond interactions, three π–alkyl bond interactions, a π–sulphur interaction, and one metal acceptor interaction with Mpro. GC376 exhibited H-bond interactions with Phel40, Hisl63, and Glul66 of the S1 pocket residues. In the S1′ pocket, hydrophobic π–alkyl interaction was observed with Leu27 and Cysl45, while H-bonding was observed with Serl44. With the S2 pocket residues Met49 the π–sulphur interaction was observed and with residue Glnl89 the H-bond interactions were observed. In the S3 pocket residue Metl65, hydrophobic interaction π–alkyl was observed while with residue Thrl90 metal acceptor interaction was observed.
 | ||
| Fig. 12 Two-dimensional (2D) interaction plot (a) synthesized compound (12l) (b) referenced drug GC376. | ||
Comparative molecular modeling and surface analysis of the intermediate (8h) and final compound (12l)
Our molecular modeling studies revealed that intermediate 8h, while capable of engaging some key residues, does not comprehensively interact with all four binding pockets of Mpro. It fails to fully engage the NGSC motifs in the S1′ pocket and does not extend into the S3 pocket (Fig. 13). | ||
| Fig. 13 Interaction of 8h with the Mpro binding sites: the S3 pocket is not occupied by 8h. | ||
In contrast, the final product 12l incorporates a trifluoromethyl-triazole moiety that facilitates additional interactions across the S1, S1′, S2, and S3 pockets as shown in Fig. 14. This modification enhances both hydrogen bonding and halogen interactions with residues across all pockets, resulting in the optimal alignment of compound 12l within the Mpro binding clefts. This improvement is reflected in its high inhibitory potency, with an IC50 of 0.054 μM, when compared to the intermediate compound 8h. Overall, our docking analysis via surface diagram confirms that the structural modification introduced in 12l is critical for achieving comprehensive engagement of Mpro's binding pockets and, consequently, for its enhanced antiviral activity.
 | ||
| Fig. 14 Comprehensive engagement of 12l with the Mpro binding sites. | ||
Molecular dynamic simulations (MDS)
Molecular dynamic simulations of protein and protein–ligand complex were performed to understand the inhibition of our compound at the molecular level and verify the docking results in possible body conditions. Compound inhibition was analyzed in terms of how it changes or controls the protein overall and regional dynamics as well as their conformation changes. The RMSD graph (Fig. 15) shows that target protein Mpro (PDB Id = 6XHM) shows considerable stability in the first 20 ns and after 20 ns there are so many conformational changes during the simulation which indicates their active conformation changes. The frequency of conformation changes has been reduced in the last 20 ns. The overall RMSD of protein Mpro is more than 4 Å. On the other hand, after the binding of the ligand, the protein–ligand complex shows complete stability with RMSD below 2.5 nm throughout the simulation. | ||
| Fig. 15 The root means square deviation (RMSD) graph of protein (blue) and protein–ligand (12l) complex (brown). | ||
The root means square fluctuations (RMSF) (Fig. 16) of target protein 6XHM and protein–ligand complex are almost similar, with few exceptions in the complex RMSF plot.
 | ||
| Fig. 16 The root means square fluctuations (RMSF) graph of protein (6XHM) (blue) and protein–ligand (12l) complex (brown). | ||
The RMSF graph for both is between 0 and 0.6 nm, mostly below 0.2 nm, except for four peaks, which show the flexibility of protein in the ranges of 1–10, 49–55, 190–200, and 290–300. The protein–ligand complex shows more fluctuations at 49–55 and 190–200. Hydrogen bonding analysis was performed to analyze the interaction between the target protein (6XHM) and ligand (12l).
The histogram (Fig. 17) shows that the minimum number of hydrogen bonds in every frame throughout the simulation is three and the maximum is five, which is evident in the strong inhibition of our ligand.
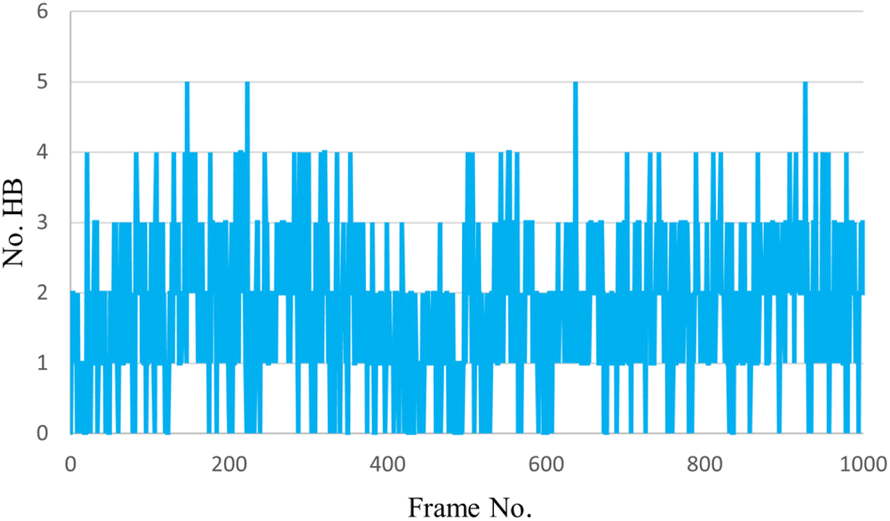 | ||
| Fig. 17 The H-bonding interactions histogram of protein (6XHM) and ligand (12l). | ||
Pharmacokinetic predictions
Pharmacokinetic properties of the most potent derivatives (12a, 12j, 12k, 12l) were evaluated using in silico tools to predict their potential for Blood–Brain Barrier (BBB) penetration, Human Intestinal Absorption (HIA), and AMES toxicity.The SMILES strings of these synthesized compounds were submitted to the online admetSAR server (https://lmmd.ecust.edu.cn/admetsar2/) for prediction.
As summarized in Table 1, the results indicate that all tested compounds are likely to penetrate the BBB, exhibit significant absorption in the intestine, and are predicted to be non-AMES toxic. These predictions suggest favourable pharmacokinetic profiles, underscoring their potential for further development as therapeutic agents against SARS-CoV-2 Mpro.81
| Comp. no. | BBB | HIA | AMES toxicity |
|---|---|---|---|
| 12a | −(0.9399) | +(0.8739) | Non-toxic (0.5949) |
| 12j | −(0.9205) | +(0.6844) | Non-toxic (0.5488) |
| 12k | −(0.8224) | +(0.5774) | Non-toxic (0.5496) |
| 12l | −(0.9665) | +(0.8197) | Non-toxic (0.6035) |
Conclusions
The study harnessed the dihydropyrimidine-2-thiones as a scaffold due to its extensive applications in various medications and the availability of an in-house library within our research group. Among diverse derivatized dihydropyridines-2-thiones, 6a, 6b, and 6c DHPM's based intermediate emerged as potent leads subsequently optimized through Structure Activity Relationship (SAR) and computational docking interaction analysis through AutoDock4 version (v4.2.6). These intermediates were also docked and subjected to in vitro analysis. Conclusively the study identifies fifteen potent dihydropyrimidine-2-thione-based compounds (12a–l) as potential inhibitors of SARS-CoV-2 Mpro. These compounds underwent biological assay against the Mpro confirming significant inhibitory activity. Notably, compounds (12a), 12j, 12k, and 12l exhibited superior inhibition compared to the standard drug GC376. Conversely, compounds (12b), (12c), (12e), (12f), (12g), and (12i) demonstrated less inhibitory efficacy. Molecular docking and Molecular Dynamic (MD) simulation revealed significant coherence with biological assay and confirmed the ligand (12l)–protein complex (6XHM) stability respectively. The cytotoxic profile of compounds 12c, 12i, 12j, 12k, and 12l were evaluated, with CC50 values confirming an acceptable safety margin for human consumption. Furthermore, the oral toxicity of compounds 12j, and 12l were predicted and these were found inactive to the oral toxicities. Collectively designating these compounds as promising non-covalent inhibitors for Mpro, compound 12l exhibited exceptional potency against the Mpro of SARS-CoV-2.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Umer Rashid conceived, designed, and supervised this study. He was involved in all the phases (from synthesis to pharmacological evaluation and manuscript writing/editing) that led to the manuscript's completion. Anees Saeed synthesized the compounds. In vitro experiments were performed by Anees Saeed, Fahad Hussain, and Abdul Sadiq. Docking studies were performed by Anees Saeed. Docking results were analyzed and written by Ayesha Tahir and UR. MD simulations were performed by Muhammad Shah. The manuscript was drafted by Anees Saeed. Umer Rashid reviewed and edited the drafts. All the authors have read the manuscript and approved it for publication.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research is financially supported by a Project Grant from the Higher Education Commission Pakistan to Umer Rashid (PI) under the National Research Program for Universities (NRPU) (20-14513/NRPU/R&D/HEC/2021 2021).References
- A. R. Fehr and S. Perlman, Coronaviruses: Methods and Protocols, 2015, pp. 1–23 Search PubMed.
- W. S. T. Consortium, N. Engl. J. Med., 2021, 384, 497–511 CrossRef PubMed.
- L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. De Jesus, P. Teriete and M. V. Hull, Nature, 2020, 586, 113–119 CrossRef CAS PubMed.
- D. R. Owen, C. M. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo and K. J. Coffman, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- P. V’kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef PubMed.
- M. M. Lamers and B. L. Haagmans, Nat. Rev. Microbiol., 2022, 20, 270–284 CrossRef CAS PubMed.
- D. Cucinotta and M. Vanelli, Acta Bio Med. Atenei Parmensis, 2020, 91, 157 Search PubMed.
- E. Mahase, BMJ, 2020, 368, m1036 CrossRef PubMed.
- D. S. Hui, E. I. Azhar, T. A. Madani, F. Ntoumi, R. Kock, O. Dar, G. Ippolito, T. D. Mchugh, Z. A. Memish and C. Drosten, Int. J. Infect. Dis., 2020, 91, 264–266 CrossRef CAS PubMed.
- F. G. Hayden, R. B. Turner, J. M. Gwaltney, K. Chi-Burris, M. Gersten, P. Hsyu, A. K. Patick, G. J. Smith III and L. S. Zalman, Antimicrob. Agents Chemother., 2003, 47, 3907–3916 CrossRef CAS PubMed.
- Y. Kim, H. Liu, A. C. Galasiti Kankanamalage, S. Weerasekara, D. H. Hua, W. C. Groutas, K.-O. Chang and N. C. Pedersen, PLoS Biol., 2016, 12, e1005531 Search PubMed.
- H. Yang, W. Xie, X. Xue, K. Yang, J. Ma, W. Liang, Q. Zhao, Z. Zhou, D. Pei and J. Ziebuhr, PLoS Biol., 2005, 3, e324 CrossRef PubMed.
- W. Dai, B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng and F. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- Y. Chen, Q. Liu and D. Guo, J. Med. Virol., 2020, 92, 418–423 CrossRef CAS PubMed.
- S. Hussain, J. a. Pan, Y. Chen, Y. Yang, J. Xu, Y. Peng, Y. Wu, Z. Li, Y. Zhu and P. Tien, J. Virol., 2005, 79, 5288–5295 CrossRef CAS PubMed.
- R. Ramajayam, K.-P. Tan and P.-H. Liang, Biochem. Soc. Trans., 2011, 39, 1371–1375 CrossRef CAS PubMed.
- K. Anand, G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21, 3213–3224 CrossRef CAS PubMed.
- H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye and H. Pang, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS PubMed.
- S. Zhang, M. Krumberger, M. A. Morris, C. M. T. Parrocha, A. G. Kreutzer and J. S. Nowick, Eur. J. Med. Chem., 2021, 218, 113390 CrossRef CAS PubMed.
- K. Świderek and V. Moliner, Chem. Sci., 2020, 11, 10626–10630 RSC.
- M. Bartlam, H. Yang and Z. Rao, Curr. Opin. Struct. Biol., 2005, 15, 664–672 CrossRef CAS PubMed.
- D. Stern and B. Sefton, J. Virol., 1982, 44, 804–812 CrossRef CAS PubMed.
- M. A. Marra, S. J. Jones, C. R. Astell, R. A. Holt, A. Brooks-Wilson, Y. S. Butterfield, J. Khattra, J. K. Asano, S. A. Barber and S. Y. Chan, Science, 2003, 300, 1399–1404 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski and L. Y. Li, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu and T. Suto, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- J. D. Tyndall, J. Med. Chem., 2022, 65, 6496–6498 CrossRef CAS PubMed.
- S. G. V. Rosa and W. C. Santos, Rev. Panam. Salud Públic, 2020, 44, e40 Search PubMed.
- N. Drayman, J. K. DeMarco, K. A. Jones, S.-A. Azizi, H. M. Froggatt, K. Tan, N. I. Maltseva, S. Chen, V. Nicolaescu and S. Dvorkin, Science, 2021, 373, 931–936 CrossRef CAS PubMed.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang and C. Peng, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty and Y. Chen, Cell Res., 2020, 30, 678–692 CrossRef CAS PubMed.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang and P. Niu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- P. Aparoy, K. Kumar Reddy and P. Reddanna, Curr. Med. Chem., 2012, 19, 3763–3778 CrossRef CAS PubMed.
- A. Saeed, S. A. Ejaz, A. Khalid, P. A. Channar, M. Aziz, Q. Abbas, T. A. Wani, N. A. Alsaif, M. M. Alanazi and A. M. Al-Hossaini, Int. J. Mol. Sci., 2022, 23, 13164 CrossRef CAS PubMed.
- A. Elmaghraby, I. Mousa, A. Harb and M. Mahgoub, Int. Scholarly Res. Not., 2013, 2013, 706437 CAS.
- F. Sánchez-Sancho, M. Escolano, D. Gaviña, A. G. Csáky, M. Sánchez-Roselló, S. Díaz-Oltra and C. Del Pozo, Pharmaceuticals, 2022, 15, 948 CrossRef PubMed.
- I. S. Zorkun, S. Saraç, S. Çelebi and K. Erol, Bioorg. Med. Chem., 2006, 14, 8582–8589 CrossRef CAS PubMed.
- R. Kaur, S. Chaudhary, K. Kumar, M. K. Gupta and R. K. Rawal, Eur. J. Med. Chem., 2017, 132, 108–134 CrossRef CAS PubMed.
- G. C. Rovnyak, K. S. Atwal, A. Hedberg, S. D. Kimball, S. Moreland, J. Z. Gougoutas, B. C. O'Reilly, J. Schwartz and M. F. Malley, J. Med. Chem., 1992, 35, 3254–3263 CrossRef CAS PubMed.
- S. M. D. Rizvi, S. Shakil and M. Haneef, EXCLI Journal, 2013, 12, 831 Search PubMed.
- R. Quiroga and M. A. Villarreal, PLoS One, 2016, 11, e0155183 CrossRef PubMed.
- A. A. Jabbar, R. A. Mothana, M. A. Abdulla, F. O. Abdullah, K. A.-A. Ahmed, R. R. Hussen, M. F. Hawwal, O. I. Fantoukh and S. Hasson, Saudi Pharm. J., 2023, 31, 101850 CrossRef CAS PubMed.
- M. A. Abu-Zaied, G. H. Elgemeie and N. M. Mahmoud, ACS Omega, 2021, 6, 16890–16904 CrossRef CAS PubMed.
- N. P. Marques, C. S. Lopes, N. C. T. Marques, L. Cosme-Silva, T. M. Oliveira, C. Duque, V. T. Sakai and J. A. C. Hanemann, Laser Med. Sci., 2019, 34, 465–471 CrossRef PubMed.
- C. Shivanika, D. Kumar, V. Ragunathan, P. Tiwari and A. Sumitha, J. Biomol. Struct. Dyn., 2022, 40, 585–611 CrossRef CAS PubMed.
- E. Radchenko, A. Dyabina, V. Palyulin and N. Zefirov, Russ. Chem. Bull., 2016, 65, 576–580 CrossRef CAS.
- B. H. Sarvaiya, P. I. Vaja, N. A. Paghdar and S. M. Ghelani, J. Heterocycl. Chem., 2024, 61, 1325–1348 CrossRef CAS.
- Z. Wang, H. Zhang, Z. Gao, Z. Sang, E. De Clercq, C. Pannecouque, D. Kang, P. Zhan and X. Liu, Acta Pharm. Sin. B, 2024, 14, 1257–1282 CrossRef CAS PubMed.
- C. Gege, F. Hahn, C. Wangen, S. Häge, A. Herrmann, N. Uhlig, V. Eberlein, L. Issmail, R. Klopfleisch and T. Grunwald, ChemMedChem, 2024, e202400292 CrossRef CAS PubMed.
- A. M. Elshamsy, T. F. Ali, M. Osman and N. A. El-Koussi, Journal of Advanced Biomedical and Pharmaceutical Sciences, 2023, 6, 114–123 CrossRef.
- D. S. E. Sayed and E.-S. M. Abdelrehim, BMC Chem., 2022, 16, 82 CrossRef CAS PubMed.
- D. C. Schultz, R. M. Johnson, K. Ayyanathan, J. Miller, K. Whig, B. Kamalia, M. Dittmar, S. Weston, H. L. Hammond and C. Dillen, Nature, 2022, 604, 134–140 CrossRef CAS PubMed.
- E. Mansour, A. M. Sayed and S. I. Elewa, Polycyclic Aromat. Compd., 2024, 1–26 Search PubMed.
- R. Xiong, L. Zhang, S. Li, Y. Sun, M. Ding, Y. Wang, Y. Zhao, Y. Wu, W. Shang and X. Jiang, Protein Cell, 2020, 11, 723–739 CrossRef CAS PubMed.
- U. Rashid, S. F. Hassan, S. Nazir, A. Wadood, M. Waseem and F. L. Ansari, Med. Chem. Res., 2015, 24, 304–315 CrossRef CAS.
- U. Rashid, R. Sultana, N. Shaheen, S. F. Hassan, F. Yaqoob, M. J. Ahmad, F. Iftikhar, N. Sultana, S. Asghar and M. Yasinzai, Eur. J. Med. Chem., 2016, 115, 230–244 CrossRef CAS PubMed.
- G. Ahmad, N. Rasool, K. Rizwan, A. A. Altaf, U. Rashid, M. Z. Hussein, T. Mahmood and K. Ayub, Molecules, 2019, 24, 2609 CrossRef CAS PubMed.
- M. J. Ahmad, S. F. Hassan, R. U. Nisa, K. Ayub, M. S. Nadeem, S. Nazir, F. L. Ansari, N. A. Qureshi and U. Rashid, Med. Chem. Res., 2016, 25, 1877–1894 CrossRef CAS.
- F. Iftikhar, F. Yaqoob, N. Tabassum, M. S. Jan, A. Sadiq, S. Tahir, T. Batool, B. Niaz, F. L. Ansari and M. I. Choudhary, Bioorg. Chem., 2018, 80, 99–111 CrossRef CAS PubMed.
- M. Bibi, N. A. Qureshi, A. Sadiq, U. Farooq, A. Hassan, N. Shaheen, I. Asghar, D. Umer, A. Ullah and F. A. Khan, Eur. J. Med. Chem., 2021, 210, 112986 CrossRef CAS PubMed.
- M. A. Javed, N. Ashraf, M. Saeed Jan, M. H. Mahnashi, Y. S. Alqahtani, B. A. Alyami, A. O. Alqarni, Y. I. Asiri, M. Ikram and A. Sadiq, ACS Chem. Neurosci., 2021, 12, 4123–4143 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- C.-H. Zhang, E. A. Stone, M. Deshmukh, J. A. Ippolito, M. M. Ghahremanpour, J. Tirado-Rives, K. A. Spasov, S. Zhang, Y. Takeo and S. N. Kudalkar, ACS Cent. Sci., 2021, 7, 467–475 CrossRef CAS PubMed.
- A. Thakur, G. Sharma, V. N. Badavath, V. Jayaprakash, K. M. Merz Jr, G. Blum and O. Acevedo, J. Phys. Chem. Lett., 2022, 13, 5776–5786 CrossRef CAS PubMed.
- S. Tomar, M. L. Johnston, S. E. S. John, H. L. Osswald, P. R. Nyalapatla, L. N. Paul, A. K. Ghosh, M. R. Denison and A. D. Mesecar, J. Biol. Chem., 2015, 290, 19403–19422 CrossRef CAS PubMed.
- J. Mondal, P. Tiwary and B. Berne, J. Am. Chem. Soc., 2016, 138, 4608–4615 CrossRef CAS PubMed.
- H. H. Chan, M. A. Moesser, R. K. Walters, T. R. Malla, R. M. Twidale, T. John, H. M. Deeks, T. Johnston-Wood, V. Mikhailov and R. B. Sessions, Chem. Sci., 2021, 12, 13686–13703 RSC.
- C. Zhang, ACS omega, 2022, 7, 18206–18212 CrossRef CAS PubMed.
- C. Ma, Y. Hu, J. A. Townsend, P. I. Lagarias, M. T. Marty, A. Kolocouris and J. Wang, ACS Pharmacol. Transl. Sci., 2020, 3, 1265–1277 CrossRef CAS PubMed.
- W. Vuong, M. B. Khan, C. Fischer, E. Arutyunova, T. Lamer, J. Shields, H. A. Saffran, R. T. McKay, M. J. van Belkum and M. A. Joyce, Nat. Commun., 2020, 11, 4282 CrossRef CAS PubMed.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS PubMed.
- I. Y. Akbayrak, S. I. Caglayan, L. Kurgan, V. N. Uversky and O. Coskuner-Weber, Curr. Res. Struct. Biol., 2022, 4, 349–355 CrossRef CAS PubMed.
- A. Chandra Manivannan, A. Malaisamy, M. Eswaran, A. Meyyazhagan, V. A. Arumugam, K. R. Rengasamy, B. Balasubramanian and W.-C. Liu, Front. Mol. Biosci., 2022, 9, 918101 CrossRef PubMed.
- A. M. Shaqra, S. N. Zvornicanin, Q. Y. J. Huang, G. J. Lockbaum, M. Knapp, L. Tandeske, D. T. Bakan, J. Flynn, D. N. Bolon and S. Moquin, Nat. Commun., 2022, 13, 3556 CrossRef CAS PubMed.
- M. Macchiagodena, M. Pagliai and P. Procacci, Chem. Phys. Lett., 2020, 750, 137489 CrossRef CAS PubMed.
- C. Lin, H. Jiang, W. Li, P. Zeng, X. Zhou, J. Zhang and J. Li, Structure, 2023, 31, 1016–1024.e3 CrossRef CAS PubMed.
- H. P. Shao, T. H. Wang, H. L. Zhai, K. X. Bi and B. Q. Zhao, Chem.-Biol. Interact., 2023, 371, 110352 CrossRef CAS PubMed.
- L. Alzyoud, M. A. Ghattas and N. Atatreh, Drug Des., Dev. Ther., 2022, 16, 2463 CrossRef PubMed.
- Q. Hu, Y. Xiong, G. H. Zhu, Y. N. Zhang, Y. W. Zhang, P. Huang and G. B. Ge, MedComm, 2022, 3, e151 CrossRef CAS PubMed.
- M. D. Sacco, C. Ma, P. Lagarias, A. Gao, J. A. Townsend, X. Meng, P. Dube, X. Zhang, Y. Hu and N. Kitamura, Sci. Adv., 2020, 6, eabe0751 CrossRef CAS PubMed.
- J. Shen, F. Cheng, Y. Xu, W. Li and Y. Tang, J. Chem. Inf. Model., 2010, 50, 1034–1041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08449g |
| This journal is © The Royal Society of Chemistry 2025 |