
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective cyclocondensations with thiobarbituric acid: spirocyclic and azocine products, X-ray characterization, and antioxidant evaluation†
Efraín Polo-Cuadrado *a,
Karen Acosta-Quirogab,
Cristian Rojas-Peñab,
Yeray A. Rodriguez-Nuñezc,
Edgard Fabián Blanco-Acuñad,
Jhon J. Lopeza,
Iván Britoe,
Jonathan Cisternaef,
Joel B. Aldereteg and
Margarita Gutiérrez*h
*a,
Karen Acosta-Quirogab,
Cristian Rojas-Peñab,
Yeray A. Rodriguez-Nuñezc,
Edgard Fabián Blanco-Acuñad,
Jhon J. Lopeza,
Iván Britoe,
Jonathan Cisternaef,
Joel B. Aldereteg and
Margarita Gutiérrez*h
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad de Concepción, Concepcion, Chile. E-mail: epolo@udec.cl
bDoctorado en Quimica, Departamento de Quımica Organica y Fisicoquımica, Universidad de Chile, Santiago, Chile
cUniversidad Andrés Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Laboratorio de Síntesis y Reactividad de Compuestos Orgánicos, Santiago 8370146, Chile
dGrupo de Investigación en Ciencias Basicas (NUCLEO), Facultad de Ciencias e Ingenieria, Universidad de Boyacá, Tunja, Boyacá 150003, Colombia
eDepartamento de Química, Facultad de Ciencias Básicas, Universidad de Antofagasta, Avda, Universidad de Antofagasta, Campus Coloso, Antofagasta 02800, Chile
fDepartamento de Química, Facultad de Ciencias, Universidad de Católica del Norte, Sede Casa Central, Av. Angamos, Antofagasta, 0610, Chile
gInstituto de Química de Recursos Naturales, Universidad de Talca, Casilla 747, Talca 3460000, Chile
hLaboratorio Síntesis Orgánica y Actividad Biológica (LSO-Act-Bio), Instituto de Química de Recursos Naturales, Universidad de Talca, Casilla 747, Talca 3460000, Chile. E-mail: mgutierrez@utalca.cl
First published on 19th March 2025
Abstract
Multicomponent cyclocondensations of 5-amino-3-methyl-1-phenyl-1H-pyrazole (AMPZ), thiobarbituric acid, and p-formaldehyde under conventional thermal heating or ultrasonic irradiation were studied. Treatment of the reaction mixture in ethanol in an ultrasonic bath for 3 h produced azocine compound 4b, while the same mixture in ethanol under reflux conditions for 15 h produced spiro compound 4a. This work encompasses intricate experimental details, X-ray diffraction measurements, and multifaceted computational analyses employing methods such as the density functional theory and Hirshfeld surface analysis. Crystallographic investigations revealed the molecular structure of the compound and clarified its interactions involving hydrogen bonds and weak intermolecular forces. This article describes the synthesis and characterization of a novel spirocyclic compound. The study also evaluated the antioxidant potential in vitro using the DPPH and ABTS methods. The results showed that these compounds showed the best free radical scavenging ability, even in very small amounts, and that even at very low concentrations, these compounds showed excellent radical scavenging potential. Surprisingly, these compounds exhibited strong (ABTS+) radical scavenging activities, mainly attributed to the HAT mechanism, indicating their potential as therapeutic agents. Facile multipurpose, three-component selective procedures for new spiroheterocycles have been proposed, presenting intriguing perspectives in the field of medicine, particularly in the field of antioxidants. The geometric values of the computationally optimized structure were calculated using the density functional theory in LC-BLYP/6-31(d), aligned with the X-ray diffraction data, reinforcing the precision of our findings.
1. Introduction
Alkaloids are nitrogenous organic compounds that constitute an important class of secondary metabolites produced by numerous terrestrial and marine organisms (plants, fungi, and bacteria), and are of utmost importance in the defense, proliferation, and reproduction of the species that contain them.1,2 These secondary metabolites are usually characterized by many biological activities; therefore, they are of great interest to researchers in organic and pharmacological chemistry. Among alkaloids, the most studied compounds have been characterized as N-heterocycles with five, six, and seven members (indoles, imidazoles, pyridines, azepines, and their respective variants); however, eight-membered alkaloids have not been extensively studied.1The structural blocks of 8-membered alkaloids that stand out are azocines and azocanes, which are usually found in fungi such as Penicillium dimorphosporum, Penicillium simplicissimum and Laccaria proxima, as well as in plants like Campylospermum flavum and some marine sponges, such as the Okinawan sea sponge Pellina sp.3–7 Azocines and azocanes have been shown to be important biological centers, acting as insecticides,7–10 neuroprotective agents,11–14 antibacterials,3–6,15 antifungals,4,6,16 antimalarials,17–19 antivirals,20 anticancer21,22 and anti-inflammatory.23,24
In contrast, within the group of azocines and azocanos derivatives with bridges (see Fig. 1) have proven to be interesting scaffolds at the pharmacological level; for example, alstomarine B and C molecules have shown significant cytotoxicity against four lines of human osteosarcoma tumor cells (Saos-2, Mg-63, U2-OS, and SOSP-9607).25 Likewise, the Uleine compound inhibits enzymes such as acetylcholinesterase, butyrylcholinesterase, and β-secretase and was patented in 2010 for the prevention and/or treatment of infectious diseases such as HIV.26,27 In contrast, FR901483 plays a key role in exerting strong immunosuppressive activity in vitro and prolonging skin graft survival time in a mouse allograft model.28 It is still important to highlight that, to date, no studies have reported the antioxidant capacity of bridged azocines, and even less so that they have an antioxidant capacity comparable to that of a reference antioxidant such as ascorbic acid, so this is an interesting area of research to provide a new therapeutic approach for this type of molecule.
 | ||
| Fig. 1 The derivatives of azocine and azocane have interesting biological properties. | ||
Thus, there has been a growing interest in the synthesis of new compounds derived from bridged azocanes and azocines to obtain molecules that are potentially applicable to medicine. However, obtaining these compounds can be challenging for organic chemists because of the difficulty involved in obtaining an eight-membered ring and effectively assembling the bridge.29,30 Mohammad et al. conducted the synthesis of a new series of dipyrazolo[1,5]diazocine-3,8-diones by the one-pot reaction of 3-aminopyrazolone with substituted benzaldehydes in the presence of catalytic p-toluenesulfonic acid.31 Also, Vachan et al. recently reported the synthesis of 2,6-methanobenzo[d][1,3]diazocines via the reaction between primary amine beta-ketoesters and 2-aminoarylaldehydes under reflux for 1 h32 (Scheme 1). To the best of our knowledge, there are no reports on the synthesis of 1,3-diazocines fused with pyrazole derivatives. Furthermore, in this work, a multicomponent synthesis is presented, which, depending on the energy source, can favor the formation of the azocine derivative or a spirone derivative, which together with the pyrazolopyrimidine are two other types of scaffold widely studied both in terms of reactivity, as well as in a wide range of promising biological properties.33–43
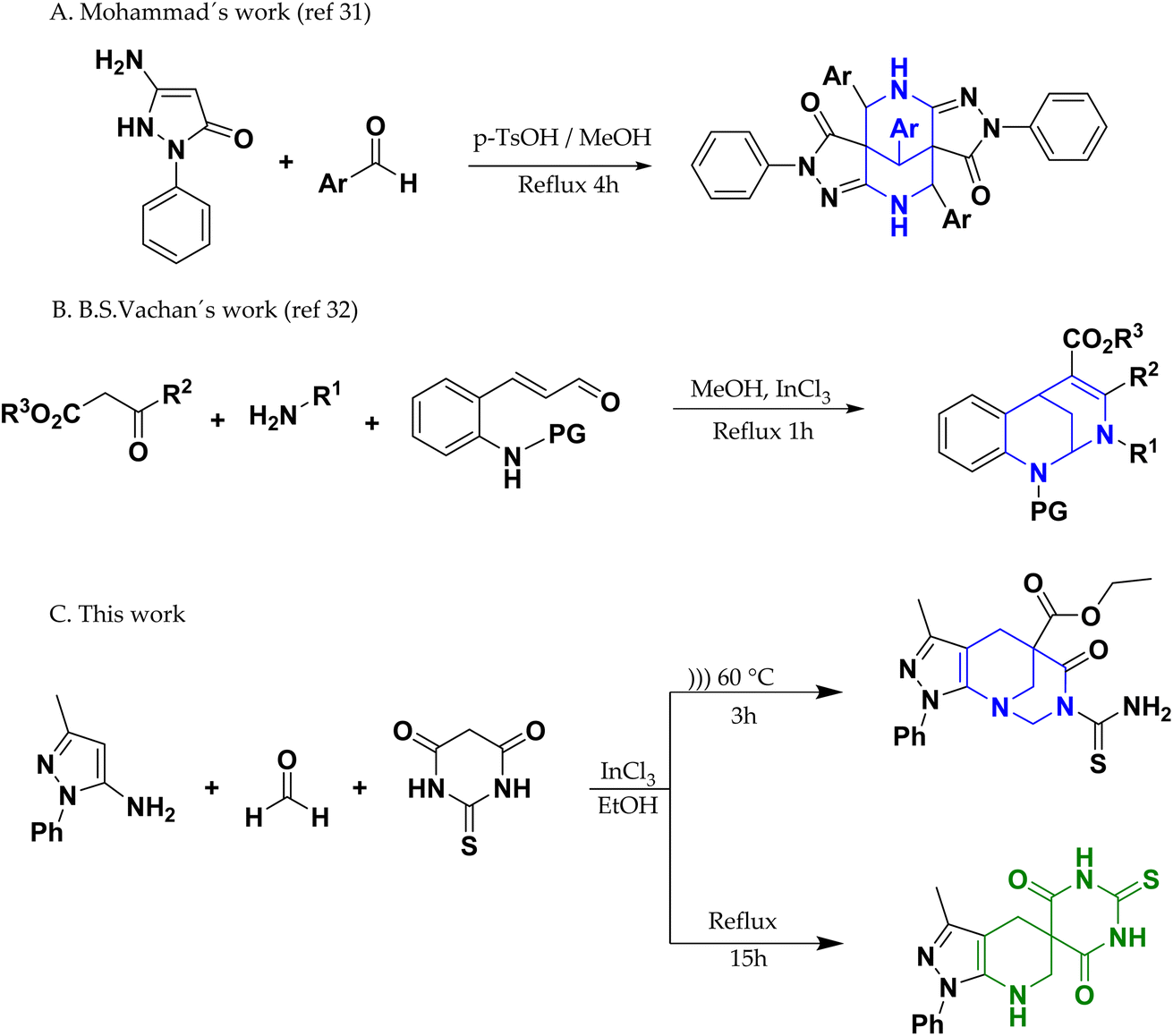 | ||
| Scheme 1 Synthesis of bridged-azocine. | ||
Taking the above into account, in the present work we synthesized, crystallized, and characterized a new nucleus derived from the bridged diazocine system using the experimental techniques XRD, NMR, FT-IR, and HRMS. Likewise, confirmation of the stable crystal structure has been based on quantum chemistry results, such as geometry optimization, Hirshfeld surface analysis, and energy framework calculations of the frontier molecular orbitals. Motivated by the great potential of bridged azocine derivatives, we determined the antioxidant capacity of the compound using DPPH and ABTS assays.
2. Results and discussion
2.1. Chemistry
In the present work, we have synthesized compounds 4a (62% yield) and 4b (33% yield) through the implementation of reflux and ultrasound as energy sources, respectively (Scheme 2). The general mechanism proposed for the syntheses of 4a and 4b (Scheme 3)44 involves the formation of a Knoevenagel product (step A) between thiobarbituric acid (1) and paraformaldehyde (3). Continuing this mechanism, the formation of an imine (step B) was observed because of the reaction between compound 2 and a molecule of paraformaldehyde (3). On the other hand, the imine formed reacts with the Knoevenagel product of step A via aza-Diels–Alder (ADA) reaction, generating the intermediate of step C, which due to proton transfer is transformed into compound 4a (step D). Subsequently, the reaction continues, adding a molecule of ethanol to 4a (step E), generating an esterification reaction that allows the formation of the intermediate of step F, which attacks a formaldehyde molecule through the nitrogen of the piperidine group, generating the addition of a methanol molecule (step G). Finally, cyclization occurs through dehydration, promoted by the attack of the carbamothioylacetamide nitrogen on the hydroxyl carbon, which leads to the formation of product 4b. | ||
| Scheme 2 Conditions and synthesis of spiro-pyrazolopyridine (4a) and 1,3-diazocine (4b). | ||
 | ||
| Scheme 3 Proposed mechanism for the synthesis of 4a and 4b. | ||
2.2. FT-IR spectra
Fig. 2 shows the experimental and simulated infrared (IR) spectra obtained using the LC-BLYP/6-31G* method for 4a and 4b. On the other hand, in Table 1, the resulting vibrational frequencies for the optimized geometries are shown from highest to lowest, along with the proposed vibrational assignments and IR intensities. Comparisons of the theoretical and experimental IR spectra indicated that the strong vibrations in the experimental spectrum were also strong in the theoretical spectrum. | ||
| Fig. 2 IR spectra calculated using DFT (B3LYP/6-31G*) (A and B) and experimental (C and D) IR spectra for 4a and 4b. | ||
| Normal mode | LC-BLYP/6-31G* | Experimental in this study | Freq. (cm−1) | Approximate assignments |
|---|---|---|---|---|
| Freq. (cm−1) | Intensity (km mol−1) | |||
| 4a | ||||
| 1 | 3442 | 0.27 | 3465 | Symmetric stretching N–H |
| 2 | 3079 | 0.06 | 3074 | Symmetric stretching C–H sp2 |
| 3 | 2967 | 0.08 | 2972 | Symmetric stretching C–H sp3 |
| 4 | 2924 | 0.06 | 2941 | Asymmetric stretching C–H sp3 |
| 5 | 1714 | 0.60 | 1689 | Stretch C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O × 2 O × 2 |
| 6 | 1593 | 0.14 | 1597 | Stretching CC |
| 7 | 1489 | 1.00 | 1506 | Rocking amide N–H |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| 4b | ||||
| 1 | 3537 | 0.30 | 3371 | Asymmetric stretching N–H2 |
| 2 | 3347 | 0.33 | 3234 | Symmetric stretching N–H2 |
| 3 | 3071 | 0.06 | 3059 | Symmetric stretching C–H sp2 |
| 4 | 2984 | 0.14 | 2974 | Symmetric stretching C–H sp3 |
| 5 | 2924 | 0.10 | 2918 | Asymmetric stretching C–H sp3 |
| 7 | 1731 | 0.46 | 1724 | Stretch CO (ester) |
| 8 | 1662 | 0.27 | 1660 | Stretch CO (amide) |
| 9 | 1593 | 0.15 | 1583 | Stretching CC |
| 10 | 1559 | 1.00 | 1508 | Scissoring N–H2 |
| 11 | 1222 | 0.81 | 1257 | Stretching C–O |
The IR signals of 4a and 4b allowed the identification and characterization of their functional groups (see Table 1). Both present signals for C sp2–H, C sp3–H, and CC. However, 4b showed more signals owing to the presence of thioamide and ester groups.
In 4a, a signal was observed for N–H (stretching) at 3465 cm−1 and one for CO (stretching) at 1689 cm−1, and there was no signal for C–O. In contrast, 4b presents two signals for N–H (asymmetric and symmetric stretching) at 3371 and 3234 cm−1, two for CO (ester and thioamide stretching) at 1724 and 1660 cm−1, and one for C–O (ester) at 1257 cm−1.
As shown in Table 1, the frequencies calculated using the LC-BLYP/6-31G* method were in good agreement with the experimentally obtained results. Therefore, this computational method is valuable for identifying important functional groups to characterize the molecules studied here.45
2.3. Frontier molecular orbital (FMOs) analysis and molecular reactivity
To obtain information on the reactivity and stability of compounds 4a and 4b, descriptors obtained from density functional theory were studied: HOMO and LUMO energies, HOMO–LUMO gap, electron affinity (EA), ionization energy (IE), electronegativity (χ), chemical hardness (η), chemical softness (s), and electrophilicity (w) (see Table 2 and Fig. 3). It was found that for molecule 4a the values were −784.81, −23.10, 761.74, 83.08, 676.61, 379.85, 296.77, 0.0034 and 243.09, while, for molecule 4b, the values were −797.83, 88.10, 885.92, 0.12, 756.41, 378.14, 0.0026 and 189.19 kJ mol−1, respectively.| 4a | 4b | |
|---|---|---|
| EA | 83.08 | 0.12 |
| IE | 676.61 | 75.41 |
| χ | 379.85 | 378.27 |
| η | 296.77 | 378.14 |
| s | 0.0034 | 0.0026 |
| w | 243.09 | 189.19 |
| Ra | 0.49 | 0.26 |
| Rd | 1.47 | 1.33 |
 | ||
| Fig. 3 The frontier molecular orbitals and related energies of 4a and 4b are expressed in kJ mol−1. | ||
The HOMO energy defines the susceptibility of a compound to donate electrons and undergo electrophilic addition. In contrast, the LUMO energy provides information about the susceptibility of a compound to accept electrons, undergoes nucleophilic attack, and is related to electronegativity (tendency to attract electron density) and electron affinity (ability to accept electrons), whereas the HOMO–LUMO gap provides information about chemical reactivity and kinetic stability. A molecule with a high energy gap is associated with low chemical reactivity and high kinetic stability and vice versa (see Fig. 3). Finally, chemical hardness and softness are related to the polarizability of a molecule; that is, the greater the hardness, the lower the polarizability, and the greater the softness, the greater the polarizability.
Fig. 3 shows the molecular frontier orbitals of compounds 4a and 4b. For the 4a molecule, it is evident that the HOMO orbital shows a high electronic density located on the pyrazole and benzene rings, whereas in the case of the LUMO orbital, the highest probability region is found on the thiobarbituric acid ring, which agrees with the proposed mechanism because it is a zone with the capacity to accept electrons and therefore suffers from nucleophilic attack. On the other hand, for 4b, the HOMO orbital shows electron density on the thioamide, while the LUMO orbital shows that the highest probability region is located both in the thioamide and the two carbonyl groups present in the molecule.
In general, the 4a molecule has a smaller HOMO–LUMO gap, lower chemical hardness, greater chemical softness, greater electronegativity, greater electronic affinity, and lower ionization energy than the 4b molecule, for which the 4a molecule presents greater reactivity, less kinetic stability, greater polarizability, and greater ability to attract electron density, as well as to accept electrons.
2.4. Biological activity
We used two diverse assays to measure the in vitro antioxidant activity: (a) the interaction with the stable free radical DPPH and (b) the interaction with the water-soluble azo compound ABTS+, where ascorbic acid was used as a positive control. These radicals are not biologically relevant but are often used as indicator compounds to test the hydrogen transfer capacity, which is related to antioxidant activity. Antioxidant properties were expressed as EC50 values (Table 4).In the DPPH assay, the EC50 values of 4a and 4b were <100 μg mL−1. Both compounds showed significant ABTS+ free radical scavenging activity (EC50: 0.0967 and 1.6820 μM) compared to the commercially available antioxidants ascorbic acid, quercetin, and Trolox (EC50 > 2 μM).
The radical scavenging activity of pharmacophore leads depends on electron-donating groups such as hydroxyl (–OH) and –NH groups, independent of their attachment, which can easily abstract free radicals and potentially convert highly reactive free radicals to their non-reactive forms.47
In general, it has been described that antioxidants can act through two mechanisms on radicals, the transfer of a hydrogen atom known as (HAT) and the transfer of a single electron (SET). However, both ABTS and DPPH radicals can be stabilized by hydrogen atom transfer (HAT) or electron transfer (ET) mechanisms, but the reactivity patterns and mechanism are difficult to interpret.48
Thus, we calculated IE, EA, w (related to the SET spectra), and the hydrogen bond dissociation energy BDE (related to the HAT mechanism) at the DFT level to determine the possible mechanism of action of the compounds 4a and 4b. IE is the minimum energy required to extract an electron from a gaseous atom in its ground state; therefore, the lower the ionization energy of the antioxidant, the easier it is for it to donate an electron and neutralize a radical. On the other hand, EA is the energy released when a gaseous atom in its ground state accepts an electron to form a negative ion. The higher the EA of the antioxidant, the easier it is for it to accept an electron from a radical and neutralize it. W measures the ability of a molecule to attract electrons, thereby allowing it to neutralize them. Finally, BDE is the energy needed to break a hydrogen bond, and the lower it is, the greater is the antioxidant potential of the compound.
In addition, we drew a donor–acceptor map, DAM, as proposed by Martínez.49,50 This map, normalized with F as a good electron acceptor and Na as a good electron donor, is a useful graphical indicator. Therefore, this comparison was performed using computational values for the F and Na atoms at the same theoretical level for the molecules studied.
If Ra = 1, L is a compound with an electron acceptor efficiency like F. If Ra > 1, L is a more effective electron acceptor than F. Finally, if Ra < 1, L is a less effective electron acceptor that F. Similarly, if Rd = 1, L is an electron donor with an efficiency similar to that of Na. If Rd > 1, then L is a less efficient electron donor than Na. Similarly, if Rd < 1, L is a more efficient electron donor than Na is. Plotting Ra and Rd is the correct way to visualize the antioxidant scheme using MAP (Fig. 4).
 | ||
| Fig. 4 Donor–acceptor map of the molecules studied in the gas phase. | ||
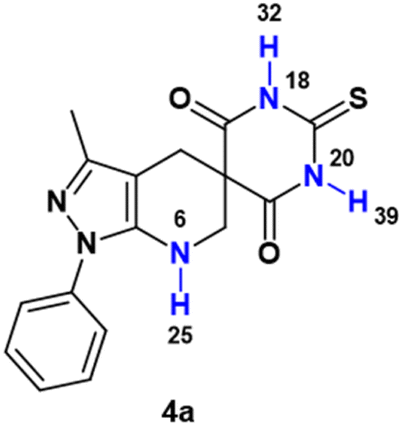
Compound 4a has three sp-hybridized –NH protons, which can be transferred to free radicals (e.g., the N-centered hydrazinyl free radical of ABTS+) by hydrogen atom transfer (HAT), with BDE of 228.01, 220.26 and 220.17 kJ mol−1 for the hydrogens H25, H32 and H39 respectively, while molecule 4b has a –NH2 group, with BDE of 294.80 and 269.73 kJ mol−1 for the hydrogens H41 and H42, respectively (see Table 3). These results suggest, on the one hand, that the most labile hydrogen in molecule 4a is 39, while the most labile hydrogen in molecule 4b is hydrogen 42, so these may be the main responsible for a possible neutralization of the radicals studied by HAT. Likewise, a comparison of the values of the most labile hydrogens in each molecule (H39 for 4a and H42 for 4b) shows that molecule 4b has a higher BDE of 49.56 kJ mol−1 with respect to 4a, which agrees with the experimental antioxidant results obtained, where 4a presents a greater antioxidant capacity.
|
|
||
|---|---|---|---|
| Entry | BDE (kJ mol−1) | Entry | BDE (kJ mol−1) |
| N6–H25 | 228.01 | N21–H41 | 294.80 |
| N18–H32 | 220.26 | N21–H42 | 269.73 |
| N20–H39 | 220.17 | ||

| Compound | Antioxidant assay EC50a (μM) | |
|---|---|---|
| DPPH assay | ABTS+ assaya | |
| a Values are expressed as mean ± SEM of three parallel measurements (p < 0.05).b Reference compound. | ||
| 4a | >292.921 | 0.0967 ± 0.001 |
| 4b | >294.577 | 1.6820 ± 0.020 |
| Ascorbic acidb | 8.517 ± 0.2 | 156.824 ± 3.5 |
| Quercetin46 | 8.688 ± 0.02 | 15.487 ± 0.04 |
| Trolox47 | — | 11.798 ± 0.05 |
In contrast, the IE, EA, and w results indicate that 4a has a lower ionization potential, greater electronic affinity, and greater electrophilicity than 4b. Furthermore, compounds 4a and 4b presented values of 0.49 and 0.26 for Ra and 1.47 and 1.33 for Rd, respectively. All of the above results allow us to infer that 4a has greater availability to accept and donate electrons than 4b, so the latter has less potential as an antioxidant. Likewise, it is important to mention that, in general, the values obtained for BDE compared to those for ionization potential are more than 400 kJ mol−1, which is lower for both molecules, so it is possible that both act mainly by the HAT mechanism.
2.5. Crystallographic studies
The molecular structure of compound 4b corresponded to ethyl 7-carbamothioyl-3-methyl-6-oxo-1-phenyl-1,2,3,4,7,8-hexahydro-5,9-methanopyrazolo[3,4-b]azocine 5(6H)-carboxylate. This compound crystallizes in a monoclinic cell system with space group P21/c (Z = 4). The molecular structure of the compound agrees with spectroscopic characterization and the proposed structure, and both show a centrosymmetric setting with normal bond distances and angles51 (see Fig. 5). The dihedral angle between the mean planes of the phenyl ring and the 5-membered heterocycle is 19.49(9)°, which is almost coplanar. Additionally, the two fused rings exhibited a fold angle of 91.01(7)°. According to the Cremer & Pople parameters,52 the two fused rings in the title compound have a half-chair conformation, (QT = 0.541(2)/0.558(2) Å; θ = 5.0(3)/51.3(2)°; ϕ = 331.8(3)/277.1(3)°).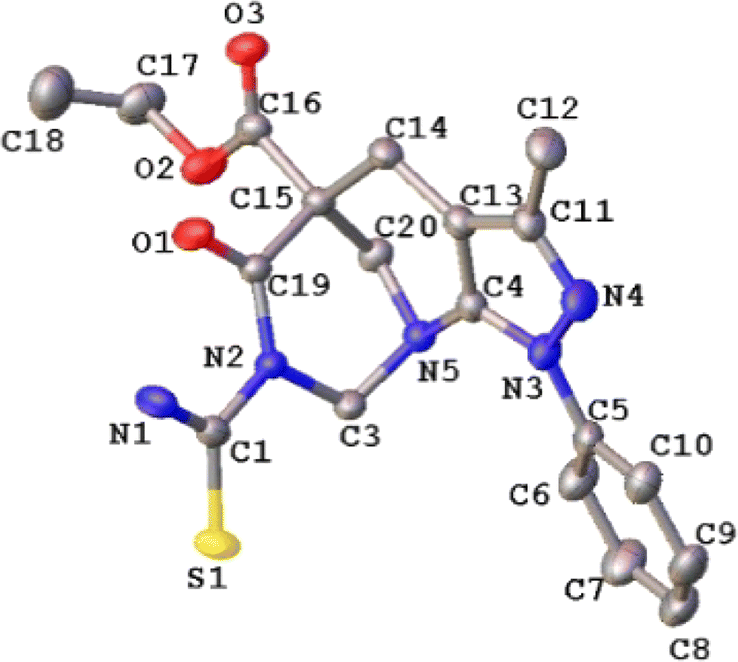 | ||
| Fig. 5 ORTEP plot of the 4b. Thermal ellipsoids were drawn at a 30% of probability. The H atoms were omitted for simplicity. | ||
All the hydrogen atoms were placed at their calculated positions, assigned fixed isotropic thermal parameters, and constrained to ride on their parent atoms. All geometrical calculations were performed using Platon software.53 A summary of the crystal data, the collection of which is presented in Table 5, and additional crystallographic details are provided in the CIF file. ORTEP views were drawn using OLEX2 software.54
| Empirical formula | C20H22N4O3S | F(000) | 840.0 |
| Formula weight | 398.47 | Crystal size/mm3 | 0.19 × 0.17 × 0.02 |
| Temperature/K | 297.75 | 2θ range | 7.086 to 56.76 |
| Crystal system | Monoclinic | Index ranges h,k,l | −11/11, −26/27, −14 ≤ l ≤ 14 |
| Space group | P21/c | Reflections collected | 22466 |
| a/Å | 8.6243(6) | Independent reflections | 4675 [Rint = 0.0372, Rsigma = 0.0265] |
| b/Å | 20.6113(13) | Comp. qmax (%) | 99.5 |
| c/Å | 10.7734(7) | Max/min transmission | 0.746, 0.669 |
| β/° | 102.261(2) | Data/restraints/parameters | 4675/0/255 |
| Volume/Å3 | 1871.4(2) | Goodness-of-fit on F2 | 1.057 |
| Z | 4 | Final R indexes [I ≥ 2σ(I)] | R1 = 0.0542, wR2 = 0.1572 |
| ρcalc g cm−3 | 1.414 | Final R indexes [all data] | R1 = 0.0671, wR2 = 0.1689 |
| μ/mm−1 | 0.203 | CCDC number | 2200261 |
Additionally, the crystal packing of the title compound does not present geometrical parameters corresponding to classical hydrogen bonding55 and is stabilized by intra- and intermolecular non-conventional hydrogen bond-like interactions, N–H⋯X (X = O and S) and C–H ⋯O. In the title compound, two intramolecular hydrogen bonds can be described using the graph-set motif S(6). Likewise, intermolecular hydrogen bond interactions generate R43(13) and R44(22) ring motifs along the [100] direction, as a consequence of the 21 screw axis. Additionally, the C11(4) and C11(8) graph set motifs (symmetry operations = +X, 1/2 − Y, −1/2 + Z, and 1 − X, 1 − Y, 1 − Z, respectively).56 Form extended chains running along the [001] and [100] directions (see Fig. 6 and Table 6).
 | ||
| Fig. 6 Crystal packing of 4b. | ||
2.6. Hirshfeld surface analysis
To observe other intermolecular contacts across the crystal structure, Hirshfeld surface analysis was performed to complement XRD analysis. The intermolecular interactions were represented by N–H⋯X (X = O and S). The contacts are shown as red (dnorm < vdW radii), white (dnorm = vdW radii), and blue (dnorm > vdW radius) spots on the dnorm surfaces for all compounds. Moreover, there is evidence of another interesting weak contact in the crystal structures of all compounds. The reciprocal contacts, their respective contributions, and all fingerprint plots with dnorm (where dnorm = di + de) surfaces for their intermolecular contacts are depicted in Fig. 7. Additionally, for H⋯H contacts in each compound, does not generate a significant effect on molecular packing in the crystal structure stabilization because their contacts are di + de > 2.4 Å, in other words, these contacts are slightly longer than the sum of the vdW radii for these atoms,57 which cannot support the crystal packing in the title compound. | ||
| Fig. 7 Overall, the Hirshfeld surface (left) of 4b and the fingerprint plot (right) are shown. | ||
4b shows the same interaction as that described above, where hydrogen-bond-like interactions are observed on the dnorm surface. Another type of weak interaction was observed when the Hirshfeld surface was analyzed. For example, aromatic π contact was verified over the title compound (Fig. SI-5†) a with a shape-index surface. This was verified using the shape index surface, which allowed us to determine the presence of these weak interactions. The yellow-orange spots show surface subsidence owing to the proximity of the neighboring moieties, and the blue-green spots show the reciprocal contacts of the moieties that generate the subsidence. The contact is between the ethyl and phenyl fragments in the title compound, with a contribution of approximately 16.8% and a de + di of ≈3.0 Å, as a clamp pattern in the fingerprint plot.
A C⋯S chalcogen bond interaction is also observed, and this was verified over the carbonyl and carbamothioyl fragments (Fig. SI-6†) with a contribution of around 0.6% with de + di ≈ 3.4 Å, which works as electron donor in a σ-hole noncovalent bond.58,59
Finally, the energy framework60 was analyzed to better understand the packing and topology of the crystal structure and supramolecular rearrangement. This method allows the calculation and comparison of different energy components, that is, repulsion (E_rep), electrostatic (E_ele), dispersion (E_dis), polarization (E_pol), and total (E_tot) energies, based on the anisotropy of the topology of pairwise intermolecular interaction energies (see Fig. SI-7 and Table SI-1†). The thickness of the cylinder radius shows the grade of interactions, is related to the energy magnitude, and provides information regarding the stabilization of the crystal packing.61 Based on the tube direction, it can be concluded that the formation of the framework is directed by the translational or centrosymmetric elements. However, this rearrangement allowed the formation of weak interactions in the crystal structure.
The results of the calculations revealed that the dispersion interactions exhibited a distorted zigzag ladder-shaped topology in the title compound. The significant difference between E_ele and E_pol is due to the absence of classical hydrogen bond interactions.
3. Experimental section
3.1. General information
Ultrasound irradiation was performed using a Branson model 1510, 115v, 1.9 L ultrasonic bath, time machine (continuous hold for 60 min), and switch heater (47 kHz). 1H and 13C NMR spectra (400 MHz for protons and 100 MHz for carbon) were recorded on an AM-400 spectrometer (Bruker, Rheinstetten, Germany) using CDCl3 and DMSO-d6 as the solvents. Tetramethylsilane (TMS) was used as an internal standard. IR spectra (KBr granules, 500–4000 cm−1) were recorded on a NEXUS 670 FT-IR spectrophotometer (Thermo Nicolet, Madison, WI, USA). ESI-MS and ESI-MS/MS high-resolution mass spectrometry were performed on a high-resolution (Q) hybrid quadrupole time of flight (TOF) mass spectrometer (Waters/Micromass Q-TOF Micro, Manchester, UK) with a constant nebulizer temperature of 100 °C. Melting points (uncorrected) were measured using an IA9100 electrothermal melting point device (Stone, Staff, UK). The progress of the reaction was evaluated by thin-layer chromatography (TLC) using silica gel 60 (Merck, Darmstadt, Germany). All the reagents and solvents were purchased from Merck or Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. The final purification of all the products for the analysis was conducted by column chromatography using silica gel 60 (Merck 7734) with a particle size that fluctuated between 0.063 and 0.200 mm, as eluent phase ether mixtures of petroleum: ethyl acetate in increasing polarity.3.2. Chemistry
:ethyl acetate in increasing polarity as eluant phase to afford the compound target 4a in 62% yield as a white solid, IR (KBr, cm−1): 3465, 3074, 2972, 2941, 1689, 1597, 1506; 1H NMR (400 MHz, DMSO-d6) 2.07 (s, 3H), 2.87 (s, 2H), 3.36 (s, 2H), 5.90 (t, J = 6.5 Hz, 1H), 7.19 (t, J = 7.3 Hz, 1H), 7.40 (t, J = 8.0 Hz, 2H), 7.67 (t, J = 8.0 Hz, 2H), 11.18 (s, 2H); 13C NMR (100 MHz, DMSO-d6) 12.6 (CH3), 23.8 (CH2), 47.8 (C), 51.2 (CH2), 100.2 (C), 120.5 (2× CH), 125.2 (CH), 129.4 (2× CH), 139.9 (C), 142.8 (C), 146.0 (C), 150.9 (C), 171.7 (2× C); HRMS (ESI, m/z): calculated for C16H17N5O2S [M + 2H]+ 343.1103 found 343.8190.:ethyl acetate in increasing polarity as eluant phase, producing compound (4b) in 33% yield as a green crystal; IR (KBr, cm−1): 3371, 3234, 3059, 2974, 2918, 1724, 1660, 1583, 1508, 1257; 1H NMR (400 MHz, CDCl3) 0.99 (t, J = 7.1 Hz, 3H), 2.21 (s, 3H), 3.20 (q, J = 7.1 Hz, 2H), 3.61 (s, 2H) 4.08 (s, 2H), 4.36 (s, 2H), 7.28 (t, J = 7.5 Hz, 1H), 7.40 (t, J = 7.8 Hz, 2H), 7.60 (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) 12.1 (CH3), 14.8 (CH3), 22.1 (CH2), 49.5 (CH2), 64.1 (CH2), 81.4 (CH2), 102.6 (C), 124.1 (2× CH), 127.2 (CH), 129.1 (2× CH), 139.2 (C), 142.4 (C), 146.4 (C); HRMS (ESI, m/z): calculated for C17H18N5O3S [M–C2H4 + H]+ 372.1125 found 372.9570 (McLafferty rearrangement).3.3. Biological
| Percentage of free radical removal DPPH˙+ = 100 × (1 − AE/AD), | (1) |
| Percentage of free radical removal ABTS˙+ = [(A0 − As)/A0] × 100 | (2) |
3.4. X-ray diffraction measurements
Diffraction data were collected at 297 K on a Bruker D8 Venture diffractometer equipped with a bi-dimensional CMOS Photon 100 detector using graphite monochromated Mo Kα (λ = 0.71073 Å) radiation. The diffraction frames were integrated using the APEX3 package64 and corrected for absorption SADABS.65 The structure of 4b was solved by intrinsic phasing66 using OLEX2 software54 and refined with full-matrix least-squares methods based on F2 (SHELXL).67 Non-hydrogen atoms were refined using anisotropic displacement parameters.3.5. Computational details
We employed Orca software for optimization and frequency calculations, and the Gaussian 09 software package was used to determine the Wiberg bond indices.68,69 The first molecular geometries were generated using the Avogadro molecular editor70 to create the corresponding input file. All molecular structures were optimized using LC-BLYP with the 6-31G(d) basis set applied uniformly to all the investigated chemical species (including non-radicals, anions, and cations). The ionization energy (IE) and electron affinity (EA) were calculated adiabatically,71 where IE = Ecation − Eneutral and EA = Eneutral − Eanion. The calculation of bond dissociation energy (BDE) followed the method outlined by Rakiba Rohman et al., resulting in BDE as a product of η and the Wiberg bond index (BIWiberg).72The determination of electrophilicity involves consideration of electronegativity and hardness. In a system forming N electrons subjected to an external potential v(r) and having a total energy E, the electronegativity is defined as the partial derivative of the energy concerning the number of electrons at a constant potential. In a finite-difference approximation, this corresponds to half the sum of ionization energy (IE) and electron affinity (EA):73

Chemical hardness was calculated according to the definition proposed by Parr and Pearson, involving the differentiation of the chemical potential with respect to the number of electrons at a constant energy potential. The latter can be approximated as half the difference between ionization energy (IE) and electron affinity (EA).74 To achieve symmetry with and to ensure electronegativity symmetry, the product is multiplied by half, as highlighted by Pearson:75

The electrophilicity index is calculated as follows:
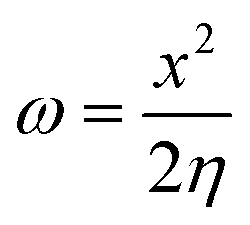
The ionization energy (IE) and electron affinity (EA) can be used to compute the electrondonor (ω−) and electronacceptor (ω+) indices, as suggested by Gázquez and colleagues.76 Electrondonating power gauges the tendency to donate charge and is defined by the following equation:

The definition of the electronaccepting power or inclination to accept electrons (ω+) is as follows:
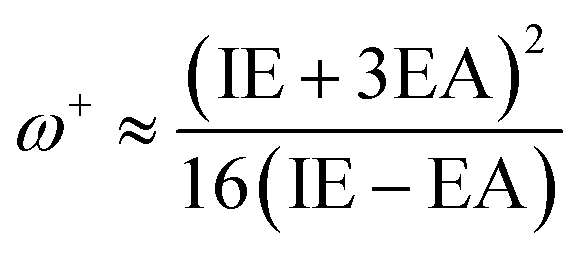
Reduced ω− values signify an increased ability to donate charge, whereas elevated ω+ values indicate a heightened ability to accept charge.
For any given compound L, the electron acceptance index is defined as follows:

Similarly, the electron donation index is defined as follows:
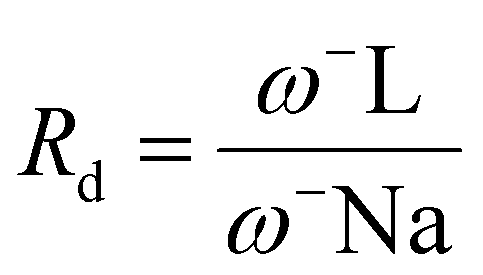
The graph has four central regions; the worst antiradical activity was found within the zone with poor donor and acceptor capacities. Two regions with good antiradical behavior correspond to bad acceptors, but good donors, and suitable acceptors, but bad donors. Finally, the best antiradical zone represents both a suitable acceptor and donor (see Fig. 8).
 | ||
| Fig. 8 Donor–acceptor map (DAM) diagram. These four regions were distinguished and described in detail by Martinez et al. The dashed lines separating the regions indicate only image clarification. | ||
3.6. Hirshfeld surface analysis details
CrystalExplorer 21.3 software77 was used to calculate the Hirshfeld78 and associated 2D-fingerprint plots79 of 4b using the crystallographic information file (CIF) as input for the analysis. The normalized contact distance dnorm, defined in terms of the de, di, and vdW radii of the atoms, was calculated using eqn (3), where the distance from the Hirshfeld isosurface to the nearest nucleus is external (de) or internal (di), and vdW is the van der Waals radii of atoms taken from the literature.80,81
 | (3) |
Hirshfeld surface analysis was performed using the 6-31G(d,p) basis set at the B3LYP level of theory over the range of ±0.002 au78 using the TONTO computational package integrated into the CrystalExplorer program.82 The bond lengths of the hydrogen atoms involved in the interactions were normalized to standard values obtained from neutron diffraction measurements (C–H = 1.083 Å, N–H = 1.009 Å, and O–H = 0.983 Å).83 The intermolecular forces between pairs of molecules in the crystal packing were calculated at the B3LYP/6-31G(d,p) level of theory in clusters with a 3.8 Å radius around the molecules.84,85
4. Conclusions
In this study, we synthesized a new spiro derivative (4a) and a new azocine derivative (4b) from a mixture of 5-aminopyrazole, p-formaldehyde, thiobarbituric acid, and InCl3 using reflux and ultrasound, respectively. Compound 4a was characterized by means of spectroscopic studies: FT-IR, HRMS, 1H NMR and 13C NMR; while for compound 4b the spectroscopic studies were used: FT-IR, HRMS and X-ray crystallography. The X-ray findings showed that 4b has two fused rings in a half-chair conformation and crystallizes in the monoclinic system with a P21/c space group, Z = 4, and unit cell parameters a = 8.624(6) Å, b = 20.611(7) Å, c = 10.773(7) Å, β = 102.261(14)°, and V = 1871.4(11) Å3. In general, good agreement was found between all the investigated theoretical properties (structural, electronic, and spectroscopic) and the experimental results. Analysis of the FMOs and chemical reactivity descriptors revealed that 4a was more reactive and less stable than 4b was. Hirshfeld surface analysis of the crystal structure indicated that the greatest contribution to stabilization was due to unconventional intermolecular interactions, similar to hydrogen bonds N–H⋯X (X = O and S) and C–H⋯O. Likewise, compounds 4a and 4b presented good antioxidant activities in the ABTS assay, with EC50 of 0.033 and 0.571 μg mL−1 respectively, being better than the positive controls, therefore, in general, both compounds have interesting antioxidant activities that give them good therapeutic potential. Finally, based on the DFT results, it was possible to show that compound 4a had greater ease in accepting and donating electrons and lower hydrogen bond dissociation energies (BDE) than compound 4b, which justifies its improved antioxidant capacity. Furthermore, when comparing the ionization energies (IE) of both compounds with their respective BDEs, it is evident that both 4a and 4b stabilized the radicals studied via the hydrogen atom transfer (HAT) mechanism.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
The authors acknowledge the Research Group of the Laboratory of Organic Synthesis and Biological Activity at the University of Talca. E. P.-C. thanks FONDECYT Post-Doctoral Fellowship No. 3220681. Y. A. R.-N. thanks FONDECYT project no. 11241068, Fondecyt Project 1200531. The authors also acknowledge to FONDEQUIP program (EQM 130021 and 180024).References
- S. Lee and J. Sperry, Bioorg. Med. Chem., 2022, 54, 116560 CrossRef CAS PubMed.
- Natural Products, ed. K. G. Ramawat and J.-M. Mérillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013 Search PubMed.
- O. I. Zhuravleva, S. S. Afiyatullov, V. A. Denisenko, S. P. Ermakova, N. N. Slinkina, P. S. Dmitrenok and N. Y. Kim, Phytochemistry, 2012, 80, 123–131 CrossRef CAS PubMed.
- H. Nakamura, S. Deng, J. Kobayashi, Y. Ohizumi, Y. Tomotake, T. Matsuzaki and Y. Hirata, Tetrahedron Lett., 1987, 28, 621–624 CrossRef CAS.
- J. T. Ndongo, M. Shaaban, J. N. Mbing, D. N. Bikobo, A. D. T. Atchadé, D. E. Pegnyemb and H. Laatsch, Phytochemistry, 2010, 71, 1872–1878 CrossRef CAS PubMed.
- H. Schrey and P. Spiteller, Chem.–Eur. J., 2019, 25, 8035–8042 CrossRef CAS PubMed.
- H. Hayashi, T. Fujiwara, S. Murao and M. Arai, Agric. Biol. Chem., 1991, 55, 3143–3145 CAS.
- S. Murao, H. Hayashi, K. Takiuchi and M. Arai, Agric. Biol. Chem., 1988, 52, 885–886 CAS.
- Y. Shiono, K. Akiyama and H. Hayashi, Biosci. Biotechnol. Biochem., 2000, 64, 1519–1521 CrossRef CAS PubMed.
- H. Hayashi, K. Takiuchi, S. Murao and M. Arai, Agric. Biol. Chem., 1989, 53, 461–469 CAS.
- M. Hamann, D. Alonso, E. Martín-Aparicio, A. Fuertes, M. J. Pérez-Puerto, A. Castro, S. Morales, M. L. Navarro, M. Del Monte-Millán, M. Medina, H. Pennaka, A. Balaiah, J. Peng, J. Cook, S. Wahyuono and A. Martínez, J. Nat. Prod., 2007, 70, 1397–1405 CrossRef CAS PubMed.
- O. I. Zhuravleva, A. S. Antonov, V. T. D. Trang, M. V. Pivkin, Y. V. Khudyakova, V. A. Denisenko, R. S. Popov, N. Y. Kim, E. A. Yurchenko, A. V. Gerasimenko, A. A. Udovenko, G. von Amsberg, S. A. Dyshlovoy and S. S. Afiyatullov, Mar. Drugs, 2021, 19, 32 CrossRef CAS PubMed.
- J. Li, J. Li, Y. Xu, Y. Wang, L. Zhang, L. Ding, Y. Xuan, T. Pang and H. Lin, Nat. Prod. Res., 2016, 30, 800–805 CrossRef CAS PubMed.
- J. Peng, S. Kudrimoti, S. Prasanna, S. Odde, R. J. Doerksen, H. K. Pennaka, Y. M. Choo, K. V. Rao, B. L. Tekwani, V. Madgula, S. I. Khan, B. Wang, A. M. S. Mayer, M. R. Jacob, L. C. Tu, J. Gertsch and M. T. Hamann, J. Med. Chem., 2010, 53, 61–76 CrossRef CAS PubMed.
- A. Z. A. Abouem, T. N. Joseph, D. S. Bikobo and J. L. Nkot, Int. J. Pharm. Pharmaceut. Sci., 2014, 6, 252–256 Search PubMed.
- J. N. I. Kobayashi, D. Watanabe, N. Kawasaki and M. Tsuda, J. Org. Chem., 1997, 62, 9236–9239 CrossRef CAS.
- H. Zhang, S. Qiu, P. Tamez, G. T. Tan, Z. Aydogmus, N. Van Hung, N. M. Cuong, C. Angerhofer, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, Pharm. Biol., 2002, 40, 221–224 CrossRef CAS.
- K. K. H. Ang, M. J. Holmes, T. Higa, M. T. Hamann and U. A. K. Kara, Antimicrob. Agents Chemother., 2000, 44, 1645–1649 CrossRef CAS PubMed.
- Z. E. D. S. Torres, E. R. Silveira, L. F. R. E Silva, E. S. Lima, M. C. De Vasconcellos, D. E. D. A. Uchoa, R. B. Filho and A. M. Pohlit, Molecules, 2013, 18, 6281–6297 CrossRef PubMed.
- J. R. Palem, M. Mudit, S. C. V. Hsia and K. A. E. Sayed, Z. Naturforsch., C: J. Biosci., 2017, 72, 49–54 CrossRef CAS PubMed.
- S. P. Gunasekera, G. Cordell and N. R. Farnsworth, Phytochemistry, 1980, 19, 1213–1218 CrossRef CAS.
- A. K. Singh, V. Raj and S. Saha, Eur. J. Med. Chem., 2017, 142, 244–265 CrossRef CAS PubMed.
- Y. L. Zhao, Z. F. Yang, B. F. Wu, J. H. Shang, Y. P. Liu, X. H. Wang and X. D. Luo, J. Ethnopharmacol., 2020, 259, 112949 CrossRef CAS PubMed.
- J. H. Shang, X. H. Cai, T. Feng, Y. L. Zhao, J. K. Wang, L. Y. Zhang, M. Yan and X. D. Luo, J. Ethnopharmacol., 2010, 129, 174–181 CrossRef CAS PubMed.
- T. L. Yan, D. X. Han, J. Hu, X. Y. Huang and H. K. Wang, J. Asian Nat. Prod. Res., 2017, 19, 550–556 CrossRef CAS PubMed.
- C. Seidl, C. d. M. Santos, A. De Simone, M. Bartolini, A. Weffort-Santos and V. Andrisano, Curr. Alzheimer Res., 2017, 14, 1 CrossRef PubMed.
- V. L. de Almeida, C. G. Silva, A. F. Silva, P. R. V. Campana, K. Foubert, J. C. D. Lopes and L. Pieters, J. Ethnopharmacol., 2019, 231, 125–140 CrossRef CAS PubMed.
- K. Sakamoto, E. Tsujii, M. Miyauchi, T. Nakanishi, M. Yamashita, N. Shigematsu, T. Tada, S. Izumi and M. Okuhara, J. Antibiot., 1993, 46, 1788–1798 CrossRef CAS PubMed.
- X. Ma, X. Zhang, G. Xie, J. M. Awad and W. Zhang, Tetrahedron Lett., 2019, 60, 151127 CrossRef CAS.
- W. Ouyang, J. Rao, Y. Li, X. Liu, Y. Huo, Q. Chen and X. Li, Adv. Synth. Catal., 2020, 362, 5576–5600 CrossRef CAS.
- M. R. Khodabakhshi, F. M. Moghaddam and M. Kiamehr, Tetrahedron Lett., 2018, 59, 4503–4508 CrossRef CAS.
- B. S. Vachan, M. Karuppasamy, G. Jan, N. Bhuvanesh, C. U. Maheswari and V. Sridharan, J. Org. Chem., 2020, 85, 8062–8073 CrossRef CAS PubMed.
- K. Acosta-Quiroga, C. Rojas-Peña, L. S. Nerio, M. Gutiérrez and E. Polo-Cuadrado, RSC Adv., 2021, 11, 21926–21954 RSC.
- E. Polo-Cuadrado, C. Rojas-Peña, K. Acosta-Quiroga, L. Camargo-Ayala, I. Brito, J. Cisterna, F. Moncada, J. Trilleras, Y. A. Rodríguez-Núñez and M. Gutierrez, RSC Adv., 2022, 12, 33032–33048 RSC.
- B. Basu and B. Banerjee, Multicomponent Synthesis: Bioactive Heterocycles, 2023, pp. 1–433 Search PubMed.
- D. M. Patel, P. J. Patel and H. M. Patel, Eur. J. Org. Chem., 2022, 2022, e202201119 CrossRef CAS.
- M. P. Parmar, R. M. Vala and H. M. Patel, ACS Omega, 2023, 8, 1759–1816 CrossRef CAS PubMed.
- A. J. S. Alves, N. G. Alves, C. C. Caratão, M. I. M. Esteves, D. Fontinha, I. Bártolo, M. I. L. Soares, S. M. M. Lopes, M. Prudêncio, N. Taveira and T. M. V. D. Pinho e Melo, Curr. Top. Med. Chem., 2019, 20, 140–152 CrossRef PubMed.
- F. S. Almeida, G. L. S. Sousa, J. C. Rocha, F. F. Ribeiro, M. R. de Oliveira, T. C. S. de Lima Grisi, D. A. M. Araújo, M. S. Michelangela, R. N. Castro, I. P. G. Amaral, T. S. L. Keesen and R. O. de Moura, Bioorg. Med. Chem. Lett., 2021, 49, 128289 CrossRef CAS PubMed.
- P. Das, S. Boone, D. Mitra, L. Turner, R. Tandon, D. Raucher and A. T. Hamme, RSC Adv., 2020, 10, 30223–30237 RSC.
- C. Du, X. Yang, Y. Long, X. Lang, L. Liu, Y. Xu, H. Wu, Y. Chu, X. Hu, J. Deng and Q. Ji, Eur. J. Med. Chem., 2023, 255, 115388 CrossRef CAS PubMed.
- A. H. Abdel-Rahman, E. M. Keshk, M. A. Hanna and S. M. El-Bady, Bioorg. Med. Chem., 2004, 12, 2483–2488 CrossRef CAS PubMed.
- A. Donaire-Arias, A. M. Montagut, R. P. de la Bellacasa, R. Estrada-Tejedor, J. Teixidó and J. I. Borrell, Molecules, 2022, 27, 2237 CrossRef CAS PubMed.
- N. Ma, B. Jiang, G. Zhang, S. J. Tu, W. Wever and G. Li, Green Chem., 2010, 12, 1357–1361 RSC.
- E. Polo-Cuadrado, C. Rojas-Peña, K. Acosta-Quiroga, L. Camargo-Ayala, I. Brito, J. Cisterna, F. Moncada, J. Trilleras, Y. A. Rodríguez-Núñez and M. Gutierrez, RSC Adv., 2022, 12, 33032–33048 RSC.
- F. Sonmez, Z. Gunesli, B. Z. Kurt, I. Gazioglu, D. Avci and M. Kucukislamoglu, Mol. Divers., 2019, 23, 829–844 CrossRef CAS PubMed.
- K. Acosta-Quiroga, C. Rojas-Peña, L. S. Nerio, M. Gutiérrez and E. Polo-Cuadrado, RSC Adv., 2021, 11, 21926–21954 RSC.
- D. C. Christodouleas, C. Fotakis, A. Nikokavoura, K. Papadopoulos and A. C. Calokerinos, Food Anal. Methods, 2015, 8, 1294–1302 CrossRef.
- D. A. Hernandez, J. G. Rodriguez-Zavala and F. J. Tenorio, Struct. Chem., 2020, 31, 359–369 CrossRef CAS.
- A. Martínez, J. Phys. Chem. B, 2009, 113, 4915–4921 CrossRef PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- D. Cremer, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 498–500 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- E. J. Lenardão, C. Santi and L. Sancineto, in New Frontiers in Organoselenium Compounds, Springer International Publishing, Cham, 2018, pp. 157–183 Search PubMed.
- W. Dong, Q. Li and S. Scheiner, Molecules, 2018, 23, 1681 CrossRef PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- H. A. Khamees, K. Chaluvaiah, N. A. El-Khatatneh, A. Swamynayaka, K. H. Chong, J. P. Dasappa and M. Madegowda, Acta Crystallogr. E, 2019, 75, 1620–1626 CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT, 1995, 28, 25–30 CrossRef CAS.
- S. Khwaja, K. Fatima, Hassanain, C. Behera, A. Kour, A. Singh, S. Luqman, J. Sarkar, D. Chanda, K. Shanker, A. K. Gupta, D. M. Mondhe and A. S. Negi, Eur. J. Med. Chem., 2018, 151, 51–61 CrossRef CAS PubMed.
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, SADABS, Software for Empirical Absorption Corrections, University of Göttingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeerschd, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- D. A. Hernandez, J. G. Rodriguez-Zavala and F. J. Tenorio, Struct. Chem., 2020, 31, 359–369 CrossRef CAS.
- R. Rohman, R. Nath and R. Kar, Comput. Theor. Chem., 2023, 1223, 114097 CrossRef CAS.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Pearson, J. Chem. Sci., 2005, 117, 369–377 CrossRef CAS.
- J. L. Gázquez, A. Cedillo and A. Vela, J. Phys. Chem. A, 2007, 111, 1966–1970 CrossRef PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Gromwood, P. R. Spackman and D. Jayalitaka, Crystal Explorer 17.5, https://hirshfeldsurface.net/ Search PubMed.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theor. Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2200261. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra07966c |
| This journal is © The Royal Society of Chemistry 2025 |