DOI:
10.1039/D4RA08347D
(Paper)
RSC Adv., 2025,
15, 6593-6633
B@LGTs of Nd, Eu, Er, and Yb lanthanides with physicochemical interfacing for enhanced photocatalytic reduction of fluorescent dyes, transition metal ions, and quinonoid phenolphthalein†
Received
25th November 2024
, Accepted 13th January 2025
First published on 3rd March 2025
Abstract
In this work, lanthanide sulphide nanorods (LSNRs) of neodymium (Nd2S3), europium (Eu2S3), erbium (Er2S3), and ytterbium (Yb2S3) were prepared with a LnCl3·6H2O salt, sodium metal, and H2S gas through a crash reaction methodology (CRM) at NTP. The LSNRs were doped with gadolinium ions (Gd3+) and coated with graphene oxide (GO) to prepare bimetallic LSNRs (B@LSNRs) and GO templates (B@LGTs), respectively. LSNRs, B@LSNRs, and B@LGTs were characterised using XRD, FT-IR spectroscopy, BET analysis, UV/vis spectroscopy, HR-TEM, SEM, TGA/DTG, XPS, Raman spectroscopy, and elemental analysis. The B@LGTs as interstitial photocatalysts photocatalytically reduced the Coomassie brilliant blue red (BBR) dye, transition metal ions (TMIs), and quinonoid phenolphthalein (QHIn) in aqueous solutions under visible light. Experimental parameters including pollutant concentrations, B@LGT dosages, physicochemical properties (PCPs), and pH were optimized for achieving monodispersion and maximum PCR. The PCPs like density, viscosity, sound velocity, surface tension, friccohesity, and isentropic compressibility have predicted the spontaneity and sustainability at 288.15, 298.15, and 310.15 K with photocatalysing medium. B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO with 2.23, 2.28, 2.38, and 1.88 eV bandgaps (Eg) and −670.14, −829.18, −767.39, and −925.57 J mol−1 activation energies (Ea) having −0.9869, 0.8843, −1.4011, and −1.2102 J mol−1 entropies (ΔS) photocatalytically reduced a dye with 96.35, 96.67, 97.60, and 99.17% quantum yields (Φ), respectively. The above-mentioned data indicated that the B@LGTs are robust photocatalysts that photocatalytically reduce BBR in 48 and 30 times shorter duration than LSNRs and B@LSNRs, respectively. It was found that 0.01 g% B@LGTs photocatalytically reduced 40 ppm BBR and 20 ppm CrCl3, NiCl2, CuCl2, and QHIn in 30, 20, 40, 35, and 15 min, respectively. Kinetic rate constants of 3.25 × 10−2, 3.21 × 10−2, 3.18 × 10−2 and 3.15 × 10−2 min−1 for BBR were in the order of B@Nd2S3:GO > B@Er2S3:GO > B@Eu2S3:GO > B@Yb2S3:GO with 4f3e, 4f11e, 4f6e, and 4f13e electrons, respectively. This indicated first-order reaction similar to QHIn and TMIs. Furthermore, the B@LGTs exhibited favourable stabilities, with 58.1–68.05% PCR efficiencies after 10 cycles of reduction experiments.
1. Introduction
Designing quantum materials for emitting phonons of longer wavelengths to harness solar energy1 has been a thrust area in nanoscience and technology for reducing wastewater effluents2 without any sophisticated infrastructure.3 Generally, the effluents discharged from various industries are intrinsically soluble in water and are difficult to separate through mechanical ways due to strong coulombic interactions.4 The phonons noted as negative (e−) and positive (h+) holes of longer wavelengths reach charged centres to counterbalance the chemical linkages5 naturally, enabling separation without the adoption of expensive methodology.6 The over-harnessing of the natural resources poses a serious challenge7 to a sustainable ecosystem.8 Resources such as electricity, fuel,9 and infrastructure are usually used to convert these wastes into second-generation pollutants.10 The wastewater treatment plants, incinerators, resinous filtration, thin films, and landfill technologies have been used for recycling these wastes.7 The discharge of waste dyes and heavy metals from the textile and metallic industrial establishments11 causes nuisance in the environment.12 However, conventional approaches have been on the corrective modes through research activities that are initiated to weaken the interactions of modern-day effluents. Hence, the need for smart photocatalysts has arisen, and their physicochemical properties are applied for wide-ranging applications like adsorption. The wastewater treatment technologies have been indispensable for recycling the wastewater effluents.13 Among several nanomaterials,14 the lanthanide-driven quantum materials have been found to trap the photons (hν) from solar radiation that have generated the negative and positive holes noted as intensive redox cycles (ROCs) to photocatalyze the effluents.15 LSNRs have been effective core nanomaterials to be doped and coated with various functional and photosensitive thermodynamically stable molecules such as GO. However, the synthesis of Ln2S3 at temperatures >800 °C has restricted scalability, limiting their wide applications in photocatalysis16 (Table 1). Their poorly shielded 4f electrons (4f@e) restricted the synthesis at NTP.29 Ln3+ aligns the residual positive charges holding crystalline water strongly as follows:| | |
Ln3+ = 4d105s25p6 (core) 4f@e5d06s0 (free electron)
| (1.0) |
Table 1 Comparative study about lanthanide sulfide synthesis at higher T/K
| Synthesis methods |
Temp. (°C) |
References |
| Hydrothermal |
750 |
17 |
| Hydrothermal |
400–1150 |
18 and 19 |
| Hydrothermal |
800–1300 |
20 and 21 |
| Solvothermal |
750–800 |
22–25 |
| Hydrothermal |
600–1000 |
16, 21 and 26–28 |
| Crash reaction methodology |
NTP |
This work |
The residual positive charge with a stronger Ln3+·6H2O ion-dipole has formed a thick hydration sheath.30 The Ln2S3 synthesis at a high temperature disrupts the crystal growth, agglomeration,31 phase inhomogeneity, impurity, and the particle size and shape.32 This study reports the reduction of LnCl3·6H2O with Na metal for LSNR synthesis at NTP.28,33 The synthesis of LSNRs or their doping with Ln3+ at NTP has not yet been reported (Table 1). At NTP, the reaction kinetics precisely tailors the particle size, shape, and phase composition by minimizing the thermal agitation of atoms for an ordered lattice.34 The Ln2S3 synthesis at NTP is aimed to enhance the optical conductivity, electrical conductivity, and magnetic coercivity untapped by a high temperature.26,30 The B@LGTs with minimum QEB and Eg have avoided electron–electron collisions and electron–hole recombination.35 The B@LGTs with the GO lattice with oxygenated functional groups (FGs) have enhanced the surface continuity that receives the hν to a larger extent and transforms to holes for expeditious photocatalysis without rebouncing.36 The B@LGTs have three interstitial tiers to promulgate the quantum tunnelling effect (QTE) and the least quantum energy barrier (QEB) via friccohesity resonating energy transfer (FRET). The tiers have enabled the B@LGTs for an expeditious PCR of the effluents. A lot of electricity, infrastructure,37 manpower, and water are used in recycling the wastes38 through the conventional methods.39 Thus, the B@LGT photocatalysts, on equipartitioning the potential energy (PE) to kinetic energy (KE), have enhanced the surface area, allowing for the excitation of electrons from the valence band (VB) to the conduction band (CB) upon exposure to solar energy.33 Since 2016 to date, only a few research works have been reported, as given in Table S28.† This is the first reported study, wherein lanthanides are used as dopants. The sulfides of lanthanum or cerium have produced H2 and O2 by splitting wastewater using sunlight or electricity.40 The B@LGTs could intensify the cyclic voltammetry to transform the hν to holes, which seems incomparable to the f-GO reported previously.41 The B@LGTs have split the BBR, TMIs, and QHIn wastewater pollutants discharged from the textile,23 food, paper, printing,42 leather, and cosmetic industries.43 The conjugated water-soluble textile and non-biodegradable Coomassie BBR dyes,12,44 TMIs, and QHIn lower the biological and chemical oxygen demands upon light exposure.33,45 It splits O2 into free radicals30 as BBR + hν → BBR* + O2 → BBR + 2O˙, causing a reactive oxygen stress. The B@LGT with 1.88–2.38 eV Eg could act as a cracking catalyst in the petroleum industry replacing the transition metals.46,47 The B@LGTs with the interstitial tiers of different electronic energies seem to align as a monolayer with adherence for enhancing the frictional force with least surface tension (γ). Therefore, the energy of B@LGTs is integrated with  through the dopant and the GO upon exciting 4f@e (ψ4f) and FGs (ψFGs) seems to transform the hν into the redox energy9 for PCR. The B@LGT s exhibit enhanced surface resonance owing to oxidation (Ln3+ or ψLn3+) and reduction (S2− or ψS2−) potentials aimed at boosting the surface plasmonic resonance (SPR).48 The B@LGTs could advance the thin-film technology, digital ink, quantum dot MRI and tomography22,49 with a contrasting activity caused by Gd3+. The B@LGTs could be used as complementary nanomaterials for spintronics,50,51 electrical and thermal conductivies,33 piezoelectric devices, thermoelectric devices, and biocompatibility.52,53 The B@LGTs for solid oxide fuel cells could serve as the redox catalysts and electrode nanomaterials for enhanced lifespan to split water.54 The anodic B@LGT aligns to a cathodic dye with a higher viscosity and a lower surface tension, favouring the energy storage and supercapacitors.55,56 The higher viscosity with stronger frictional forces and lower surface tension comfortably aligns the B@LGT to develop a stable double layer of the negative and positive charges (Fig. 16). The study of PCPs have acted as the prerequisite database to interface the photocatalysis. The monodispersions of aq-B@LGTs predicted with the PCPs have improved the PCR of the BBR, TMIs, and QHIn on subsequent adsorption.
through the dopant and the GO upon exciting 4f@e (ψ4f) and FGs (ψFGs) seems to transform the hν into the redox energy9 for PCR. The B@LGT s exhibit enhanced surface resonance owing to oxidation (Ln3+ or ψLn3+) and reduction (S2− or ψS2−) potentials aimed at boosting the surface plasmonic resonance (SPR).48 The B@LGTs could advance the thin-film technology, digital ink, quantum dot MRI and tomography22,49 with a contrasting activity caused by Gd3+. The B@LGTs could be used as complementary nanomaterials for spintronics,50,51 electrical and thermal conductivies,33 piezoelectric devices, thermoelectric devices, and biocompatibility.52,53 The B@LGTs for solid oxide fuel cells could serve as the redox catalysts and electrode nanomaterials for enhanced lifespan to split water.54 The anodic B@LGT aligns to a cathodic dye with a higher viscosity and a lower surface tension, favouring the energy storage and supercapacitors.55,56 The higher viscosity with stronger frictional forces and lower surface tension comfortably aligns the B@LGT to develop a stable double layer of the negative and positive charges (Fig. 16). The study of PCPs have acted as the prerequisite database to interface the photocatalysis. The monodispersions of aq-B@LGTs predicted with the PCPs have improved the PCR of the BBR, TMIs, and QHIn on subsequent adsorption.
 |
| | Fig. 1 (a) Photocatalysis interfacing with PCPs. (b) PCR of effluents with time. | |
2. Materials and methods
2.1. Materials
Graphite flake (Gt < 20 mm), H2SO4 (99.9%), KMnO4 (>99.9%), H2O2 (30%), HCl, NaOH, hexahydrate chloride salts of neodymium, europium, erbium, and ytterbium, gadolinium nitrate hexahydrate (>98%), sodium metals (>99%), FeS (98%), BBR, phenolphthalein, CrCl3, NiCl2, and CuCl2 were procured from Sigma-Aldrich. Petroleum ether (40–60) was obtained from Sisco research laboratory and absolute alcohol from SCVUS Mandli India. The solutions were prepared with Milli-Q water.
2.2. Crash reaction methodology (CRM) for LSNR synthesis
LSNRs were synthesised by our previously reported method.33 The hexahydrate of NdCl3, EuCl3, ErCl3, and YbCl3 (0.001 M) was crashed with Na metal (0.006 M) individually by CRM. Nd(OH)2, Eu(OH)2, Er(OH)2, and Yb(OH)2 were produced, along with NaCl, NaOH, and H2 gas as side products. The side products were confirmed with AgNO3, phenolphthalein, and gas chromatography.30 Ln(OH)2 was rinsed with water, centrifuged and dried at 60 °C. Nd(OH)2, Eu(OH)2, Er(OH)2, and Yb(OH)2 were individually mixed with H2O and sonicated for 2 h to monodisperse on testing. The H2S gas was passed (10 min) to monodisperse the lanthanide hydroxides (Fig. 2(I)). The LSNRs were obtained and dried at 50 °C for 12 h in an oven, and the reaction is described as follows:| |
 | (1.1) |
2.3. H2 gas generation
First, 73.17 mL H2 gas was generated from 0.001 M LnCl3·6H2O with 0.006 M Na during CRM. Na has dissociated the crystalline H2O into H2 flame as follows:| |
 | (1.2) |
The n = 6 infers the H2O crystalline molecules of LnCl3 that split to H2 gas that had intensified a flame. Thus, the 2H2 + O2 = 2H2O, ΔH = −572 kJ mol−1 combustion is noted as clean energy process achieved at ∼500 °C through 350 °C + ∼150 °C. The yellow flame was sharpened from NdCl3 to YbCl3 on increasing their 4f electron from 4 to 13 respectively. Na reduced H2O to H2 gas in a shorter duration with YbCl3 of 4f13e than ErCl3. Then 3s1 Na by competing with 4f6e spins detached 6H2O from LnCl3. ErCl3 with 4f11e has turmoiled a flame with H2 clouds (Fig. 2(II)). The sustainable H2 fuel could be used to generate electricity,57 ammonia for fertilizer, and methanol58 and remove sulfur from fuels. H2 with CO2 could be used to synthesise several hydrocarbons for combustion engines.59
 |
| | Fig. 2 (I) CRM for LSNR via CRM at NTP. (II) Energy storage model and H2 Flames. | |
2.4. Synthesis of B@LSNRs
B@LSNRs were synthesised by refluxing aq-Gd(NO3)3 with aq-LSNR in 1/4 ratio at 650 rpm and 90 °C for 12 h. The product was settled and washed with water to remove the undoped Gd3+ followed by centrifugation. The initial filtrate had a decolourised pink colour of QHIn (indicator) owing to formation of HNO3 during B@LSNRs synthesis; however, it was removed after washing 3 times. Excess HNO3 has indicated Gd3+ doping in larger amounts, as revealed by XPS and elemental analysis60 (Fig. 3, 13 and Table 5). The Gd3+ had entered the lattice of the LSNRs and developed its own hydration spheres with those of the LSNRs. That has facilitated a doping due to in situ HNO3 formation with chemical potential (μi) demonstrated as.| | |
Ln2S3 + Gd(NO3)3 → Ln2S3 ≈ Gd3+ (aq-B@LSNR)
| (1.3) |
| |
 | (1.4) |
The wavefunction of H2O monomer (ψH2O) with rotational, vibrational, and translational (rovitronic) motions of Gd(NO3)3 has encapsulated Gd3+ with LSNR, and the activities (ai) are as follows:
| |
 | (1.5) |
Gd
3+ has entered the S
2− and Ln
3+ cavities of LSNRs with Δ
GLSNR ≠ Δ
GNHS zone spontaneities due to different redox potentials of S
2− and Ln
3+. The S
2− of LSNR has enabled a doping with the Gd
3+ on broadening a size of NRs by developing the moderate interstitial van der Waals forces (VWFs) (
Fig. 9).
 |
| | Fig. 3 LSNR doping with Gd3+ for B@LSNR synthesis. | |
2.5. Synthesis of graphene oxide (GO) and B@LGTs
GO was synthesized according to a previous report with slight modifications.61 The Gt flakes and KMnO4 were taken in an RB flask in 1/6 ratio and H2SO4 (100 mL) was poured dropwise keeping on an ice bath with continuous stirring. It was refluxed at 650 rpm at 55 °C in an oil bath for 12 h. Later, cold water and 3 mL H2O2 (30%) were added under stirring for 10 min to obtain a GtO (eqn (1.5)). The resulting product was centrifuged at 8000 rpm for 10 min and washed with normal water, HCl, and absolute alcohol until the pH reaches 7. NaCl and Na2SO4 were removed in 5 sequential washings, which was confirmed by testing with AgNO3 and BaCl2. The GtO mixture on sonication at 28 kHz for 3 h has formed a brownish GtO dispersion on exfoliating to GO sheets62 after the addition of petroleum ether for separation and drying at 60 °C. Brownish GO sheets were reproduced61 (Fig. S1†), and after sonication for 3 h, they were mixed with B@LSNR in 1/6 ratio and refluxed at 650 rpm at 90 °C for 12 h. After 12 h, the resulting dark product was settled and washed with H2O, followed by centrifugation at 8000 rpm for 8 min. The GO sheets could have competitively interlinked with Gd3+ and Ln3+ via S2− or ψS2− of tentropic cavities aligning the B@LGT as astral geometry, as elucidated by HR-TEM (Fig. 9).| |
 | (1.6) |
3. Results and discussion
B@LGTs have transformed the hν into holes of longer wavelengths to photocatalytically reduce the pollutants. The tiers have oscillated in the same phase aligned with the specific NHS of water dipoles shortening the PCR time. The tiers have synchronized the oscillations of each constituent to a single-valued wavefunction by avoiding electron bouncing to expeditiously cross the Fermi energy barrier or QEB. The equipartitioning of the surface charges stabilized to the electrostatic dipoles has been predicted with the Raman spectra with a sharper intensity of D and G bands (Fig. 11). The tier with a least interfacial energy barrier and Ea has enabled the B@LGTs to photocatalytically reduce the effluents. The tier structures have enhanced reusability as the reduced effluents did not coagulate rather adhered to the interstities with moderate VWFs as per Schrödinger and Le Chatelier spontaneities. The B@LGTs favour adherence and coherence by minimizing the collisions for ROCs (Fig. 17). Fig. 4 elucidates the morphology of B@LGTs with the 1st tier for LSNR, 2nd tier for Gd3+, and 3rd tier for GO that have photocatalytically reduced BBR, CrCl3, NiCl2, CuCl2, and QHIn in 30, 20, 40, 35, and 15 min, respectively. The PCPs and thermodynamic parameters have predicted a role of each tier enhancing the sustainable photocatalytic activities. The B@LGTs as superphotocatalysts or photomultipliers on monolayer adhesion with LSNR–Gd3+–GO interfacial templates photocatalytically reduced the effluents in 15–40 min with 90–99.8% Φ (Fig. 19 and 22). The Φ value from 4f3e to 4f13e (x-axis) is polynomially increased with a linear decrease in Ea, while Eg initially increased from 4f3e to 4f11e, but from 4f11e to 4f13e, it is decreased (Fig. 1a). The increasing Φ inferred an access of phonons to BBR on decreasing Ea. The B@LGTs have acted as anodic site to receive the hv for exciting the VB electrons to CB on storing the solar energy. The e− and h+ holes have access to cathodic BBR and others with a longer λ. Fig. 1a predicts the robust anodic and cathodic interfacing for harnessing solar energy via 4f3e to 4f13e for photocatalytic reduction.
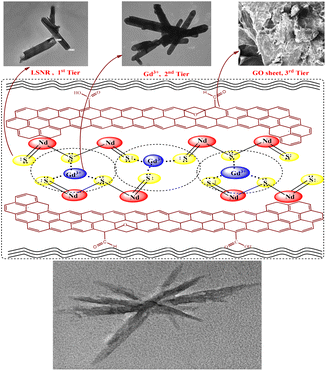 |
| | Fig. 4 B@LGT with three-tier interstities in a same phase for higher functionality. | |
3.1. Temperature-dependent PCPs as PCR sensors
The temperature-dependent surface properties of 0.01 g% B@LGTs at 288.15, 298.15, and 310.15 K have predicted the monodispersion, while the regression coefficients of PCP data from 288.15 to 310.15 K were used to calculate the respective PCPs at 283.15 and 313.15 K. The temperature variation has inferred a thermal stability of aq-B@LGTs on absorbing a maximum hv at 298.15 K than others. At 298.15 K, an infinitesimal decrease in surface tension indicates monodisperse B@LGTs for hv capturing (Fig. 19). No coagulation, no dimerization, and no clustering with least QEB have optimized the PCR at 298.15 K. The T/K below and above 298.15 K is being pursued for determining the temperature dependence of PCR and physisorption activities of B@LGT for reusability. The surface tension (γ), viscosity (η), friccohesity (σ), density (ρ), and isentropic compressibility have predicted the monodispersion for ordering the e− and h+ holes w.r.t. the anodic B@LGT and cathodic effluents. The tier-like structure of the B@LGT with a higher surface area and a moderate surface tension acted as a superphotocatalyst because a longer λ opted to the quantum tunnelling effect. The lower Ea, ΔH, ΔG, and ΔS values have predicted the spontaneous reorienting activities of the B@LGT to receive the hν. Lower γ with higher η values for aq-B@LGT than aq-LSNR and B@LSNR at 298.15 K have inferred on aligning the hν receiving dipoles. The BBR, TMIs, and QHIn were photocatalytically reduced for the first time to recycle wastewater. The PCPs and thermodynamic parameters of B@LGTs have acted as a prerequisite database for receiving the hv consistently without coagulation. The hv receiving sites get reoriented to bulk with a higher γ value, while the lower γ with higher η values align at surfaces (Fig. 18 and 19). γ and η both interfaced via σ have depicted the spatial orientation of B@LGTs aligning along the effluents in situ. The B@LGTs have absorbed the maximum hv, intensifying the ROCs at 298.15 K with surface continuity due to a lower surface tension with a higher viscosity for higher Φ. The fraction of occupied Fermi surface (θ) with a vacant surface (1 − θ) have facilitated to receive the hν and also a transfer of holes to the effluents as k1(1 − θ)hν = k2θ steady state.63 The k1 and k2 values infer the hν receptance and transfer of holes in the first order with energy distribution for generating the holes as follows:| |
 | (1.7) |
‘n’ refers to the number of energy ensembles, total energy NE and ne could have been NE ≈ ∑ne, equipartitioned (eqn (1.7)). The receptance of the least numbers of hν excited the functional sites of B@LGT for a higher surface energy (KE ≈ PE) for PCR as follows:| |
 | (1.8) |
The B@LGT due to aligned64 interstities generated holes with ψ interfacing 4f@e along Ln3+ and S2− oscillating with the wavefunctions (ψLn3+) and (ψS2−) as ψLn3+ ≠ ψS2− (eqn (1.8)). |ψLn3+| × |ψS2−| ≠ 0 of LGT reduced the dyes in 60 min, as reported previously.33 The oscillating charges (ψLn3+ ≠ ψS2−) of B@LGT have received hν with a higher relaxation time (eqn (1.9)). The minimum internal entropy (ds/dt) and topographical entropy (tentropy, ΔST) seem to equally spin the electrons of B@LGT at position ‘x’ nm at time ‘t’ with V(x, t) PE. The GO ≈ NHS and GO ≈ LSNR linkages of aq-B@LGTs spin in the same phase with entropy as follows:
| | |
ΔSB@LGT ± ΔSNHS ≈ ΔSB@LGT–NHS, ΔSB@LGT ± ΔSNHS ≤ 0 and ΔSB@LGT–NHS
| (1.9) |
The ΔSB@LGT ± ΔSNHS > 0 > ΔSB@LGT ± ΔSNHS trends have weakened and strengthened the B@LGT ≈ NHS out of B@LGT ≈ B@LGT and NHS ≈ NHS linkages. Higher friccohesity infers the ψB@LGT–NHS(x, t) at ‘x’ ≠ 0 position between B@LGT and NHS at ‘t’ relaxation time in the same phase (φ). Thermograms have also predicted a streamlining of thin-layer 2D whitish laces with energy as follows:
| | |
EB@LGT–NHS = ECorner + EFGs at opposite plane + EFGs at width
| (2.0) |
Eqn (2.0) infers a plane area being occupied from the corner and width of aq-B@LGT upon increasing the γ values and decreasing the η values (Fig. 18 and 19). Aq-B@LGTs with monolayer NHS adhesion moderately align. The water dipoles aligned with core LSNRs, dopant, and GO of B@LGTs to communicate mutually with comparatively high γ values (Fig. 9). The B@LGTs with lower Eg could upgrade nano-thin films,65 fibres, optics, catheters, optoelectronics,66 digital inks, and nanomaterials.67 The B@LGT could be doped with I2 of induced dipoles with the lowest reduction potential widening energy domains68 similar to our earlier study33 on LSNR doped with GO and without doping (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). This deals with the coating of B@LSNR with GO unlike LGT for B@LGTs. The Oδ− and 2Hδ+ of H2O with metallic (TMI, Ln3+) dopants lowered the Eg solar energy.61,69 The B@LGTs had aligned the tentropic cavities upon the scintillation of electrons from the VB to the CB as |ψDipole–DOS|2 > ψDipole ≈ ψDOS hybridizing |ψDipole–DOS|2 ≈ 80%. BET has predicted the stabilized dipoles with lower Eg and higher surface area (Fig. 6). Terminals of S
:4). This deals with the coating of B@LSNR with GO unlike LGT for B@LGTs. The Oδ− and 2Hδ+ of H2O with metallic (TMI, Ln3+) dopants lowered the Eg solar energy.61,69 The B@LGTs had aligned the tentropic cavities upon the scintillation of electrons from the VB to the CB as |ψDipole–DOS|2 > ψDipole ≈ ψDOS hybridizing |ψDipole–DOS|2 ≈ 80%. BET has predicted the stabilized dipoles with lower Eg and higher surface area (Fig. 6). Terminals of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ln–S–LnS aligned via ψS2− and ψLn3+ eigenenergy with the NHS zone (Fig. 4). The ψGd3+, ψS2−, and ψLn3+ of B@LGT in continuity had a higher viscous flow time (vft) and surface energies as the tiers also adhere the water dipoles with a higher shear stress to stabilising the holes (Tables 11–13).
Ln–S–LnS aligned via ψS2− and ψLn3+ eigenenergy with the NHS zone (Fig. 4). The ψGd3+, ψS2−, and ψLn3+ of B@LGT in continuity had a higher viscous flow time (vft) and surface energies as the tiers also adhere the water dipoles with a higher shear stress to stabilising the holes (Tables 11–13).
3.2. Water binding and heat holding abilities of B@LGTs for PCR
The 0.002 g% aq-Gd@Nd2S3-GO was boiled in 45 min at 103 °C@5 °C than the same amount of water in 25 min at 100 °C@5 °C due to stronger interaction of Gd@Nd2S3-GO. It was found that aq-Gd@Nd2S3-GO has acquired an extra heat at 682.543 kJ °C min−1 at a sharper boiling point than that of water alone. The water dipoles adhered to interstities on aligning more strongly than the hydrogen bonding of water (Fig. 5). Their constituents with water dipoles develop oscillating interfaces that detain higher heat than water alone and predict the energy storing ability of the B@LGTs. The heat holding ability of aq-Gd@Nd2S3-GO is useful for various thermal applications in biophysical, biochemical, and biothermodynamic fields. The stronger heat holding capacity is also attained on stronger GO binding depicted with TGA and BET (Fig. 6 and 15). It has intensified photoluminescence (PL) due to ψ in single phase by darkening a colour on stacking by producing nanoclusters of similar sizes with sharper emittance after 2 h at RT (Fig. 5c). On heating, the water molecules got escaped from interstities to phase out nanoclustered sheets. The monodispersion with ηaq-B@LGT < ηH2O and γaq-B@LGT < γH2O has caused PL without rebouncing the hν(Fig. 5c, 18, and 19). The aq-B@LGTs were realigned to a hydrophilic head and a hydrophobic tail with brighter PL70,71 on forming the symmetric bipolar electrostatic stabilisation due to needful η and comparatively lower γ values. With the needful Nernst potential, 4f@e and π-conjugation enhanced the photocatalytic activity61 (Fig. 5d). The thermal energy generated PL, as [O] was unable to functionalize all the π-conjugated GO and responded differently upon heating as follows:| |
 | (2.1) |
H2O dipoles with the wavevector of Gd3+ 4f7e within SLn–S–LnS have aligned to intrinsic tentropic cavities (ΔST) correlated with the moment of inertia (I) as follows:| |
 | (2.2) |
ν is the vibrational frequency of the bond based on atomic mass, υ the vibrational quantum number of electrons, m the atomic mass and l the length of NRs. The rotational activities with the hν could shorten PCR time on mLnlLn2 ≠ mSlS2 ≠ mGdlGd2. The alignment of solvated B@LGT stabilised the chemical bond vibrational frequency vis-a-vis the vibrational quantum number of 4f electrons. The aq-Gd@Nd2S3-GO had generated a whitish PL on storing energy with similar wavevector (K-space) without causing electronic collisions. This was attained on distributing the hν energy within the interstitial constituents of Gd@Nd2S3-GO through the energy distributing factor 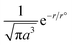 , the r°, Bohr and r, radii for lattice ‘a’. The B@LGT combined with aq-BBR, TMIs, and QHIn to boost the PL (Fig. 16). B@LGT at r > r° receives hν with least permittivity (ε) and mutually shares it with effluents, as predicted via PCPs. The r = r° infers no reduction and no PCPs without ψ1 ≠ ψ2 ≠ ψ3… ≠ ψn conjugation.72 Fig. 5 depicts the single lattice elongating a wavelength73 on thinner 2D GO sheets similar to those reported previously.61 XRD, HR-TEM, and BET predicted that the synchronized electronic clouds of dopants, and coating agents of B@LGT have intimately hybridized without twisting on aligning interstities similar to their individual angular momentums. The La2S3, Ce2S3, Tb2S3, and Ho2S3 as core LGTs as the two tiers have slightly modulated the electrostatic interacting activities of aq-BSA protein with mild unfolding dynamics reported in an earlier study.33 Noticeably, polyvinylpyrrolidone (PVP)-capped rod-shaped europium (Eu3+)-doped gadolinium oxide (Gd2O3) nanoparticles (PVP@Gd2O3:Eu3+ NPs) have been effective to tune electrostatic charges of salt bridges of proteins.24 Thereby, the B@LGTs could further elaborate the charges to intensify the interacting activities with the peptides and intercalate the base pairs of dsDNA which are being pursued. The aligned B@LGTs have generated the e− and h+ holes of a longer mean free path that photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn within 15–40 min (Fig. 22).
, the r°, Bohr and r, radii for lattice ‘a’. The B@LGT combined with aq-BBR, TMIs, and QHIn to boost the PL (Fig. 16). B@LGT at r > r° receives hν with least permittivity (ε) and mutually shares it with effluents, as predicted via PCPs. The r = r° infers no reduction and no PCPs without ψ1 ≠ ψ2 ≠ ψ3… ≠ ψn conjugation.72 Fig. 5 depicts the single lattice elongating a wavelength73 on thinner 2D GO sheets similar to those reported previously.61 XRD, HR-TEM, and BET predicted that the synchronized electronic clouds of dopants, and coating agents of B@LGT have intimately hybridized without twisting on aligning interstities similar to their individual angular momentums. The La2S3, Ce2S3, Tb2S3, and Ho2S3 as core LGTs as the two tiers have slightly modulated the electrostatic interacting activities of aq-BSA protein with mild unfolding dynamics reported in an earlier study.33 Noticeably, polyvinylpyrrolidone (PVP)-capped rod-shaped europium (Eu3+)-doped gadolinium oxide (Gd2O3) nanoparticles (PVP@Gd2O3:Eu3+ NPs) have been effective to tune electrostatic charges of salt bridges of proteins.24 Thereby, the B@LGTs could further elaborate the charges to intensify the interacting activities with the peptides and intercalate the base pairs of dsDNA which are being pursued. The aligned B@LGTs have generated the e− and h+ holes of a longer mean free path that photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn within 15–40 min (Fig. 22).
 |
| | Fig. 5 (a–c) Thermally nucleated photoluminescence analysis on heating the 0.002 g% of aq-Gd@Nd2S3-GO for elucidating thermal stability. (d) HR-TEM image of B:Nd2S3-GO. | |
3.3. Brunauer–Emmett–Teller (BET) analysis
With the BET technique, a solid material measured the adsorption of inert gases such as N2 upon increasing the relative pressure (P/P0) at NTP. Thus, the amounts of adsorbed gas by LSNR, B@LSNR, and B@LGT of Nd2S3 determined their surface area, pore volume, and pore size at 77 K (Fig. 6). B:Nd2S3@GO adsorbed a higher amount of N2 and also desorbed at the same rate. The similar rates infer the distribution of the similar sized pores of similar dimensions with 100% physisorption. The higher surface area, larger pore size, and pore volume and physisorption enabled the use of B@LGTs for 10 fresh dye samples of 40 ppm for PCR with 10 times reusability. Each time, the B@LGTs were washed, used for adsorbing the reduced effluents and desorbed for subsequent cycles. The N2 adsorptions with B:Nd2S3@GO are fitted with standard isotherms as per the IUPAC classification.9 The similar rates of adsorption and desorption with B@LGTs have predicted the mesopores of sharper and broader edges.42 Nd2S3 and B@Nd2S3 of 13.131 and 15.80 m2 g−1 surface area with 0.009 and 0.019 cc g−1 pore volume respectively indicate a lower N2 adsorption than B@LGTs due to coating. The coating seems to have transformed the cohesive to adhesive forces with two different centres of electronic energy well that exchanged their energies through FRET. Thus, a surface area of B:Nd2S3@GO has been increased with higher hν receiving and e− and h+ hole generating abilities. The 2D GO sheets might have equipartitioned their energies normalizing the pore sizes than the uncoated. The 61.480 m2 g−1 area of B:Nd2S3@GO is higher than Nd2S3, and B@Nd2S3 by 4.60, and 3.89 times respectively supported with SEM and HR-TEM (Fig. 9 and 14). Therefore, this research opens the novel gateways to choose several coating agents to enhance the surface area. The Barrett–Joyner–Halenda and Villarroel–Barrera–Sapag (VBS) methods have been fitted for their pore size distribution with monolayer to multilayer N2 adsorption at a lower pressure with ∼2–50 nm pore diameter (Fig. 6). The first-order adsorption to desorption with B:Nd2S3@GO with almost the same slope values has inferred equal distribution of residual charges for equally reorienting the pore sizes uniformly without much compression at high pressures and low temperatures (Fig. 6a and b). The compressibility and pore volume as aq-B:Nd2S3@GO > GO > B@Nd2S3 > Nd2S3 infer a higher surface energy with B:Nd2S3@GO for expeditious access to BBR, TMIs, and QHIn (Tables S26 and S27†). Thus, the B@LGT could be extended to several other wastewater material to recycle them through the PCR and then with a subsequent adsorption. B:Nd2S3@GO with a longer wavelength (λ) of holes overcomes a QEB than others. B:Nd2S3@GO is reusable for five cycles to harness the hν as hν + B@LGT = B@LGT* + (e−, h+)* → PCR supported with PCPs (Fig. 17). The slopes as B:Nd2S3@GO < B@Nd2S3 < Nd2S3 < GO have inferred stronger N2 absorbing ability with B:Nd2S3@GO than with GO (Fig. 6e). The least N2 adsorption rate with GO infers stronger VWFs at constant temperatures and pressures at fixed compositions. Thus, similar to N2 entry to the interstities, the water dipoles were also entered that had equipartitioned the potential to kinetic energy.
 |
| | Fig. 6 (a and b) Isotherms and (c and d) pore size of Nd2S3, B@Nd2S3, GO, and B:Nd2S3@GO. (e) Slopes obtained during the adsorption of N2 by B:Nd2S3@GO. | |
3.4. UV/vis analysis
Aq-LSNRs have consolidated the transitions at λmax = 214 nm with abs = 0.4 unlike λmax = 302 nm and abs = 0.6–0.9 for B@LSNR while Gd3+ has caused a transition on a dive in abs = 0.32–0.48 at λmax = 268 nm. The dive abs infers engaging the electrostatic Ln3+ and S2− domains by weakening the transitions of B@LSNR at λmax = 268 nm, and it is elucidated by morphology and BET analysis (Fig. 6 and 9). Abs = 0.32–0.48 and 0.24–0.56 diffused transitions at 228 and 312 nm for GO and B@LGT on π → π* and n → π* respectively indicate a longer wavelength with B@LGT. Thus, the n → π* energies have been further enrooted to the dopant and LSNRs in the same phase to enhance the resulting λ. The coating has diffused the transitions within 300–475 nm λ with abs = 0.10–0.34 on uniform GO sheet distribution with B@LSNR. The B@LGTs with a similar lattice have indicated an interlink with Gd3+ on elongating the wavelength compared to B@LSNRs (Fig. 7c). The B@LSNR with the dopant aligned the water dipoles without leaching out the dopant and coating agents on sonication. The quantum mechanically flexible GO with the quantized energies of FE-LSNR, FG-LSNR, FE-Gd-LSNR, and FG-Gd-LSNR each might have developed uncounted ensembles. The B@LSNRs have integrated the energies to eigenenergy with a single-valued wavefunction (ψsv). The π → π* transition for CC and n → π* for C–O–C of GO have intensified the distinguished activities.74 E228 = (hc/228) and E312 = (hc/312) from 228 to 312 nm with B@LGT integrated the holes on VB → CB to photocatalytically reduce BBR in 30 min. The B@LGTs have aligned the H2O dipoles in their interstities with a higher surface tension and longer λ than GO. Absorbance as Yb2S3 > Nd2S3 > Er2S3 > Eu2S3 with 4f13e, 4f3e, 4f11e, and 4f6e respectively was occurred at λmax = 214 nm. That had predicted the similar fermi energy levels for the Yb2S3, Nd2S3, Er2S3, and Eu2S3 because their electrons are filled in 4f orbitals. It differs from 300–500 nm with B@LGTs as the electrons of dopant and FGs of coating agent have shared their angular momentum. That have shifted the λmax = 300–500 nm to absorb energy from sunlight (Fig. 7d). The Yb2S3 could have detained a higher energy than Nd2S3. Their 4fe could not affect the λmax = 214 nm except absorption, as their energy levels remain the same except the degeneracies. The 4f13e and 4f3e shared by S2− of LSNR matrices75 synchronized the surface area of tentropic cavities (Fig. 7c). Thus, the abs as B@Yb2S3 > B@Nd2S3 > B@Er2S3 > B@Eu2S3 have shortened the PCR time. The B@LGTs with CC have induced the π → π* transition at λmax = 228 nm compared to LSNR at λmax = 214 nm and B@LSNR at λmax = 302 nm that have realigned the ψ as follows:| |
 | (2.3) |
The p2/2m, KE and q+q−/4πεr2 are PEs.76. |ψ|B@LGT2 > |ψ|2 < |ψ|dye2 ≠ |ψ|B@LGT2 ≈ |ψ|2 ≈ |ψ|dye2 did not photocatalytically reduce compared to |ψ|B@LGT2 ≈ |ψ|2 ≈ |ψ|dye2 at 312 nm, which photocatalytically reduced in a shorter time. The B@LSNR has produced the new peaks at λmax = 268 and 302 nm than 214 nm with the bare LSNR due to energy equipartitioning on doping (Fig. 7b). Thus, the doping interstities also gained a geometry with their own energies. B@LSNR268 nm − LSNR214 nm = 3.6 × 10−25 J is the doping energy on streamlining 4f@e. Gd3+ has reduced Eg with longer λmax, the Gd3+⋯S2− attraction and Gd3+⋯Ln3+ repulsion might have synchronized the holes at λmax = 302 nm with B@LSNR302 nm − LSNR214 nm = 2.25 × 10−25 J, for a shorter PCR time (Fig. 7b).
 |
| | Fig. 7 UV/vis absorption spectra of (a) LSNR, (b) B@LSNR and (c) B@LGT. | |
λmax = 228for B@LGT, 214 nm for the LSNR, while 268 and 302 nm for B@LSNR, which enhanced the PCR rate. The B@LSNR302 − LSNR214 = 2.25 × 10−25 J and B@LSNR302 − LSNR268 = 5.8 × 10−25 J energies are noted as coating energy that had decreased the Eg from 4.86 to 1.88 eV. The higher Eg with shorter λ also induces the electron–electron collisions while lower Eg with longer λ = 302 nm gains stronger quantum tunnelling effects on doping and coating by partitioning the cohesive to adhesive forces to oscillate with longer λ. For (μ) with B@LGT, a partial Gibbs energy (e−Δμ/RT) for electronic excitement is as follows:
| | |
μ = μ° + RTlnGO, Δμ = RTlnGO and aGO = eΔμ/RT, aB@LGT+dye = eB@LGTΔμ/RT + edyeΔμ/RT
| (2.4) |
The GO with intensified Raman band |ψ|Constructed2 = |ψ|D2 ≈ |ψ|G2 has accessed BBR, TMIs, and QHIn. TMI-GO, LGTs, and B@LGTs have distinguished a role of 3d and 4f electrons on GO coating.61 The B@LGTs photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively but TMI-GO and LGT in 60 and 120 min on partially filled 3d and 4f electrons.33,61 The B@LGTs have developed the interstities with the reduced transition metals encapsulated in 2D GO sheets interfaced with the dopant and core LSNR.
3.5. Bandgap energy (Eg) from the Tauc plots
Eg indicates the energy range from the top of VB to the bottom of CB, which excites an 4f electron within a Fermi energy gap. The resulting VB electrons or holes in the lattices of B@LGTs move freely to approach the effluents for PCR. The B@LGT lattices with partially unfilled 4f electrons in the VB move to the CB within lattices as charge carriers. The B@LGTs with Eg = 1.88 eV have acted as semiconductors on forming the continuous energy bands to intensify the PCR. The B@LGT dimension affects the Fermi energy gaps where the electrons with LSNRs of ∼4.70 eV are forbidden. The electrons of B@LGTs have jumped from the VB to the CB with infinite redox cycles with intense cyclic voltammetry for PCR. For excitation, the electrons gain an energy by absorbing hν from light or heat analysed with PCPs at variable T/K (Fig. 18–20). The Eg value of B@LGTs was tuned with compositions of the dopant and coating GO. The interstities with B@LGTs on doping and coating have caused the interfacial linkages as molecular epitaxy that could have acted as heterojunctions for receiving the hν from light. The LSNRs with >4.70 eV have acted as the mildest semiconductor (Fig. 8a). The Eg value has decreased on increasing T/K as the amplitude of atomic vibrations was increased with a larger interatomic spacing as follows:77| |
 | (2.5) |
The E0g value is extrapolated to zero; α and β (β = 1/kBT, J−1) are constants of B@LGTs; the lattice vibrations increase on increasing T/K, so their PCPs are studied from 288.15 to 315.15 K. Their interaction with water dipoles might have interlinked their lattice phonons, free electrons, and holes that could have affected Eg to a smaller extent. Thus, the pressure generated on their dipolar interactions might have also affected the electronic structure. The B@LGTs have energy equipartitioning, so the Eg value is fixed with a quantum confinement effect. The B@LGTs have a direct Eg value due to stable lattices attained on doping and coating (Fig. 8c). The energy split occurs at the Brillouin zone edge of B@LGTs for 1D on a weak periodic potential producing a gap between bands. The XRD analysis depicts the symmetric planes where the dopant and GO coating have further strongly aligned the VB to CB transitions with Nernst potential through stable lattice. The VB electrons are directly excited into the CB by hν having a larger energy than the bandgap as a direct bandgap tends to have stronger light emission and absorption.78 The electronic transitions are directly connected to Eg with maximum charge carriers as transition states detained by B@LGTs at λ = 660 nm calculated from λ = hc/Eg (Fig. 7). The Planck constant h = 6.623 × 10−34 J s, light velocity c = 3 × 108 m s−1, and Eg = 1.88 eV × 1.602 × 10−19 = 3 × 10−19 J. The B@LGTS have detained 3 × 10−19 J, as hν has excited VB electrons to reach the CB space.79 The 3 × 10−19 J energy with longer λ = 660 nm have predicted an intimate interfacing with the BBR, TMIs, and QHIn to PCR. Their Eg value is calculated using the Tauc plot, which directly reflects the density of energy detained by them as follows:61
| |
 | (2.6) |
Eqn (2.6) indicated that
Eg ∝ (1/
λ), where
α is the absorption coefficient,
Eg the optical bandgap, B the band tailing parameter,
hν the photon, and
n = 1/2 for direct
Eg. The
Eg value was calculated from (
αhν)
2 vs. hν (
eqn (2.6)). The
Eg value for Yb
2S
3, Er
2S
3, Eu
2S
3, and Nd
2S
3 is 4.67, 4.75, 4.71, and 4.86 eV, respectively. Gd
3+@Yb
2S
3, Gd
3+@Er
2S
3, Gd
3+@Eu
2S
3, and Gd
3+@Nd
2S
3 have two
Eg values in the ranges of 4.64–1.90, 4.62–2.41, 4.59–2.43, and 4.50–2.50 eV respectively (
Fig. 8a and b).
Two Eg values infer the VB electrons of Gd3+ and Ln3+, individually reaching the respective CBs as 18S2− = 1s22s22p53s23p6 has fully filled orbitals. LSNRs doped with Gd3+ (4f7e) have produced two Eg values caused due to two separate intrinsic VB and CB energy domains despite synchronization. This was reduced on coating with photosensitive GO sheets unlike the pristine lattice of LSNRs and GO (Fig. 8b and c). The two bandgap values of zinc-doped Co3O4 nanoparticles calculated by the Tauc plot method are reported.80,81
The 2nd Eg values of 1.88, 2.38, 2.28, and 2.23 eV for B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO have predicted the effects of covalent bond of Ln3+ with S2−, including the secondary bond of Gd3+. The electrons of 4f orbitals of Ln3+ and also of Gd3+ individually absorb the hν from light and reach the respective CB spaces developing two Eg values. Therefore, the doping has generated a mild band with low Eg values with longer λ that contributed to the PCR in a shorter time. The Gd3+ split Eg from VBLSNR to CBLSNR and VBGd3+LSNR to CBGd3+LSNR for Ln3+ and Gd3+ respectively.
 |
| | Fig. 8 Eg spectra from the Tauc plots for (a) LSNR, (b) B@LSNR and (c) GO and B@LGTs. | |
B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO have Eg values in the range of 4.05–2.23, 4.02–2.28, 4.03–2.38, and 3.97–1.88 eV respectively, which altered the intramolecular activities for PCR (Fig. 8c). Thus, the doping has favourably integrated the resulting holes to access the dye.82
3.6. HR-TEM analysis
The HR-TEM images elucidated the S2− and Ln3+ sites of LSNRs on integrating the wavevector. B@LSNR with ψGd3+ and ψS2− align in bunches of 100 nm size as the adhesive ≥ cohesive forces is depicted with the PCPs in Fig. 9 and 18–20. Thon ring pattern via Fourier transform has predicted the equalisized surface charges of terminal S2− by aligning them through the consistent cohad points (CCPs) for receiving the hν. The B@LGTs have streamlined to astral morphology via central internodes due to enlarged surface area. This depicted that the 2D GO sheets probably align along the NR to occupy an adequate space for oscillations for getting synchronized, where each coated NR seem to synchronize the hv receptance and aligning the resultant holes by gaining the least Ea to avoid the electron–electron collisions, avoiding the recombination. This mechanism has also generated the higher surface area depicted with the BET supporting a monolayer adherence of solvated dye and adsorption of its reduced form. Thus, the friccohesity for 0.014 to 0.012 S cm−1 of LSNR to B@LSNR indicated the clubbed NRs with longer λmax (Fig. 20). The B@LGTs are confined to a central point from 20 to 100 nm. Selected area electron diffraction (SAED) depicts the 2D electron diffraction patterns of Nd2S3, B@Nd2S3, and B@Nd2S3:GO NRs with concentric rings indexed as (211), (310), (321), (001), and (420) lattices to elucidate the uniform wavevector (Fig. 9a(i), b(i), and c(i)). This observation has inferred that the dopant and coating have been conjugated as a single structure. The π → π* of FGs and FEs seem to get fitted with the oxidation and reduction potential of Ln3+ and S2− respectively. The Gd3+ dopant has induced the oscillations by attracting the S2− from primary bond with the Ln3+. The sharper SAED patterns of Nd2S3, B@Nd2S3, and B@Nd2S3:GO NRs than others have predicted the sharing of their lattices with dopants as new wavevectors. The dopant has developed new lattice planes, depicting resolved NRs with a higher surface area, and GO coating has further enhanced the surface area with highly systematized planes, depicted with the BET sharper SAED (Fig. 6). The HR-TEM has predicted the highly resolved surface area because the Ea values of LSNR and dopant both excite their electronic transitions within 1.88 eV energy domain to attain the same phase. Spatial lattice patterns are sharpened for Nd2S3, B@Nd2S3, and B@Nd2S3@GO as dopants and coating have reoriented their lattices proven with their SAED. From the HR-TEM images of Nd2S3, doped, and coated, the coated B@Nd2S3@GO has developed a prominent node (Fig. 9c). Based on the HR-TEM and XRD input, the possible structural linkages are developed (Fig. 10). Gd3+ seems to attract the terminal S2− to join with this, while the NRs seem to wrap with the GO by intensifying the concentric arrangement of electrons. The angular momentums for 4f3e of Nd2S3 might have intermixed to adjoin a centre of internode. The B@LGTs with the optimized lattice in 20–100 nm have expressed the distinctive hν transforming activities83 to holes (Fig. 9). The sharper edges anchor a direct conduction due to a relatively high surface area, improved hole absorption, and chemical sensing abilities. B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO have all the constituents common except increasing 4f electrons that have aligned the overall morphology (Fig. 9f, i, and l). Such critical 4f electron-driven interstitial interfacing has never been reported yet where the spins of increasing 4f electrons with B@LGTs have been expressing the molecular activities. The B@Eu2S3:GO has aligned divergently on optimizing its lattices while the B@Er2S3:GO as tiny divergence occurred due to difference in their 4f electrons. The B@Yb2S3:GO has inferred the sharper and cloudy dimensional edges due to closely spaced alignment (Fig. 9l). The 4f electrons from the VB to the CB face collisions despite the same energy levels and such collisions occur with B@Yb2S3:GO than others. B@Eu2S3:GO with infinite collisions optimized the energy to synchronize to the same energy states (Fig. 9f).
 |
| | Fig. 9 HR-TEM images of (a–c) Nd2S3, B@Nd2S3, and B@Nd2S3:GO with (a(i), b(i), c(i)) their SAED pattern; (d–f) Eu2S3, B@Eu2S3, and B@Eu2S3:GO; (g–i) Er2S3, B@Er2S3, and B@Er2S3:GO. (j–l) Yb2S3, B@Yb2S3, and B@Yb2S3:GO, respectively. | |
3.7. XRD analysis
XRD analysed the crystalline structures (Fig. 10). The d-spacings of Nd2S3 are 0.85, 0.61, 0.55, 0.49, 0.38, 0.32, 0.30, 0.28, 0.22, and 0.18 nm corresponding to the planes (011), (022), (012), (112), (013), (123), (004), (033), (025), and (245) at 10.47°, 14.28°, 15.94°, 18.00°, 23.5°, 27.5°, 28.78°, 32°, 40.4°, and 50.5 respectively. The d-spacings of Eu2S3 with 0.93, 0.55, 0.31, 0.30, 0.27, 0.22, 0.20, and 0.18 nm correspond to (011), (012), (123), (004), (033), (025), (225), and (245) at 9.44°, 16.07°, 28°, 29.3°, 32.5°, 41°, 43.44°, and 50.5° respectively. The d-spacings of Er2S3 with 0.93, 0.30, 0.26, 0.18, and 0.15 nm correspond to (011), (004), (124), (045), and (445) at 9.46°, 29.28°, 34°, 48.75°, and 57.9°, respectively. The d-spacings of Yb2S3 with 0.93, 0.46, and 0.30 nm correspond to (011), (134), and (004) at 9.50°, 18.99°, and 29.07°, respectively (Fig. 10). The d-spacing values for LSNRs have been found within a common range to critically authenticate the similar lattice framework with the similar wavevector generating a non-shifting bandgap. The peaks at 2θ values as per 4f electron consolidate the crystallographic planes with the corresponding d-spacing indexed as 1D NRs (Table S12†). The positions of XRD peaks are slightly changed with few diffraction lines and are close to the standard JCPD cards. The XRD profiles of Nd2S3, Eu2S3, Er2S3, and Yb2S3 are in agreement with the standard JCPD cards (01-081-0924),84, (JCPDF#26-1419),85, (00-021-0324)86,87 and (65–2373)88,89 respectively. The energies of 4f3e and 4f6e with Nd2S3 and Eu2S3 split 90 into more peaks than those of Er2S3 and Yb2S3 to distinguish a role of their more unfilled suborbital and more filled suborbital expressed as ψn as nNd2S3 > nEu2S3.21,91 The sharpened cps from VB to CB had predicted the infinite nodes of the similar frequencies used to calculate resultant energy as| |
 | (2.7) |
Nd2S3 and Eu2S3 with sharper peaks indicate almost equal eigen energies unlike the hedgy split of nearly filled 4f11e and 4f13e of Er2S3 and Yb2S3 respectively92 (Fig. 10a). Enhanced peak intensities with B@LSNR lattice planes compared to the LSNRs indicated chargewise intensified and stabilized hole-generating sites (Fig. 10b). B:Nd2S3@GO, B:Eu2S3@GO, B:Er2S3@GO, and B:Yb2S3@GO have all the constituents common with 4f3e, 4f6e, 4f11e, and 4f13e respectively. This predicts that a variation in 4f electrons has substantially aligned the overall morphologies due to enhancing spins. B:Yb2S3@GO is a flower petal-like structure, in which the NRs have been aligned divergently. B:Er2S3@GO developed a tiny structure, while B:Yb2S3@GO developed a cloudy morphology with sharper diverging NRs. The 4f@e from the VB to the CB also faces collision despite the same 4f@e energy levels. Such partial collisions are nil with B:Yb2S3@GO while B:Eu2S3@GO seems to face much collisions, optimizing the energy to synchronize the same energies. Their spin intensity predicts the nodal plan (n) and its values are used to calculate ψn using eqn (2.7). At a higher d spacing with Nd2S3, there are prominent planes and Eu2S3 also developed the (001) plane on shifting from 4f3e to 4f6e (Fig. 10a and b). These predict the role of contraction effect, where increasing the electrons in the same orbital lowered their atomic radii. However, no such data have been reported yet, but their behaviour is observed similar to the transition metals (Table S28†). On increasing the 4f@e from Nd2S3 to Eu2S3 has generated the corresponding planes at specific 2θ values. That had predicted an influence of nuclear charge being applied on the Ln3+ and S2−. These results are also supported by the TGA and UV/vis spectroscopy (Fig. 7 and 15). Upon increasing the number of 4f electrons within the same shell, a more nuclear charge is applied on them (Table 2). Thus, for estimating the values of atomic radii of LSNRs from Nd3+ to Yb3+, the coulombic forces are generated as follows:| |
 | (2.8) |
r = Bohr radii = 0.0529 nm, ε = 78.4 for H2O at 25 °C. The higher is the attractive force, the lower is the radii (1/attractive force). The Er2S3 and Yb2S3 have developed the same planes except (012) that appeared with Nd2S3 due to a slightly higher nuclear charge, affecting an overall lattice wavevector (Fig. 10a). The planes with Yb2S3 appeared at 9.50°, 18.99°, and 29.07° compared to others (Fig. 10a).
Table 2 Coulombic forces on increasing the 4f electrons
| Ln3+ |
# Electrons |
# Protons |
Coulombic force |
| Nd3+ |
57 |
60 |
48410.51 |
| Eu3+ |
60 |
63 |
53506.35 |
| Er3+ |
65 |
68 |
62565.63 |
| Yb3+ |
67 |
70 |
66387.51 |
Probably, Yb2S3 with 4f13e developed a stronger nuclear charge on Yb3+ strongly affecting the position of S2− in the Yb2S3 lattice. Second, no peaks appeared at higher 2θ values with Yb2S3 as Yb3+–S2− seems to have the strongest potential energy. Thus, no multiple rotation, vibration, and translations are possible at higher 2θ values. The [Yb3+]2[S2−]3/4πr2 with Yb2S3 have observed the maximum peaks at lower 2θ values while at higher 2θ values no peaks. Thus, the no collisions have occurred due to one UPE of Yb2S3 than the 4f14e fully 4f field orbital. Fundamentally, the same energy levels for the 4f orbitals with degeneracies on increasing the electrons could be compared with the XRD intensity and d spacing pattern (Fig. 10c). Thus, 0.88 nm d spacing of GO compared to 0.34 nm of Gt indicates the interfacial forces working among their primitive cells or lattices. The d spacing of Gt is almost 3 times that of GO, as Gt has the C–C framework of primitive cells with stronger PE > KE without oscillating its energy than that of GO sheets with equal FG distribution (Fig. 10c). The FGs of different electron densities are bonded, which are mutually shared with the main lattice; hence, they expand with the covalent bonds increasing the d spaces. Similarly, an increase in the 4f electrons has resulted in an increase in the nuclear charges within 4f orbitals. The functionalisation of GO has increased the d spacing with a wider distribution compared to the sharper one with Gt (Fig. 10c). Gt has a uniform lattice with 100% equipartition that responds to a laser light without collisions with ψsv. However, GO with 2θ from 11° to 21° values synchronized the oscillation of the FGs, where the O atom dominates (Fig. 10c). Such lattice elements have synchronised the d spacing to 0.88 nm with 10900 cps against 85000 with Gt. Gd3+ with Er2S3 and Yb2S3 has slightly changed the peaks unlike Nd2S3 and Eu2S3 on equipartitioning the lattice of LSNRs. The XRD data for rhodium sulfide and its doping with Cu, Fe, Co, and Ni with some extra peaks are reported.93 Nearly fully filled 4f electrons have effective LSNR matrices with Gd3+ due to a 4f7 half-filled suborbital. The shift in peak position has occurred with a lower angle on strain in doped nanomaterials, as reported elsewhere.94 Hence, Gd3+ doping with the LSNRs could not disrupt a native lattice of LSNRs except enhancing the intensity by ∼3000 cps (Fig. 10a and b). Thus, Gd3+ has been accommodated with lattices of respective LSNRs as the FGs become the constituents of the main lattice of GO. Though the Gd3+ did not destabilize the main lattice of LSNRs, the Ln3+ and S2− have developed stronger covalent bonds, while Gd3+ has been the competent dopant to dent their PE with a new state of B@LSNRs. The increments in the intensities of lattices indicated that the repulsion between Gd3+ and Ln3+ is equal to the attraction between the 2Gd3+ and 3S2− ions. This had maintained the consistency in the lattice planes without generating a new lattice with no effect in d spacing. Therefore, Gd3+ has effectively aligned the LSNRs by attracting the terminal S2− (Fig. 9 and 12). The GO coating reoriented and aligned the lattice planes of B@LSNRs, the GO plane with their 0.88 nm d spacing was the main plane at 2θ = 10.52–11.24° (Fig. 10d). However, the intensities have been reduced to half values. The structuredness expressed as manifolds state was attained with the energy partitioning. Their intensities are as B:Er2S3@GO > B:Yb2S3@GO > B:Eu2S3@GO > B:Nd2S3@GO with 4f11e, 4f13e, 4f6e, and 4f3e. The variable factors with XRD intensities and d-spacing are the 4f electrons that have substantially synchronised the planes of the B@LSNRs, to the plane of GO (Fig. 10c and d). The said synchronisation of their planes predicts that the FGs and FE both with −ve and +ve centres of B@LSNRs have substantially interacted to split their residual energies (Fig. 15).
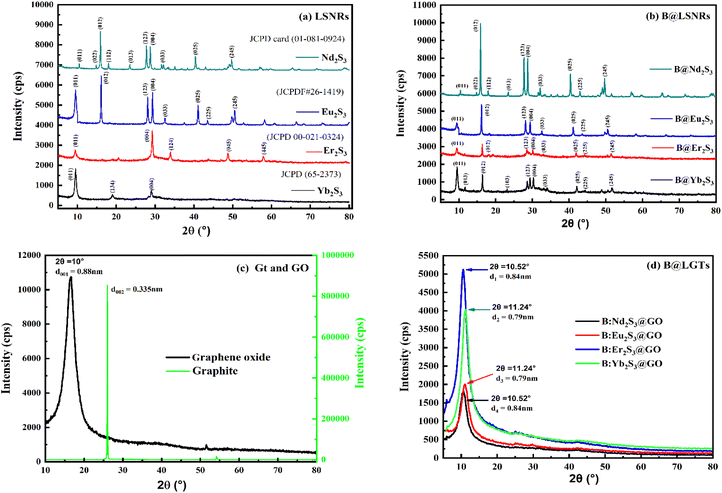 |
| | Fig. 10 XRD spectra elucidating the d spacing and diffraction planes at 2θ values for (a) LSNR, (b) B@LSNR, (c) GO and Gt and (d) B@LGTs. | |
3.8. Raman spectroscopy analysis
Raman laser elucidates the structural defects of B@LGTs that have variable intrinsic energies synchronized as intense D and G bands, unlike the pristine matrices.61 Laser becomes relevant to analyse an equipartitioning of energies caused on aligning Gd3+ and GO in the matrices of LSNRs expressed as follows:| |
laser ![[radiolysis arrow - arrow with voltage kink]](https://www.rsc.org/images/entities/char_e116.gif) B@LGT → ID + IG + I2D B@LGT → ID + IG + I2D
| (2.9) |
The intensities (I) of D, G, and 2D bands with GO at 1373, 1597, and 2750 cm−1 respectively have inferred a consolidation of the electronic activities of FG and π conjugation (sp2 → sp3). That had rationalized as ID/IG = 0.86 < 1 along with a mild modulation bump of 2D band against D and G bands61 (Fig. 10). The integrated G band with E2g induced the electronic activities from sp2 → sp3 with an apex as disordered GO with a sharper D band on localized synchronization among the tiers. The B@LGTs with higher ID/IG = 0.93 < 1 than GO has further inferred an induction of more energy domains integrated to sharper D bands than a pristine GO8 (Fig. 11 and Table 3). B@Er2S3:GO with sharper D and G intrinsic intensities detected with Raman laser are correlated with IR stretching of S2− and bonded with Ln3+ in matrices of LSNR via double and single bonds. Er3+ of B@Er2S3:GO has 4f11 electrons, i.e. neither half nor a fully filed orbital causing the prominent individualistic energy domain within a structure responsible for effective photocatalysis and physicochemical interactions, While GO despite being a single 2D sheet has also developed the spontaneities due to localized FG and FE activation, which could be due to twisting or distortions but these are negligible compared to B@Er2S3:GO and others (Fig. 11b). Therefore, a GO coating has tremendously consolidated the electronic excitations in response to the Raman laser with robust phonon generation of the specific frequencies integrated to overall single energy value (Table 3). Such response of Raman laser could be aligned with the FT-IR spectra where the Fingerprint region is slightly intensified over stretching on coating due to the streamlining of the surface charges. The 4f13e of B@Yb2S3:GO with lower intensities and large d-spacings strengthened synchronization with GO aligning the charges (Fig. 10d and 12b). The surface energies of B@Er2S3:GO are enhanced, causing inelastic incoherent neutron scattering depicted by symmetric IR with the moderate Raman activity. B@Nd2S3:GO and B@Eu2S3:GO upon integrating to synchronized interstities photocatalytically reduced BBR, TMIs, and QHIn in a shorter time (Fig. 11b). Raman spectra with incident laser have predicted a stronger communication of 4f@e with GO oscillations. Thus, the incident laser indicates a stronger dipole moment for B@Er2S3:GO with maximum D and G intensities as surface plasmonic resonance (SPR).95 The D and G bands with B@LGTs infer Rayleigh scattering due to the aligned GO sheet. However, B@LGTs with interstitial tiers on the aligned matrices of variable 4f@e by minimizing the hν bouncing photocatalytically reduced BBR, TMIs, and QHIn in 15–40 min. Considering the expeditious photocatalytic activities of B@Eu2S3:GO and B@Yb2S3:GO, the possibility could be explored to replace Gd3+ with Pd, Ag, Pt, and Au noble metals.96 These metals could intensify the sharper D and G bands of B@Er2S3:GO and B@Nd2S3:GO enhancing 4f@e interactions with FGs on stabilizing the dipoles (Fig. 11b). The 4f6e and 4f13e of B@Eu2S3:GO and B@Yb2S3:GO distract alignment on electronic collisions unlike 4f11e and 4f3e of B@Er2S3:GO and B@Nd2S3:GO respectively. The FGs with 4f@e regulated on quenching the undesired stretching with higher polarizability (Fig. 11b). Nd2S3 at 200 and 300 cm−1 has attributed to E2g and A1g bands,97 while Er2S3 and Yb2S3 have broader bands at 390 and 400 cm−1 respectively. The Raman laser has excited the 4f electrons to oscillating intensities within 200–1200 cm−1, where the prominent intensities with Nd2S3 at 196, 392, 831, and 984 cm−1 and also at 196 and 392 cm−1 have attributed to E2g and A1g bands respectively.97 Eu2S3 has shown maximum intensities at 217, 469, and 846 cm−1 while Er2S3 and Yb2S3 at 370, 1000, and 450, 1000 cm−1 respectively. The more oscillating intensities at variable Raman shifts are prominent with Nd2S3 and Eu2S3 having less than half-filled 4f electrons. Er2S3 and Yb2S3 have broader peaks at 370 and 450 cm−1, where Yb2S3 from 316 to 510 cm−1 predict that the single 4f unpaired electron could not cause robust spins and collisions (Fig. 11a). A similar behaviour is predicted with B@Yb2S3:GO that had intensified a maximum PCR rate among the B@LGTs (Fig. 11). The LSNRs doped and coated have produced the well-defined D and G bands unlike LSNRs alone. The D and G bands predicted that the 4f electrons of Ln and dopant might have synchronized the oscillating activities of the GO sheet. The doping and coating of LSNRs have been fitted with surface-enhanced FRET to consolidate the electronic oscillations w.r.t. the structural constituents FGs and FE of the GO sheet.30
Table 3 Raman shifts with D and G bands and ratios for GO and B@LGTs
| LSNR@GO |
D-shift (cm−1) |
G-shift (cm−1) |
ID/IG |
| GO |
13238 |
15462 |
0.86 |
| B:Nd2S3@GO |
19532 |
20470 |
0.95 |
| B:Eu2S3@GO |
12498 |
13546 |
0.92 |
| B:Er2S3@GO |
12614 |
13448 |
0.94 |
| B:Yb2S3@GO |
34406 |
37386 |
0.92 |
 |
| | Fig. 11 Raman spectra for (a) LSNRs and (b) GO and B@LGTs elucidated the (c) D and G band intensities with ID/IG < 1. | |
3.9. FT-IR analysis
FT-IR have tracked the 1071, 1400, 1250, 1750, and 1626 cm−1 bending and stretching frequencies for C–O–C, –CHO, >CO, –COOH, and –OH of GO and B@LGTs respectively.61 This predicted that the said groups assist GO coating by aligning along B@LSNR. The broader peak for –OH at 3428 cm−1 infers free water molecules adhered with GO and B@LGTs (Fig. 12a). The peaks at 500–623 cm−1 and 697 cm−1 ascribed to Ln–S or Gd–S metal sulfide,84 1071–1400 cm−1 ascribed to C–O–C, –CHO, and >CO of the GO sheet and the bending at 3428 cm−1 for –OH, respectively33 indicated the functionalisation of B@LGTs (Fig. 12b). B@LGT with aligned interstitial charges has absorbed maximum IR radiation for an energy storage with inelastic ROCs.23 The interfacing of the B@LGT with lithium or Ti metal could enhance the energy storage performance by further reducing the Eg with aligned electrostatic dipoles.98 The B@LGTs by adsorbing H2 could lower their ignition point for use as clean fuel. The B@LGT have intensified the bending and stretching in the fingerprint region30 compared to GO with longer λ in the same φ (Fig. 12b). This also reoriented the H2O dipoles with higher shear and surface tension (Fig. 18 and 19). The Oδ− of H2O could have competitively realigned either with Gd3+ or Ln3+ sharpening the bending by lowering the stretching and stabilizing the electrostatic dipoles of interstitial tiers. The FGs of GO have been prominently stretching with negligible bending, as a fingerprint domain with GO is plain compared to B@LGTs (Fig. 12a). Thus, the 2D GO sheet could not be rotated, vibrated, and translated except stabilising the hv receiving sites to oscillate with undesired activities and was chosen for coating. However, the stretching and bending patterns of LSNRs were harmonised compared to GO (Fig. S11a†). The GO stretching domains are active while the LSNRs normalised with bending unlike confined to a single value. However, the huge charge in bending on coating the B@LSNRs with GO seems to be the broader and undecided stretching despite that sharper with LSNRs (Fig. 11b). Hence, both the stretching and bending are largely modified on coating with GO, due to the spontaneities of both the GO and LSNRs for interfacing them (Fig. 12). Moreover, stretching and bending of B@LSNRs indicate a novel interface than LSNRs. The Gd3+ dopant has functionalised them where the stretching is broadened from 3600 to 3000 cm−1 (Fig. S11b†). Thus, the stretching of S2− of LSNRs has become most active to stretch as well as to induce the exponential undistinguished bending credited to the response of S2− to the Gd3+, and at the same time, accommodating the covalent bond with Ln3+. The dopant has substantially transformed the PE to eigenenergy elucidated with the HR-TEM and TGA (Fig. 9 and 15). The bending indicates the optimized B@LSNRs due to the energy distribution. However, the intrinsic stretching frequency of LSNRs from 1638 to 1405 cm−1 prove shifting via S2− to accommodate Gd3+.
 |
| | Fig. 12 FT-IR spectra Illustrating stretching and bending frequencies with (a) GO and (b) B@LGT with the increase in their 4f electrons. | |
The sharper bending pattern of manifolds from 500 to 1200 cm−1 appeared due to the metal sulfide bonding of LSNRs (Fig. S11a†). The LSNRs bending on doping with Gd3+ has also acted as the constituents for bending the B@LSNRs. The bending patterns of Gd3+ dopants with B@LSNRs have never been reported yet. Both S2− and Ln3+ have shared their electron densities with Gd3+ unlike LSNRs alone. The 1700–2700 cm−1 indicate almost the normalised behaviour of B@LSNRs, which may be due to repulsion and attraction, where [Gd3+][S2−]/4πr2 as ra = rr (Fig. S11b†). These bending and stretching have introduced valuable ψsv to photocatalytically reduce the effluents. The frequencies with B@LSNRs broadened from 3600 to 2700 cm−1 than a straight line for LSNRs with 100% abs. It has predicted that the terminal S2− and −S2− both are connected to the Gd3+ and have acquired almost similar KEs oscillating with similar frequencies. This is noted as  normalised than
normalised than  orthogonal due to different intramolecular activities or oscillations synchronised with Gd3+. Gd3+ has functionalised the fingerprint region as well as the stretching domain of B@LSNRs (Fig. S11b†). The broader stretching frequency and fingerprint region have further been sharpened on coating because GO has established communication with Ln3+ and S2− as well as Gd3+ on energy equipartition. Highly functionalised GO with stronger covalent bonds is not disrupted upon coating. Therefore, it has further sharpened the broader stretching domain of LSNRs as well as B@LSNRs (Fig. S11b†). The integrated GO sheets with FGs and FE equally resonate to transfer energy intimating the dopant and coating agents with intimated adherence with LSNRs. This has caused the stronger FRET generating ψsv on energy equipartitioning through the GO due to a sharper frequency at 1430 cm−1 and 1680 cm−1. The GO has stabilised the molecules by normalising the bending frequencies due to a stronger link with the LSNR and Gd3+ to integrate the overall bending pattern as follows:
orthogonal due to different intramolecular activities or oscillations synchronised with Gd3+. Gd3+ has functionalised the fingerprint region as well as the stretching domain of B@LSNRs (Fig. S11b†). The broader stretching frequency and fingerprint region have further been sharpened on coating because GO has established communication with Ln3+ and S2− as well as Gd3+ on energy equipartition. Highly functionalised GO with stronger covalent bonds is not disrupted upon coating. Therefore, it has further sharpened the broader stretching domain of LSNRs as well as B@LSNRs (Fig. S11b†). The integrated GO sheets with FGs and FE equally resonate to transfer energy intimating the dopant and coating agents with intimated adherence with LSNRs. This has caused the stronger FRET generating ψsv on energy equipartitioning through the GO due to a sharper frequency at 1430 cm−1 and 1680 cm−1. The GO has stabilised the molecules by normalising the bending frequencies due to a stronger link with the LSNR and Gd3+ to integrate the overall bending pattern as follows:
| |
 | (3.0) |
The bending energy pattern as B:Eu2S3@GO > B:Nd2S3@GO > B:Er2S3@GO > B:Yb2S3@GO indicates B:Yb2S3@GO as highly normalised due to 4f13e than B:Eu2S3@GO with 4f6e. The sharpening at 1450 cm−1 indicates 100% equipartitioning as B:Nd2S3@GO, B:Eu2S3@GO, B:Er2S3@GO, and B:Yb2S3@GO have attained the undistinguishable energy states with instantaneous PCR without the energy ingredients (Fig. 12b). The stabilised bending has been supported by the Raman spectra, especially B:Er2S3@GO with a stronger D band at 35000 cps. B@LGTs have gained normalised bending at 3100 cm−1 promoting the PCR and PCP activities with effluents than LSNRs and B@LSNRs (Fig. S11†).
3.10. XPS analysis
The XPS spectra of Er2S3, B@Er2S3, and B@Er2S3:GO analysed the oxidation state, elemental composition, and lattice energy. Their survey spectra indicate excitation of 4f11e electrons shared with S2− like Er3+ in Er2S3, while the 2p electrons resisted inner electronic activities as per the Compton effect.99 Er2S3 with 2p1/2 and 2p3/2 states of the S at 162.58 and 164.58 eV respectively indicate optimized stability (Fig. 13f). The S 2p states (Table 4) have confirmed the structural stability of Er2S3 w.r.t. the core–shell electrons of lanthanides. The 4f electrons confined towards terminal sulfur atoms form double covalent bonds with specific binding energy.30 Ln3+ is covalently bonded with S2− while Gd3+ with a secondary bond on aligning the 4f@e and 2p core electrons. Gd3+ with B@Er2S3 has broadened the 2p1/2 and 2p3/2 peaks at 177 and 173 eV respectively for the S atom than Er2S3 alone (Fig. 13g). The broadening inferred that their spins noted as angular momentum could have been shared through repulsion and attractions charges in the LSNR matrices. Thus, the 2p1/2 and 2p3/2 energy states for S2− increased with Gd3+ on realigning the matrices of Er2S3 and on increasing the unpaired 4f@e with a higher binding energy of S2− with 18–60% wt loss at 100–500 °C (Fig. 15).100 The interstities of B@Er2S3 have broadened the peaks with a higher electron density than that of Er2S3 alone. The sharper peaks of lower intensity with B@Er2S3:GO are noticed. The FGs with ψGd3+, ψLn3+, and ψS2− interstities have equalized the lattices. B@Er2S3:GO with aligned charges enhanced the binding energy of S2− from 164.58 to 177 and 162.58 to 173 eV of 2p1/2 and 2p3/2 respectively due to Gd3+ (Fig. 13h). Gd3+ has enhanced the peak intensity of B@Er2S3 with least Eg and a higher surface area than Er2S3 analysed with BET (Fig. 6 and 8). The higher electron density of B@Er2S3:GO decreased the binding energy from 177 to 161.12 and 173 to 159 eV of 2p1/2 and 2p3/2, respectively (Fig. 13 g and h). The C 1s spectra of B@Er2S3:GO at ∼284, 285, and ∼286.7 eV for C–C, C–O, and C–O–C respectively with stronger stability of respective bonds indicate intense hybridization61 (Fig. 13d). The O K-edge with B@Er2S3:GO indicate C–OH, C–O–C, and CO groups at ∼530.7, ∼531.8, and 533.5 eV respectively due to stronger O linkages than the GO alone61 (Fig. 13d). The reduction of 532 eV of O 1s with GO to 530 eV with B@Er2S3:GO implied the O participation due to its electronegativity with the oxidation potential of dopant Gd3+ (Fig. 13e). The electron density of O with B@Er2S3:GO is higher with a broader peak for C–O–C at 531.8 than with GO.30 Thus, the GO interactions have conjugated it with B@LGT than GO alone due to a higher PE. The higher binding energy for O of B@Er2S3:GO indicates the interfacing of FGs with LSNR matrices and dopants (Fig. 11). Thus, the C 1s and O 1s spectra have split at 530 eV due to FGs and 4f@e vis-à-vis Gd3+ 4f7e on strengthening a nuclear charge than GO alone30 that agrees to literature values.71,101 From 164.58 to 177 eV for Er2S3 of B@Er2S3 indicated the conjugation of Gd3+ with Er2S3 matrices via S2− as the primary bond with Ln3+ and secondary with Gd3+ (Fig. 13f and g). The survey scans have elucidated the elemental compositions with various binding energies (Fig. 13a and c). The O 1 s with GO and B@Er2S3:GO at 530.86, 532.64, 533.88, and 530.7, 531.8, 533.5 eV for C–OH, C–O–C, and CO respectively30 indicate interstitial lattice with slightly less electron densities (Fig. 13d). Energies of 168.05 and 171 eV for 4d5/2 and 4d3/2 respectively for Er of B@Er2S3:GO than ∼1227 and ∼1219 eV for 3d3/2 and 3d5/2 respectively for Gd3+ have predicted the integrated interstities (Fig. 13j). Gd3+, a dopant, has shared the PE in attraction for S2− and repulsion for Ln3+ of matrices. The 3d and 4d electron spins attain stable interstities with dopants and GO responds to the hv for hole generation.30 The spectra for Er3+ and Gd3+ with 4d and 3d for B@Er2S3:GO with distinct binding energies of respective core electrons have exponentially enhanced their spins on sharpening the D and G bands supported by Raman intensities78,102 (Fig. 11). The sharp peaks of Gd at ∼1227 and ∼1219 eV for 3d3/2 and 3d5/2 states, respectively, with B@Er2S3:GO indicate spin orbital coupling71,101 (Fig. 13j). The spectra for Er3+ and Gd3+ with 4d and 3d with the B@Er2S3:GO resemble as dopant harmonized the electronic state of Er2S3.78
| ΔBEB@Er2S3–Er2S3 = ΔBE2–1, ΔBEB@Er2S3:GO–B@Er2S3 = ΔBE3–2 |
Table 4 Binding energies of the S atom for Er2S3, B@Er2S3, and B@Er2S3:GO
| S (O.S) |
LSNR |
B@LSNR |
ΔBE2–1 |
B@LGT |
ΔBE3–2 |
| S 2p1/2 |
164.58 |
177 |
12.42 |
161.12 |
−15.88 |
| S 2p3/2 |
162.58 |
173 |
10.42 |
159 |
−14.00 |
 |
| | Fig. 13 XPS spectra: (a–c) survey scan for B@Er2S3 and B@Er2S3:GO; (d) C 1s and (e) O 1s of B@Er2S3:GO; (f–h) S 2p states; (i) Er 4d3/2 and 4d5/2 and (j) Gd 3d3/2 and Gd 3d5/2. | |
3.11. SEM analysis
SEM images have depicted the LSNRs and GO constituent integrated as B@LGT as a lace-like thin sheet of 10 and 100 μm morphology with a higher surface area with oscillating interlinks. The LSNR with uniform surface charges differ from B@LSNR of 2–10 μm via oxidation potential-driven linkages attained due to Gd3+ dopants (Fig. 14a and b). B@LGT on receiving the hν with oscillating intensified interstities (I) profiled oscillating energies as follows:| | |
I = [ψLn3+ × ψS2−]+ [ψh+ × ψe−] and |ψLSNR|2 and |ψGO|2
| (3.1) |
The monodisperse B@LTs were aligned similarly while the rough surfaces elucidated the uniform GO multilayers compared to GO alone (Fig. 14c and d). SEM elucidates the continuous networking of LSNR and GO sheets while the B@LGTs have been organized as a prominent integrated surface area in an intense smooth dimension. This established a close contact with the effluents for photocatalytic reduction, after PCR effluents get adsorbed onto B@LGTs. The B@LGT as an NR fibril network with activated surfaces has enhanced the photocatalytic reduction of BBR, TMIs, and QHIn. The integrated smooth sheet-like structure predicted that the interstities might have been staggered to intimate aligning despite the different energies but in the same φ. The square-like morphology of B@LGTs compared to the wire-type network distinguishes the role of the GO sheets, which have surrounded the B@LSNR. The morphological expression has been the function of the internal structural network in close contact on equipartitioning the energy through FRET (Fig. 14).
 |
| | Fig. 14 SEM images of (a) LSNR, (b) B@LSNR, (c) GO and (d) B@LGT morphologies. | |
3.12. Elemental analysis
The C, H, and S elements with GO and B@LGTs both indicate that the coating is a chemical process on developing the uniform interstities with energy equipartitioning. Their values were detected using an elemental analyser and an XPS analyser (Fig. 13). Table 5 presents higher C% with GO alone than in the doped state in 1.5/1.0 ratio. Thus, almost 50% GO was doped when B@LSNR:GO = 1:6. Interestingly, B@LGTs from Nd2S3@Gd:GO to Yb2S3@Gd:GO have similar C% and H% with ±0.5% and ±0.2% variations respectively, though this variation with S is ±0.1% (Table 5). Therefore, all the B@LSNRs have doped almost a similar GO amount due to similar interacting sites. It is also depicted with their similar N2 adsorbing and desorbing rates with BET analysis (Fig. 6). Thus, their structural geometry is thermodynamically and quantum mechanically stable, and hence, they are used as photocatalysts with manifold reusability.
Table 5 Elemental analysis of GO and B@LGTs
| Systems |
C (%) |
H (%) |
S (%) |
Wt (mg) |
| GO |
60.558 |
3.306 |
— |
1.0752 |
| Nd2S3@Gd:GO |
39.808 |
3.147 |
0.890 |
1.004 |
| Eu2S3@Gd:GO |
41.629 |
2.818 |
0.936 |
1.234 |
| Er2S3@Gd:GO |
40.978 |
3.098 |
0.797 |
1.365 |
| Yb2S3@Gd:GO |
41.698 |
2.381 |
1.045 |
1.380 |
3.13. ICP-OES of LSNRs, B@LSNRs, and B@LGTs
First, 2 mg/100 mL of each LSNR, B@LSNR, and B@LGT was sonicated at 28 kHz in a conical flask for 2 h. It was washed twice with warm water and centrifuged, and later, a homogenized solution was prepared for ICP-OES analysis. Initially, 2, 4, 6, 8, and 10 ppm standard solutions were prepared by mixing 0.2, 0.4, 0.6, 0.8, and 1 mL of each of NdCl3, EuCl3, ErCl3, YbCl3, and GdCl3 salts out of their 1000 ppm stock solutions for calibration.
The intensity vs. amount of respective metals with 1st order obtained on regression with residual coefficient (R2) values between 0.973964 and 0.992382 (Fig. S15†) have inferred accuracy. The Gd3+ doping amounts were decreased on increasing the 4f@e of lanthanide metals in B@LSNR. These were also estimated in the interpretation of PCPs as 4f7e of Gd3+ had caused a repulsion with the respective chosen lanthanide metals. The intensities of Gd are increased from Nd2S3 to Er2S3 while decreased for Yb2S3 due to 4f13e with milder electronic collisions. The intensities of Gd with B@LGTs are decreased due to the intimate communication of 2D GO sheets. The intensities and amounts of Nd2S3 to Eu2S3 are increased due to less than half-filled 4f@e, while from Er2S3 to Yb2S3, the intensities are decreased. The LSNRs in the range of 4.223–4.783 mg L−1 elemental compositions indicate their equivalent stoichiometric ratios. The elemental compositions of respective metals and Gd dopants in B@LSNRs are in the range of 3.066–4.150 and 1.162–1.763 mg L−1 respectively. While with B@LGTs, the elemental compositions of respective metals and Gd are in the range of 2.216–2.502 and 1.111–1.750 mg L−1 respectively (Table 6). The database obtained from ICP-OES analysis indicates that Gd3+ and GO were adequately doped and coated with LSNRs. While their nuclear sizes, the number of 4f@e, and FGs of exfoliated GO sheet have proven the B@LGTs as the robust photocatalysts.
Table 6 ICP-OES analysis of LSNRs, B@LSNRs, and B@LGTs
| Analyte quantity |
Elements |
Corresponding intensity (g) |
Prep. vol.: conc. (calib) (mg L−1) |
Conc. (sample) (mg L−1) |
| Nd2S3 (406.109) |
Nd |
1334.4 |
4.513 |
4.513 |
| Eu2S3 (381.967) |
Eu |
1694409.6 |
4.429 |
4.429 |
| Er2S3 (337.27) |
Er |
74385.78 |
4.223 |
4.223 |
| Yb2S3 (328.937) |
Yb |
1543.1 |
4.783 |
4.783 |
| Nd2S3@Gd (406.109) |
Nd |
3806 |
4.150 |
4.150 |
| Gd |
9831.9 |
1.763 |
1.763 |
| Eu2S3@Gd (381.967) |
Eu |
1623457.4 |
3.974 |
3.974 |
| Gd |
21394.0 |
1.341 |
1.341 |
| Er2S3@Gd (337.27) |
Er |
17697.5 |
3.822 |
3.822 |
| Gd |
36522.8 |
1.212 |
1.212 |
| Yb2S3@Gd (328.937) |
Yb |
44130.3 |
3.066 |
3.066 |
| Gd |
1527.9 |
1.162 |
1.162 |
| Nd2S3@Gd:GO (406.109) |
Nd |
101043.6 |
2.326 |
2.326 |
| Gd |
212602 |
1.639 |
1.639 |
| Eu2S3@Gd:GO (381.967) |
Eu |
644080.9 |
2.502 |
2.502 |
| Gd |
10172 |
1.750 |
1.750 |
| Er2S3@Gd:GO (337.27) |
Er |
73521.4 |
2.451 |
2.451 |
| Gd |
8703.4 |
1.439 |
1.439 |
| Yb2S3@Gd:GO (328.937) |
Yb |
2027.4 |
2.216 |
2.216 |
| Gd |
290.3 |
1.111 |
1.111 |
The B@LGTs have enhanced the PCR of BBR, TMIs, and QHIn compared to GO, LSNRs, and B@LSNRs. The elemental compositions confirmed the substantial chemical binding of dopants and coating agents with the angular momentum of LSNRs. The detail of the ICP-OES analysis is given in Table S29.†
3.14. Thermal stability via TGA, DTA, and DTG
TGA indicated the structural stability and heat holding ability of samples in terms of wt loss. Thus, B@LGTs have gained uniform thermal energy and Gt of the C–C skeleton also lost wt in the 1st order due to strong VWFs (Fig. 15a). The stronger heat holding capacities with the Gt → GO → LGT→ and LSNR → B@LSNR → B@LGT indicate the structural stability.30 The Gt with no wt loss for moisture accounts for the stronger VWFs than GO with 15.24% wt loss at 106 °C. The LSNRs and Gt due to compact structures with stronger VWFs could not hold much water, while GO with manifold FGs and FEs having several energy ensembles almost in the same phase showed sharper 88.76% wt loss at 194 °C. Thus, the structural activities of GO could be expressed as  shifting to
shifting to  from Gt to GO respectively with ψm ≠ ψn, as their initial m and final n coefficients differ at two temperatures due to alignment (Fig. 15a and b). The LGTs were split at ∼250 °C with hedgy oscillations, while Nd2S3, Er2S3 and Yb2S3 have quenched the oscillations by synchronization and Eu2S3 was stabilized. Eu2S3 with hedgy oscillations is shifted to an empty 4f suborbital than other LSNRs. The resonating B@LSNRs get detached103 from the lattice, as Gd3+ from 0 to 250 °C accelerated the
from Gt to GO respectively with ψm ≠ ψn, as their initial m and final n coefficients differ at two temperatures due to alignment (Fig. 15a and b). The LGTs were split at ∼250 °C with hedgy oscillations, while Nd2S3, Er2S3 and Yb2S3 have quenched the oscillations by synchronization and Eu2S3 was stabilized. Eu2S3 with hedgy oscillations is shifted to an empty 4f suborbital than other LSNRs. The resonating B@LSNRs get detached103 from the lattice, as Gd3+ from 0 to 250 °C accelerated the  and ψGd3+ ≈ ψS2− and ψGd3+ ≈ ψLn3+ to shorten the PCR time. The spins of the 4f@e could boost the efficiencies of sensors, acoustivity,61 TFT, and contrasting activities16 in the CT scan.32 Nd2S3@Gd has lost 28.03% wt at 287.49 °C on oscillation as
and ψGd3+ ≈ ψS2− and ψGd3+ ≈ ψLn3+ to shorten the PCR time. The spins of the 4f@e could boost the efficiencies of sensors, acoustivity,61 TFT, and contrasting activities16 in the CT scan.32 Nd2S3@Gd has lost 28.03% wt at 287.49 °C on oscillation as  to ψGd3+ ≈ ψS2− (Fig. S4†). B@LSNR with specific frequencies could exchange a coded message104 similar to receiving the hν transforming into the holes compared with the standard known quantum materials. Gd3+ equipartitioned Nd2S3 at 750 °C, while DTA for Eu2S3 indicated uniform charge distribution compared to other LSNRs. The B@LSNR lost wt at 254 and 436 °C along endothermic and exothermic peaks at 750 °C. The Gd3+⋯S2− and Ln3+−S2−⋯Gd3+ strings could have aligned Eu2S3 with Gd3+. The Gd3+ 4f7e could not induce major oscillations with Eu2S3 except mass transitions at 254.82, 436.6, and 800 °C with 37.04, 27.54, and 35.42% wt losses respectively (Fig. S3 and S4†). Gd3+ with 2Eu3+ and 3S2− of Eu2S3 enhanced the holes via ψLn3+ ≈ ψS2−
to ψGd3+ ≈ ψS2− (Fig. S4†). B@LSNR with specific frequencies could exchange a coded message104 similar to receiving the hν transforming into the holes compared with the standard known quantum materials. Gd3+ equipartitioned Nd2S3 at 750 °C, while DTA for Eu2S3 indicated uniform charge distribution compared to other LSNRs. The B@LSNR lost wt at 254 and 436 °C along endothermic and exothermic peaks at 750 °C. The Gd3+⋯S2− and Ln3+−S2−⋯Gd3+ strings could have aligned Eu2S3 with Gd3+. The Gd3+ 4f7e could not induce major oscillations with Eu2S3 except mass transitions at 254.82, 436.6, and 800 °C with 37.04, 27.54, and 35.42% wt losses respectively (Fig. S3 and S4†). Gd3+ with 2Eu3+ and 3S2− of Eu2S3 enhanced the holes via ψLn3+ ≈ ψS2−  ) with tetrahedron tentropic cavities (eqn (3.2)). B@LGT supersedes the Schottky and Frankel defects to regulate a current flow105 via ψLn3+ and ψGd3+ in a photocatalysing medium to calculate the energies as follows:
) with tetrahedron tentropic cavities (eqn (3.2)). B@LGT supersedes the Schottky and Frankel defects to regulate a current flow105 via ψLn3+ and ψGd3+ in a photocatalysing medium to calculate the energies as follows:| |
 | (3.2) |
ψS2−–Gd3+ mutually attracts while ψGd3+ × ψLn3+ repels with exothermic and endothermic energy cycles respectively to store energy. B@LGT with ψS2−–Gd3+ contraction and ψGd3+ × ψLn3+ expansion cycles are aimed to advance piezoelectric sensors, bionics, and, spintronics with ½ filled 4f@e.98 Er2S3@Gd3+ at 220 °C and 310 °C has been reoriented with a sharper mass transition than Eu2S3@Gd3+. Yb2S3@Gd3+ with hedgy oscillations credited to equidistribution with a similar value of electron defined probability |ψYb2S3@Gd3+|2 as Yb3+ has 4f13e (Fig. S3 and S4†). The B@LGTs with ψLn3+–S and ψGd3+–S have induced 1st mass transition at 50 °C than at 210 °C with LGT98 (Fig. 15c–f and S3†). B:Er2S3@GO has lost 23.7% at 129 °C compared to 28% wt loss from Eu2S3@Gd3+ at 143.8 °C, indicating higher stability for B:Er2S3@GO. The wt loss with B:Yb2S3@GO is 38.76% at 246.9 °C than 34.79, 18.42, and 23.79% at 258.3, 252.8, and 265 °C for B:Nd2S3@GO, B:Eu2S3@GO, and B:Er2S3@GO respectively, indicating the higher stability of B:Yb2S3@GO. The Peaks with B:Eu2S3@GO are sharpened at 210 °C upon aligning with Gd3+ (Table S13†). ψYb3+ and ψGd3+ of B:Yb2S3@GO might have induced a drift velocity of holes/charges via tentropic cavities (Fig. 15c–f). The DTG peaks with Gt are resonating with the same frequencies except with 15 μg min−1, 17 μg min−1, and −2.5 g min−1 at 250 °C, 450 °C and 730 °C respectively due to intimate sheet alignment with the stronger VWFs. Each sheet with a stronger FRET could not localize heat, while with GO a single DTG signal appeared as all the FGs and FEs of GO had equally oscillated with the same heat contents (Fig. 15b). It has predicted an equal FG distribution throughout a GO sheet without energy gradient and was chosen for coating. The DTG for B:Nd2S3@GO, Eu2S3@Gd3+, B:Er2S3@GO, and B:Yb2S3@GO with sharper structural changes at 258.3 °C, 252.8 °C, 265.0 °C, and 246.79 °C with 34.79, 18.42, 23.79%, and 38.79% wt loss have predicted a common lattice equally integrated with GO sheets (Fig. 15c–f).
 |
| | Fig. 15 TGA, DTA and DTG of (a) Gt, (b) GO and (c–f) B@LGTs. | |
4. PCPs interfacing photocatalysis at different T/K values
The monodisperse B@LGT surfaces oscillating with ψ in the same phase as of hν from visible light have allowed negligible rebouncing. Therefore, the PCPs have predicted stabilized surfaces generating resonating wavefunctions in the same phase as of hν. Thus, the interstitial surfaces have been proven asset to resonating the wavefunctions of photocatalyzing surfaces with that of the light sources. Fig. 16A illustrates the overall intimate density of energy states to manifold the emitted ψ accessing a cathodic dye. The viscosity with adequate adherence has predicted a surface partitioning with intimate continuity without causing the energy gradients that hurdle hν receptance. Basically, a medium of highest friccohesity economically allows each B@LGT molecule to participate for the PCR of a dye. Therefore, the concentrations of B@LGT were cautiously chosen to avoid nanoclustering that could have damaged an operative string depicted in Fig. 16. B@LGTs with the lowest sonication time have developed an intense photoluminescence on generating the resonating wavefunction w.r.t. visible light, which have photocatalytically reduced a dye in 30 min. The monolayer solvent adherence with a higher viscosity and the strongest surface continuity with adequate surface tension have been the asset to receive the hv.
 |
| | Fig. 16 (A) Wavefunctions of the surface in the same phase to receive maximum hν emitting phonons of robust ψ to photocatalytically reduce (B) Aq-LSNR, B@LSNR, and B@LGT. (C) PCR vs. sonication time. | |
For initiating interactions with the dipoles of water, 0.01 g% LSNR, B@LSNR, and B@LGT were sonicated at 28 kHz for 5, 3, and 2 h. The dipoles get aligned at the electrostatic sites of LSNRs for developing monolayer adherence with adequate coherence via an anticoagulating friccohesity mechanism (Fig. 16a). The order of B@LGT > B@LSNR > LSNR inferred monodispersion stability for 2 h, 3 h, and 10 months respectively. The order and time of stability have predicted the sharing of PE of LSNRs with dopants via repulsion with Ln3+ but attraction with S2−. Such infinite charge matrices could have been generated in a most symmetric pattern aligning the charges in electrostatic dipoles to flow the electron in response to the hν. Therefore, the coating with a 2D GO sheet with uniform distribution of FGs and FEs made the surfaces receptive for hν. At the same time, GO has equipartitioned the surface energies. Therefore, water monomers have entered the interstitial tiers of B@LGTs on adhering around the charged sites regaining the PE of same strengths. Such arrangement of charges has also attracted the dipoles of H2O to enhance the surface continuity in the flow of e− and h+ holes. The water monomers gained on disrupting the hydrogen bonded (HB) water have been depicted with the viscosity, surface tension, friccohesity and thermodynamic parameters like activation energy (Ea) (Fig. 21). The monodispersion and Ea have predicted the intimate adherence of B@LGT photocatalysts with BBR, TMIs, and QHIn for PCR (Fig. 16). Thermograms of the B@LGTs after the PCR of a dye developed a hedgy DTG pattern due to the adsorption of reduced dye with an intimate interfacial layer that exchanges energies through the FRET. Shifts in HR-TEM and FT-IR stretching of B@LGTs with reduced BBR predicted their adsorptions (Fig. 30 and 31). This is also proven with the BET analysis of B@LGTs that have adsorbed inert N2 gas upon increasing the relative pressure. The adhered water dipoles with B@LGTs are replaced with the reduced dye to separate. The unreduced dye and water interactions are stronger, while the reduced dye did not interact with the water dipoles. There is a novel interfacial science work with the reduced dyes, as these did not coagulate and did not get settle. Under this condition, a reduced dye opts to enter the interstitial cavities of the B@LGTs where water has stronger affinity to interact with the water molecules. Therefore, η, γ, σ, and thermodynamic properties explained the adherence of adequate H2O molecules without mixing the effluents. It had replaced the adhered H2O molecule with the reduced effluents having the affinities to be adhered with the interstities of B@LGTs, while their adhered water due to stronger dipolar interaction go together as the water phase pushes out the reduced effluents to the interstities. The reduced dye increased the density of B@LGTs and got settled. The PCPs such as η, γ, and σ affect this replacement (Fig. 16). The aq-B@LGT with dynamic property η elucidated the normalization of surfaces to cohere H2O dipoles with moderate adherence (Fig. 16f). The B@LGTs with monolayer water adherence to each site of interstities aligned similarly with slightly higher γ values. The interstitial strings LSNR–NHS–NHS–LSNR and B@LGT–NHS–NHS–B@LGT developed stronger interfacial continuity (Fig. 16f). The PCPs for 0.01 g% at 288.15, 298.15, and 310.15 K were measured, while at 283.15 and 313.15 K, they are calculated from the regression constants obtained from the above-mentioned temperature range (Tables 8–13). The monolayer water adhesion to aq-B@LGTs balanced by frictional and cohesive forces noted as friccohesity predicted energy equipartitioning.33 B@LGTs photocatalytically reduced BBR, TMIs, and QHIn in 15–40 min as NHS aligned around the charged sites that acted as molecular switches and linkers for reorienting to receive the hν to generate the holes. The Eg values of aq-B@LGT from 298.15 to 310.15 K decrease attaining a suitable reorienting state with uniform H2O adherence with a distinguished PL. Nd2S3 with 4f3e mildly realigned the H2O dipoles compared to Eu2S3, Er2S3, and Yb2S3 while B@LGTs sharpened PL to harness solar energy (Fig. 16B).
4.1. Recycling of wastewater effluents and recovery
A new pedagogy of B@LGTs as a novel research methodology for recycling wastewater effluents has been highly economic for getting rid of nuisance of polluting effluents. Initially, their PCPs have predicted an entry of H2O dipolar molecules at NTP, generating certain internal pressure due to interactions with BBR on PCR. Thus, the PCPs have been substitution of BET to analyse structural stability and adsorption abilities at variable T/K. Since, the H2O dipoles adhered to each interstities have softened the functional surface area. As before PCR, no solvated effluents could have entered the adhered H2O. While with time, the reduced effluents have been aside by the cohesive force of H2O moving in two ways notably the post-PCR dynamics for a recovery of the reduced effluents. The mutual communication of B@LGTs and effluents via FRET predicts spontaneity and sustainable PCR mechanism. Fig. 17c depicts the wastewater recycling potential of interstitial B@LGT initiated by monodispersing BBR in a common solvent, which elucidated the gradients in interacting activities of unreduced and reduced effluents represented with PCPs as a key prerequisite database for extensive recycling studies. The energy gradients signify the higher friccohesity values on unreduced effluents which with time decrease on phasing out a water from B@LGT and effluents both, it forms a pure water phase with lower friccohesity values. At the end, the effluents have replaced the water dipoles, which upon gaining density get settled with shorter size.
Thus, their monodispersion has occupied a higher surface area with higher adhesion by remaining suspended in a thermodynamically stable solution with their stronger FRET. The hν has reduced the effluents that had acted as the cathode by causing a gradient in energy, separating them (Fig. 17).
 |
| | Fig. 17 (a and b) CCPs for LSNRs with NHS monodispersing photocatalysts for symmetric Brillion zones avoiding hν annihilation. (c) PCPs elucidating monodispersion with an intimate FRET for recycling effluents and clean water through photocatalysis. | |
4.2. Viscosity (η, mPa s) for molecular alignments
Rovitronic motions with specific rheological resistance with adequate relaxation time to mimic ψsv to photocatalytically reduce BBR, TMIs, and QHIn. The twisting or deforming of solvated B@LGTs during PCR disrupts the interfacial continuity that hampers receiving of hν. Therefore, monolayer adhesion with moderate cohesion has predicted continuity and is measured with friccohesity. η is a prerequisite of friccohesity interfacing a photocatalyst to capture hν, which is calculated as follows:33| |
 | (3.3) |
where ρ is the density of the solution, ρ° the density of water, η° the viscosity of water, ηr the relative viscosity, and vft the relaxation time (τ). The shear rate with certain heat capacity has stabilized the charge centres tuned by T/K (Tables 8 and 9). The consistent shear stress and rate are both inversely proportional to the uniform lattice of wavevector ‘k’ (−π/a, 0, π/a), and are analysed by DTG and fitted with the Schrödinger equation transforming PE to KE as follows:33| |
 | (3.4) |
V(x, t) = 0 energy at position x at time ‘t’ aligns the B@LGTs to receive the hν with higher η and lower surface tension106 (Fig. 18). The 4f@e have behaved like an upper critical (UC) solution to increase the PCR rate shortening the PCR at 298.15 K. The 4f@e spins of B@LGTs have enhanced the consistent interacting activities107 noted as cohad consistency points (CCPs) aligning the adhesive and cohesive energies as follows:| |
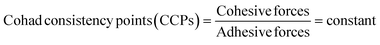 | (3.5) |
NHS–NHS, NHS–LSNR, and NHS–LSNR–NHS with B@LGTs have been aligned by the cohesive, adhesive, and cohad forces respectively (Fig. 17 and 18). The η values for aq-Nd2S3 and Yb2S3 have coincided at CCPs with equilibrated cohad forces at 288.15 K. The dopant has shifted the CCPs values to 298.15 and 300 K while a coating agent GO to the 291.15 and 298.15 K, respectively (Fig. 18a and b). The B@LGTs of Nd2S3 and Eu2S3 with partially while the Er2S3 and Yb2S3 fully filled 4f orbitals have coincided with CCPs respectively (Fig. 18c). Gd3+ and Ln3+ both have attracted the Oδ− of H2O interfacing the S2− with adhesion = cohesion of solvent competitively. The CCPs, the novel interfacing with PCPs in the lanthanide chemistry, have advanced the PCR.108 The NHS with dopant and matrices have generated the CCPs around the B@LGTs.61 The B@LGTs of Nd2S3, Eu2S3, Er2S3, and Yb2S3 at 291.15 and 298.15 K respectively have produced the CCPs while the LGTs of La2S3, Ce2S3, Tb2S3, and Ho2S3 at 283.15, 298.15, and 315.15 K, as reported previously 33 (Fig. 18c). The origin of the CCPs infers energy with ψsv of a longer wavelength. The CCPs for a series agree at comparable T/K on a common alignment. LSNR and B@LGTs both might have generated the vibrational (Ev) and rotational (Erot) energies as Ev = Erot via NHS at 315.15 T/K (Fig. S9†). The H2O alone on monolayer adhesion has fastened the fluid flow with η as Nd2S3 > Yb2S3 > Eu2S3 > Er2S3 > H2O at 298.15 than Yb2S3 > Eu2S3 > Er2S3 > Nd2S3 > H2O at 310 K. The η values at 300 K infer constant cohad forces with similar CCPs due to the LSNR–NHS linkages (Fig. S12†). The NHS around S2− and Ln3+ have synchronized the spins for the similar activities predicted from the CCPs (Fig. 17). The η values for H2O differ from that of the aq-LSNR as the NHS remained the same with CCPs at 315.15 K (Fig. S12†). The NHS adhesion to Eu2S3, Er2S3, and Yb2S3 has interfaced with that of the Gd3+ by avoiding their deposition including of cholesterol.109 The competitive alignment of electronegative O2− of H2O with either the Ln3+ or the Gd3+ has truly created an energy distributing ingredients. As Ln3+ is bonded with S2− through the primary bond, Gd3+ is bonded through the secondary bond. These have been functional interstities to align the 2D GO sheet for generating a higher surface area and the frictional forces depicted with the η. That were obtained on aligning the water dipoles for PCR with robust FRET. Because the said charge intricacies are ultrasensitive to thermal energy and hence PCPs were studied at 283.15, 288.15, 298.15, 310.15 K and 313.15 K. The B@LGTs could be an alternate quantum material for the CT scan for body X-rays22 with more contrast110 than Gd3+ alone generating ψsv of longer λ. The η value of B@LSNRs as B@Er2S3 > B@Yb2S3 > H2O > B@Nd2S3 > B@Eu2S3 has predicted the effective adherence of water around B@LSNRs than others with least Ea for the PCR. The higher η values for Er2S3 and Yb2S3 than Nd2S3 and Eu2S3 have inferred the monolayer adhesion of water dipoles. The Nd2S3 and Eu2S3 with the CCPs at 300 K than Er2S3 and Yb2S3 at 298.15 K with higher η values have normalized the spins unlike Nd2S3 and Eu2S3 with less ½ filled 4f electrons (Fig. 18b). Nd2S3 with stronger adhesion at 288.15 K has responded with robust FRET than composition (Fig. 18 and S12a†). Nd2S3, Eu2S3, Er2S3, and Yb2S3 have reoriented the solvated state with a common shear but are narrowed down to 315.15 K (Fig. S12a†). Gd3+ has streamlined a shear stress with extra CCP at 300 K, while a common CCP for Er2S3, and Yb2S3 at 298.15 K has higher η values. The dopant has partially equilibrated ψS2− and ψLn3+ of B@LGT on aligning the NHS dipoles with different CCPs at 291.15 and 298.15 K.
 |
| | Fig. 18 Viscosities for (a) aq-LSNR, (b) aq-B@LSNR and (c) aq-B@LGT plotted vs. temperatures with higher resolution for B@LSNRs, while the values of B@LGTs are in a narrow range. | |
4.3. Surface tension (γ, mN m−1)
Since, the Ln3+⋯H2O and Gd3+⋯H2O interactions have equipartitioned their energies where the Ln3+ is covalently bonded with S2− while the Gd3+ with the secondary. These charge wavevectors along GO have developed the electrostatic dipolar alignments of H2O at the air-aq-B@LGTs interfaces. This directly influenced the receptance of hv from solar energy. The surface energy noted as surface tension (γ) directly affects the hv interactions. Lower γ values with small sized pendent drop number (pdn) were formed on NHS adherence. The pdn were used to calculate γ values with eqn (3.6), the lower γ values have enhanced a PCR.61| |
 | (3.6) |
where γ°, n°, ρ° and γ, n, ρ are the surface tension, pdn, and density of water and solution respectively. The γ ∼78 mN m−1 for LSNR and B@LSNR infer realigned H2O with Ln3+ and with S2− with stronger cohesive forces at 288.15 and 298.15 K (Fig. 19). The B@LSNRs have decreased multilayer adhesion upon shortening the PCR time (Fig. 18a and Tables 10, 11). aq-LSNR with 65–77.28 and B@LSNR with 51.98–76.64 mN m−1 photocatalytically reduced BBR, TMIs, and QHIn in a longer time than that of B@LGTs (Fig. 19b). The B@LGTs have weakened the dipolar attraction than LSNR and B@LSNR by partitioning the NHS. The higher γ values at 288.15 than at 298.15 K infer the manifold NHS with a lower surface energy, while after 310 K, a decrease in the γ value on monolayer water adherence is observed (Fig. S13†). The NHS and Gd3+ have aligned ψS2− and ψLn3+ decreasing γ from 78 to 77.6 mN m−1 at 298.15 K (Fig. 19). The γGd3+ = γLSNR − γB@LSNR infers a reorientation of ψS2− and ψLn3+ with a slightly higher relaxation time on equipartitioning the NHS. The η3+Gd = ηB@LSNR − ηLSNR is linearly decreased (Fig. 11 and 18) upon lowering γ shifting from LSNR to B@LGTs by reducing BBR in a shorter time, in agreement with Fermi energy (eqn (3.7)):| |
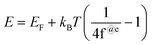 | (3.7) |
Gd3+ with E–EF synchronized both γ and η as B@LSNRs have slightly scattered the γ values at higher T/K than at 298.15 K due to the competitive Gd3+–NHS and Ln3+–NHS interactions without ψGd3+ = ψS2− = ψLn3+ at 298.15 K. The ψGd3+ value has enhanced the surface energy for B@LSNR as γLSNR > γB@LSNR at 298.15 K (Fig. 19a and b). The spins of Ln3+ are sensitive to T/K compared to composition while S2− with shared orbitals is not so sensitive to oscillate.
 |
| | Fig. 19 Surface tension of (a) aq-LSNR, (b) aq-B@LSNR and (c) aq-B@LGT plotted against temperatures with a higher resolution. | |
The aq-B@LSNR with higher friccohesity than aq-LSNR have enhanced ψS2−, ψLn3+ and |ψLSNR|2 to |ψGd–LSNR|2 on attraction and repulsion respectively enhancing the PCR rate. The B@LGTs have synchronized the deficient sites of BBR, TMIs, and QHIn with higher UV-vis absorption111 (Fig. 12 and 26). The B@LGTs with Eg = 1.88 eV with intense wave and charge sensors may tune the confirmatory state of protein or intercalation of DNA.112 Higher γ values for LSNR and B@LGT are coincided at 298.15 and 310 K than lower with B@LSNR at 298.15 K equipartitioning an energy (eqn (3.8)):
| | |
E = Ee− + ENu+ + EEER + Ee–Nu + ENu–Nu
| (3.8) |
E is the vital line oscillation on increasing
T/K
via moderate shear and uniform surface energy to track
hν. The B@LGTs have quantized the laminar flow and surface energies expressed as friccohesity (
Fig. 20).
4.4. Friccohesity (σ, S cm−1)
The friccohesity basically aligns the photocatalysts avoiding deformation and wrinkling of surfaces, which cause surface twisting that inhibit the receiving of hν. The solvent has monodispersed the B@LGTs developing the thin hydration spheres attained via interstities. Thereby, the adequate adherence of H2O dipoles from monolayer to multilayer generates a protective mechanism noted as cohesive force with sufficient PE to avoid twisting or coagulating, coalescence, of B@LGTs, where hv is received by generating the favourable rovitronic activities. The monodispersion have been tracked with the vft and pdn values calculated with eqn (3.9):| |
 | (3.9) |
where σ and σ0 are the friccohesity of solution and solvent, respectively, ±B/t is the kinetic correction and 0.0012(1 − ρ) is the buoyancy correction, and other symbols are depicted as usual.113 The corrections of 1 × 10−7 order are omitted and the Man Singh constant (Mc) depicts the shear stress and surface energy coefficient of aq-B@LGTs (eqn (3.7)). The σ values have predicted the surface forces (Tables 11 and 12) and laminar layer on getting the rovitronic motions of monolayered adhesion of water around B@LGTs (Fig. 17). Because the energies and minimum entropies of B@LGTs on getting reoriented are highly interfacial and mutual tuned with electronic oscillation to receive hν. Therefore, the values of γ and η individually could elucidate the intimate intricacies of the ultrasensitive interfacial hairline marked with the friccohesity. The σ realigns the NHS dipoles in compliance with Gd3+ for PCR in a longer time. The γ values have distinguished the surface energies confined to more than two CCPs than the σ at variable T/K (Fig. 20). The CCPs have predicted a monolayered NHS–B@LGT validated with stretching and bending frequencies with the FT-IR spectra (Fig. 12). ψS2− and ψLn3+ with |ψLSNR|2 ≠ 0 seem to align the NHS dipoles with ψS2− separating the CCPs as T/K sensitive. The η values infer ψLSNR, ψB@LSNR, and ψB@LGT interfacing the surface charges of the effluents33 to intensify the CCPs with different γ and friccohesity values at 288.15 and 298.15 K with an integrating fluid dynamics104 (Fig. 20). The friccohesity for aq-B@LGT photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively.
 |
| | Fig. 20 Friccohesity of (a) aq-LSNR, (b) aq-B@LSNR and (c) aq-B@LGT plotted against temperatures with higher resolution for LSNR and B@LSNRs, while the values of B@LGTs are in a narrow range. | |
4.5. Thermodynamic parameters derived from PCPs for PCR
Thermodynamic parameters (Ea, ΔH, ΔG, and ΔS) derived from the viscosity database predicted the spontaneity of a photocatalysing medium.61 The ΔH value ranging from −0.6 to −6.7 kJ mol−1 with ΔG < 0 on decreasing linearly on raising T/K indicates monodispersion fastening the PCR of BBR, Cr3+, Ni2+, and Cu2+, and QHIn with B@LGT in 30, 20, 40, 35, and 15 min respectively. ΔH, ΔG, and ΔS with aq-B@LGTs at lower T/K predict a spontaneity for the PCR than at higher T/K (Fig. 21). The photocatalysing medium with ΔH < 0 < TΔS at lower T/K than higher shortened the PCR time, calculated as follows:33| |
 | (4.0) |
The ΔG ≪ 0 ≫ ΔS has lowered the PCR rate and plot of ΔG vs. ΔS is nonlinear due to unaligned functional sites at higher T/K. Gd3+@Eu2S3 has shifted a common T/K point to 310 than 308 K with Eu2S3 as the Ea of dopant and LSNR matrices get aligned at the cost of restricted collisions (Fig. S7 and S8†). The common T/K point is supplemented with ΔS and B@LSNRs have lowered a common point to 298.15 K on equal energy distribution. The Gd3+@Er2S3 and Gd3+@Nd2S3 have been aligned with nearly half and fully filled 4f orbitals with ΔG ≥ ΔS in the same ratio25 at lower T/K (Fig. 21). The Gd3+@Yb2S3 and Gd3+@Eu2S3 with similar ΔG, ΔS, and ΔH trends are credited to the number of electrons in the 4f orbital. The ΔG, ΔH, and least ΔS with B@LGTs have photocatalytically reduced BBR, TMIs, and QHIn in 15–40 min, and Ea was calculated with log(η) vs. (1/T) with eqn (4.1):33
| |
 | (4.1) |
The ΔH, ΔS, and ΔG values were calculated using Ea as follows:33
| |
 | (4.2) |
B@LGT with ΔH < 0 and Ea from −670.14 to −925.57 J mol−1 with B:Nd2S3@GO to B:Yb2S3@GO indicates a spontaneous photochemical reaction (Fig. 21 and Tables S22–25†). The ΔS < 0 < ΔS up to 312 K is noted as the effluent saturation point33 on being weakly adsorbed by B@LGT by releasing the ΔH. The B@LGTs with least ΔS with ΔG < 0 > ΔH have PCR a BBR in 30 min (Fig. 21). The ΔG < 0 values were obtained on receiving the hν that had enhanced the π → π* favouring the PCR.
4.5.1. Ea from fluid dynamics (Ea,η) and UV/vis (Ea,UV). The higher negative Ea values for undoped LSNR than the B@LSNR are decreased on enhancing the interacting activities as KE ≫ PE (Tables S14–S21†). The B@LSNR with strongly aligned water dipoles closely interfaces to photocatalytically reduce the effluents in a shorter time than LSNR. Thus, no charges were originated out of the Ln3+⋯H2O and Gd3+⋯H2O matrices due to energy equipartitioning that were missing with the LSNRs.Ln3+⋯H2O and Gd3+⋯H2O are missing with LSNRs. The lower Ea values with aq-B@LGTs than B@LSNR and LSNR determined from their respective UV/vis abs and fluid dynamics have enhanced the PCR activities. Ea,UV in the range of −3.95 to −30.12 J mol−1 for Nd2S3@GO, Eu2S3@GO, Er2S3@GO, and Yb2S3@GO than Ea,η from −210.62 to −700.25 J mol−1 for Nd2S3, Ee2S3, Er2S3, and Yb2S3 consumed more energy in thermodynamic reorientations than UV transition (Tables S7–S10 and S14–S17†). Ea,η for B@Nd2S3, B@Eu2S3, B@Er2S3, and B@Yb2S3 decreased from −361.47 to −765.88 J mole−1 upon entering water dipoles to their interstities causing the reorientational motions with continuity in capillary flow (Tables S18–S21†). The hν of UV light excited electrons from VB to CB of unreduced BBR by overcoming a fermi energy resistance to interact with BBR. The B@LGTs with monolayer NHS in the excited state expeditiously got reoriented for transferring the holes to BBR by avoiding a mismatch in spatial occupancies. Ea,η acquired from a system is more negative while an increasing T/K has compensated Ea,η at a constant concentration by avoiding the interactions with self for receiving the maximum hν. Ea,UV of aq-B@LGTs and BBR with time was compensated with Ea,η on increasing T/K. The T/K increments have controlled the rovitronic motions in fluid dynamics for optimizing the state. The molecular reorientations have gained much energy, while UV absorption has overcome the resulting motions, Ea,η > Ea,UV. The Ea,UV and Ea,η interfacing has predicted the monodisperse photocatalysts for receiving more hν for PCR buffered with water dipoles (Tables S7–S25†). The B@LGTs acquire less Ea,UV and Ea,η with least Eg to PCR the effluents in a shorter time than others, which is given as follows (Fig. 21e):
| |
 | (4.3) |
KPCP predicts the spontaneity of B@LGTs that photocatalytically reduced BBR in 30 min in 1st order with the residual coefficient (
R2) from 0.87 to 0.98. The B@LGTs could not adsorb an unreduced dye on stronger water adhesion while a reduced dye has replaced H
2O to get adsorbed onto B@LGTs.
| | |
B@LGTs + hv → B@LGTs* → B@LGTs + holes → holes + BBR → reduced BBR
| (4.4) |
Low Ea,UV and Ea,η allowed aq-B@LGTs to adsorb reduced BBR as B@LGTs-BBR acquire energy to come out of NHS sheath. The B@LGT molecules on getting reoriented have aligned a BBR for PCR while the reduced BBR was adsorbed by B@LGTs predicted with PCPs.
| | |
hv + B@LGTs = holes = BBR ≅ B@LGTs − BBR + ΔH = −ve
| (4.5) |
Hence, the exchange of adhered H2O to B@LGTs before the PCR of effluents has different energetics than a condition when adhered H2O is replaced with reduced effluents.
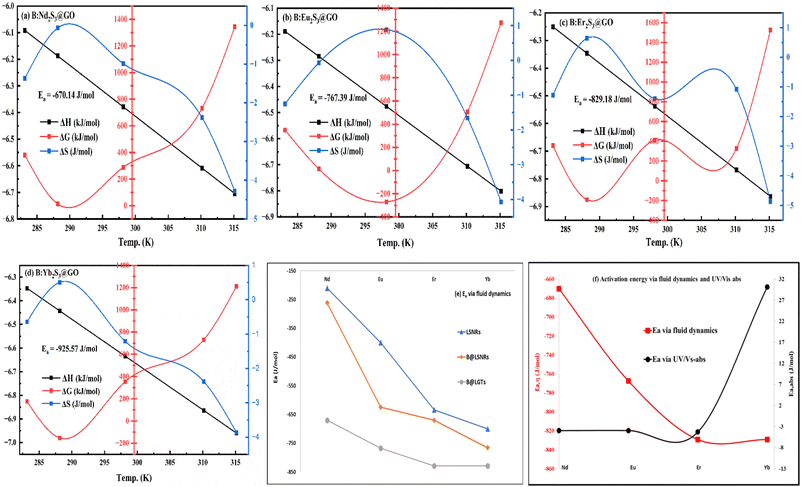 |
| | Fig. 21 (a–d) For B@LGTs, dH linearly decreased while dG and dS oppositely vary upon release of heat content on increasing temperature with reorientations and (e) Ea for photocatalysis. (f) Ea calculated from fluid dynamics and UV/vis-abs. | |
5. PCR for BBR, heavy metal ions, and QHIn on subsequent adsorption
To begin with, the B@LGTs photocatalytically reduced the pollutants by neutralizing their electronic-rich and -deficient sites to disconnect them from wastewater molecules. The B@LGTs have received the hν to generate the e− and h+ holes approaching their respective sites along the splitting of H2O. The pollutants had developed stronger linkages with the H2O dipoles through their respective N+–, SO3−, active methyl groups, delocalized π conjugation sites, and Cr3+, Ni2+, and Cu2+ did not allow adsorption (Fig. 22a and 28h). The QHIn with phenolic groups developed the linkages with stronger ionic interactions without adsorbing with B@LGTs. Thus, the B@LGTs could not adsorb them without PCR due to their stronger linkages with H2O dipoles than the B@LGTs. The B@LGTs supplemented with the hν had generated the e− and h+ holes to attack their linkages with the H2O dipoles by reducing their electronic sites responsible for linkages. Thus, B@LGTs did not directly adsorb the pollutants, and no direct adsorption isotherm/equilibrium is possible as the PCR and adsorption had worked simultaneously. Thus, the aim of the study was not to establish an adsorption but to photocatalytically reduce those, which were adsorbed at the same rate from a homogeneous solution to recycle wastewater. Before PCR, the BBR dye and other pollutants had emitted colour on receiving the hν where their electronically rich and deficient sites oscillated. The PCR had neutralized them, making them unable to respond to the light, and resulting in colourless solutions. No fragmentations of their structures were aimed except PCR and separation from wastewater, focusing on recycling wastewater. Thus, the physicochemical and PCR studies of B@LGTs are directly connected to excite the VB to CB electrons. The sole moto to study PCPs at different T/K with the same composition was to investigate the kinetic behaviour of 4f electrons. Thus, the PCR experiments designed with B@LGTs of Nd, Eu, Er, and Yb metals upon increasing their 4f electrons were correlated with the PCPs. The density of energy states of Gd and GO both had caused to receive the maximum hν as the Fermi energy gap between their VB and CB was less. This had favoured to excite the VB electrons to the CB without collisions and recombination. The adsorbed pollutants were analysed by FT-IR spectroscopy, and their sites did not respond to IR except resembling to B@LGTs114 (Fig. 23f). The λmax value of effluents did not change in the darkness, while on receiving hv they have generated hole pairs avoiding their recombination (Fig. 22). The UV-vis band spectra were reduced at specific λmax due to single electronic response of unfragmented effluents (Fig. 23 and 27). The FT-IR spectra of BBR has a vibrant fingerprint region on robust bending but after PCR, its quaternary nitrogen (N+–) was reduced, which neither responded to IR nor the H2O dipoles. The electron was shifted towards N+– with stronger electron finding probability enhancing its interaction with H2O dipoles. On PCR, BBR has reduced the vibrancy of bending and adsorbed without structural fragmentation (Fig. 23f). The conductivities of 0.01%g aq-B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO as 30.24, 25.50, 27.23, and 24.50 μs cm−1 are produced with 2.23, 2.28, 2.38, and 1.88 eV Eg, respectively (Fig. 8 and 32c). For aq-B @Nd2S3:GO, the lower the Eg, the higher the conductivity. The B@Yb2S3:GO have generated Eg = 1.88 eV with 24.50 μs cm−1 conductivity credited to 4f13 electrons of Yb3+. The one unpaired electron out of the 4f13 electrons has reached to CB without causing much collisions. The B@Yb2S3:GO being large in size has low conductivity (Fig. 32c). This research work established a relation among the conductivities and Eg and the number of 4f electrons of LSNRs coated with photosensitive materials such as GO (Fig. 24). The PCR performance was correlated with conductivity, bandgap energy, and BBR quantum yield, as the electronic configuration of the respective B@LGTs has exhibited variable electronic spins due to nuclear charge applied on increasing the 4f electrons in the same period of periodic table. The variable spins of Nd3+, Eu3+, Er3+, and Yb3+ of Nd2S3, Eu2S3, Er2S3, and Yb2S3 respectively align the 4f7e of Gd3+ on equipartitioning the overall energies, which are subsequently consolidated with FGs and FE of GO. Thus, in the 1st step, a dopant reduced the Fermi energy gap that was further integrated with GO to respond to the external electronic potential with certain mobility noted as conductivities. The least permittivity between the VB and CB has enhanced the mobility of respective ions in the conductometer. A similar mechanism seems to follow during PCR in transforming the hole pairs supported with PCPs and thermodynamic parameters (Fig. 17). The exponential redox cycles without dissociating Ln3+ and S2− from LSNRs, nor Gd3+ dopant, leached out except narrowing down the Fermi energy gap. These mechanisms have safeguarded optically active GO and equally adhered with B@LSNRs upon generating a high frequency of FRET confining energy to a shorter Eg rather than dissipating. Thus, the magnitudes of PCPs such as conductivity, viscosity, and surface tension promote the PCR of effluents. Therefore, the conductivity and other PCPs of B@LGTs have analysed a mobility of holes for PCR as well as their interacting activities with dipolar water as a protective mechanism. Moreover, a decrease in their conductivities upon increasing the PCR time indicated the adsorption of reduced BBR, TMIs, and QHIn species (Fig. 32c). The respective charge centres of these species had stronger interactions with water dipoles with higher mobility,115 but the reduced species could not interact with water and got adsorbed with B@LGTs. Thus, their charge centres, which were individually interacting with water with higher mobility, were now occupied mutually, and the water was phased out with stronger cohesive forces with lower mobility or conductivity. The sizes as Yb2S3 > Er2S3 > Eu2S3 > Nd2S3 with the 4.67, 4.75, 4.71, and 4.86 eV Eg values than the Gd3+@Yb2S3 > Gd3+@Er2S3 > Gd3+@Eu2S3 > Gd3+@Nd2S3 B@LSNR have generated the two Eg values 4.64–1.90, 4.62–2.41, 4.59–2.43, and 4.50–2.50 eV respectively (Fig. 8). The Eg values of LSNR are increased with the increase in 4f electrons from 13, 11, 7, and 3 for Yb3+, Er3+, Eu3+, and Nd3+ as their collisions on occupying 4f orbitals are minimized. Gd3+ has partitioned Eg from VBLSNR to CBLSNR and VBGd3+@LSNR to CBGd3+@LSNR for Gd3+ and Ln3+ respectively, where S2− has a similar electronic state except orienting effects. The B @Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO have two Eg, such as 3.97–1.88, 4.03–2.38, 4.02–2.28, and 4.05–2.23 eV, respectively, for completing PCR in 15–40 min. Gd3+ has shared the PE of LSNR enhancing a KE on reducing the Eg values. Thereby, with more KE, the Gd3+ has reduced the size of B@LSNR to B@LGT from 100 to 20 nm. From B@LSNR to B@LGTs, the ∼62 to 78 mN m−1 within the 100 to 20 nm size respectively have PCR the BBR, TMIs, and QHIn in shorter time. The decrease in the size of B@LGTs with the increase in γ values directly predicts the alignment of Ln3+⋯H2O and Gd3+⋯H2O, though S2− bonded with the Ln3+ through the covalent bond seems to work similarly. Both Ln3+⋯H2O and Gd3+⋯H2O have addition effects on aligning H2O mutually interconnecting together, similar to the HB away from the bare H2O. The B@LGTs have catalysed an alignment of water dipoles on increasing the dipolar forces at an air–liquid interface. The B@LGTs w.r.t. water dipoles with stabilized charges might have generated
the synchronized holes that had shortened a PCR time along monodisperse the effluents. The B@LGTs having 1.88–2.38 eV Eg with lower permittivity on intimate continuity116 indicate the stabilized dielectric dipoles for PCR activity fitted using Schrödinger eqn (4.6) as follows:| |
 | (4.6) |
where pis the momentum, m is the mass, V(x, t) is PE at x position and t time, and r, nm is the distance between B@LGTs and effluents. At t = 0 (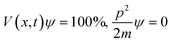 while changing t from 5 to 30 min, the
while changing t from 5 to 30 min, the  is >90% and V(x, t)ψ is <10%, as predicted via PCPs database with B@LGTs (Fig. 22). H2O dipoles adhered with the B@LGTs were replaced by reduced BBR, TMIs, and QHIn and have restricted the stretching and bending causing hedgy oscillations despite a lower surface tension than the aq-B@LSNR (Fig. 17 and 19). The uniform intense oscillating patterns produced with TGA after PCR than a sharper before PCR due to equidistribution of BBR with interstitities of B@LGTs (Fig. 31). Their interfacing with effluents is accountable for stronger physisorption with localized FRET exchanging their mutual energies. The PCPs have revealed their unique response on lowering γ and relaxation time on specific adherence and coherence. The Gd3+ dopant and GO due to the spins of 4f electrons and stretching of FGs respectively have accommodated the water dipoles (Fig. 22). Thus, an unreduced effluent did not enter the interstities of BL@LGTs except H2O dipoles to lower the γ. The DTA analysis of B@LGT before and after PCR predicted a uniform energy distribution (Fig. 31). The BBR has been uniformly aligned with synchronized interstities of B@LGT depicted with PCPs like surface properties (Fig. 18–20). The B@LGTs could form the oxygen-based superoxide radicals (O2˙−) on splitting the wastewater effluents.115 O2˙− on protonation seems to produce hydroperoxyl radicals (HO2˙−) and free hydroxyl radicals (OH˙).43 The BBR after PCR adsorbed by B@LGT with higher density and negligible VWFs with water get settled at bottom (Fig. 22 and 23). The aq-B@LGTs with slightly higher coherence on aligned H2O dipoles have enhanced the mobility of holes and photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively. The B@LGTs have absorbed the hν at 228 and 312 nm similar to LGTs via FRET.33 Their spins at 302 nm resonated with the BBR dye at 550 nm λ to overcome QEB117 (Fig. 7a and b). The PCR activities with B@LGTs of Nd (4f4), Eu (4f7), Er (4f12), Yb (4f14) have shortened the PCR time by 50% compared to the TMI-GO of Ni (3d8), Zn (3d10), Cd (4d10) Cr (3d5), Mn (3d5), and Fe (3d6).61,118 The B@LGT with Eg (1.88–2.30 eV) harnessed solar energy two times than TMI-GO (2.12–4.47 eV) and 4 times than GO33,61 as 4f@e have generated holes expeditiously than 3d electrons. The B@LGTs have reduced BBR, TMIs, and QHIn in 15–40 min as B:Yb2S3@GO > B:Er2S3@GO > B:Eu2S3@GO > B:Nd2S3@GO with 1.88 < 2.38 < 2.28 < 2.23 eV Eg respectively. Their resonating constituents have higher KE with longer λ with least permittivity.
is >90% and V(x, t)ψ is <10%, as predicted via PCPs database with B@LGTs (Fig. 22). H2O dipoles adhered with the B@LGTs were replaced by reduced BBR, TMIs, and QHIn and have restricted the stretching and bending causing hedgy oscillations despite a lower surface tension than the aq-B@LSNR (Fig. 17 and 19). The uniform intense oscillating patterns produced with TGA after PCR than a sharper before PCR due to equidistribution of BBR with interstitities of B@LGTs (Fig. 31). Their interfacing with effluents is accountable for stronger physisorption with localized FRET exchanging their mutual energies. The PCPs have revealed their unique response on lowering γ and relaxation time on specific adherence and coherence. The Gd3+ dopant and GO due to the spins of 4f electrons and stretching of FGs respectively have accommodated the water dipoles (Fig. 22). Thus, an unreduced effluent did not enter the interstities of BL@LGTs except H2O dipoles to lower the γ. The DTA analysis of B@LGT before and after PCR predicted a uniform energy distribution (Fig. 31). The BBR has been uniformly aligned with synchronized interstities of B@LGT depicted with PCPs like surface properties (Fig. 18–20). The B@LGTs could form the oxygen-based superoxide radicals (O2˙−) on splitting the wastewater effluents.115 O2˙− on protonation seems to produce hydroperoxyl radicals (HO2˙−) and free hydroxyl radicals (OH˙).43 The BBR after PCR adsorbed by B@LGT with higher density and negligible VWFs with water get settled at bottom (Fig. 22 and 23). The aq-B@LGTs with slightly higher coherence on aligned H2O dipoles have enhanced the mobility of holes and photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively. The B@LGTs have absorbed the hν at 228 and 312 nm similar to LGTs via FRET.33 Their spins at 302 nm resonated with the BBR dye at 550 nm λ to overcome QEB117 (Fig. 7a and b). The PCR activities with B@LGTs of Nd (4f4), Eu (4f7), Er (4f12), Yb (4f14) have shortened the PCR time by 50% compared to the TMI-GO of Ni (3d8), Zn (3d10), Cd (4d10) Cr (3d5), Mn (3d5), and Fe (3d6).61,118 The B@LGT with Eg (1.88–2.30 eV) harnessed solar energy two times than TMI-GO (2.12–4.47 eV) and 4 times than GO33,61 as 4f@e have generated holes expeditiously than 3d electrons. The B@LGTs have reduced BBR, TMIs, and QHIn in 15–40 min as B:Yb2S3@GO > B:Er2S3@GO > B:Eu2S3@GO > B:Nd2S3@GO with 1.88 < 2.38 < 2.28 < 2.23 eV Eg respectively. Their resonating constituents have higher KE with longer λ with least permittivity.
 |
| | Fig. 22 (a) Aligned lattice interstities for hole generation without recombination. (b) Relationship between the bandgap energy and the quantum yield (%) of BBR dye. | |
 |
| | Fig. 23 (a–d) PCR of BBR with B@LGTs and (e) B@LSNRs. (f) FT-IR spectra before and after PCR. (g) Relation between Eg and Φ. | |
 |
| | Fig. 24 Negative holes of B@LGT (anodic) approaches towards the quaternary nitrogen of BBR (cathodic) under sunlight and resemble the energy storage of supercapacitors. | |
The amount of dye photocatalyzed with time t noted as Ct was calculated from initial amount (C0) expressed as (C0−Ct). The plot (Ct/C0) vs. time gave 2nd order, while the ln(C0/Ct) vs. time, indicated 1st order kinetics to PCR with a residual coefficient R2 = ∼95–99 with 96.35–99.17% Φ (Fig. S5† and Table 7). 40 ppm of BBR was mixed with 0.01 g% of B@LGT for PCR and decreasing UV/vis absorption with time has indicated the PCR of the dye. The absorption vs. time plot was used for calculating Ea when electronic transitions occurred, while the viscosity vs. T/K plot was used for determining when the B@LGTs underwent rovitronic motions. Thus, the electronic and molecular motions have connected the Eg and Ea values mutually plotted with time (eqn (S1)†). The 96.35, 96.67, 97.60, and 99.17% with B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO in 30 min while the 42.11, 42.22, 42.33, 43.51% Φ with B@Nd2S3, B@Eu2S3, B@Er2S3, and B@Yb2S3 respectively have predicted the significance of doping and coating both (Table 7, S7–S10 and 32b†). The decrease in Eg has increased Φ (%), and Gd3+ has equipartitioned the PE to KE as per Schrödinger eqn (4.6). Gd3+ with half-filled stability oscillating with longer λ with QTE avoided the electronic collisions and recombination. The holes got strengthened to reach CB channelized to effluents. The lower Eg value with expeditious excitation of 4f electrons has photocatalytically reduced in a shorter time producing maximum Φ (%) without recombining the holes (Fig. 22b and Table 7). The BBR was reduced with B@LGTs from 5 to 30 min at 550 nm with almost similar slope values in the 1st order. The 1st order has predicted that BBR had received the holes at the same rate with each B@LGT being also adsorbed in the first order without any reaction (Fig. S5a†). Thus, the B@LGTs have been reused 10 times to photocatalytically reduce the 10 fresh samples each of 40 ppm BBR than B@LSNR. The constant slope with B@LGTs inferred that the doping and coating have uniformly engaged the LSNR matrices with almost similar charges. The novelty of B@LGTs was to photocatalytically reduce a dye under visible light at 660 nm λ. The fingerprint region of FT IR for BBR before and after PCR remains almost the same except the variation in transmittance. The overall trends of stretching and bending remain the same as no functional sites are occupied permanently during chemisorption with BBR, but the physisorption works well (Fig. 23f). The physisorption of reduced BBR by B@LGTs attained as the water dipoles adhered with unreduced BBR were phased out. Thus, the PCR of charged sites phased out the water dipoles from the dye as the reduced dye phased out water dipoles from B@LGTs, which is noted as a greentech to recycle water (Fig. 22). These changes accompanied infinitesimally small changes in thermodynamic parameters, which predicted phasing of water dipoles from dye to water phase while to dye from water, in case of B@LGTs.
Table 7 Kinetics at pH 7, 0.01 g% B@LGTs, 40 ppm BBR with 1st order
| B@LGT |
Eg (eV) |
Ea (J mol−1) |
PCR (%) |
K (min−1) |
R2 |
| B@Nd2S3:GO |
2.23 |
−670.14 |
96.35 |
0.0325 |
0.9975 |
| B@Eu2S3:GO |
2.28 |
−767.39 |
96.67 |
0.0318 |
0.9561 |
| B@Er2S3:GO |
2.38 |
−829.18 |
97.60 |
0.0321 |
0.9818 |
| B@Yb2S3:GO |
1.88 |
−925.57 |
99.17 |
0.0315 |
0.9736 |
Table 8 Viscosities (mPa S) of aq-LSNR and aq-B@LSNR solutions at different temperatures
| Temp. (K) |
Nd2S3 |
Eu2S3 |
Er2S3 |
Yb2S3 |
Nd2S3@Gd |
Eu2S3@Gd |
Er2S3@Gd |
Yb2S3@Gd |
| 283.15 |
1.0152 |
1.0000 |
0.8726 |
1.0000 |
0.9050 |
0.8539 |
0.8705 |
0.8514 |
| 288.15 |
1.0555 |
0.9701 |
0.9702 |
1.0590 |
1.0763 |
0.9325 |
1.2536 |
1.0213 |
| 298.15 |
1.1939 |
1.0126 |
0.9930 |
1.0442 |
0.8628 |
0.8590 |
1.1860 |
1.1860 |
| 310.15 |
0.7240 |
0.8223 |
0.8114 |
0.8732 |
0.8950 |
0.7386 |
0.6585 |
0.9569 |
| 315.15 |
0.6379 |
0.5990 |
0.5833 |
0.5650 |
0.7679 |
0.5930 |
0.5833 |
0.5650 |
Table 9 Viscosity (mPa S) of B@LGTs at different temperatures
| Temp. (K) |
B:Nd2S3@GO |
B:Eu2S3@GO |
B:Er2S3@GO |
B:Yb2S3@GO |
| 283.15 |
0.8506 |
0.8625 |
0.8596 |
0.9281 |
| 288.15 |
0.9943 |
0.9940 |
1.0828 |
1.0658 |
| 298.15 |
0.8904 |
1.1151 |
0.8472 |
0.8669 |
| 310.15 |
0.7531 |
0.8222 |
0.8805 |
0.7531 |
| 315.15 |
0.5990 |
0.6148 |
0.5586 |
0.6301 |
Table 10 Surface tension values (mN m−1) of aq-LSNRs and aq-B@LSNRs at different temperatures
| Temp. (K) |
Nd2S3 |
Eu2S3 |
Er2S3 |
Yb2S3 |
Nd2S3@Gd |
Eu2S3@Gd |
Er2S3@Gd |
Yb2S3@Gd |
| 283.15 |
65.97 |
63.82 |
66.01 |
65.56 |
73.58 |
70.91 |
70.90 |
68.367 |
| 288.15 |
76.64 |
76.01 |
76.64 |
76.02 |
77.30 |
77.28 |
76.64 |
76.64 |
| 298.15 |
72.79 |
72.79 |
72.79 |
72.79 |
72.20 |
72.21 |
72.21 |
72.21 |
| 310.15 |
71.63 |
72.18 |
72.19 |
72.19 |
72.75 |
72.18 |
72.19 |
72.18 |
| 315.15 |
51.98 |
66.52 |
60.56 |
62.15 |
70.47 |
72.09 |
67.92 |
65.58 |
Table 11 Surface tension values (mN m−1) of aq-B@LGTs at different temperatures
| Temp. (K) |
B:Nd2S3@GO |
B:Eu2S3@GO |
B:Er2S3@GO |
B:Yb2S3@GO |
| 283.15 |
70.90 |
72.53 |
70.90 |
68.38 |
| 288.15 |
76.66 |
76.64 |
76.01 |
76.64 |
| 298.15 |
72.78 |
72.79 |
72.79 |
72.79 |
| 310.15 |
76.36 |
72.18 |
72.18 |
72.18 |
| 315.15 |
67.95 |
68.00 |
66.01 |
70.51 |
Table 12 Friccohesity values (cm S−1) of aq-LSNRs and aq-B@LSNRs at different temperatures
| Temp. (K) |
Nd2S3 |
Eu2S3 |
Er2S3 |
Yb2S3 |
Nd2S3@Gd |
Eu2S3@Gd |
Er2S3@Gd |
Yb2S3@Gd |
| 283.15 |
0.0154 |
0.0141 |
0.0132 |
0.0130 |
0.0123 |
0.0120 |
0.0123 |
0.0129 |
| 288.15 |
0.0138 |
0.0128 |
0.0127 |
0.0139 |
0.0139 |
0.0121 |
0.0164 |
0.0133 |
| 298.15 |
0.0164 |
0.0139 |
0.0136 |
0.0143 |
0.0111 |
0.0096 |
0.0164 |
0.0164 |
| 310.15 |
0.0101 |
0.0117 |
0.0112 |
0.0125 |
0.0123 |
0.0102 |
0.0091 |
0.0133 |
| 315.15 |
0.0122 |
0.0090 |
0.0096 |
0.0091 |
0.0109 |
0.0082 |
0.0086 |
0.0086 |
Table 13 Friccohesity values (cm S−1) of aq-B@LGTs at different temperatures
| Temp. (K) |
B:Nd2S3@GO |
B:Eu2S3@GO |
B:Er2S3@GO |
B:Yb2S3@GO |
| 283.15 |
0.0120 |
0.0119 |
0.0121 |
0.0149 |
| 288.15 |
0.0130 |
0.0130 |
0.0142 |
0.0139 |
| 298.15 |
0.0122 |
0.0153 |
0.0116 |
0.0119 |
| 310.15 |
0.0099 |
0.0117 |
0.0126 |
0.0108 |
| 315.15 |
0.0088 |
0.0090 |
0.0084 |
0.0047 |
5.1. pH effect on photocatalysis
The wastewater discharged from textile,50 ink, coating, pulp and paper, and other industries contains various dyes, ions, and even acidic and alkaline contents.49 The ionic pollutants remain intimately suspended with the water dipoles with stronger dipolar interactions,119 and hence, a chemical method fails to recycle the water. Initially, the B@LGTs have photocatalytically reduced them to phase out water for adsorbing the reduced pollutants. In this process, the H+ and OH− ions engage the holes affecting the PCR rate. The B@LGTs also split wastewater to H+ and OH− ions to balance the charge density needed to photocatalytically reduce the pollutants.120 The 0.01 g% B@LGTs photocatalytically reduced 40 ppm BBR and 20 ppm TMIs and QHIn at pH 2–12. pH values of 2 and 7 are favourable for the reduction of BBR and 20 ppm TMIs by 35.5 and 99.8% Φ respectively in 15–40 min (Fig. 25a). The pH 2 has formed the H3O+ by engaging the water dipoles that had affected the hv capaturing of B@LGTs. The B@LGTs at pH < 7 have generated the positive charges on surfaces either by adsorbing H+ or H2O + H+ = H3O+ with electrostatic repulsion between the B@LGTs and BBR with a lower PCR activity (Fig. 25a). At pH > 7, the surfaces of B@LGTs with OH− have attracted the h+ holes121 on lowering Φ by 35–50% and B@LGTs could have aggregated (Fig. 16). The B@LGTs from 2 to 12 pH have absorbed the UV/vis up to 35% while at pH 7, a 99.8% on counterbalancing the H+ and HO− mutually without engaging the holes (Fig. 25a). While the e− hole seems to interact with 2H+ + e− = H2 in acidic media, the h+ hole interacts with 4HO− + h+ = 2H2O + O2 in alkaline media.
5.2. Monodispersibility for photocatalysis
The 0.01 g% B@LGTs have PCR the 10 to 80 ppm BBR and 20 ppm of each TMIs and QHIn. The @PCR for 80 ppm is lower than that for 40 ppm and 20 ppm BBR and TMIs respectively due to aggregation122 (Fig. 25c). The 0.008 to 0.012 g% B@LGTs (@0.01 g%) did PCR the BBR and TMIs in 30–45 min. The 0.01 g% B@LGTs have PCR the 99.8% while for >0.01 g%, the @PCR was slightly lowered. The higher B@LGT concentration got aggregated (Fig. 25b). The >0.01 g% B@LGTs have weakened the H2O–B@LGT interstitial linkages on strengthening the B@LGT–B@LGT interactions. A monolayer NHS aligned around B@LGT favoured PCR (Fig. 16). The water dipoles adhere along the Gd3+ and Ln3+ sites in situ with two separate alignments with stronger cohesive forces by weakening surface continuity predicted with PCPs and thermodynamic parameters (Fig. 18–20).
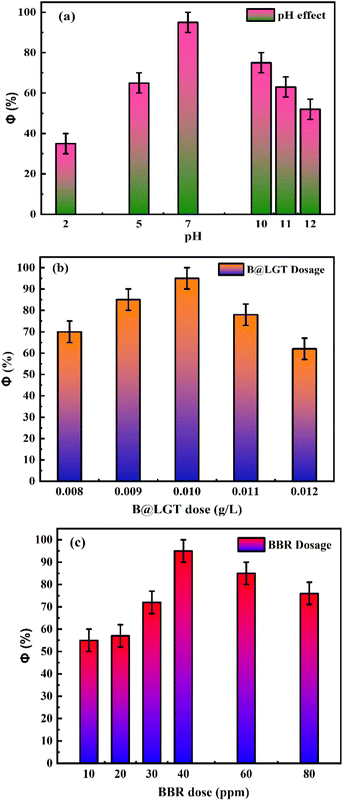 |
| | Fig. 25 Effect of (a) pH, (b) B@LGT photocatalyst dosage and (c) BBR dosage. | |
5.3. PCR of CrCl3, NiCl2, and CuCl2 under visible light on subsequent adsorption
At pH = 7, 0.01 g% aq-B@LGTs photocatalytically reduced and subsequently adsorbed 20 ppm of CrCl3, NiCl2, and CuCl2 individually in 20, 40, and 35 min respectively. The e− holes produced by the B@LGTs have reduced the 3d3 (Cr3+), 3d8 (Ni2+), and 3d9 (Cu2+) ions to 3d4 (Cr0), 3d8 (Ni0), and 3d9 (Cu0) state. The longer PCR time with NiCl2 having 3d8 electrons than 3d9 of Cu2+ and 3d3 of Cr3+ predicted the unpaired electrons enhancing the PCR rate. The coloured aq-solutions of Cr3+, Ni2+, and Cu2+ turned colourless in 10 min, while in the next 10 min, the spongy nanoclusters appeared (Fig. 27). Initially, B@LGTs with specific surface energies reduced Cr3+, Ni2+, and Cu2+ on subsequent adsorption by weakening the ionic interactions with water to zero.123 The water dipoles aligned with the respective cation tended towards Gd3+ and Ln3+ integrating the charge spontaneities.124 The ions with respective charges receive the e− and h+ holes. The B@LGTs with LSNR–Gd3+–GO have trapped the hv for PCR and physisorbed the Cr, Ni, and Cu metals and got settled (Fig. 26). The B@LGTs have oxidized Cl− to Cl2 in a two-step PCR122 process (eqn (4.7)–(4.9)). The PCR of TMIs differ from BBR as B@LGTs initially seem to align them with a brownish colour125 from their respective colours in 1–2 min (Fig. 26B). The B@LGTs competitively disrupted the ionic NHS for physisorption. The ions may get associated with water dipoles adhered126 to B@LGTs to receive the holes and get reduced (eqn (4.7)–(4.9)). The water dipoles did not interact with Cr, Ni, and Cu metals and so are adsorbed by B@LGs forming a spongy state (Fig. 26). The overall system right from receiving hv and generate the e− and h+ holes for PCR on subsequent adsorption is dynamic within a liquid phase.127 Thus, their PCPs have predicted the valuable input about avoiding the air bubbles that hurdle the mobility and continuity. Therefore, the moderate shear and surface tension both have predicted a continuity (Fig. 18 and 19).| |
 | (4.7) |
| |
 | (4.8) |
| |
 | (4.9) |
 |
| | Fig. 26 PCR of 20 ppm CrCl3 (A), NiCl2 (B), and CuCl2 (C) on subsequent adsorption using 0.01 g% of B@LGTs under visible light. | |
 |
| | Fig. 27 (a–c) UV-vis spectra for the PCR of CrCl3, NiCl2, and CuCl2 and (d) QHIn using 0.01 g% B@LGTs. | |
5.4. PCR of QHIn to HIn
First, 2 mg HIn and 1 mg NaOH dissolved in 100 mL water formed pink-coloured QHIn at pH > 9. Keeping 2 mL of 0.01 g% aq-B@LGTs with 30 mL aq-QHIn under visible light showed discolouration in 15 min upon reducing QHIn to HIn (Fig. 27d). The B@LGTs have split H2O into H+ and OH− as follows:| |
 | (4.10) |
Excess e− holes reoriented the quinonoid benzene ring of QHIn, while Na+ oxidised an anion of HIn as a sodium benzoate salt of HIn as intermediate.128 The B@LGT solution has faded a QHIn pink stint at t = 15 min on sharing the electronic charges due to charged sites. The B@LGT with spontaneities has intimately adhered QHIn to initiate a reduction. The colour could not disappear with LSNR and B@LSNR while the B@LGTs have aligned OH−, Na+, and quinonoid ring to interact with holes in infinite populations. The B@LGTs with respective charges/holes have turned the native pink stint of QHIn on reducing OH− to O2 and H2 (eqn (4.10) and (Fig. 28b)). OH− and Na+ could have generated stronger affinities for water dipoles.129 The partial positive charge could have interacted with OH−, Na+, and quinonoid ring for PCR in certain area (Fig. 28). The quinonoid benzene ring could have reacted with Na+ while OH− entered B@LGT interstities gaining higher density to get settled at bottom with time. Initially, the salts and QHIn were monodispersed with B@LGT on moderate electrostatic interactions with robust oscillating activities.
 |
| | Fig. 28 (a–f) Disappearance of the colour followed by the reduced QHIn settling at the bottom. (f) Recycled clean water. (g) Proposed mechanism under visible light. | |
5.5. PCR of methyl orange (MO) dye
The 0.01 g% aq-B@LGTs with 20 ppm MO turned into a dark red colour solution after 2 h. The residual charges of 4-(dimethylamino)phenyl, diazenyl, and sodium benzenesulfonate could not resonate the energies. The wavefunction ψ–CH3 of ERG –CH3 could not interface ψ–phenyl and ψdizenyl of 4-(dimethylamino)phenyl and diazenyl that have disintegrated benzenesulfonate (Fig. 29a). Thus, their mismatched ψ counterbalance mutually on occupying their own functional sites deflecting the holes unlike the BBR, TMIs, and QHIn (Fig. 29d). The B@LGTs could not photocatalytically reduce a conjugated aligned dye due to a stronger azo (–NN–) group. The e− and h+ holes have responded SO3− and partially got settled after 5 h as delocalization of MO could have been tracked by the FGs of GO in coated B@LGT (Fig. 29f). The (–N(CH3)3) and (–NN–) constituents of MO have mutually mismatched their respective angular momentums with least electron finding probability expressed as orthogonal 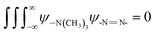 that could not receive the holes except minor adsorption (Fig. 29). Its transition dipole moment (μ = 0) within the VB to CB space (dτ = dxdydz) along the x, y, and z dimensions is almost nil as follows:
that could not receive the holes except minor adsorption (Fig. 29). Its transition dipole moment (μ = 0) within the VB to CB space (dτ = dxdydz) along the x, y, and z dimensions is almost nil as follows:| | |
Transition dipole moment = ∫(ψ–N(CH3)3)μ × (ψ–NN–)dτ
| (4.11) |
Thus, the MO needs stronger photocatalysts than B@LGTs to photocatalytically reduce the quinonoid, quaternary nitrogen (N+–), and TMIs to receive the holes. The B@LGTs have acted as photosensors for the effluents containing quinonoid, N+–, and TMIs to photocatalytically reduce except azo groups such as MO (Fig. 29).
 |
| | Fig. 29 (a and b) Plausible mechanism for MO dye to favour PCR (c) original MO (d) dye with B@LGTs (e) solution after 2 hours and (f) solution after 5 hours, indicated the B@LGTs did not PCR but partially adsorbed the MO dye under visible light owing to the presence of highly stable azo group with a high potential energy. | |
5.6. Photocatalyst reusability
It was found that 0.01 g% B@LGTs have reduced 40 ppm BBR and 20 ppm TMIs in the 1st cycle. For the 2nd cycle, the used B@LGTs were washed with H2O to photocatalytically reduce fresh 40 ppm BBR. It continues to reuse the B@LGTs after washing for reducing the 3rd fresh sample of 40 ppm BBR. Thus, 0.01 g% B@LGTs after washing were reused to photocatalytically reduce 10 fresh BBR samples in 30, 35, 40, 45, 50, 60, 75, 90, 105, and 120 min respectively with 68–98.9% Φ (Fig. 32a). The B@LGTs have physisorbed BBR, TMIs, and QHIn within the interstitial tiers with weaker VWFs. The B@LGT with adsorbed dye seem to have the two energy ensembles within tiers with milder VWFs up to 10 reuse cycles. After 5 subsequent PCR cycles, the B@LGTs were analysed with the FT-IR spectra, TEM images and TGA curves (Fig. 30 and 31). Their hedgy thermograms predicted physisorption that resulted in an acoustic structure on exchanging energies mutually. Reduced BBR, TMIs, and QHIn with stronger interaction have differed in bending and stretching frequencies. Thus, the reduced effluents with their residual charges due to different electronic configurations of their constituents have interacted with the B@LGTs apart from the PCR of quaternary nitrogen. Thermally stable B@LGT could advance the science in areas of nano thin films, piezoelectric devices, and photoluminescent sensors.98 The B@LGTs on reuse for 10 cycles seem to have been reinforced with enhanced durability and networking55 similar to smart resin with GO and metallic nanoparticles.130 The FT-IR spectra, TGA curves, and HR-TEM images predicted dye-reinforced B@LGT (DRB) rather than bare ones by detaining the same frameworks (Fig. 30 and 31). The HR-TEM images predicted a slightly flattened morphology compared to the bare ones due to residual forces98 generated on exposing to hv. The dopant and coating agents have developed a quasistatic structure of B@LGTs with symmetric interstities oscillating in the same phase. That had excellently worked as the reversible state on receiving the photons and developing the holes without undergoing structural transitions. Thus, the B@LGTs could act as photoluminescent to sharpen the nano thin films on window glass, vehicle glass, and solar panels.39,131
 |
| | Fig. 30 (a and b) HR-TEM images of B@LGT elucidating the stable morphology before and after the PCR of the pollutants. | |
 |
| | Fig. 31 (a and b) Varying TGA, DTG and DTA patterns differentiating the spinning of increasing 4f electrons for B@LGT before and after the PCR of pollutants. | |
 |
| | Fig. 32 Spectra elucidating (a) Φ of BBR, (b) 10 times reusability of B@LGTs, (c) conductivity during the PCR and (d) B@LGTs negligibly undergoing PCR rather than partially adsorbing the MO dye in an acidic medium due to higher potential energy. | |
5.7. MO + B@LGTs treated in acidic and basic media separately
The B@LGTs had photocatalytically reduced BBR, TMIs, and QHIn as their native colours got disappeared while MO in an acidic solution (with HCl) showed colour fading negligibly with negligible PCR. The holes from B@LGTs have oxidized Cl− to Cl2 and split water, while the holes could not interact with (–N(CH3)3) and (–NN–) eqn (4.12). The holes from B@LGTs 300–400 nm could not access highly stable groups of MO and no colour faded (Fig. 32d). The B@LGTs to photocatalytically reduce MO are functionalized further with highly π conjugated molecules such as flavonoids. The B@LGTs in an acidic medium form H3O+ and have split water to O2 and H2 with robust effervescences, generating holes under visible light that acted as an indicator to control the pH (Fig. 32d). The B@LGTs have oxidized Cl− into Cl2 with 2h+ holes to scavenge Cl− that could equally scavenge in biomedical, biophysical, dyeing, and water treatment.| |
 | (4.12) |
The B@LGTs have scavenged OH− in alkaline solutions and H+ in acidic solutions, respectively, as pH scavenger and antidots for controlling the acidity and alkalinity of the solutions.23
6. Conclusion
Lanthanide sulphide nanorods of Nd, Eu, Er, and Yb have been synthesized by reducing 0.001 M LnCl3·6H2O with 0.006 M Na metal at NTP for generating ∼73.71 mL H2 gas by CRM. On passing the H2S gas to aq-Ln(OH)3, LSNRs have been produced. LSNRs were doped and coated with Gd3+ and GO for B@LSNRs and B@LGTs respectively. Then 0.01 g% of B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, and B@Yb2S3:GO have separately photocatalytically reduced 40 ppm of BBR and 20 ppm of CrCl3, NiCl2, and CuCl2, and QHIn individually with 96.35–99.17% quantum yield in 30, 20, 40, 35, and 15 min respectively under visible light. B@Yb2S3:GO and B@Er2S3:GO have proven to be robust photocatalysts by performing PCR in 30 min with thermodynamic stability predicted with PCPs. The PCPs such as density, viscosity, sound velocity, surface tension, and friccohesity have predicted their thermodynamically stable monodispersions at 288.15, 298.15, and 310.15 K. Their PCPs have elucidated the functional surfaces to receive the hν and to interface with BBR, TMIs, and QHIn without aggregation. The GO and Gd3+ tiers have enhanced PCR with 10 times reusability. The intimate interfacing of BBR dye, TMIs, and QHIn with B@LGTs via quantum tunnelling effects for photosensitization was examined with PCPs and thermodynamic parameters. The higher friccohesity of 0.01 g% B@LGTs have confirmed eigenenergies to align water dipoles with a higher surface area on strengthening adhesive forces. B@Nd2S3:GO, B@Eu2S3:GO, B@Er2S3:GO, B@Yb2S3:GO, with 2.23, 2.28, 2.38, and 1.88 eV Eg respectively and with higher surface energies could be used as the heat sensor, functional group detector, chemical sensor, and energy storage. The B@LGTs could substitute the piezoelectric sensors, acoustics, and routine adsorbents due to aligned interstities. The reusable trifunctional B@LGT photocatalysts could remediate other wastewater effluents than BBR, metal salts, and QHIn.
Abbreviation
| GO | Graphene oxide |
| LSNRs | Lanthanide sulfide nanorods |
| B@LSNRs | Bimetallic lanthanide sulfide nanorods |
| NRs | Nanorods |
| B@LGT | Bimetallic lanthanide GO template |
| 4f@e | Number of 4f electrons |
| BBR | Brilliant blue red |
| TMIs | Transition metal ions |
| QHIn | Quinonoid phenolphthalein |
| PCPs | Physicochemical properties |
| FRET | Friccohesity resonance energy transfer |
| CRM | Crash reaction methodology |
| FT-IR | Fourier transform infrared |
| Rovitronic motions | Rotational, vibrational, and translational motions |
| Gt | Graphite |
| GtO | Graphite oxide |
| IFTs | Interfacial templates |
| UPE | Unpaired electron |
| MFP | Mean free path |
| RBF | Round-bottom flask |
| RPM | Revolution per minute |
| aq | Aqueous |
| PDN | Pendant drop number |
| VFT | Viscous flow time |
| Cohad | Cohesive and adhesive forces |
| CCP | Consistency cohad point |
| TC | Tentropic cavity |
| wt | Weight |
| FE | Functional edges |
| RE | Resonating energy |
| SL | Sunlight |
| PCR | Photocatalytic reduction |
| TGA | Thermogravimetric analysis |
| DTG | Differential thermal gravimetry |
| ΔH | Enthalpy |
| ΔEa | Activation energy |
| ΔSt | Tentropy |
| ψsv | Single-valued wavefunction |
Data availability
The data supporting this article have been included as part of the ESI file.†
Author contributions
Krishan Kumar conducted the bench work, calculations, some discussions, and figure plotting, and wrote up the experimental observations and conclusions. Prof. Man Singh supervised the research work and participated in discussions, explored new models to explain experimental observations and derived new scientific insights out of the research.
Conflicts of interest
The authors confirm that there is no conflict of interest.
Acknowledgements
The authors are thankful to the Central University of Gujarat, India, for infrastructure and instrumental facilities. Mr Krishan Kumar is thankful to UGC-DAE CSR Indore (MP) for financial support (Ref. CRS/ 2021-22/379) and Dr Mukul Gupta as a Principal Collaborator, Dr Vasant Sathe, UGC-DAE CSR Indore, acknowledged for providing Raman facility. The authors are thankful to Dr Mukul Gupta, Dr Ram Janay Chaudhary, and Dr N. P. Lalla for XRD, XPS, and HR-TEM analysis respectively, UGC-DAE CSR Indore. Mr Mahesh, CSMCRI-CSIR, Bhavnagar was acknowledged for BET and MNIT, Jaipur for XPS analysis.
References
- M. E. Borges, M. Sierra, E. Cuevas, R. D. García and P. Esparza, Sol. Energy, 2016, 135, 527–535 CrossRef CAS.
- X. Chen, P. Zhang, D. H. Kuo, Q. Wu, A. B. Abdeta, B. Wu, Z. Su, L. Chen, O. A. Zelekew and J. Lin, J. Mater. Chem. A, 2023, 11, 19091–19106 RSC.
- Z. Wu, X. Yuan, G. Zeng, L. Jiang, H. Zhong, Y. Xie, H. Wang, X. Chen and H. Wang, Appl. Catal. B Environ., 2018, 225, 8–21 CrossRef CAS.
- T. A. Aragaw, F. M. Bogale and B. A. Aragaw, J. Saudi Chem. Soc., 2021, 25(8), 101280 CrossRef CAS.
- P. Zhang, H. Wang, Y. Lai, Y. Xu, L. Chen, Q. Wu, D.-H. Kuo, D. Lu, M. T. Mosisa, J. Li, J. Lin and X. Chen, J. Water Process Eng., 2024, 58, 104820 CrossRef.
- T. A. Saleh, Environ. Technol. Innov., 2021, 24, 101821 CrossRef CAS.
- R. Koutavarapu, M. R. Tamtam, M. C. Rao, S. G. Peera and J. Shim, Chemosphere, 2021, 272, 129849 CrossRef CAS PubMed.
- H. Wu, L. Li, K. Chang, K. Du, C. Shen, S. Zhou, G. Sheng, W. Linghu, T. Hayat and X. Guo, J. Environ. Chem. Eng., 2020, 8(4), 103882 CrossRef CAS.
- Y. Li, M. Fu, R. Wang, S. Wu and X. Tan, Chem. Eng. J., 2022, 444, 136567 CrossRef CAS.
- M. Sun, J. Ma, M. Zhang, Y. Xiao, Y. Zhu and S. Zhang, Mater. Chem. Phys., 2020, 241, 122450 CrossRef CAS.
- P. Zhang, Y. Lai, X. Dai, Y. Xu, X. Wu, B. Yang, D.-H. Kuo, D. Lu, Q. Wu, M. T. Mosisa, J. Lin and X. Chen, J. Environ. Chem. Eng., 2024, 12, 111831 CrossRef CAS.
- D. Saini, R. Aggarwal, S. R. Anand and S. K. Sonkar, Sol. Energy, 2019, 193, 65–73 CrossRef CAS.
- P. Zhang, Q. Wu, H. Wang, Y. Xu, Y. Lai, P. Li, D.-H. Kuo, T. Huang, H. Zhang, M. T. Mosisa, J. Li, J. Lin, X. Chen and D. Lu, Mater. Today Chem., 2024, 37, 102028 CrossRef CAS.
- X. Chen, H. Sun, O. A. Zelekew, J. Zhang, Y. Guo, A. Zeng, D.-H. Kuo and J. Lin, Appl. Surf. Sci., 2020, 525, 146531 CrossRef CAS.
- M. T. Mosisa, P. Zhang, B. Wu, L. Chen, Z. Su, P. Li, H. Zhang, A. Farooq, T. Huang, A. B. Abdeta, O. A. Zelekew, D.-H. Kuo, J. Lin, X. Chen and D. Lu, J. Environ. Chem. Eng., 2024, 12, 113383 CrossRef CAS.
- V. V. Klepov, K. A. Pace, L. S. Breton, V. Kocevski, T. M. Besmann and H. C. Zur Loye, Inorg. Chem., 2020, 59, 1905–1916 CrossRef CAS PubMed.
- Z. Liu, X. Sun, S. Xu, J. Lian, X. Li, Z. Xiu, Q. Li, D. Huo and J. G. Li, J. Phys. Chem. C, 2008, 112, 2353–2358 CrossRef CAS.
- X. Luo, M. Zhang, L. Ma and Y. Peng, J. Rare Earths, 2011, 29, 313–316 CrossRef CAS.
- A. V Ruseikina, O. V Andreev, E. O. Galenko and S. I. Koltsov, J. Therm. Anal. Calorim., 2017, 128, 993–999 CrossRef.
- I. Valsamakis and M. Flytzani-Stephanopoulos, Appl. Catal. B Environ., 2011, 106, 255–263 CAS.
- R. Mauricot, P. Gressier, M. Evain and R. Brec, J. Alloys Compd., 1995, 223, 130–138 CrossRef CAS.
- S. A. Osseni, S. Lechevallier, M. Verelst, P. Perriat, J. Dexpert-Ghys, D. Neumeyer, R. Garcia, F. Mayer, K. Djanashvili, J. A. Peters, E. Magdeleine, H. Gros-Dagnac, P. Celsis and R. Mauricot, Nanoscale, 2014, 6, 555–564 RSC.
- S. R. Sanivarapu, J. B. Lawrence and G. Sreedhar, ACS Omega, 2018, 3, 6267–6278 CrossRef CAS PubMed.
- N. Vashistha, A. Chandra and M. Singh, New J. Chem., 2020, 44, 14211–14227 RSC.
- C. M. Marin, L. Wang, J. R. Brewer, W. N. Mei and C. L. Cheung, J. Alloys Compd., 2013, 563, 293–299 CrossRef CAS.
- I. L. Ikhioya and A. C. Nkele, Arabian J. Sci. Eng., 2023, 49, 1217–1225 CrossRef.
- S. Roméro, A. Mosset, J. C. Trombe and P. Macaudière, J. Mater. Chem., 1997, 7, 1541–1547 RSC.
- H. Tang, L. N. Sacco, S. Vollebregt, H. Ye, X. Fan and G. Zhang, J. Mater. Chem. A, 2020, 8, 24943–24976 RSC.
- A. Roca-Sabio, M. Regueiro-Figueroa, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas and C. Platas-Iglesias, Comput. Theor. Chem., 2012, 999, 93–104 CrossRef CAS.
- S. Dev and M. Singh, Solid State Sci., 2022, 134, 107045 CrossRef CAS.
- N. Shukla, D. K. Verma, A. K. Singh, B. Kumar, Kavita and R. B. Rastogi, ACS Appl. Nano Mater., 2020, 3, 8012–8026 CrossRef CAS.
- M. L. Redígolo, D. S. Koktysh, K. van Benthem, S. J. Rosenthal and J. H. Dickerson, Mater. Chem. Phys., 2009, 115, 526–529 CrossRef.
- K. Kumar, B. Sahoo, T. C. Meghwal and M. Singh, Energy Adv., 2023, 3, 198–225 RSC.
- P. Glaser, O. Stewart, R. Atif, D. R. C. Asuigui, J. Swanson, A. J. Biacchi, A. R. Hight Walker, G. Morrison, H. C. zur Loye and S. L. Stoll, Angew. Chem., Int. Ed., 2021, 60, 23134–23141 CrossRef CAS PubMed.
- A. Kumar, S. Kumar, A. Bahuguna, A. Kumar, V. Sharma and V. Krishnan, Mater. Chem. Front., 2017, 1, 2391–2404 RSC.
- M. T. Mosisa, P. Zhang, Z. Su, B. Wu, L. Chen, Y. Liao, A. Farooq, D. Lu, A. B. Abdeta, D.-H. Kuo, J. Lin and X. Chen, J. Environ. Chem. Eng., 2024, 12, 112111 CrossRef CAS.
- X. Chen, D.-H. Kuo and D. Lu, Chem. Eng. J., 2016, 295, 192–200 CrossRef CAS.
- X. Chen, D.-H. Kuo, D. Lu, Y. Hou and Y.-R. Kuo, Microporous Mesoporous Mater., 2016, 223, 145–151 CrossRef CAS.
- J. M. Coronado, F. Fresno, M. D. Hernández-Alonso and R. Portela, Design of Advanced Photocatalytic Materials for Energy and Environmental Applications, 2013, vol. 71 Search PubMed.
- L. Hu, Y. Li, X. Peng, W. Zheng, W. Xu, J. Zhu, L. Y. S. Lee, P. K. Chu and K. Y. Wong, Chem. Eng. J., 2021, 417, 127900 CrossRef CAS.
- T. P. Gompa, A. Ramanathan, N. T. Rice and H. S. La Pierre, Dalton Trans., 2020, 49, 15945–15987 RSC.
- H. Wu, J. Peng, H. Sun, Q. Ruan, H. Dong, Y. Jin, Z. Sun and Y. Hu, Chem. Eng. J., 2022, 432, 134339 CrossRef CAS.
- P. Borthakur and M. R. Das, J. Colloid Interface Sci., 2018, 516, 342–354 CrossRef CAS PubMed.
- X. Chen, H. Sun, J. Zhang, Y. Guo and D.-H. Kuo, J. Mol. Liq., 2019, 273, 50–57 CrossRef CAS.
- F. Uddin, Cellulose, 2021, 28, 10715–10739 CrossRef CAS.
- P. Bai, U. J. Etim, Z. Yan, S. Mintova, Z. Zhang, Z. Zhong and X. Gao, Catal. Rev., 2018, 61(3), 333–405 CrossRef.
- L. Alcaraz, O. R. Largo, F. J. Alguacil, M. Á. Montes, C. Baudín and F. A. López, Metals, 2021, 13 Search PubMed.
- C. Wang, M. Chen, J. Wu, F. Mo and Y. Fu, Anal. Chim. Acta, 2019, 1086, 66–74 CrossRef CAS PubMed.
- B. N. Joshi, H. Yoon, S. H. Na, J. Y. Choi and S. S. Yoon, Ceram. Int., 2014, 40, 3647–3654 CrossRef CAS.
- M. Ubaidullah, A. M. Al-Enizi, T. Ahamad, S. F. Shaikh, M. A. Al-Abdrabalnabi, M. S. Samdani, D. Kumar, M. A. Alam and M. Khan, J. Energy Storage, 2021, 33, 102125 Search PubMed.
- S. Fahrendorf, N. Atodiresei, C. Besson, V. Caciuc, F. Matthes, S. Blügel, P. Kögerler, D. E. Bürgler and C. M. Schneider, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- X. Zeng, Y. Liu, T. Zhang, J. C. Jin, J. L. Li, Q. Sun, Y. J. Ai, M. L. Feng and X. Y. Huang, Chem. Eng. J., 2021, 420, 127613 Search PubMed.
- O. P. Kumar, M. N. Ashiq, M. Ahmad, S. Anjum and A. ur Rehman, J. Mater. Sci. Mater. Electron., 2020, 31, 21082–21096 Search PubMed.
- I. Valsamakis, R. Si and M. Flytzani-Stephanopoulos, J. Power Sources, 2010, 195, 2815–2822 CrossRef CAS.
- J. Chang, S. Zang, W. Liang, D. Wu, Z. Lian, F. Xu, K. Jiang and Z. Gao, J. Colloid Interface Sci., 2021, 590, 114–124 CrossRef CAS PubMed.
- A. Khaligh and Z. Li, IEEE Trans. Veh. Technol., 2010, 59, 2806–2814 Search PubMed.
- A. G. Stern, Int. J. Hydrogen Energy, 2018, 43, 4244–4255 CrossRef CAS.
- V. J. Reddy, N. P. Hariram, R. Maity, M. F. Ghazali and S. Kumarasamy, World Electr. Veh. J., 2023, 14(12), 349 CrossRef.
- X. Chen, H. Sun, D.-H. Kuo, A. B. Abdeta, O. A. Zelekew, Y. Guo, J. Zhang, Z. Yuan and J. Lin, Appl. Catal. B Environ., 2021, 287, 119992 CrossRef CAS.
- R. Kumari and M. Singh, ACS Omega, 2020, 5, 23201–23218 CrossRef CAS PubMed.
- K. Kumar, R. P. Dave, S. Dev and M. Singh, RSC Adv., 2022, 12, 29734–29756 RSC.
- G. Avashthi and M. Singh, New J. Chem., 2021, 45, 5463–5483 RSC.
- R. Jain, M. Mathur, S. Sikarwar and A. Mittal, J. Environ. Manage., 2007, 85, 956–964 Search PubMed.
- Q. Liang, J. Jin, M. Zhang, C. Liu, S. Xu, C. Yao and Z. Li, Appl. Catal. B Environ., 2017, 218, 545–554 CrossRef CAS.
- A. B. Abdeta, Q. Wu, D.-H. Kuo, P. Li, H. Zhang, T. Huang, J. Zhang, J. Lin and X. Chen, J. Catal., 2022, 413, 1056–1069 CrossRef CAS.
- X. Chen, T. Huang, D.-H. Kuo, H. Sun, P. Li, O. A. Zelekew, A. B. Abdeta, Q. Wu, J. Zhang, Z. Yuan and J. Lin, Appl. Catal. B Environ., 2021, 298, 120542 CrossRef CAS.
- J. P. Guggenbichler, M. Böswald, S. Lugauer and T. Krall, Infection, 1999, 27, S16–S23 CrossRef CAS PubMed.
- M. Salavati-Niasari, M. R. Loghman-Estarki and F. Davar, Inorg. Chim. Acta, 2009, 362, 3677–3683 CrossRef CAS.
- T. Charinpanitkul, A. Soottitantawat, N. Tonanon and W. Tanthapanichakoon, Mater. Chem. Phys., 2009, 116, 125–128 CrossRef CAS.
- P. Malik, S. S. Maktedar, G. Avashthi, T. K. Mukherjee and M. Singh, Robust Curcumin-Mustard Oil Emulsions for Pro to Anti-oxidant Modulation of Graphene Oxide, King Saud University, 2020, vol. 13 Search PubMed.
- W. C. Yang, C. W. Wang, J. C. Wang, Y. C. Chang, H. C. Hsu, T. E. Nee, L. J. Chen and J. H. He, J. Nanosci. Nanotechnol., 2008, 8, 3363–3368 Search PubMed.
- G. Viola, J. Chang, T. Maltby, F. Steckler, M. Jomaa, J. Sun, J. Edusei, D. Zhang, A. Vilches, S. Gao, X. Liu, S. Saeed, H. Zabalawi, J. Gale and W. Song, ACS Appl. Mater. Interfaces, 2020, 12, 34643–34657 Search PubMed.
- M. S. Longair, Philos. Trans. R. Soc., A, 2008, 366, 1685–1696 CrossRef CAS PubMed.
- S. Saxena, T. A. Tyson, S. Shukla, E. Negusse, H. Chen and J. Bai, Appl. Phys. Lett., 2011, 99, 67–70 CrossRef.
- N. Sakai and T. Saito, Polyhedron, 2004, 23, 2611–2614 CrossRef CAS.
- A. R. Altaf, H. Teng, M. Zheng, I. Ashraf, M. Arsalan, A. U. Rehman, L. Gang, W. Pengjie, R. Yongqiang and L. Xiaoyu, J. Environ. Chem. Eng., 2021, 9, 105313 Search PubMed.
- Y. P. Varshni, Physica, 1967, 34, 149–154 Search PubMed.
- E. Asikuzun, O. Ozturk, L. Arda, A. T. Tasci, F. Kartal and C. Terzioglu, Ceram. Int., 2016, 42, 8085–8091 Search PubMed.
- X. Chen, Q. Wu, D. H. Kuo, A. B. Abdeta, H. Zhang, P. Li, T. Huang, O. A. Zelekew and J. Lin, J. Mater. Chem. A, 2023, 11, 4126–4141 RSC.
- S. Faisal, F. A. Jan, S. Saleem, R. Ullah, R. Wajidullah, N. Ullah and N. Salman, Nanotechnol. Environ. Eng., 2022, 7, 675–689 CrossRef CAS.
- P. Mountapmbeme, A. El, L. Wu, M. Waqas and Z. Tian, J. Taiwan Inst. Chem. Eng., 2018, 1–9 Search PubMed.
- Q. Wu, A. B. Abdeta, D. H. Kuo, H. Zhang, Q. Lu, J. Zhang, O. A. Zelekew, M. T. Mosisa, J. Lin and X. Chen, J. Mater. Chem. A, 2022, 10, 5328–5349 RSC.
- Z. Quan, Z. Wang, P. Yang, J. Lin and J. Fang, Inorg. Chem. ACS, 2007, 46, 1354–1360 CrossRef CAS PubMed.
- M. Mahar, G. Khuram, S. Ahmad, L. Almanqur, A. Guy and T. Suliman, J. Appl. Electrochem., 2024, 54, 257–273 CrossRef.
- M. A. Harrison, S. Somarajan, S. V Mahajan, D. S. Koktysh, K. Van Benthem and J. H. Dickerson, Mater. Lett., 2011, 65, 420–423 CrossRef CAS.
- M. M. Gul, K. S. Ahmad, S. A. Alderhami, A. G. Thomas, Y. T. Alharbi and L. Almanqur, Mater. Sci. Eng. B, 2023, 297, 116776 CrossRef CAS.
- G. Physics, A. Abdullayeva and I. Bakhtiyarly, Glass Phys. Chem., 2020, 60, 7 Search PubMed.
- S. B. Ubale, S. B. Kale, V. J. Mane, P. P. Bagwade and C. D. Lokhande, J. Solid State Electrochem., 2021, 25, 1753–1764 CrossRef CAS.
- S. B. Ubale, S. B. Kale, V. J. Mane, U. M. Patil and C. D. Lokhande, J. Electroanal. Chem., 2021, 897, 115589 CrossRef CAS.
- N. Sato, M. Odori, M. Skrobian, M. Saito, T. Fujino and N. Masuko, Shigen Sozai, 1994, 110, 869–874 CrossRef CAS.
- S. Anjana, Dalton Trans., 2019, 48, 2926–2938 RSC.
- S. H. Han, K. A. Gschneidner and B. J. Beaudry, J. Alloys Compd., 1992, 181, 463–468 CrossRef CAS.
- N. Singh, M. Gordon, H. Metiu and E. Mcfarland, J. Appl. Electrochem., 2016, 46, 497–503 CrossRef CAS.
- M. Kumar, P. Rajput, P. K. Singh, A. C. Yadav, S. A. Khan, S. N. Jha, F. Singh and A. C. Pandey, RSC Adv., 2016, 6, 108523–108529 RSC.
- H. Taz, R. Ruther, A. Malasi, S. Yadavali, C. Carr, J. Nanda and R. Kalyanaraman, J. Phys. Chem. C, 2015, 119, 5033–5039 CrossRef CAS.
- S. Wang, L. Xu, L. Wang, A. Liang and Z. Jiang, Luminescence, 2013, 28, 842–846 CrossRef CAS PubMed.
- M. Abdullah, M. M. Alanazi, S. A. M. Abdelmohsen, S. D. Alahmari, S. Aman, A. Sadaf, A. G. Al-Sehemi, A. M. A. Henaish, Z. Ahmad and H. M. T. Farid, Mater. Sci. Eng. B, 2024, 301, 117207 CrossRef CAS.
- C. H. Lai, M. Y. Lu and L. J. Chen, J. Mater. Chem., 2012, 22, 19–30 RSC.
- M. J. Neves, J. B. De Oliveira, L. P. R. Ospedal and J. A. Helayël-Neto, Phys. Rev. D, 2021, 104, 15006 CrossRef CAS.
- G. Tai, T. Zeng, J. Yu, J. Zhou, Y. You, X. Wang, H. Wu, X. Sun, T. Hu and W. Guo, Nanoscale, 2016, 8, 2234–2241 RSC.
- G. Vijayaprasath, I. Habibulla, V. Dharuman, S. Balasubramanian and R. Ganesan, ACS Omega, 2020, 5, 17892–17899 CrossRef CAS PubMed.
- A. A. Dakhel and M. El-Hilo, J. Appl. Phys., 2010, 107, 123905 CrossRef.
- P. Macaudie, J. Mater. Chem., 1997, 7, 1541–1547 RSC.
- Z. Yang, H. Peng, W. Wang and T. Liu, J. Appl. Polym. Sci., 2010, 116, 2658–2667 CrossRef CAS.
- J. J. Carey and M. Nolan, Solid State Ionics, 2017, 307, 51–64 CrossRef CAS.
- M. I. Marković and A. D. Rakić, Opt. Laser Technol., 1990, 22, 394–398 CrossRef.
- M. Singh, J. Chem. Thermodyn., 2007, 39, 240–246 CrossRef CAS.
- H. J. Zhao, L. H. Li, L. M. Wu and L. Chen, Inorg. Chem., 2010, 49, 5811–5817 CrossRef CAS PubMed.
- J. H. Qiao, V. Mishra, M. C. Fishbein, S. K. Sinha and T. B. Rajavashisth, Exp. Mol. Pathol., 2015, 99, 654–662 CrossRef CAS PubMed.
- J. Kurawa, M. A. Jaafar and J. Bayero, Pure Appl. Sci., 2021, 14, 64–70 Search PubMed.
- R. V. Blasques, M. A. A. Pereira, A. M. R. V. Mendes, N. E. M. Filho, W. C. Gomes, L. T. Arenas, J. L. Marty, M. I. P. Gurgo, G. S. Nunes and P. C. M. Villis, Mater. Chem. Phys., 2020, 243, 122255 CrossRef CAS.
- R. P. Dave and M. Singh, Int. J. Biol. Macromol., 2023, 231, 123243 CrossRef CAS PubMed.
- A. Chandra, P. Malik, S. Singh, A. Roy, N. Sahoo and M. Singh, Chem. Eng. Process., 2022, 174, 108870 CrossRef CAS.
- M. P. Rayaroth, U. K. Aravind and C. T. Aravindakumar, Chemosphere, 2015, 119, 848–855 CrossRef CAS PubMed.
- Z. Su, X. Wu, D.
H. Kuo, B. Yang, B. Wu, L. Chen, P. Zhang, J. Lin, D. Lu and X. Chen, J. Mater. Chem. A, 2024, 12, 10494–10506 RSC.
- T. S. Sreeprasad and V. Berry, Small, 2013, 9, 341–350 CrossRef CAS PubMed.
- P. Roy and S. K. Srivastava, CrystEngComm, 2015, 17, 7801–7815 RSC.
- S. Dev and M. Singh, J. Phys. Chem. Solids, 2020, 139, 109335 CrossRef CAS.
- P. Zhang, L. Chen, D. H. Kuo, B. Wu, Z. Su, D. Lu, Q. Wu, J. Li, J. Lin and X. Chen, J. Mater. Chem. A, 2024, 12, 7163–7177 RSC.
- H. Karimi, H. R. Rajabi and L. Kavoshi, J. Photochem. Photobiol., A, 2020, 397, 112534 CrossRef CAS.
- B. Li, T. Liu, Y. Wang and Z. Wang, J. Colloid Interface Sci., 2012, 377, 114–121 CrossRef CAS PubMed.
- A. Reghioua, D. Barkat, A. H. Jawad, A. S. Abdulhameed, S. Rangabhashiyam, M. R. Khan and Z. A. ALOthman, J. Polym. Environ., 2021, 29, 3932–3947 CrossRef CAS.
- A. B. Abdeta, H. Sun, Y. Guo, Q. Wu, J. Zhang, Z. Yuan, J. Lin and X. Chen, Adv. Powder Technol., 2021, 32, 2856–2872 CrossRef CAS.
- Z. Su, B. Wu, L. Chen, M. T. Mosisa, P. Zhang, Q. Wu, D.-H. Kuo, D. Lu, O. A. Zelekew, J. Lin and X. Chen, J. Sci.:Adv. Mater. Devices, 2023, 8, 100645 CAS.
- A. Barde, U. V. Agbogo and A. I. Lawal, Int. J. Multidiscip. Res. Growth Eval., 2022, 1–8 Search PubMed.
- Y. Guo, O. A. Zelekew, H. Sun, D.-H. Kuo, J. Lin and X. Chen, Sep. Purif. Technol., 2020, 242, 116769 CrossRef CAS.
- Q. Wu, X. Wang, J. Fu, O. A. Zelekew, A. B. Abdeta, D.-H. Kuo, J. Zhang, Z. Yuan, J. Lin and X. Chen, J. Sci.:Adv. Mater. Devices, 2021, 6, 578–586 CAS.
- P. Yin, Y. Chen, Y. Wang, Y. Chi, B. Li, M. Xu, H. Song and C. Chen, J. Environ. Chem. Eng., 2023, 11, 110374 CrossRef CAS.
- X. Li, L. Chen, P. Zhang, A. B. Abdeta, Q. Wu, B. Wu, J. Lin, D.-H. Kuo, Y. Zhang and X. Chen, J. Sci.:Adv. Mater. Devices, 2023, 8, 100598 CAS.
- T. Zhang, T. Ma and W. Li, J. Macromol. Sci., Part B:Phys., 2015, 54, 992–1000 CrossRef CAS.
- V. G. Rajeshmon, C. S. Kartha, K. P. Vijayakumar, C. Sanjeeviraja, T. Abe and Y. Kashiwaba, Sol. Energy, 2011, 85, 249–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of TGA, PXRD, kinetics, thermodynamic parameters, FT-IR spectra, and tables of primary data of PCPs. See DOI: https://doi.org/10.1039/d4ra08347d |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Man Singh
and
Man Singh through the dopant and the GO upon exciting 4f@e (ψ4f) and FGs (ψFGs) seems to transform the hν into the redox energy9 for PCR. The B@LGT s exhibit enhanced surface resonance owing to oxidation (Ln3+ or ψLn3+) and reduction (S2− or ψS2−) potentials aimed at boosting the surface plasmonic resonance (SPR).48 The B@LGTs could advance the thin-film technology, digital ink, quantum dot MRI and tomography22,49 with a contrasting activity caused by Gd3+. The B@LGTs could be used as complementary nanomaterials for spintronics,50,51 electrical and thermal conductivies,33 piezoelectric devices, thermoelectric devices, and biocompatibility.52,53 The B@LGTs for solid oxide fuel cells could serve as the redox catalysts and electrode nanomaterials for enhanced lifespan to split water.54 The anodic B@LGT aligns to a cathodic dye with a higher viscosity and a lower surface tension, favouring the energy storage and supercapacitors.55,56 The higher viscosity with stronger frictional forces and lower surface tension comfortably aligns the B@LGT to develop a stable double layer of the negative and positive charges (Fig. 16). The study of PCPs have acted as the prerequisite database to interface the photocatalysis. The monodispersions of aq-B@LGTs predicted with the PCPs have improved the PCR of the BBR, TMIs, and QHIn on subsequent adsorption.
through the dopant and the GO upon exciting 4f@e (ψ4f) and FGs (ψFGs) seems to transform the hν into the redox energy9 for PCR. The B@LGT s exhibit enhanced surface resonance owing to oxidation (Ln3+ or ψLn3+) and reduction (S2− or ψS2−) potentials aimed at boosting the surface plasmonic resonance (SPR).48 The B@LGTs could advance the thin-film technology, digital ink, quantum dot MRI and tomography22,49 with a contrasting activity caused by Gd3+. The B@LGTs could be used as complementary nanomaterials for spintronics,50,51 electrical and thermal conductivies,33 piezoelectric devices, thermoelectric devices, and biocompatibility.52,53 The B@LGTs for solid oxide fuel cells could serve as the redox catalysts and electrode nanomaterials for enhanced lifespan to split water.54 The anodic B@LGT aligns to a cathodic dye with a higher viscosity and a lower surface tension, favouring the energy storage and supercapacitors.55,56 The higher viscosity with stronger frictional forces and lower surface tension comfortably aligns the B@LGT to develop a stable double layer of the negative and positive charges (Fig. 16). The study of PCPs have acted as the prerequisite database to interface the photocatalysis. The monodispersions of aq-B@LGTs predicted with the PCPs have improved the PCR of the BBR, TMIs, and QHIn on subsequent adsorption.


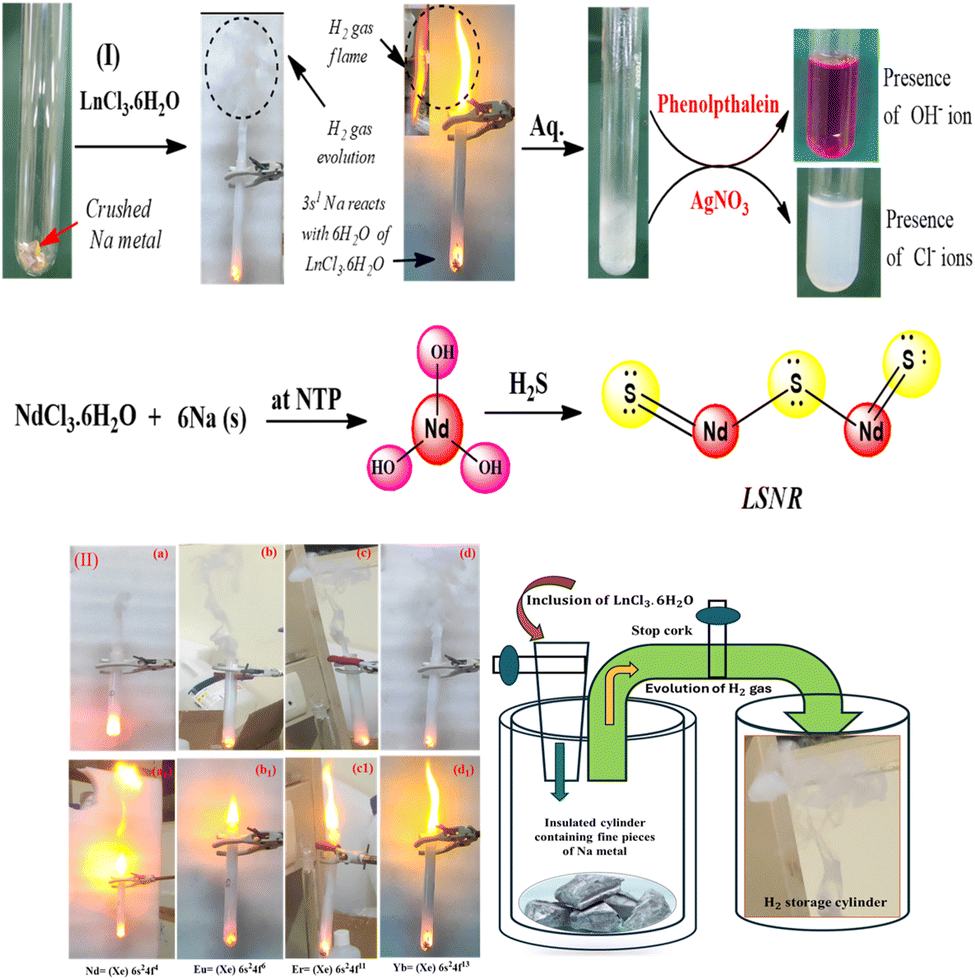









 , the r°, Bohr and r, radii for lattice ‘a’. The B@LGT combined with aq-BBR, TMIs, and QHIn to boost the PL (Fig. 16). B@LGT at r > r° receives hν with least permittivity (ε) and mutually shares it with effluents, as predicted via PCPs. The r = r° infers no reduction and no PCPs without ψ1 ≠ ψ2 ≠ ψ3… ≠ ψn conjugation.72 Fig. 5 depicts the single lattice elongating a wavelength73 on thinner 2D GO sheets similar to those reported previously.61 XRD, HR-TEM, and BET predicted that the synchronized electronic clouds of dopants, and coating agents of B@LGT have intimately hybridized without twisting on aligning interstities similar to their individual angular momentums. The La2S3, Ce2S3, Tb2S3, and Ho2S3 as core LGTs as the two tiers have slightly modulated the electrostatic interacting activities of aq-BSA protein with mild unfolding dynamics reported in an earlier study.33 Noticeably, polyvinylpyrrolidone (PVP)-capped rod-shaped europium (Eu3+)-doped gadolinium oxide (Gd2O3) nanoparticles (PVP@Gd2O3:Eu3+ NPs) have been effective to tune electrostatic charges of salt bridges of proteins.24 Thereby, the B@LGTs could further elaborate the charges to intensify the interacting activities with the peptides and intercalate the base pairs of dsDNA which are being pursued. The aligned B@LGTs have generated the e− and h+ holes of a longer mean free path that photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn within 15–40 min (Fig. 22).
, the r°, Bohr and r, radii for lattice ‘a’. The B@LGT combined with aq-BBR, TMIs, and QHIn to boost the PL (Fig. 16). B@LGT at r > r° receives hν with least permittivity (ε) and mutually shares it with effluents, as predicted via PCPs. The r = r° infers no reduction and no PCPs without ψ1 ≠ ψ2 ≠ ψ3… ≠ ψn conjugation.72 Fig. 5 depicts the single lattice elongating a wavelength73 on thinner 2D GO sheets similar to those reported previously.61 XRD, HR-TEM, and BET predicted that the synchronized electronic clouds of dopants, and coating agents of B@LGT have intimately hybridized without twisting on aligning interstities similar to their individual angular momentums. The La2S3, Ce2S3, Tb2S3, and Ho2S3 as core LGTs as the two tiers have slightly modulated the electrostatic interacting activities of aq-BSA protein with mild unfolding dynamics reported in an earlier study.33 Noticeably, polyvinylpyrrolidone (PVP)-capped rod-shaped europium (Eu3+)-doped gadolinium oxide (Gd2O3) nanoparticles (PVP@Gd2O3:Eu3+ NPs) have been effective to tune electrostatic charges of salt bridges of proteins.24 Thereby, the B@LGTs could further elaborate the charges to intensify the interacting activities with the peptides and intercalate the base pairs of dsDNA which are being pursued. The aligned B@LGTs have generated the e− and h+ holes of a longer mean free path that photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn within 15–40 min (Fig. 22).






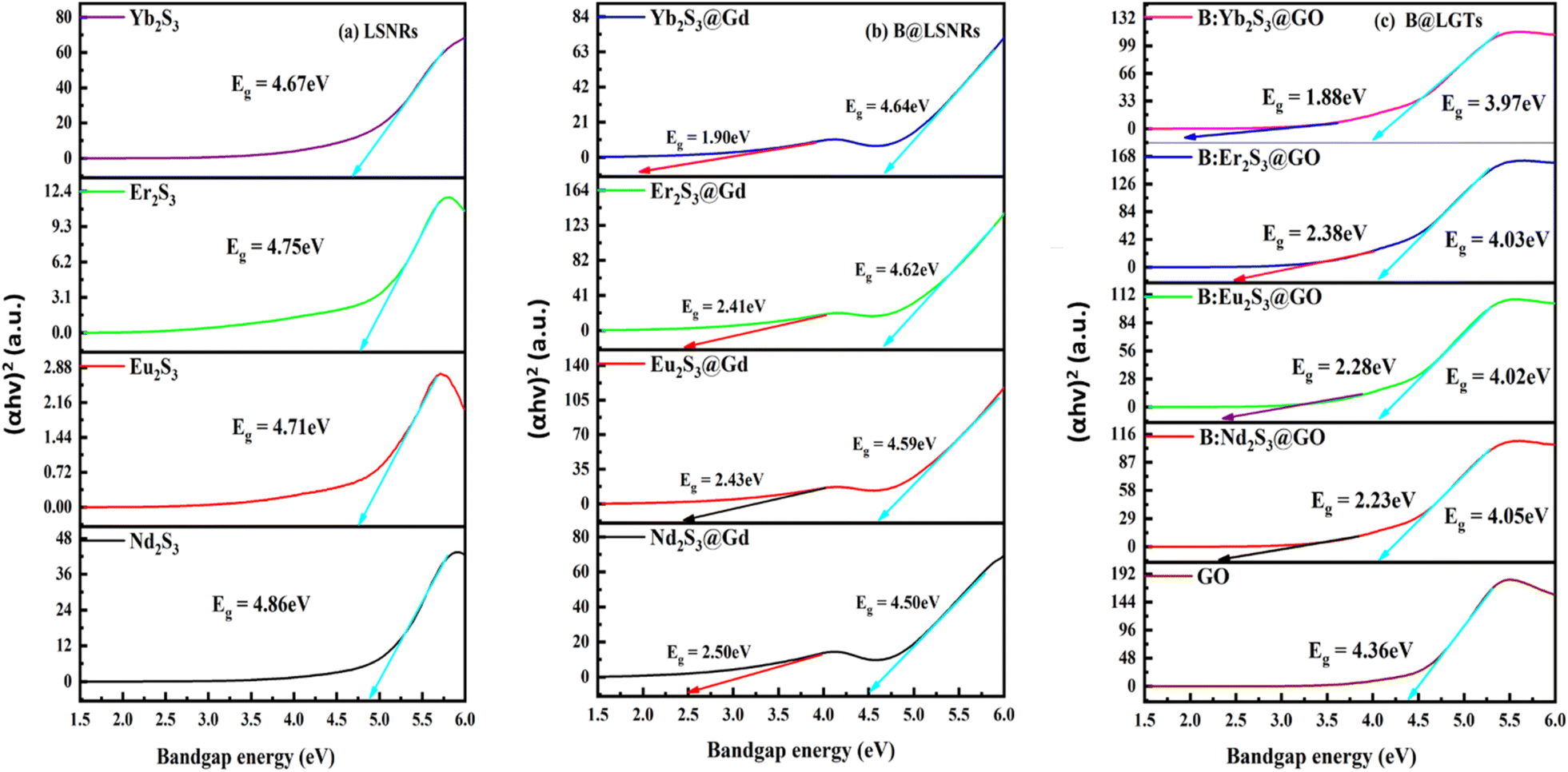


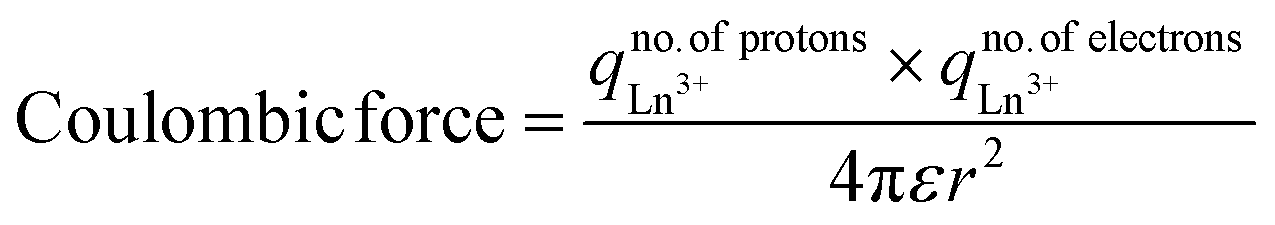
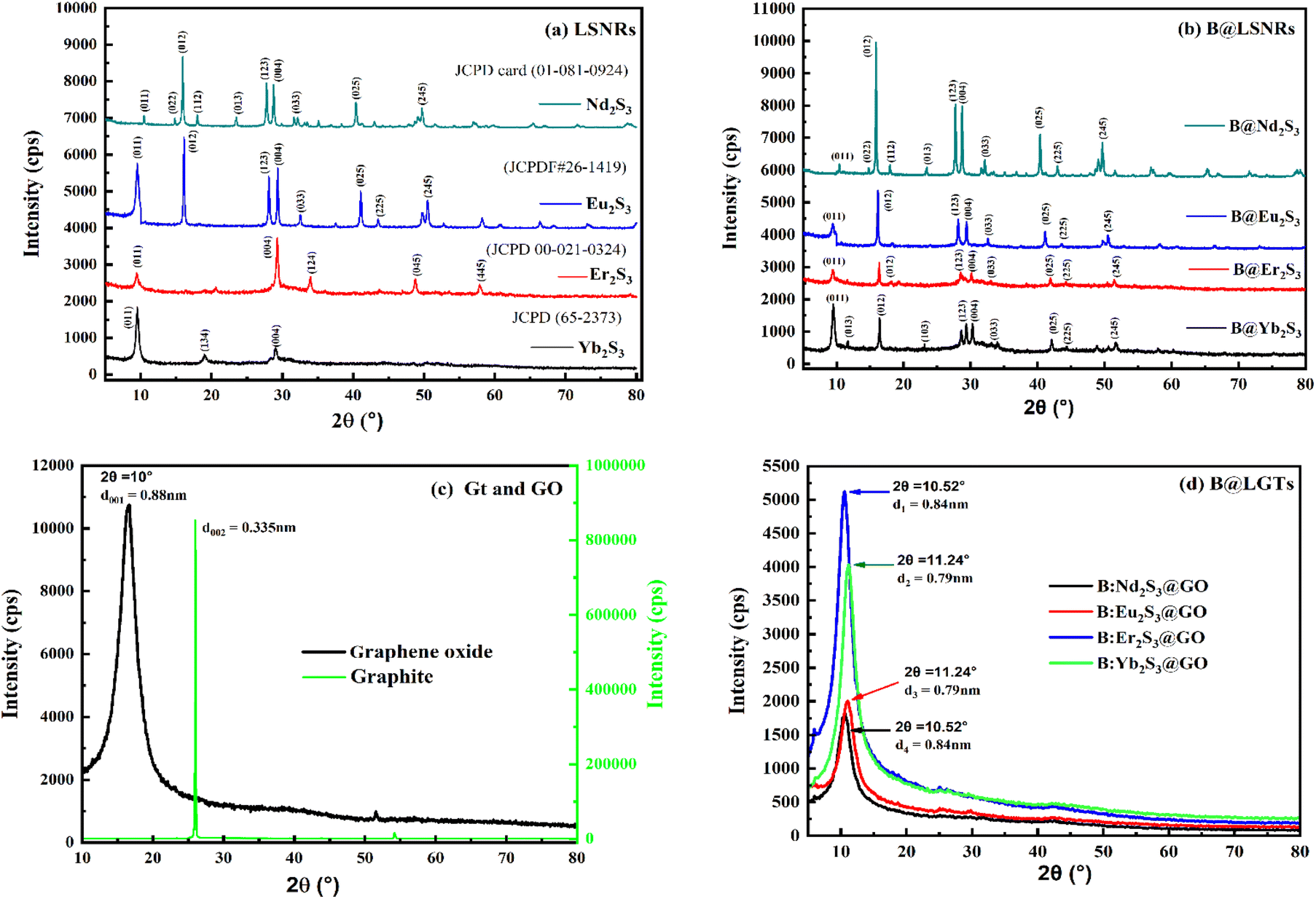


 normalised than
normalised than 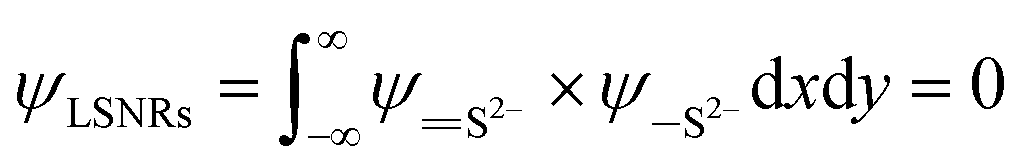 orthogonal due to different intramolecular activities or oscillations synchronised with Gd3+. Gd3+ has functionalised the fingerprint region as well as the stretching domain of B@LSNRs (Fig. S11b†). The broader stretching frequency and fingerprint region have further been sharpened on coating because GO has established communication with Ln3+ and S2− as well as Gd3+ on energy equipartition. Highly functionalised GO with stronger covalent bonds is not disrupted upon coating. Therefore, it has further sharpened the broader stretching domain of LSNRs as well as B@LSNRs (Fig. S11b†). The integrated GO sheets with FGs and FE equally resonate to transfer energy intimating the dopant and coating agents with intimated adherence with LSNRs. This has caused the stronger FRET generating ψsv on energy equipartitioning through the GO due to a sharper frequency at 1430 cm−1 and 1680 cm−1. The GO has stabilised the molecules by normalising the bending frequencies due to a stronger link with the LSNR and Gd3+ to integrate the overall bending pattern as follows:
orthogonal due to different intramolecular activities or oscillations synchronised with Gd3+. Gd3+ has functionalised the fingerprint region as well as the stretching domain of B@LSNRs (Fig. S11b†). The broader stretching frequency and fingerprint region have further been sharpened on coating because GO has established communication with Ln3+ and S2− as well as Gd3+ on energy equipartition. Highly functionalised GO with stronger covalent bonds is not disrupted upon coating. Therefore, it has further sharpened the broader stretching domain of LSNRs as well as B@LSNRs (Fig. S11b†). The integrated GO sheets with FGs and FE equally resonate to transfer energy intimating the dopant and coating agents with intimated adherence with LSNRs. This has caused the stronger FRET generating ψsv on energy equipartitioning through the GO due to a sharper frequency at 1430 cm−1 and 1680 cm−1. The GO has stabilised the molecules by normalising the bending frequencies due to a stronger link with the LSNR and Gd3+ to integrate the overall bending pattern as follows:
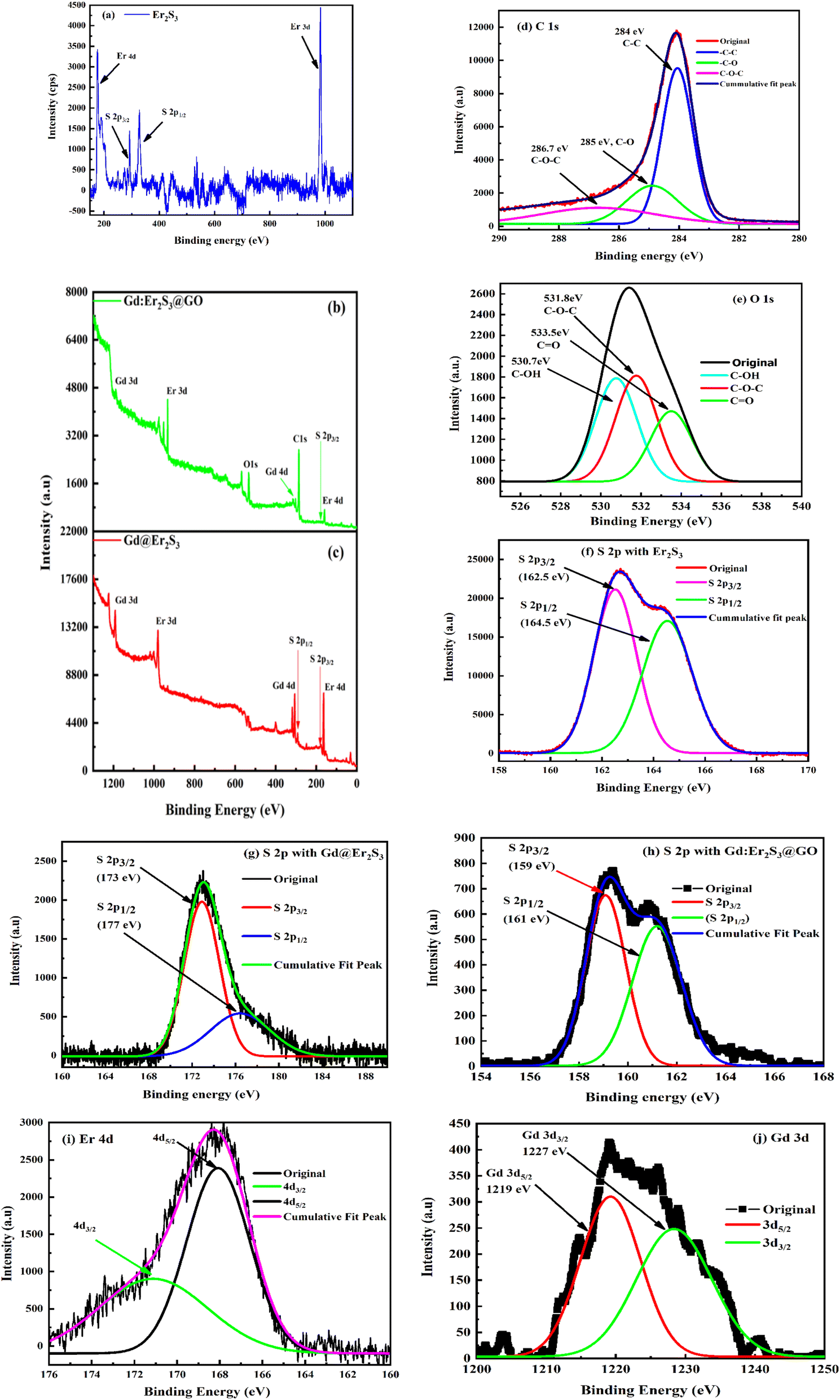

 shifting to
shifting to  from Gt to GO respectively with ψm ≠ ψn, as their initial m and final n coefficients differ at two temperatures due to alignment (Fig. 15a and b). The LGTs were split at ∼250 °C with hedgy oscillations, while Nd2S3, Er2S3 and Yb2S3 have quenched the oscillations by synchronization and Eu2S3 was stabilized. Eu2S3 with hedgy oscillations is shifted to an empty 4f suborbital than other LSNRs. The resonating B@LSNRs get detached103 from the lattice, as Gd3+ from 0 to 250 °C accelerated the
from Gt to GO respectively with ψm ≠ ψn, as their initial m and final n coefficients differ at two temperatures due to alignment (Fig. 15a and b). The LGTs were split at ∼250 °C with hedgy oscillations, while Nd2S3, Er2S3 and Yb2S3 have quenched the oscillations by synchronization and Eu2S3 was stabilized. Eu2S3 with hedgy oscillations is shifted to an empty 4f suborbital than other LSNRs. The resonating B@LSNRs get detached103 from the lattice, as Gd3+ from 0 to 250 °C accelerated the  and ψGd3+ ≈ ψS2− and ψGd3+ ≈ ψLn3+ to shorten the PCR time. The spins of the 4f@e could boost the efficiencies of sensors, acoustivity,61 TFT, and contrasting activities16 in the CT scan.32 Nd2S3@Gd has lost 28.03% wt at 287.49 °C on oscillation as
and ψGd3+ ≈ ψS2− and ψGd3+ ≈ ψLn3+ to shorten the PCR time. The spins of the 4f@e could boost the efficiencies of sensors, acoustivity,61 TFT, and contrasting activities16 in the CT scan.32 Nd2S3@Gd has lost 28.03% wt at 287.49 °C on oscillation as  to ψGd3+ ≈ ψS2− (Fig. S4†). B@LSNR with specific frequencies could exchange a coded message104 similar to receiving the hν transforming into the holes compared with the standard known quantum materials. Gd3+ equipartitioned Nd2S3 at 750 °C, while DTA for Eu2S3 indicated uniform charge distribution compared to other LSNRs. The B@LSNR lost wt at 254 and 436 °C along endothermic and exothermic peaks at 750 °C. The Gd3+⋯S2− and Ln3+−S2−⋯Gd3+ strings could have aligned Eu2S3 with Gd3+. The Gd3+ 4f7e could not induce major oscillations with Eu2S3 except mass transitions at 254.82, 436.6, and 800 °C with 37.04, 27.54, and 35.42% wt losses respectively (Fig. S3 and S4†). Gd3+ with 2Eu3+ and 3S2− of Eu2S3 enhanced the holes via ψLn3+ ≈ ψS2−
to ψGd3+ ≈ ψS2− (Fig. S4†). B@LSNR with specific frequencies could exchange a coded message104 similar to receiving the hν transforming into the holes compared with the standard known quantum materials. Gd3+ equipartitioned Nd2S3 at 750 °C, while DTA for Eu2S3 indicated uniform charge distribution compared to other LSNRs. The B@LSNR lost wt at 254 and 436 °C along endothermic and exothermic peaks at 750 °C. The Gd3+⋯S2− and Ln3+−S2−⋯Gd3+ strings could have aligned Eu2S3 with Gd3+. The Gd3+ 4f7e could not induce major oscillations with Eu2S3 except mass transitions at 254.82, 436.6, and 800 °C with 37.04, 27.54, and 35.42% wt losses respectively (Fig. S3 and S4†). Gd3+ with 2Eu3+ and 3S2− of Eu2S3 enhanced the holes via ψLn3+ ≈ ψS2−  ) with tetrahedron tentropic cavities (eqn (3.2)). B@LGT supersedes the Schottky and Frankel defects to regulate a current flow105 via ψLn3+ and ψGd3+ in a photocatalysing medium to calculate the energies as follows:
) with tetrahedron tentropic cavities (eqn (3.2)). B@LGT supersedes the Schottky and Frankel defects to regulate a current flow105 via ψLn3+ and ψGd3+ in a photocatalysing medium to calculate the energies as follows:
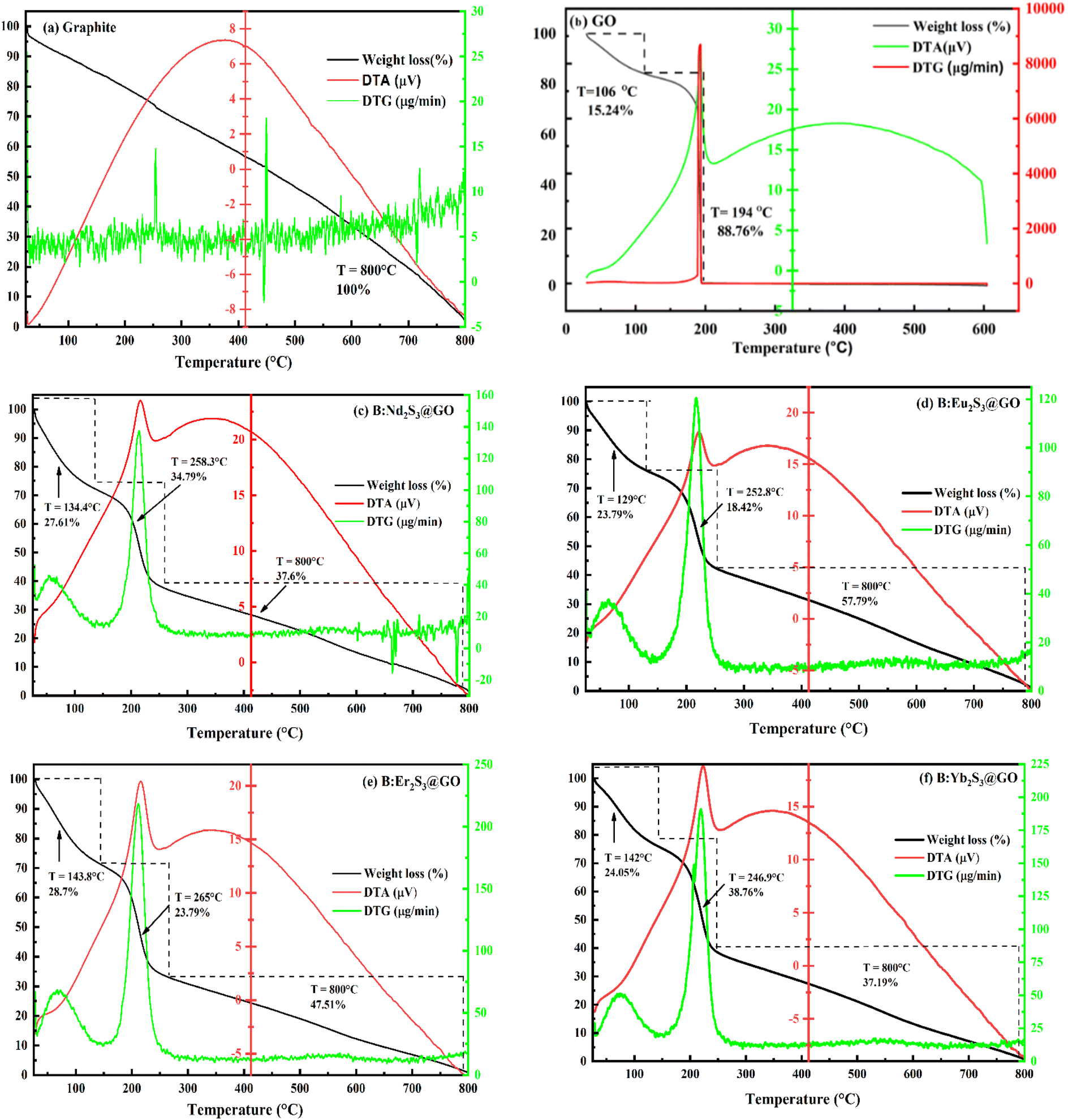

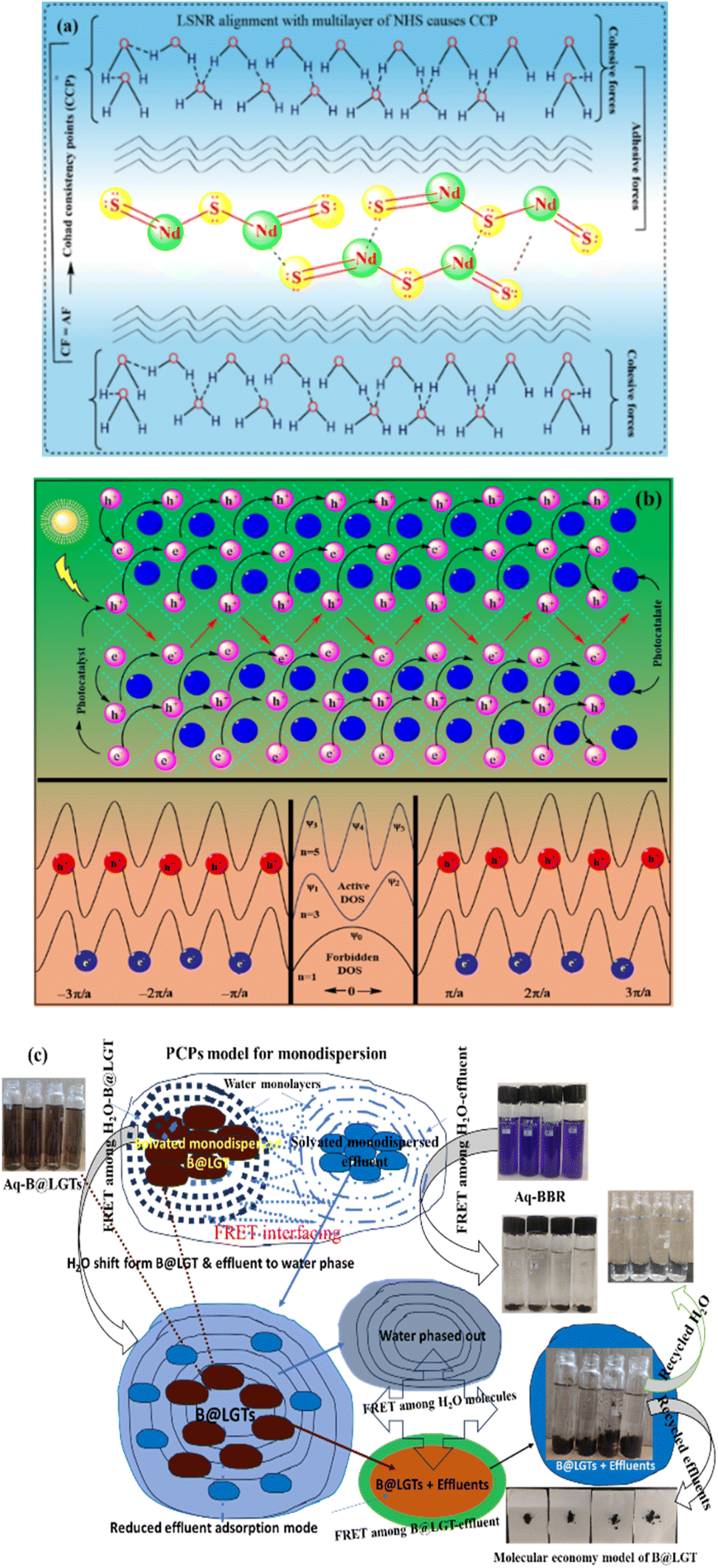
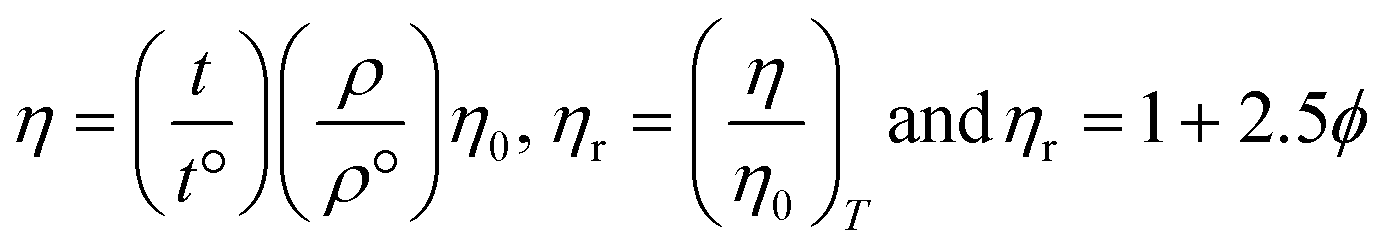






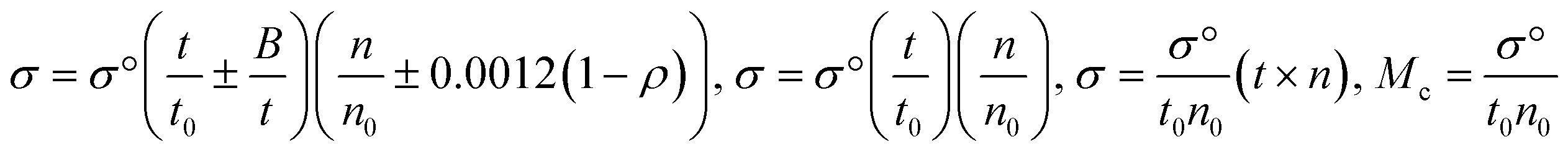
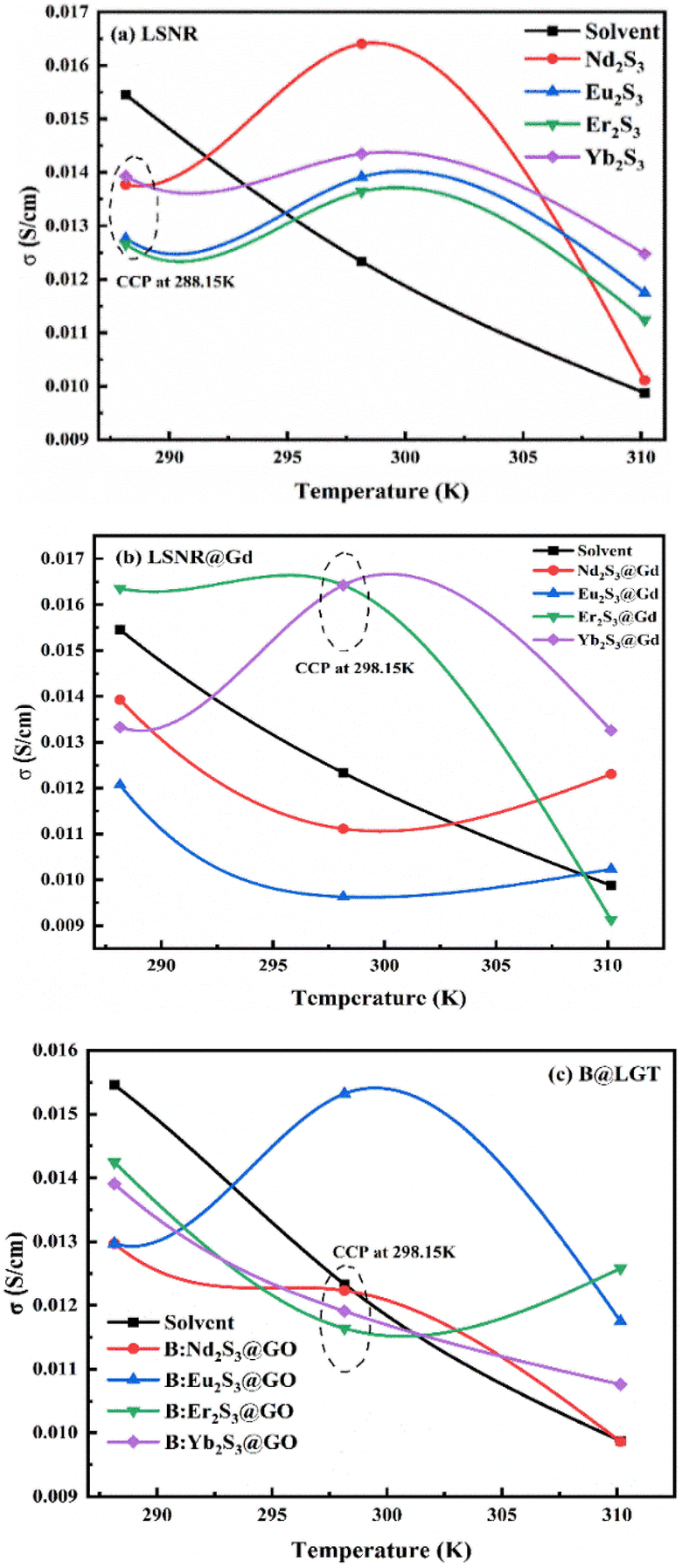

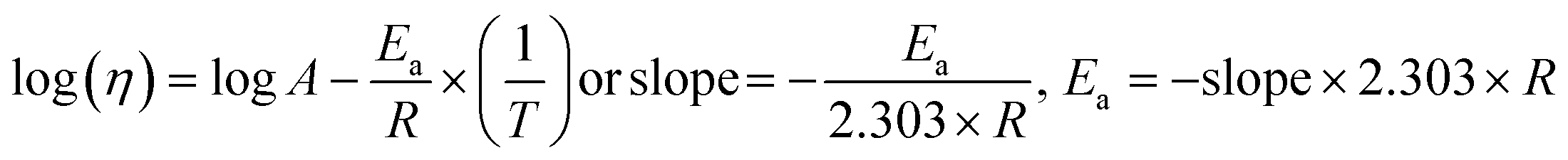




 while changing t from 5 to 30 min, the
while changing t from 5 to 30 min, the 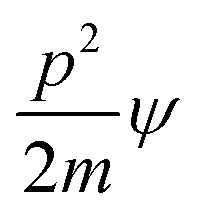 is >90% and V(x, t)ψ is <10%, as predicted via PCPs database with B@LGTs (Fig. 22). H2O dipoles adhered with the B@LGTs were replaced by reduced BBR, TMIs, and QHIn and have restricted the stretching and bending causing hedgy oscillations despite a lower surface tension than the aq-B@LSNR (Fig. 17 and 19). The uniform intense oscillating patterns produced with TGA after PCR than a sharper before PCR due to equidistribution of BBR with interstitities of B@LGTs (Fig. 31). Their interfacing with effluents is accountable for stronger physisorption with localized FRET exchanging their mutual energies. The PCPs have revealed their unique response on lowering γ and relaxation time on specific adherence and coherence. The Gd3+ dopant and GO due to the spins of 4f electrons and stretching of FGs respectively have accommodated the water dipoles (Fig. 22). Thus, an unreduced effluent did not enter the interstities of BL@LGTs except H2O dipoles to lower the γ. The DTA analysis of B@LGT before and after PCR predicted a uniform energy distribution (Fig. 31). The BBR has been uniformly aligned with synchronized interstities of B@LGT depicted with PCPs like surface properties (Fig. 18–20). The B@LGTs could form the oxygen-based superoxide radicals (O2˙−) on splitting the wastewater effluents.115 O2˙− on protonation seems to produce hydroperoxyl radicals (HO2˙−) and free hydroxyl radicals (OH˙).43 The BBR after PCR adsorbed by B@LGT with higher density and negligible VWFs with water get settled at bottom (Fig. 22 and 23). The aq-B@LGTs with slightly higher coherence on aligned H2O dipoles have enhanced the mobility of holes and photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively. The B@LGTs have absorbed the hν at 228 and 312 nm similar to LGTs via FRET.33 Their spins at 302 nm resonated with the BBR dye at 550 nm λ to overcome QEB117 (Fig. 7a and b). The PCR activities with B@LGTs of Nd (4f4), Eu (4f7), Er (4f12), Yb (4f14) have shortened the PCR time by 50% compared to the TMI-GO of Ni (3d8), Zn (3d10), Cd (4d10) Cr (3d5), Mn (3d5), and Fe (3d6).61,118 The B@LGT with Eg (1.88–2.30 eV) harnessed solar energy two times than TMI-GO (2.12–4.47 eV) and 4 times than GO33,61 as 4f@e have generated holes expeditiously than 3d electrons. The B@LGTs have reduced BBR, TMIs, and QHIn in 15–40 min as B:Yb2S3@GO > B:Er2S3@GO > B:Eu2S3@GO > B:Nd2S3@GO with 1.88 < 2.38 < 2.28 < 2.23 eV Eg respectively. Their resonating constituents have higher KE with longer λ with least permittivity.
is >90% and V(x, t)ψ is <10%, as predicted via PCPs database with B@LGTs (Fig. 22). H2O dipoles adhered with the B@LGTs were replaced by reduced BBR, TMIs, and QHIn and have restricted the stretching and bending causing hedgy oscillations despite a lower surface tension than the aq-B@LSNR (Fig. 17 and 19). The uniform intense oscillating patterns produced with TGA after PCR than a sharper before PCR due to equidistribution of BBR with interstitities of B@LGTs (Fig. 31). Their interfacing with effluents is accountable for stronger physisorption with localized FRET exchanging their mutual energies. The PCPs have revealed their unique response on lowering γ and relaxation time on specific adherence and coherence. The Gd3+ dopant and GO due to the spins of 4f electrons and stretching of FGs respectively have accommodated the water dipoles (Fig. 22). Thus, an unreduced effluent did not enter the interstities of BL@LGTs except H2O dipoles to lower the γ. The DTA analysis of B@LGT before and after PCR predicted a uniform energy distribution (Fig. 31). The BBR has been uniformly aligned with synchronized interstities of B@LGT depicted with PCPs like surface properties (Fig. 18–20). The B@LGTs could form the oxygen-based superoxide radicals (O2˙−) on splitting the wastewater effluents.115 O2˙− on protonation seems to produce hydroperoxyl radicals (HO2˙−) and free hydroxyl radicals (OH˙).43 The BBR after PCR adsorbed by B@LGT with higher density and negligible VWFs with water get settled at bottom (Fig. 22 and 23). The aq-B@LGTs with slightly higher coherence on aligned H2O dipoles have enhanced the mobility of holes and photocatalytically reduced BBR, Cr3+, Ni2+, and Cu2+, and QHIn in 30, 20, 40, 35, and 15 min respectively. The B@LGTs have absorbed the hν at 228 and 312 nm similar to LGTs via FRET.33 Their spins at 302 nm resonated with the BBR dye at 550 nm λ to overcome QEB117 (Fig. 7a and b). The PCR activities with B@LGTs of Nd (4f4), Eu (4f7), Er (4f12), Yb (4f14) have shortened the PCR time by 50% compared to the TMI-GO of Ni (3d8), Zn (3d10), Cd (4d10) Cr (3d5), Mn (3d5), and Fe (3d6).61,118 The B@LGT with Eg (1.88–2.30 eV) harnessed solar energy two times than TMI-GO (2.12–4.47 eV) and 4 times than GO33,61 as 4f@e have generated holes expeditiously than 3d electrons. The B@LGTs have reduced BBR, TMIs, and QHIn in 15–40 min as B:Yb2S3@GO > B:Er2S3@GO > B:Eu2S3@GO > B:Nd2S3@GO with 1.88 < 2.38 < 2.28 < 2.23 eV Eg respectively. Their resonating constituents have higher KE with longer λ with least permittivity.





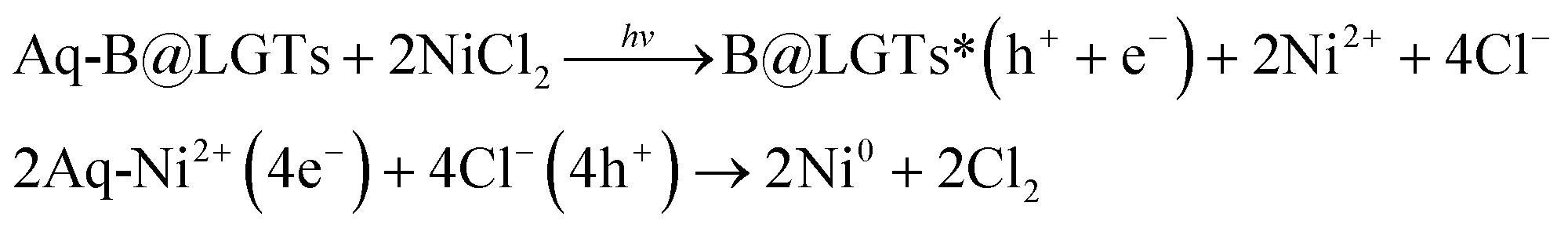


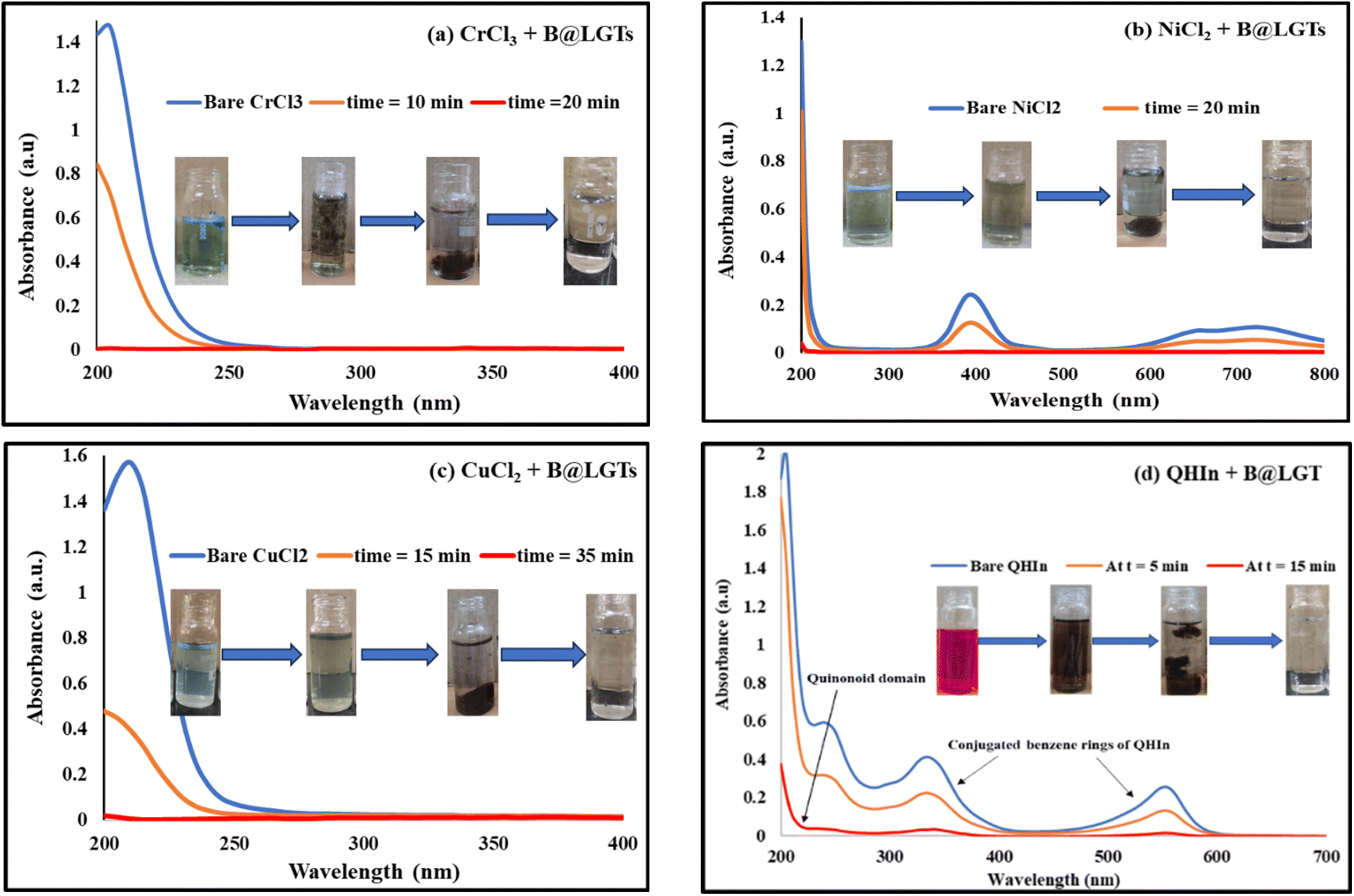


 that could not receive the holes except minor adsorption (Fig. 29). Its transition dipole moment (μ = 0) within the VB to CB space (dτ = dxdydz) along the x, y, and z dimensions is almost nil as follows:
that could not receive the holes except minor adsorption (Fig. 29). Its transition dipole moment (μ = 0) within the VB to CB space (dτ = dxdydz) along the x, y, and z dimensions is almost nil as follows:



