
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-free synthesis of bio-based N-isobutyl-5-methyloxazolidinone: an eco-friendly solvent
Antoine Fournier ,
Jean-Moïse Suisse,
Pierre de Frémont and
Jacques Andrieu*
,
Jean-Moïse Suisse,
Pierre de Frémont and
Jacques Andrieu*
Institut de Chimie Moléculaire de l'Université de Bourgogne, UMR CNRS 6302, Université de Bourgogne, 9 Av. Alain Savary, 21078 Dijon, France. E-mail: jacques.andrieu@u-bourgogne.fr
First published on 10th February 2025
Abstract
Oxazolidinones are five-membered N-heterocycle rings containing a carbamate moiety and are known for their industrial applications as antibiotics, herbicides and electrolytes in Li batteries. Considering the projected ecological transition, they have the potential to be recognized as a green solvent according to the European standards for bio-based solvents, if they can be synthesized via an eco-friendly synthetic route. Herein, a strategy is proposed for the kilogram scale synthesis of N-isobutyl-5-methyloxazolidinone (BMOX) in two steps, starting from the renewable resources from sugar industry and without using any organic solvent. The first step was the addition of bio-based isobutylamine to chloropropanol in basic aqueous solution to afford an amino-alcohol. In the second step, to this amino-alcohol, diethyl carbonate was added in the presence of a bio-based imidazolium salt catalyst to afford the desired oxazolidinone containing more than 62% bio-based carbon atoms. This study elucidates the physicochemical properties of this new bio-sourced oxazolidinone.
Introduction
Oxazolidinones are comparatively less studied than other N-heterocycles (e.g., pyridines and imidazoles). Still, since the last century, they have been industrially used as antimicrobial,1,2 antiviral, antitubercular and anticancer agents2 and in antibiotics such as linezolid,3 herbicides,4 chiral auxiliaries in synthetic chemistry,5 paint removers based on non-toxic and halide-free solvents,6 and electrolytes in electrical energy storage.7,8 In addition to these applications, we hypothesize that oxazolidinones might be sustainable solvents that will respond plainly to the requirements of European standards EN 16766:2017 Bio-based solvents.9 Bio-based solvents only account for 1.5% of the bio-based chemical sector in the EU, but their production is far from the marginal value at 75 kt per year with an expected growth of 1% per year.10 This low annual growth of only 1% is owing to the low priority given to the production of bio-sourced alternatives, despite the need to reduce non-renewable carbon resources and toxicity to humans by solvents of fossil origin while improving recyclability.11 Strong political and economic constraints, such as the lack of incentives for this change, limited availability of biomass for full deployment, and potentially higher production costs also limit the further utility of bio-based solvents.11 Although the properties of oxazolidinones are not fully examined as they lack toxicology and environmental data, they exhibit promising physicochemical properties, as highlighted in the solvent selection guide published in 2016 on the SHE (safe health and environment) criteria.12 Their boiling points are between 80 °C and 200 °C, making them suitable to achieve low emissions of harmful organic vapors (named VOCs) and an easy recycling under low energy demand. This is the case for N-isobutyl-5-methyloxazolidinone (1) reported in this work, with a bp (boiling point) of 112 °C under 120 mbar. The health potential hazard of (1) can easily be estimated through the “Classification, Labelling and Packaging” (CLP) regulation according to Global, Harmonized System (GHS).13Unsubstituted 2-oxazolidones exhibit a low toxicity limited to skin and eye irritation, with the worst hazard statement reported being H319, similar to other safe solvents such as alcohols or ketones. Besides, its low human toxicity is proved by the commercialization of another alkyl-substituted oxazolidinone named linezolid.3 However, to evaluate whether oxazolidinones can be classified as eco-friendly and safe solvents, it is important to examine the chemical and technological risks associated with the starting materials and chemical processes involved in their production.
The first syntheses of oxazolidinones involved hazardous isocyanates,14 carcinogenic catalysts,15,16 expensive aziridines,15 or propargylamines.17 Later, rhodium-catalyzed hydrogenation of amino-acids was developed to provide bio-based amino-alcohols, which further reacted with (dialkyl)-carbonates, thus promoting cyclization.16,18 Unfortunately, rhodium is toxic to humans (carcinogenic agent). It has also an abundance of 5 ppb in the earth's crust, which renders its extraction and refining prohibitive in terms of environmental costs. These important drawbacks hamper the large-scale production of amino-alcohols under rhodium catalysis. To address this matter, we described an eco-friendly synthesis of oxazolidinones19,20 starting from diethyl carbonate and amino-alcohols obtained from bio-based amino-acids under organocatalysis (Scheme 1). The diethyl carbonate was made from cheap, abundant and safe reagents such as carbon dioxide and ethanol.21,22
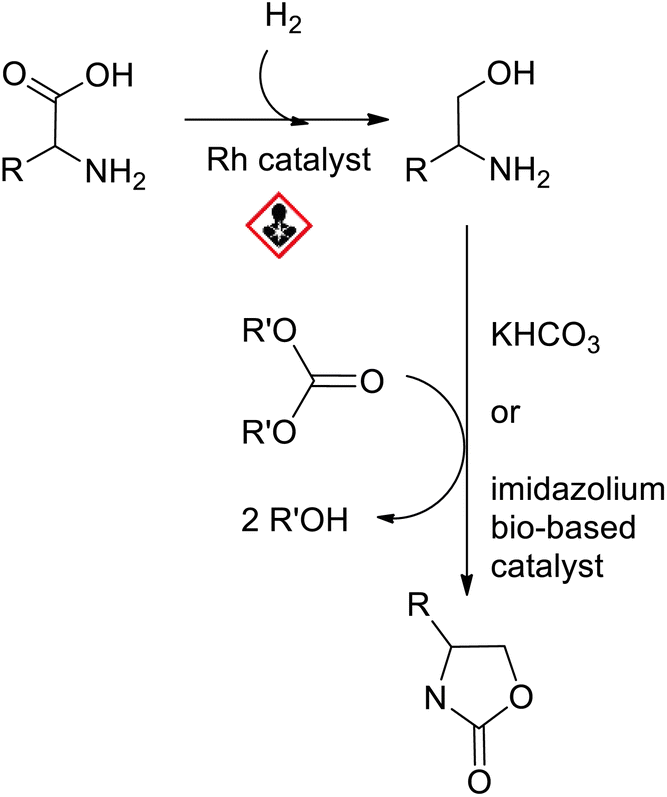 | ||
| Scheme 1 Synthetic route of oxazolidinones from amino acids via amino-alcohols. | ||
To further improve the synthesis of oxazolidinones and achieve 100% bio-based amino-alcohols, we suggest an alternative approach based on the decarboxylation of amino-acids to produce amines, which then react with propylene oxide to produce amino-alcohols (Scheme 2). Bio-sourced amines can be obtained by several pathways: (i) direct amination of bio-alcohols using ammonia,23,24 (ii) decarboxylation reactions of amino-acids with enzymes25 or with acetophenone under heating.20,26,27
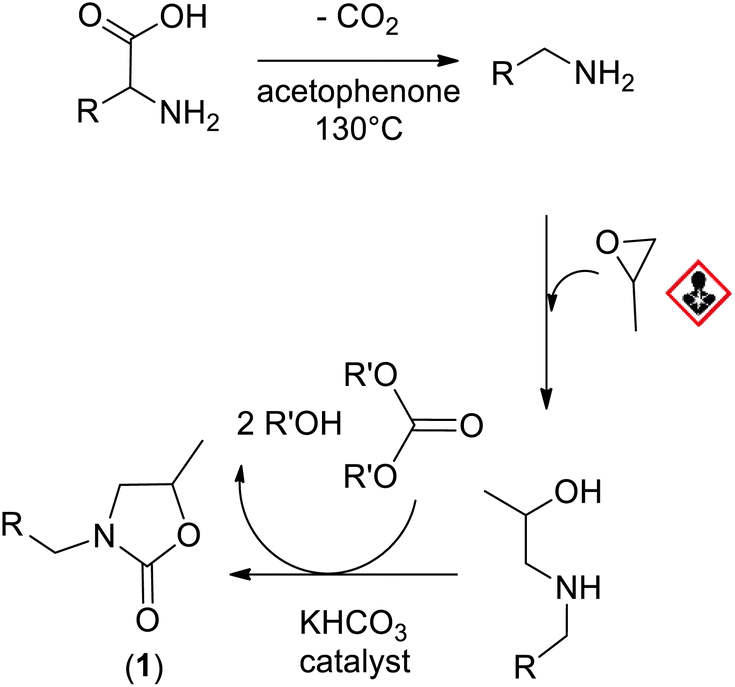 | ||
| Scheme 2 Synthesis of oxazolidones from bio-based reagents. | ||
Note that propylene oxide (PO) is also a problematic reagent because it is a flammable liquid (H224) with acute (oral, dermal and inhalation) toxicity (H302, H311, H331 and H335), mutagenic and carcinogenic properties (H340 and H350). Subsequently, it should not be directly introduced in new industrial chemical processes. Nevertheless, those fire and health hazards might be significantly reduced by generating the PO in situ by mixing chloro-alcohols with aqueous sodium hydroxide.28
Synthesis of amino-alcohol
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21 ratio. The compounds feature a boiling point at 110 °C and 166 °C, respectively, under 180 mbar pressure. They can be separated by fractional distillation, providing 46% reaction yield for 2 and 14% yield for 3, similar to the yields reported previously.29
:21 ratio. The compounds feature a boiling point at 110 °C and 166 °C, respectively, under 180 mbar pressure. They can be separated by fractional distillation, providing 46% reaction yield for 2 and 14% yield for 3, similar to the yields reported previously.29
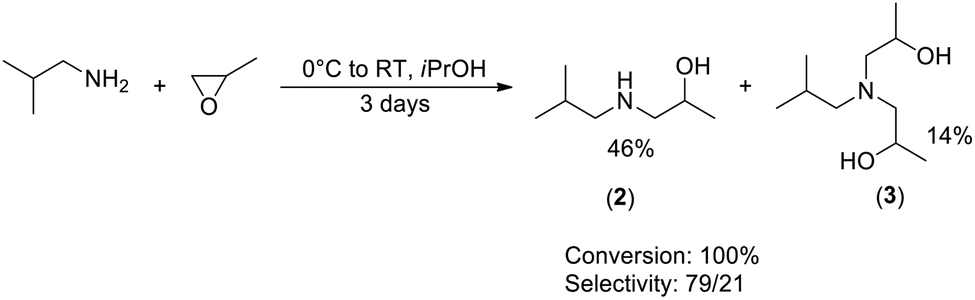 | ||
| Scheme 3 Synthesis of amino-alcohol (2) and amino-diol (3). | ||
Adding the PO at room temperature does not increase the time of the reaction while there is no sign of exothermicity. PO and isobutylamine being miscible, it is possible to perform the reaction under bulk condition, limiting the quantity of organic wastes. Interestingly, addition of a small amount of water (8% volume versus the total volume of reagents) drastically increases the rate of the reaction during the first hours, resulting in 84% conversion of PO after 6 hours (Scheme 4). However, final conversion is reached again after 3 days. The ratio of 2:3 is equal to 81:19, indicating that water does not promote the double addition of PO on the amine.
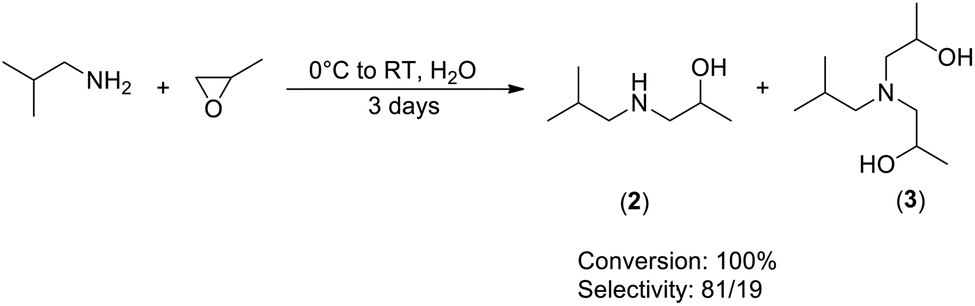 | ||
| Scheme 4 Solvent-free synthesis of amino-alcohol and amino-diol. | ||
We believe that the acidic character of water activates and stabilizes the oxygen atom of the epoxide when it undergoes ring-opening during the nucleophilic attack of the amine, forming an alcoholate and an ammonium adduct (Scheme 5). It also plays the role of a proton carrier during H-transfer from ammonium to alcoholate groups.
 | ||
| Scheme 5 Water activation mechanism of PO promoting the addition of amine. | ||
It is interesting to note that the only desired amino-alcohol 2 as well as amino-diol 3 were formed during the reaction, while another amino-alcohol isomer (N-isobutyl-propanol-2-amine), corresponding to the nucleophilic attack to the amino-group on the most hindered carbon position of propylene oxide, was not observed (Scheme 6).
 | ||
| Scheme 6 Regioselective nucleophilic attack of amine on propylene oxide. | ||
We suggest that the high electronegativity of oxygen renders the two carbon atoms of the ring more electrophilic. In addition, the methyl group creates steric hindrance along with an inductive electron-donating effect, which hinders the nucleophilic attack of the nitrogen on its neighboring substituted carbon atom. As a result, the amine selectively attacks the less substituted carbon atom, which is more electrophilic, and limits the formation of N-isobutyl-mono- and di-isopropanolamine isomers (Scheme 6). A control experiment was done with isobutylammonium hydrogenoxalate to replace isobutylamine. The bio-based ammonium salt was first solubilized in water, and propylene oxide was added dropwise to the solution at room temperature. After 22 hours of reaction, propylene oxide was fully consumed and rather converted to 1,2-propanediol by acidic hydrolysis than to amino-alcohol 2 (Scheme 7). It is therefore important to use an amine instead of an ammonium salt to synthesize the desired amino-alcohol 2 at neutral pH.
 | ||
| Scheme 7 Attempt to prepare amino-alcohol 2 from an ammonium salt. | ||
The reaction was also carried out at a larger scale (by a factor 103) going from 1.2 mL to 1.2 L of reagents. Changing the reaction scale, especially by a thousand fold, can trigger hazardous effects that need to be addressed. The addition of large amounts of PO is expected to strongly increase the temperature of the reaction medium. Thus, efficient heat exchange equipment and glassware with appropriate stirring are required, contrary to small scale reactions, which are more permissive. After several trials, we found the following optimized experimental conditions: 0.50 L of propylene oxide (or 415 g) was added dropwise, over 4 hours, to a solution of 1 equiv. of isobutylamine (0.71 L or 523 g), cooled at 0 °C, and containing a small amount of water to limit the generation of heat (10 mL, <1% vol/vol of all reagents). After one day of reaction, only 6% of the propylene oxide were consumed, indicating that the amount of water was insufficient to catalyze the reaction (Scheme 5). Thus, 90 mL of water was added dropwise (equivalent of 8% vol/total volume), and the reaction was complete after five days (Scheme 8). The 1H NMR analysis of the reaction medium confirms that all propylene oxide is converted to aminols 2 and 3 with a ratio of 78:22. After distillation under 180 mbar of the reaction mixture, the unreacted wet isobutylamine was first recovered at RT, followed by nearly 0.5 kg of the pure amino alcohol 2 at 114 °C as a colorless liquid. A second distillation at 166 °C under 180 mbar led to a third fraction of 223 g containing 89% of aminodiol 3 contaminated by 11% of 2.
 | ||
| Scheme 8 Large scale synthesis of aminols 2 and 3. | ||
As previously mentioned, propylene oxide is problematic for industrial applications. First, it is a highly flammable liquid with a flash point of −37 °C (i.e., of category 1 according to the United Nations globally harmonized system (GHS)). Second, its vapor forms explosive mixtures with air, at room temperature, within the limits from 1.9 to 38.8% volume, leading to a significant explosive atmosphere risk. Third, it is a carcinogenic compound (category 1B, i.e., potentially harmful to humans). Therefore, to limit those fire and chemical hazards in the preparation of oxazolidinones, at a large scale, we looked for another synthetic route, replacing propylene oxide by a less dangerous reagent such as its 1-chloropropan-2-ol precursor, which exhibits lower flammability, higher flash point (52 °C vs. −37 °C) and limited cytotoxicity.30
 | ||
| Scheme 9 PO formation from chloropropanol isomers in basic medium. | ||
The experimental conditions were also modified to ease the purification step by postponing the thermal treatment before the addition of a strong base. Therefore, a preliminary experiment was performed at room temperature by directly mixing chloropropanol with a slight excess of KOH in water. Since the reaction is suitable for both chloropropanol isomers, it was made from commercial blends. Monitoring the reaction mixture by 1H NMR unveils the formation of PO, within 60% conversion, after only a few minutes (Scheme 9).
At 0 °C, a similar reaction was performed with 2 equiv. of isobutylamine and 1 equiv. of chloropropanol added dropwise. First, N-isobutyl-isopropanolammonium chloride was formed, as evidenced by the presence of a small amount of white smoke. Then, a slight excess of KOH solution was slowly introduced (Scheme 10). The reaction was followed by 1H NMR, which shows that after 7 hours, 69% of chloropropanol was converted to a mixture of 81%, 5% and 14% of amino alcohol 2, amino-diol 3 and PO, respectively. The complete conversion of chloropropanol and propylene oxide was achieved after 22 hours with the ratio of 2 and 3 equal to 86:14 (Table 1, entries 1 and 2).
 | ||
| Scheme 10 Bio-based synthesis of amino alcohols with PO in situ formation. | ||
| Entry | KOH aq. (equiv. mol) | Time (h) | Conversion of chloropropanol (in mol%) | Selectivity (in mol%) | ||
|---|---|---|---|---|---|---|
| 2 | 3 | PO | ||||
| a Experimental conditions: chloropropanol (58 mmol), isobutylamine (116 mmol), 0 °C to RT. Conversion and selectivity determined by 1H NMR spectroscopy. | ||||||
| 1 | 1.1 | 7 | 69 | 81 | 5 | 14 |
| 2 | 1.1 | 22 | 86 | 94 | 6 | 0 |
| 3 | 1.57 | 8 | 100 | 91 | 9 | 0 |
| 4 | 1.41 | 6 | 100 | 92 | 8 | 0 |
| 5 | 1.29 | 24 | 98 | 91 | 9 | 0 |
The conditions of the reaction were then optimized: first, increasing the amount of KOH from 1.10 to 1.57 equiv. allows for complete conversion after 8 h instead of 22 h and the ratio of 2 and 3 was enhanced to 94:6 (Table 1, entries 2 and 3). It is worth noting that both chloropropanol isomers were totally converted even though 1-chloropropan-2-ol reacts much more rapidly than 2-chloropropan-1-ol. Indeed, the initial 1-chloropropan-2-ol/2-chloropropan-1-ol ratio of 3.76 (79% and 21% of each isomer) becomes ten times lower and equal to 0.37 (27% and 73% of each isomer) after 7 hours and ends at 0.15 (13% and 87% of each isomer) after 22 hours. We found that 1.41 equivalent of KOH solution also decreases the reaction time down to only 6 h while a lower amount of 1.29 equivalent of KOH reverses this trend dramatically (Table 1, entries 4 and 5).
These experiments clearly show that the addition of an excess of KOH decreases the reaction time (spanning from 6 and 8 h) to quantitatively produce the amino-alcohols 2 and 3, with high selectivity (92%) toward the formation of 2. After filtration to remove the excess of KOH, 2 is easily isolated, with 75% yield, by distillation under a reduced pressure. Prior to adopting these conditions, we made two minor changes to minimize the cost and the chemical risk of the reaction: first, sodium hydroxide was replaced by potassium hydroxide and then chloropropanol was added dropwise to keep the aqueous solution as cold as possible and prevent the release of gaseous propylene oxide. A large-scale synthesis was achieved by stirring for 24 h at 0 °C, and then at RT, 550 g of chloropropanol, 850 g of isobutylamine and 326 g of NaOH in water (Scheme 11). The 1H NMR analysis of the crude product features the presence of 2 and 3, exclusively, with a ratio of 92:8. Further separation by distillation between 100 and 120 °C under 180 mbar, leads to 647 grams of pure 2, corresponding to a yield of 85%. Interestingly, amino-diol 3 can be recovered and valorized as an intermediate in polyurethanes production after condensation with sebacic (also named decanoic) acid.33
 | ||
| Scheme 11 Large-scale synthesis of the bio-based amino-alcohol 2. | ||
Importantly, our scale-up synthesis is propylene oxide-free, exhibits good yields of amino-alcohols and excellent selectivity toward isomer 2.
Large scale synthesis of oxazolidinones and their physicochemical properties
As mentioned in the introduction, we demonstrated that the bio-based imidazolium hydrogen carbonate salt 4 or carbonate 5 is a catalyst capable of converting non-activated amino-alcohols to oxazolidinones in a selective manner and with high yields.19,27 In addition, it is easily recovered and recycled at the end of the runs without any loss of catalytic performance. Thus, 4 was selected for the large-scale synthesis of N-isobutyl-5-methyloxazolidinone (1). A reaction between 0.75 kg of amino alcohol 2 and 0.94 kg of diethyl carbonate was performed at 90 °C in the presence of 2 mol% of 4. The ethanol formed during the reaction was distilled in real time. The desired oxazolidinone 1 was obtained with 66% yield (equivalent to half kilogram) after distillation under reduced pressure (Scheme 12). Notably, the distillation of diethyl carbonate and ethanol in excess and the recovery of the catalyst by filtration increase the recyclability of the system, also reducing the presence of organic wastes. | ||
| Scheme 12 Large-scale synthesis of bio-based oxazolidinone 1. | ||
Another large-scale synthesis of 1 was possible in the presence of 1,3-dimethyl imidazolium carboxylate (5) (10 g, 2 mol%)27 starting from 740 g of N-isobutyl-5-methyloxazolidinone and 640 g of diethyl carbonate. It was performed at 108 °C for 5 h, providing 94% yield of oxazolidinone with same selectivity. However, catalyst 5 was petro-sourced.
Comparison of the two methods on the environmental impact
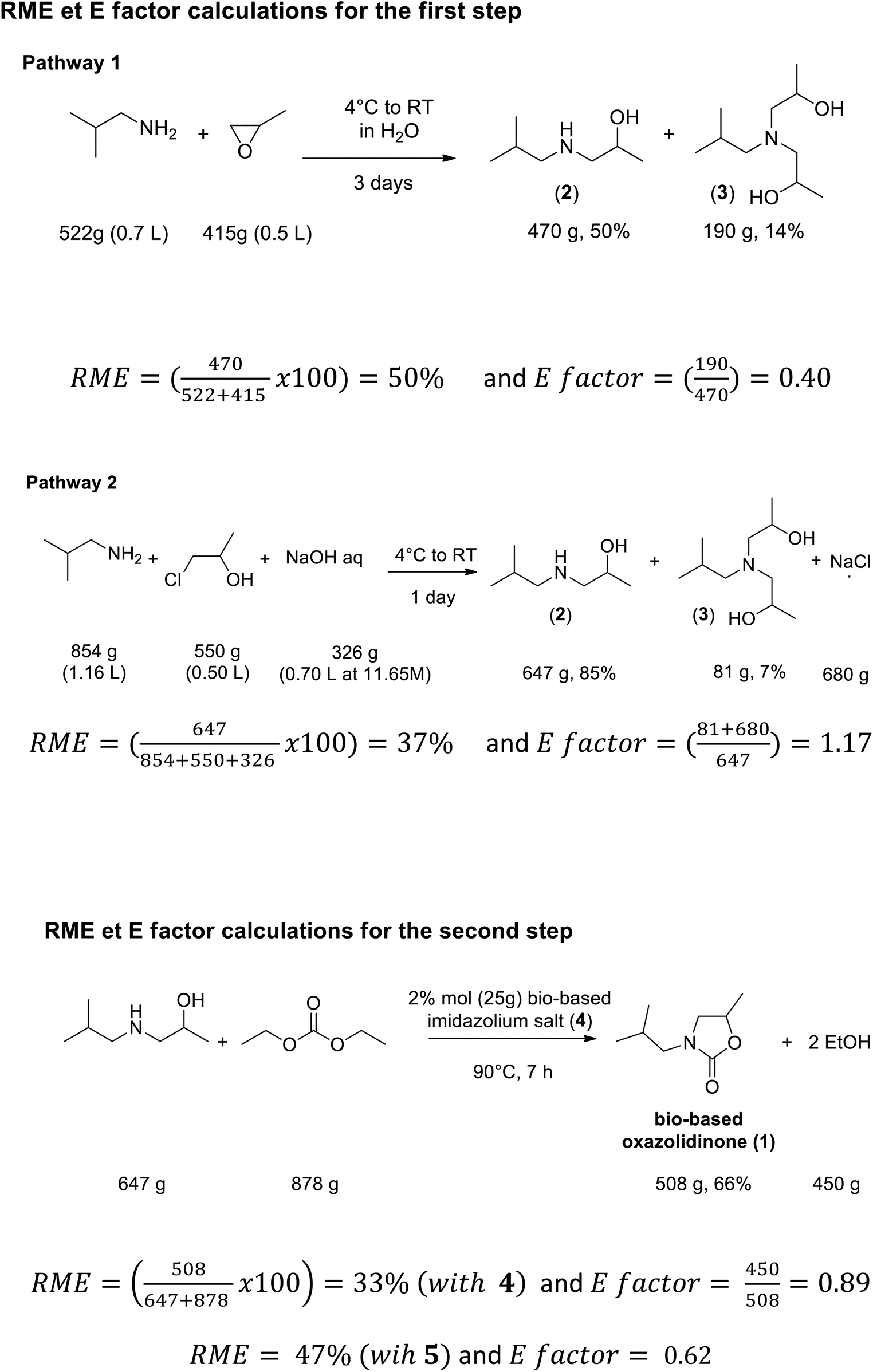
In order to compare the environmental performances of the methods using propylene oxide or chloroethanol, the atom economy AE34 or reaction mass efficiency RME (both reflect the carbon optimization introduced in the chemical process) can be evaluated as well as the environmental factor E (named E factor, which quantifies the amount of by-product or organic waste generated during the process), taking into account the selectivity toward the desired amino-alcohol.However, we prefer to use the RME35 (Reaction Mass Efficiency) instead of AE because it takes into account the yield that is specific to each synthesis and the experimental mass of reactants instead of their molecular weight. Thus, as previously reported, RME appears to be a better metric to compare different “green technologies”.35 Combined with the E factor, both metrics are particularly relevant for identifying chemical or technological eco-innovations and quantifying their environmental and economic impacts. By applying the well-known equations to calculate RME35 and E factor36 with our optimized experimental conditions and stoichiometry (Scheme 7), we find, with PO, a RME of 50% based on the complete conversion of isobutyl amine with the isolated pure product 2 and an E factor of 0.32–0.40 (Scheme 13). With chloroethanol, the synthesis proceeds in two steps. We find an RME equal to 50% for the first step and an E factor equal to 1.17 (Scheme 10 and Table 1) (Scheme 13). For the second step, we find an RME equal to 33% for the bio-based catalyst (4) and an E factor equal to 0.89. Similar synthesis performed with petro-based imidazolium (5) led to RME = 47% and E factor equal to 0.62. These comparative studies on environmental impact show comparable environmental performances between PO vs. chloroethanol in terms of reaction mass efficiency but the last synthetic method (pathway 2) increases the amount of chemical waste by 3 due to the formation of sodium chloride salt. However, chloropropanol is also more interesting when safety criteria come into play because contrary to PO, it exhibits low flammability and weak carcinogenic properties. Based on this fact, we can assume that chloropropanol is a very promising reagent for the large-scale production of bio-based amino-alcohols in compliance with the REACH obligations.
 | ||
| Scheme 13 Environmental performances of different synthetic methods for oxazolidinone. | ||
Oxazolidinones as alternative solvents for (electro)-chemistry
N-Methyloxazolidinone is a solvent widely used in industries. It can replace methylene chloride in synthetic chemistry or removal of paints6 and various organic electrolytes in lithium-ion batteries.7 Because 1 is structurally close to N-methyloxazolidinone and remains undescribed in literature, we wished to determine its potential as the first bio-based, non-toxic and halide-free solvent.For solvents, the critical properties to evaluate are the melting points, boiling temperatures and flash points because they give a range of temperatures for safe handling. We found that oxazolidinone 1 remains liquid at −18 °C and boils at 110 °C under a pressure of 180 mbar. It approximately corresponds to a boiling point of 170 °C under atmospheric pressure.
Flash point corresponds to the temperature at which vapors ignite in the presence of a spark. It makes possible to investigate the flammability risks of any compound and therefore to estimate the potential explosive atmosphere risks. Flash points are determined following the Cleveland method, described by the ISO 2592 standard of 2017.37 We applied this method by gradually heating a small quantity of oxazolidinone 1 in an open aluminum container in the presence of a flame. A slight decomposition occurred above 120 °C. Thus, at room temperature, 1 is non-volatile and non-flammable. The stability of 1 against acids and bases was studied in ethanolic solutions to provide a homogeneous environment and makes chemical degradation easier. After 7 hours under reflux in the presence of HClaq. solution at 0.1 mol L−1, the analysis of the reaction mixture by proton 1H NMR spectroscopy indicates that no degradation occurred. With similar conditions and in the presence of KOHaq. solution at 0.1 mol L−1, the analysis of the reaction mixture unveils the reformation of 5% of starting amino-alcohol 2.
Acetone, ethanol, toluene and xylene are fully miscible with 1 by opposition with water and white spirit (which contains C9–C11 alkanes), leading to two distinct liquid phases. For 1, a density of 0.997 was determined at 23 °C and a viscosity of 5.88 mPa s was measured at 25 °C with a well-known Ubbelohde viscometer.38 This value is comparable to 5.43 mPa s and 2.45 mPa s found with benzyl alcohol and N-methyl oxazolidinone, respectively.38 It is also way larger than 0.89 mPa s and 0.36 mPa s found with water and acetone, respectively. While the dielectric constant of bio-based oxazolidinone 1 was not measured, it is estimated to approximately 80 based on the value found for N-methyl oxazolidinone, which is also equal to the dielectric constants of water39 or ethylene glycol.8 In summary, the physical and chemical properties classify 1 as a solvent: (i) rather fluid, (ii) with a moderated hydrophobic character, (iii) not volatile under standard experimental conditions, (iv) stable in acidic medium, (v) odorless. Such properties might open the route to replace conventional and volatile solvents, limiting the risks of explosion this way. However, toxicologic studies are still missing to ensure that 1 is less toxic that conventional solvents, even though various commercial oxazolidinones (e.g., oxazolidinone, 3-methyloxazolidinone and 4-methyloxazolidinone) exhibit a weak health hazard, being classified as category 4 for their oral toxicity. For comparison, acetonitrile has the same oral toxicity and proven dermal and inhalation toxicities.
A preliminary electrochemical study was also made using cyclic voltammetry to evaluate the redox stability of 1 and its potential to act as an organic solvent or an electrolyte. Indeed, in electrochemistry, solvents are selected based on their available electrochemical window defining their domain of stability against oxidation and reduction potentials. The voltammograms of 1 and acetonitrile were recorded between −4.0 and +4.0 V/SCE using 0.1 M of tetrabutylammonium hexafluorophosphate to ensure electrical conductance of the solutions (Fig. 1).
 | ||
| Fig. 1 CVs of the oxazolidinone vs. acetonitrile, pure or containing 0.1 M TBAPF6 (under Ar, WE: Pt: Ø = 1 mm, ν = 10 mV s−1). | ||
As expected for a non-ionic and organic compound, pure oxazolidinone 1 does not exhibit electrochemical reactivity (red curve) due to a low electric conductivity. Upon the addition of ammonium salt, the CV of 1 (green curve) shows irreversible anodic and cathodic waves with oxidation–reduction potentials Eox and Ered of 2.0 V/SCE and 3.0 V/SCE, respectively. The products formed at extreme potentials were not analyzed. However, the oxidation and reduction peak potentials observed for BMOX are comparable to those measured for commercial N-methyloxazolidinone7 (Eox = 1.7 V/SCE and Ered = −3.0 V/SCE). The redox potentials of acetonitrile were also measured (blue curve) to be Eox = 3.3 V/SCE and Ered = −2.9 V/SCE and found consistent with the data from the literature. The bio-based oxazolidinone 1 is therefore a potential organic electrolyte. Its anodic stability, although slightly higher than that for N-methyloxazolidinone, remains too low for any application in Li-batteries.40 Notably, the stability of 1 could be significantly improved by fluorination or by the addition of another very electronegative atom, like for the reported difluorinated 3-methyloxazolidinone.40
Other bio-based oxazolidinones with weak toxicity and different physical or chemical properties could be synthesized from the amino-alcohol solvent-free protocol starting from available natural amino acids. However, the cost of these new solvents will strongly depend on the availability of the reagents from biomass. A further investigation on the life cycle assessment according to ISO 14040 will be very relevant to evaluate the environmental impact for each of them. With these remarks, only specific properties will be able to meet the expectations of manufacturers and REACH regulations.
On the other hand, the growing complexity of discovering the next generation of green solvents makes it clear that the design of new bio-based solvents should be coupled with advanced computer technologies such as artificial intelligence. Since the earliest database design methodologies by Estévez et al. in 2009,41 the SUSSOL software, developed by Hannes Sels et al. in 2020, helps identify sustainable solvent candidates using neural networks trained on a database of known solvents' physical properties.42,43 However, AI models require high-quality databases of known solvents that contain information about the desired properties such as physical and chemical, olfactive properties, ecological performance data, and economic viability. The data to be fed to AI need to be provided by the community of synthetic chemists and can be found in contributions like ours.
Conclusion
In the current environmental context, new manufacturing processes must be developed keeping their effects on the environment minimal and promoting reusability by preserving the starting material and industrial equipment. Since oxazolidinones already have several industrial or pharmaceutical applications, the quest for their new and more environmentally friendly synthetic process is therefore a necessity. The synthesis of oxazolidinone reported in this work was established from renewable resources (valine from sugar industry). Initially, a synthetic method using isobutylamine and propylene oxide was developed, resulting in the synthesis of the corresponding amino-alcohol with 50% yield on a large scale (around half kilogram). The only side-product of the reaction was the corresponding amino-diol. As propylene oxide is carcinogenic and exhibits a high explosive risk, a second method was developed to generate PO in situ from chloropropanol. This method yielded large quantity (up to kg) of amino-alcohols, with yields up to 85%. They were further engaged with diethyl carbonate and diisobutylimidazolium hydrogen carbonate (4) (as bio-sourced reagent and catalysts) to afford oxazolidinones in good yields.The evaluation of environmental performances of both synthetic protocols showed that using chloroethanol instead of PO was better in terms of green chemical process metrics, such as carbon atom optimization (RME) and undesirable organic waste (E factor). It also complied with REACH obligations and safety criteria when the carcinogenic and highly flammable propylene oxide was removed. Therefore, chloropropanol is a very promising reagent for the large-scale synthesis of bio-based amino-alcohols for producing oxazolidinones at the industrial level. The physical and electrochemical properties of 1 indicate its potential to replace other petroleum-based oxazolidinones that are used in industrial applications. Thus, this neo-green solvent will help to decarbonize industries (by the massive use of carbon from biomass, which is formed by photosynthesis from CO2) and meet the current challenges of ecological transition.
Experimental section
Synthetic procedures
The procedures to convert few grams of L-valine to the corresponding ammonium [iBuNH3](HC2O4)27 and imidazolium salts [iBu2IM](HC2O4) and [iBu2IM](HCO3) were previously reported.20,26 Bulk electrolyses were performed in a one-compartment cell with an Amel 552 potentiostat coupled with an Amel 721 electronic integrator or with an Apelex PS 304 Minipac II. Electrolyses were performed with glassy carbon beaker as the anode and stainless-steel grid or rod as the cathode.Reagents and instrumentation
Acetone (Carlo Erba, 99.8%), acetonitrile (Carlo Erba, HPLC gradient, 99.9%), acetophenone (Alfa Aesar, 99%), 1-chloro-2-propanol (TCI, 75%), diethyl carbonate (Alfa Aesar, 99%), glyoxal (WeylChem Lamotte, 100% biobased produced from bio-ethanol), isobutylamine (Acros), NaOH (Fisher Scientific, 98.5%), oxalic acid (WeylChem Lamotte, 100% biobased, produced from bio-ethanol), paraformaldehyde (Fisher Scientific, 97%), propylene oxide (Acros, 99%), toluene (Carlo Erba, 99.8%), and L-valine (Ajinomoto SAS, 98% purity, 100% biobased, produced from sugar beets). The NMR spectra were recorded on a 300, 500 or 600 MHz Bruker Advance spectrometer. The 1H NMR and 13C{1H} NMR spectra were calibrated to the deuterated solvent on the basis of the relative chemical shift of the solvent as an internal standard. The mass spectra were obtained using a Bruker Micro-ToF Q instrument in the ESI mode. Elemental analyses were performed with a Thermo Electron Flash EA 1112 Series CHNS/O analyzer.Synthesis of isobutylammonium hydrogen oxalate in large scale
The following synthesis is based on the previous preparation of isobutylammonium hydrogen oxalate,27 which was optimized to extend the production from few to hundreds of grams. A suspension of L-valine (200 g, 1.71 mol) in acetophenone (0.80 L, 6.87 mol) was introduced into a two liter flask equipped with a trap-to-trap tube connected to a one liter solution of aqueous oxalic acid at 1.71 M to neutralize vapors formed during the reaction. The reaction media was heated at 125 °C under stirring for 24 hours. After cooling to room temperature, the unreacted valine mixture was isolated by filtration (66 g). The oxalic acid solution was then added to the acetophenone solution under vigorous stirring. The acetophenone solution was separated by decantation and can be reused for a new reaction. The aqueous solution was concentrated by water evaporation using a rotary evaporator. The resulting yellow solid was washed with 250 mL plus 4 × 150 mL of acetone and dried under vacuum. The expected product was obtained as a white solid (126 g, 774 mmol, 45%, 68% based on valine conversion). 1H NMR (D2O): 0.98 (6H, d, J = 6.8 Hz, CH3), 1.94 (1H, hept, J = 6.9 Hz, CH), 2.84 (2H, d, J = 7.1 Hz, CH2). 13C NMR (D2O): 18.8 (CH3), 26.3 (CH), 46.3 (CH2), 165.6 (Cox). HRMS (ESI-MS) m/z calc. for C6H13NO4Na+ [M + Na]+: 186.07368, found: 186.07328, calc. for C10H25N2O4+ [2C + A]+: 237.18088, found: 237.18042. Satisfactory elemental analysis could not be obtained due to the stubborn traces of [NH4](HC2O4).Synthesis of biobased isobutylamine
A solution of isobutylammonium hydrogenoxalate (5 g, 30.67 mmol) and KOH (8.8 g, 157 mmol) in 50 mL of water was heated at 70 °C under stirring. The isobutylamine–water azeotrope was isolated by distillation at the expected temperature of 56 °C. 1H NMR analyses showed only the presence isobutylamine and water. The amine concentration was controlled by dosage with a hydrochloric acid solution at 0.1 mol L−1 (1.93 g, 26.4 mmol, 86%). The NMR data were consistent with those observed for the pure product purchased from Acros.Synthesis of N-isobutyl-isopropanolamine (2) and N-isobutyl-diisopropanolamine (3) in large scale
 A mixture of isobutylamine (854 g, 1.16 L, 11.70 mol) and an aqueous sodium hydroxide solution (326 g, 0.70 L at 11.65 M) was cooled at about 4 °C with an ice bath. A cold pure chloropropanol (550 g, 0.50 L, 5.82 mol) was then added dropwise for 4 hours under vigorous stirring. After stirring for 24 hours, the reaction medium led to a liquid-suspension biphasic system and was analyzed by 1H NMR spectroscopy. The proton spectrum shows the complete conversion of chloropropanol and the formation of 92% of N-isobutyl-isopropanolamine (2) and 8% of N-isobutyl-diisopropanolamine (3). After filtration to remove 680 g of sodium chloride formed during the reaction, the solution mixture was distilled under a vacuum of 180 mbar. The first fraction of 378 g distilled between 60 and 100 °C was an aqueous solution of N-isobutyl-isopropanolamine (2) (271 g, 2.07 mol, 36%). A second fraction distilled between 100 and 120 °C under vacuum (180 mbar) was a colorless liquid identified as pure N-isobutyl-isopropanolamine (2) (376 g, 2.87 mol, 49%). This corresponds to 647 g of N-isobutyl-isopropanolamine, giving a yield of 85%. Distillation at 166 °C under the same vacuum conditions led to N-isobutyl-diisopropanolamine (3) (81 g, contaminated by (2) as previously observed with pathway 1, 0.43 mol, 7%).
A mixture of isobutylamine (854 g, 1.16 L, 11.70 mol) and an aqueous sodium hydroxide solution (326 g, 0.70 L at 11.65 M) was cooled at about 4 °C with an ice bath. A cold pure chloropropanol (550 g, 0.50 L, 5.82 mol) was then added dropwise for 4 hours under vigorous stirring. After stirring for 24 hours, the reaction medium led to a liquid-suspension biphasic system and was analyzed by 1H NMR spectroscopy. The proton spectrum shows the complete conversion of chloropropanol and the formation of 92% of N-isobutyl-isopropanolamine (2) and 8% of N-isobutyl-diisopropanolamine (3). After filtration to remove 680 g of sodium chloride formed during the reaction, the solution mixture was distilled under a vacuum of 180 mbar. The first fraction of 378 g distilled between 60 and 100 °C was an aqueous solution of N-isobutyl-isopropanolamine (2) (271 g, 2.07 mol, 36%). A second fraction distilled between 100 and 120 °C under vacuum (180 mbar) was a colorless liquid identified as pure N-isobutyl-isopropanolamine (2) (376 g, 2.87 mol, 49%). This corresponds to 647 g of N-isobutyl-isopropanolamine, giving a yield of 85%. Distillation at 166 °C under the same vacuum conditions led to N-isobutyl-diisopropanolamine (3) (81 g, contaminated by (2) as previously observed with pathway 1, 0.43 mol, 7%).Diastereoisomers (R,R)/(S,S).
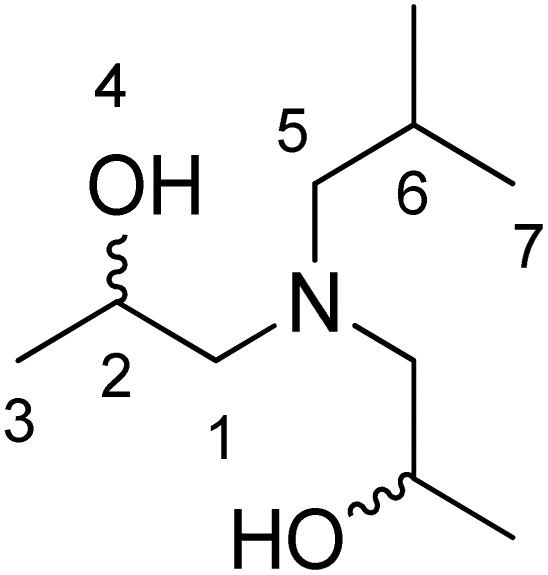 1H NMR (CDCl3): 0.81 (3H, d, J = 6.7 Hz, H7), 0.88 (3H, d, J = 6.6 Hz, H7), 1.05 (6H, d, J = 6.4 Hz, H3), 1.70 (1H, m, H6), 2.12 (1H, dd, J = 9.8 Hz, J = 12.6 Hz, H5), 2.22 (1H, dd, J = 4.9 Hz, J = 12.9 Hz, H5), 2.27 (2H, dd, J = 2.9 Hz, J = 13 Hz, H1), 2.30 (2H, dd, J = 8.7 Hz, J = 12.6 Hz, H1), 3.31 (2H, s, H4), 3.74 (2H, m, H2). 13C NMR (CDCl3): 20.3 (C3), 20.7 (C7), 21.0 (C7), 26.3 (C6), 63.2 (C1), 64.0 (C2), 64.4 (C5).
1H NMR (CDCl3): 0.81 (3H, d, J = 6.7 Hz, H7), 0.88 (3H, d, J = 6.6 Hz, H7), 1.05 (6H, d, J = 6.4 Hz, H3), 1.70 (1H, m, H6), 2.12 (1H, dd, J = 9.8 Hz, J = 12.6 Hz, H5), 2.22 (1H, dd, J = 4.9 Hz, J = 12.9 Hz, H5), 2.27 (2H, dd, J = 2.9 Hz, J = 13 Hz, H1), 2.30 (2H, dd, J = 8.7 Hz, J = 12.6 Hz, H1), 3.31 (2H, s, H4), 3.74 (2H, m, H2). 13C NMR (CDCl3): 20.3 (C3), 20.7 (C7), 21.0 (C7), 26.3 (C6), 63.2 (C1), 64.0 (C2), 64.4 (C5).
Diastereoisomers (S,R)/(R,S). 1H NMR (in CDCl3): 0.83 (6H, d, J = 6.6 Hz, H7), 1.06 (6H, d, J = 6.3 Hz, H3), 1.66 (1H, hept, J = 6.8 Hz, H6), 2.20 (2H, d, J = 7.2 Hz, H5), 2.33 (2H, dd, J = 9.8 Hz, J = 13 Hz, H1), 2.40 (2H, dd, J = 4.3 Hz, J = 13.1 Hz, H1), 3.31 (2H, s, H4), 3.73 (2H, m, H2). 13C NMR (in CDCl3) 20.6 (C3), 20.8 (C7), 26.7 (C6), 64.7 (C1), 65.2 (C5), 65.5 (C2).
For both diastereoisomers. HRMS (ESI-MS) m/z calc. for C10H24NO2+ [M + H]+: 190.18016, found: 190.17952, calc. for C10H23NO2Na+ [M + Na]+: 212.16210, found: 212.16119, calc. for C10H22NO+ [M − OH]+: 172.16959, found: 172.16902. Elemental analysis was consistent with those already reported.33
Synthesis of N-isobutyl-5-methyloxazolidinone (1)
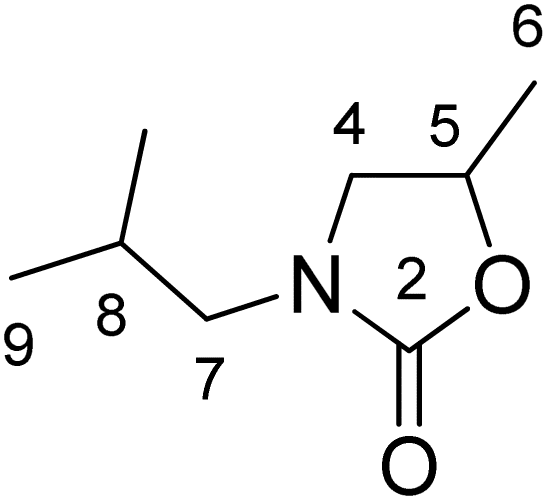 Starting from amino-alcohol aqueous solution: an aqueous solution of N-isobutyl-isopropanolamine (647 g, 4.94 mol), diethyl carbonate (0.90 L, 7.44 mol) and diisobutylimidazolium hydrogen carbonate (25 g, 103 mmol) were introduced into a two liter flask equipped with a distillation column. The reaction mixture was heated at 90 °C for 7 hours, allowing for the removal of ethanol and the diethyl carbonate–water azeotrope (bp = 82 °C as reported)44 formed during the reaction. The undistilled organic phase was separated from water by decantation and reintroduced into a one-liter flask to be heated at 124 °C for 8 hours. The expected product 1 was then isolated as a colorless and odorless liquid by distillation at 112 °C under a vacuum of 180 mbar (508 g, 3.24 mol, 66%). Compound 1 contains 8 carbon atoms and 5 coming from bio-sourced reagents (L-valine plus DMC); as a result, 1 is 62% biobased. 1H NMR (CDCl3): 0.92 (6H, d, J = 6.7 Hz, H9), 1.42 (3H, d, J = 6.2 Hz, H6), 1.87 (1H, hept, J = 6.7 Hz, H8), 3.03 (2H, m, H7), 3.10 (1H, dd, J = 6.8 Hz, J = 1.7 Hz, H4), 3.62 (1H, t, J = 8.4 Hz, H4), 4.62 (1H, m, H5). 13C NMR (CDCl3): 19.9 (C9), 20.9 (C6), 26.9 (C8), 51.7 (C7), 52.0 (C4), 69.9 (C5), 158.4 (C2). HRMS (ESI-MS) m/z calc. for C8H16NO2+ [M + H]+: 158.11756, found: 158.11723, calc. for C8H15NO2Na+ [M + Na]+: 180.09950, found: 180.09904. Elemental analysis for C8H15NO2: calc.: C, 61.12, H, 9.62, N, 8.91, found: C, 61.22, H, 10.47, N, 9.18. d (23 °C) = 0.9974.
Starting from amino-alcohol aqueous solution: an aqueous solution of N-isobutyl-isopropanolamine (647 g, 4.94 mol), diethyl carbonate (0.90 L, 7.44 mol) and diisobutylimidazolium hydrogen carbonate (25 g, 103 mmol) were introduced into a two liter flask equipped with a distillation column. The reaction mixture was heated at 90 °C for 7 hours, allowing for the removal of ethanol and the diethyl carbonate–water azeotrope (bp = 82 °C as reported)44 formed during the reaction. The undistilled organic phase was separated from water by decantation and reintroduced into a one-liter flask to be heated at 124 °C for 8 hours. The expected product 1 was then isolated as a colorless and odorless liquid by distillation at 112 °C under a vacuum of 180 mbar (508 g, 3.24 mol, 66%). Compound 1 contains 8 carbon atoms and 5 coming from bio-sourced reagents (L-valine plus DMC); as a result, 1 is 62% biobased. 1H NMR (CDCl3): 0.92 (6H, d, J = 6.7 Hz, H9), 1.42 (3H, d, J = 6.2 Hz, H6), 1.87 (1H, hept, J = 6.7 Hz, H8), 3.03 (2H, m, H7), 3.10 (1H, dd, J = 6.8 Hz, J = 1.7 Hz, H4), 3.62 (1H, t, J = 8.4 Hz, H4), 4.62 (1H, m, H5). 13C NMR (CDCl3): 19.9 (C9), 20.9 (C6), 26.9 (C8), 51.7 (C7), 52.0 (C4), 69.9 (C5), 158.4 (C2). HRMS (ESI-MS) m/z calc. for C8H16NO2+ [M + H]+: 158.11756, found: 158.11723, calc. for C8H15NO2Na+ [M + Na]+: 180.09950, found: 180.09904. Elemental analysis for C8H15NO2: calc.: C, 61.12, H, 9.62, N, 8.91, found: C, 61.22, H, 10.47, N, 9.18. d (23 °C) = 0.9974.Synthesis of diisobutylimidazolium hydrogen oxalate in large scale
A suspension of isobutylammonium hydrogen oxalate (130.9 g, 925.8 mmol), paraformaldehyde (15.27 g, 509.2 mmol) and glyoxal (64 mL, 561.0 mmol, 40% in water) in half liter of toluene was heated at 130 °C. The water formed during the reaction was removed with a Dean–Stark equipment. When water distillation was achieved, the mixture was cooled down to room temperature and toluene was removed by decantation. The addition of 100 mL of acetone under stirring afforded a brown oil, which led to a solid after the addition of 100 mL of acetone. The solid was filtered, washed with 3 × 100 mL of acetone and dried under vacuum. The expected product was obtained as a beige solid (91.2 g, 337.8 mmol, 84%).1H NMR (D2O): 0.91 (12H, d, J = 6.7 Hz, CH3), 2.14 (2H, hept, J = 6.8 Hz, CH), 4.02 (4H, d, J = 7.2 Hz, CH2), 7.49 (2H, s, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 8.80 (1H, s, NCHN). 13C NMR (D2O): 18.5 (CH3), 28.8 (CH), 56.4 (CH2), 122.7 (CH), 135.6 (NCHN), 164.4 (Cox). HRMS and elemental analysis were consistent with those already reported.26
), 8.80 (1H, s, NCHN). 13C NMR (D2O): 18.5 (CH3), 28.8 (CH), 56.4 (CH2), 122.7 (CH), 135.6 (NCHN), 164.4 (Cox). HRMS and elemental analysis were consistent with those already reported.26
Synthesis of diisobutylimidazolium hydrogen carbonate
), 8.81 (1H, s, NCHN). 13C NMR (D2O): 18.5 (CH3), 28.8 (CH), 56.4 (CH2), 122.6 (CH), 135.3 (t, J = 33.4 Hz, NCHN), 160.1 (HCO3). HRMS (ESI-MS) m/z calc. for C11H21N2+ [C]+: 181.16993, found: 181.16902, calc. for C2H3O3− [A–OH + OCH3]−: 75.0877, found: 75.00843, calc. for C15H27N2O6− [C + 2A–2OH + 2OCH3]−: 331.18746, found: 331.18624. Elemental analyses were consistent with those already reported.26Electrochemistry
All manipulations were performed using Schlenk techniques in an atmosphere of dry oxygen-free argon at room temperature, T = (20 ± 3) °C. The supporting electrolyte TBAPF6 (tetrabutylammonium hexafluorophosphate) was degassed under vacuum before use and then dissolved to a concentration of 0.1 M. Voltammetric analyses were carried out in a standard three-electrode cell, with a PGP 201 Voltamaster potentiostat, connected to an interfaced computer that employed electrochemistry Voltamaster software. The sweep rate was 10 mV s−1. The reference electrode was a saturated calomel electrode (SCE) separated from the analyzed solution by a sintered glass disk filled with the background solution. The auxiliary electrode was a platinum wire separated from the analyzed solution by a sintered glass disk filled with the background solution. For all voltammetric measurements, the working electrode was a platinum electrode disk (Ø = 1 mm), a platinum plate was the counter electrode and saturated calomel electrode was the reference electrode. In these conditions, when operating in CH3CN (0.1 M TBAPF6), the formal potential for the ferrocene (+/0) couple was found to be +0.40 V vs. SCE.Data availability
All data is available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Centre National de la Recherche Scientifique, the Conseil Régional de Bourgogne Franche-Comté (Grant ANER-CASTOR 2024-2027) and the Université de Bourgogne for their financial support. A. H. F. acknowledges a PhD support grant from Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation.References
- S. J. Brickner, D. K. Hutchinson, M. R. Barbachyn, P. R. Manninen, D. A. Ulanowicz, S. A. Garmon, K. C. Grega, S. K. Hendges, D. S. Toops, C. W. Ford and G. E. Zurenko, J. Med. Chem., 1996, 39, 673 CrossRef CAS PubMed.
- G. F. S. Fernandes, C. B. Scarim, S.-H. Kim, J. Wu and D. Castagnolo, Med. Chem., 2023, 14, 823–847 CAS.
- D. J. Diekema and R. N. Jones, Lancet, 2001, 358, 1975 CrossRef CAS.
- N. Kudo, M. Taniguchi, S. Furuta, K. Sato, T. Endo and T. Honma, J. Agric. Food Chem., 1998, 46, 5305 CrossRef CAS.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS.
- K. C. Caster and R. L. Readshaw, US Pat., US4865758A, 1988 Search PubMed.
- M. Ue, K. Ida and S. Mori, J. Electrochem. Soc., 1994, 11, 2989 CrossRef.
- L. Gzara, A. Chagnes, B. Carré, M. Dhahbi and D. Lemordant, J. Power Sources, 2006, 156, 634 CrossRef CAS.
- CEN/TC411, BS EN 16766:2017 Bio-based Solvents, Requirements and Test Methods. Standards Developed by CEN (European Standardization Institute), Technical Committee (TC 411), 2017 Search PubMed.
- N. R. D. d. Souza, M. Groenestege, J. Spekreijse, C. Ribeiro, C. T. Matos, M. Pizzol and F. Cherubini, Wiley Interdiscip. Rev.:Energy Environ., 2024, 13(4), e534 Search PubMed.
- J. Spekreijse, T. Lammens, C. Parisi, T. Ronzon and M. Vis, EUR 29581 EN, JRC112989, Publications Office of the European Union, 2019 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC.
- No 1272/2008, R.E., REGULATION (EC) No 1272/2008 of the European Parliament and Council, Official Journal of the E.U., 2008 Search PubMed.
- G. P. Speranza and W. J. Peppel, J. Org. Chem., 1958, 23(12), 1922 CrossRef CAS.
- A. W. Miller and S. T. Nguyen, Org. Lett., 2004, 6(14), 2301 CrossRef CAS.
- A. Vandekerkhove, L. Claes, F. De Schouwer, C. Van Goethem, I. F. J. Vankelecom, B. Lagrain and D. E. De Vos, Sustainable Chem. Eng., 2018, 6(7), 9218 CrossRef CAS.
- R. Maggi, C. Bertolotti, E. Orlandini, C. Oro, G. Sartoria and M. Selva, Tetrahedron Lett., 2007, 48, 2131 CrossRef CAS.
- Rhodium and its inorganic compounds, The MAK-Collection for Occupational Health and Safety ISSN 2509-2383, Wiley-VCH, 2012, vol. 13, p, p.235 Search PubMed.
- A. H. Fournier, G. de Robillard, C. H. Devillers, L. Plasseraud and J. Andrieu, Eur. J. Org Chem., 2016, 21, 3514 CrossRef.
- J. Andrieu, C. H. Devillers and G. de Robillard, WO2017009578A1, 2017.
- O. Arbelaez, A. Orrego, F. Bustamante and A. L. Villa, Top. Catal., 2012, 55, 668 CrossRef CAS.
- I. Prymak, V. N. Kalevaru, S. Wohlrab and A. Martin, Catal. Sci. Technol., 2015, 5, 2322 RSC.
- D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905 CrossRef CAS.
- P. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720 CrossRef.
- R. Médici, P. D. d. Maria, L. G. Otten and A. J. J. Straathof, Adv. Synth. Catal., 2011, 353, 2369 CrossRef.
- G. De Robillard, A. H. Fournier, H. Cattey, C. H. Devillers and J. Andrieu, Green Chem., 2017, 19, 4912 RSC.
- J. Andrieu, C. H. Devillers and G. de Robillard, WO2016001436A1, 2016.
- K. Pihlaja, K. Aaljoki, M.-R. Lyytinen, M.-L. Huusko and M. Hotokka, ARKIVOC, 2011, 5, 188 Search PubMed.
- J. E. Saavedra, G.-K. Pei and D. W. Farnsworth, Org. Prep. Proced. Int., 1988, 20, 385 CrossRef CAS.
- CPAChemMaterial Safety Data Sheet, https://www.cpachem.com/msds?num=SB39770, ref num. SB39770, safety data sheet according to 1907/2006/EC, article 31, revised june 1st, 2023 Search PubMed.
- W. Fickett, H. K. Garner and H. J. Lucas, J. Am. Chem. Soc., 1951, 73, 5063 CrossRef CAS.
- V. Zacharopoulou, E. S. Vasiliadou and A. A. Lemonidou, Green Chem., 2015, 17, 903 RSC.
- J. L. Boivin, Can. J. Chem., 1958, 36, 1405 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1472 CrossRef PubMed.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- International standard ISO 2592:2017, Petroleum and related products — Determination of flash and fire points — Cleveland open cup method Search PubMed.
- L. Ubbelohde, Ind. Eng. Chem., Anal. Ed., 1937, 9, 85 CrossRef CAS.
- M. D. Jackson and P. G. Sears, J. Chem. Eng. Data, 1979, 24, 199 CrossRef CAS.
- N. Nanbu, K. Hagiyama, M. Takehara, M. Ue and Y. Sasaki, Electrochem, 2010, 78, 450 CrossRef CAS.
- C. Estévez, Sustainable Solutions for Modern Economies, Green Chem. Series, ed. R. Höfer, 2009, vol. 10, p. 407 Search PubMed.
- H. Sels, H. De Smet and J. Geuens, Molecules, 2020, 25, 3037 CrossRef CAS PubMed.
- C. Chen, D. T. Nguyen, S. J. Lee, N. A. Baker, A. S. Karakoti, L. Lauw, C. Owen, K. T. Mueller, B. A. Bilodeau, V. Murugesan and M. Troyer, Condensed Matter - Materials Science, J. Am. Chem. Soc., 2024, 146(29), 20009 CrossRef CAS.
- J. D. Ripoll, S. M. Mejía, M. J. L. Mills and A. L. J Mol, Model, 2015, 21, 93 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |