
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organophosphonate- and dimethylarsinate-functionalized hexamolybdates(V) and their solution and gas phase properties†
Vinaya
Siby
 a,
Arun
Pal
a,
Bassem S.
Bassil
a,
Saurav
Bhattacharya
ab,
Anusree
Sundar
a,
Juliane
Oberstein
a,
Damin
Kim
a,
Rizul
Gupta
a,
Alisher
Kuanysh
a,
Jana
Hölscher
a,
Dorothea
Schmidt
a,
Nikolai
Kuhnert
a and
Ulrich
Kortz
*a
a,
Arun
Pal
a,
Bassem S.
Bassil
a,
Saurav
Bhattacharya
ab,
Anusree
Sundar
a,
Juliane
Oberstein
a,
Damin
Kim
a,
Rizul
Gupta
a,
Alisher
Kuanysh
a,
Jana
Hölscher
a,
Dorothea
Schmidt
a,
Nikolai
Kuhnert
a and
Ulrich
Kortz
*a
aSchool of Science, Constructor University, Campus Ring 1, 28759 Bremen, Germany. E-mail: ukortz@constructor.university
bDepartment of Chemistry, BITS Pilani K. K. Birla Goa Campus, 403726 Goa, India
First published on 18th February 2025
Abstract
Here we report a comprehensive study on the synthesis and characterization of dimethylarsinate-functionalized phosphomolybdates(V), [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4) and the monoanionic mixed-valent heptamolybdate [HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]−. These nine novel polyanions feature a reduced, cyclic hexanuclear {MoV6O24} core, decorated peripherally by three dimethylarsinate ligands and centrally by six different organophosphonate groups including fluorinated ones, as well as phosphate and phosphite. The polyanions were synthesized under simple one-pot aqueous solution conditions and characterized (i) in the solid state by single-crystal and powder XRD, TGA, elemental analysis, (ii) in solution by multinuclear (1H, 31P, 19F, 13C) NMR, and (iii) in the gas phase by ESI mass spectrometry.
1. Introduction
In materials science, the burgeoning domain of hybrid organic–inorganic materials carries profound implications, driving innovation in the development of multifunctional materials. The synergistic combination of the structural intricacies and electronic characteristics inherent to polyoxometalates (POMs),1a,b which are discrete, well-defined, anionic metal-oxo clusters composed of early transition metal ions (M = MoVI, WVI, VV, NbV and TaV) in high oxidation states, with the flexibility, tunability and functionality of organic or organometallic components,1c,d represent a rapidly growing field of research with unique, potential applications in areas such as catalysis,1e,f molecular nanosciences,1g,h photo (electro-) chemistry,1i,j biotechnology, and medicine.1k–m New hybrid architectures could be generated through interaction of classical lacunary POMs with organic or organometallic groups, or via in situ self-assembly between metal building blocks and O-donating organic ligands. These hybrid POMs, featuring reactive sites, offer multifaceted platforms for subsequent post-functionalization.2The conventional domains of polyoxomolybdates and -tungstates constitute the primary subsets within POM frameworks. The formation of polyoxomolybdate-organic hybrids is highly desirable due to the attractive redox properties and stability of multielectron-reduced polyoxomolybdates. Nevertheless, it poses a notable hurdle due to the increased lability and the consequent instability associated with polyoxomolybdates, resulting in limited documentation on the synthesis of molecular hybrids involving lacunary polyoxomolybdates and multi-dentate organic ligands.3 The investigation of in situ hybrid POMs gathered significant impetus after Pope and coworkers structurally characterized the organoarsenic functionalized polyoxomolybdates, [(RAs)2Mo6O24]4− (R = CH3, C6H5, p-C6H4NH2),4b [R2AsMo4O15H]2− (R = CH3, C2H5, C6H5),4c,d initially proposed by Rosenheim and co-workers.4a
The first organophosphonate derivatized heteropolyanions were the pentamolybdobisphosphonate complexes,5a [(RP)2MoVI5O21]4− (R = H, CH3, C2H5, C6H5, C2H4NH3+, p-CH2C6H4NH3+), reported by Pope's group in 1975. Subsequently, in 1976, Stalick and Quicksall elucidated the structures of [(CH3P)2Mo5O21]4− and [(NH3C2H4P)2Mo5O21]2−.5b The crystal structure of [(HP)2Mo5O21]4− was published by Sasaki's group in 1988,5c while Lyxell and Strandberg concurrently reported crystal structure of [(C6H5P)2Mo5O21]4−.5d Subsequently, Kortz's group has published a series of [(RP)2MoVI5O21]4− derivatives, featuring aliphatic phosphonocarboxylate groups (R = O2CCH2, O2CC2H4)6a and aryl carboxyphenyl phosphonate groups (R = 4-O2CC6H4, 4-O2CC6H4CH2).6b The incorporation of enantiopure aminoalkylphosphonates (R = CH3CH(NH3), CH3CH(CH3)CH(NH3)) induced the formation of self-assembled fibrillar nanostructures with heightened optically activity.6c The hexamolybdic wheel structural type, [RPMoVI6O21(O2CCH2NH3)3]2− (R = OH, CH3, C2H5, H) underwent covalent modification at two distinct sites: peripherally by glycine molecules and centrally by organophosphonate group, yielding molybdenum oxo frameworks characterized by their flexibility in both steric and electronic aspects.6d Additionally, similar structural archetype has been reported earlier, [XMo6O21(O2CRNH3)3]n− (n = 2, X = SeIV, TeIV; n = 3, X = AsIII, SbIII, BiIII; R = CH2, C2H4, C3H6, CHCH3, CH(CH2)4NH2), where lone-pair containing heteroatoms occupy the central position and are surrounded by amino acids bound peripherally via their carboxylate functions.6e
In recent years Kortz's group has reported a number of polyoxo-noble-metalate clusters functionalized by dimethylarsinate (also known as cacodylate) ligands.7 The inclusion of this functional moiety in reduced polyoxomolybdate chemistry led to the discovery of an organofunctionalized mixed-valent polyoxomolybdate wheel [MoVI18MoV12O84{AsO2(CH3)2}18]18− (Mo30) and a neutral, reduced tetramolybdate cubane cluster [MoV4O8{AsO2(CH3)2}4] (Mo4).8 The introduction of phosphorous into the same system, at elevated pH levels, resulted in a series of reduced hexanuclear, dimethylarsinate- functionalized phosphomolybdenum clusters, with general formula [RPMoV6O15(OH)3{AsO2(CH3)2}3]n−, reminiscent of the Anderson archetype, where the polyoxoanion core can be centrally modified systematically and rationally through incorporation of phosphite, phosphate and hydrolytically stable alkyl and aryl phosphonates, represented as RPO32−. This particular type of hexanuclear molybdic wheel was initially isolated by Haushalter's group as the reduced molybdenum phosphate,{P4MoV6}, where peripheral phosphates and hydroxo-bridges mutually connect {MoV2O4} dimeric structural units.9 Subsequent studies demonstrated the versatility of this prototype, where PO43− could be substituted with C6H5PO32−,10a–c C6H5AsO32−,10d CO32−,10e and SO32−.10f Thiomolybdate analogues, [X4Mo6S6O6(OH)3]5− (X = HPO4, HAsO4 as assembling groups) with dimolybdenum pairs bridged by six sulfur atoms, have also been documented,11a with a study showcasing regioselective substitution of sulfur atoms in oxothio analogue {P4Mo6S6} to obtain {P4Mo6S3O3} illustrated by NMR reported later.11b The mixed-valent MoV/MoVI molecular anion, [Mo7O16(O3PCH2PO3)3]8−{P6Mo7}, anchored by peripheral diphosphonate groups could accommodate MoVIO4 tetrahedron in the center, due to the larger aperture created from corner-sharing arrangement of the molybdenum dimers differing from the edge-sharing mode observed in {P4Mo6}.12
Now we decided to investigate the system of molybdate in the presence of cacodylate and phosphonates in aqueous solution under reducing conditions in detail, and we isolated a new family of eight functionalized polyoxomolybdates(V) with interesting solid state, solution and gas phase properties.
2. Experimental details
2.1 Synthesis of Na1.8(NH4)0.2[HOPMoV6O15(OH)3{AsO2(CH3)2}3]·0.8NH4Cl·6H2O (NaNH4-PMo6)
(NH4)6Mo7O24·4H2O (0.265 g, 0.215 mmol) and N2H4·2HCl (0.052 g, 0.5 mmol) were dissolved completely in 10 mL 0.5 M sodium dimethylarsinate buffer (prepared as follows: 0.5 M aqueous solution of dimethylarsinic (cacodylic) acid (pH 4) adjusted to pH 7 by adding NaOH pellets). To this mixture, sodium phosphate, Na3PO4 (0.041 g, 0.25 mmol) was added followed by pH adjustment to 6.8–6.9 by dropwise addition of 6M NaOH solution. The initial dark green solution, which progressively turned dark red, was heated at 80 °C for 1 hour in closed vial. Dark-red block-shaped crystals of NaNH4-PMo6 started to form within 3–4 days and were collected after 1 week and air dried. Isolated yield: 0.140 g (37% based on P). Elemental analysis (%): calcd (found) for NaNH4-PMo6: Na, 2.70 (2.68); Mo, 37.6 (38.2); As, 14.7 (14.7); P, 2.0 (2.3); C, 4.7 (4.8); H, 2.55 (2.62); N, 1.10 (1.33). FT-IR data (KBr pellet) in cm−1: 3410–3232 (s) [ν(O–H)], 3025 (w), 2926 (w) [ν(C–H)], 2091 (w), 1632 (m) [δ(O–H)], 1407 (m) [δ(C–H)], 1278 (m), 1257 (sh) [ν(As–C)], 1089 (m), 1010 (w) [ν(P–O)], 957–920 (s) [ν(Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)], 857 (sh), 825 (s) [ν(As–O)], 751 (s), 655 (w), 608, 491 (s), 459 (s) [δ(Mo–O(Mo))].
O)], 857 (sh), 825 (s) [ν(As–O)], 751 (s), 655 (w), 608, 491 (s), 459 (s) [δ(Mo–O(Mo))].
2.2 Synthesis of Na1.2H0.8[HPMoV6O15(OH)3{AsO2(CH3)2}3]·11H2O (Na-HPMo6)
The polyanion HPMo6 was prepared using the same method as that for PMo6, except that sodium phosphite, Na2HPO3·5H2O (0.054 g, 0.25 mmol) was used instead of sodium phosphate. The pH of the reaction mixture was adjusted to 6.8 with 6M NaOH before heating at 80 °C. Dark-red block crystals were isolated after 1 week and air dried. Isolated yield: 0.145 g (38% based on P). Elemental analysis (%): calcd (found) for Na-HPMo6: Na, 1.80 (1.80); Mo, 37.5 (36.8); As, 14.6 (14.3); P, 2.0 (2.1); C, 4.7 (4.8); H, 2.8 (2.6). FT-IR data (KBr pellet) in cm−1: 3400 (s) [ν(O–H)], 3019(w), 2928 (w) [ν(C–H)], 2427 (w) [ν(P–H)], 2088 (w), 1632 (m) [δ(O–H)], 1404 (m) [δ(C–H)], 1274 (w), 1257 (sh) [ν(As–C)], 1143 (sh), 1072(s) [ν(P–O)], 972 (s), 944 (s) [ν(MoO)], 852 (sh), 820 (s) [ν(As–O)], 752 (s), 656 (m), 615 (w), 540 (m), 496 (s), 460 (m) [δ(Mo–O(Mo))].
2.3 Synthesis of NaNH4[CH3PMoV6O15(OH)3{AsO2(CH3)2}3]·11H2O (NaNH4-CH3PMo6)
The polyanion CH3PMo6 was prepared similarly to PMo6, substituting methyl phosphonic acid (CH3PO3H2) (0.024 g, 0.25 mmol) for sodium phosphate. pH of the reaction mixture was adjusted to 6.8 with 6 M NaOH prior to stirring and heating at 80 °C for 1 hour. As the reaction mixture cooled to room temperature, dark-red block crystals suitable for single crystal XRD emerged within a day at the bottom of the vial. After a week of crystallization in open air, the compound was collected and characterized by IR spectroscopy, resulting in a total yield of 0.180 g (46% based on phosphorus). Elemental analysis (%): calcd (found) for NaNH4-CH3PMo6: Na, 1.5 (1.5); Mo, 36.8 (36.7); As, 14.4 (14.7); P, 2.0 (1.9); C, 5.38 (5.43); H, 3.22 (3.39); N, 0.90 (1.06). FT-IR data (KBr pellet) in cm−1: 3488 (s) [ν(O–H)], 3017 (w), 2926 (w) [ν(C–H)], 1629 (m) [δ(O–H)], 1404 (m) [δ(C–H)], 1311–1274 (w) [ν(P–C)], 1275 (m), 1257 (sh) [ν(As–C)], 1116(sh), 1051 (m) [ν(P–O)], 999 (w), 967 (s), 939 (m) [ν(MoO)], 854 (sh), 822 (s) [ν(As–O)], 778 (w), 750 (m), 655 (w), 616 (w), 552 (m), 493 (s), 461 (s) [δ(Mo–O(Mo))].
2.4 Synthesis of Na1.6(NH4)0.4[HO2CCH2PMoV6O15(OH)3{AsO2(CH3)2}3]·0.5NH4Cl·9H2O (NaNH4-HO2CCH2PMo6)
The polyanion HO2CCH2PMo6 was prepared similarly to PMo6, substituting phosphonoacetic acid (H2O3PCH2COOH) (0.035 g, 0.25 mmol) for sodium phosphate. pH of the reaction mixture was adjusted to 6.8 with 6 M NaOH and the dark red solution is heated at 80 °C for 1 hour. The dark-red block-shaped crystals of NaNH4-HO2CCH2PMo6 that formed within 2 days in the vial kept closed at room temperature, was later characterized by single crystal XRD. After 2 weeks of open-air crystallization, the compound was collected and characterized by IR. Total isolated yield: 0.125 g (31% based on P). Elemental analysis (%): calcd (found) for NaNH4-HO2CCH2PMo6: Na, 2.33 (2.43); Mo, 36.5 (37.0); As, 14.3 (14.2); P, 2.0 (2.2); C, 6.10 (5.97); H, 2.82 (3.08); N, 0.80 (0.84). FT-IR data (KBr pellet) in cm−1: 3404 (s), 3020 (w) [ν(O–H)], 2927 (w), 2085 (w) [ν(C–H)], 1635 (m) [δ(O–H)], 1581 (m) [ν(CO)], 1405 (m) [δ(C–H)], 1378 (m) [ν(P–C)], 1275 (w), 1259 (sh) [ν(As–C)], 1215 (w), 1132 (w), 1056 (m) [ν(P–O)], 967 (s), 942 (m) [ν(MoO)], 851 (sh), 822 (s) [ν(As–O)], 754 (s), 656 (w), 612 (w), 553 (m), 495 (s), 465 (s) [δ(Mo–O(Mo))].
2.5 Synthesis of NaNH4[HO2CC2H4PMoV6O15(OH)3{AsO2(CH3)2}3]·10H2O (NaNH4-HO2CC2H4PMo6)
The synthesis of HO2CC2H4PMo6 was accomplished by using a procedure identical to that of HO2CCH2PMo6, except for substituting phosphonoacetic acid with phosphonopropionic acid (H2O3PC2H4COOH) (0.039 g, 0.25 mmol). The pH of the reaction mixture was adjusted to 6.8 with 6M NaOH before heating at 80 °C for 1 hour. Dark-red block-shaped crystals started forming at the bottom of the vial within two days. Total isolated yield after 2 weeks: 0.120 g (30% based on P). Elemental analysis (%): calcd (found) for NaNH4-HO2CC2H4PMo6: Na, 1.43 (1.72); Mo, 35.9 (36.6); As, 14.0 (14.5); P, 1.9 (1.9); C, 6.74 (6.57); H, 3.14 (3.40); N, 0.87 (1.15). FT-IR data (KBr pellet) in cm−1: 3400 (s), 3247 (s) [ν(O–H)], 3008 (w), 2923 (w) [ν(C–H)], 2094 (w), 1631 (m) [δ(O–H)], 1559 (m) [ν(CO)], 1444 (sh) [ν(P–C)], 1403 (m) [δ(C–H)], 1320 (w), 1273 (w), 1254 (sh) [ν(As–C)], 1258 (w), 1213 (w), 1050 (m) [ν(P–O)], 990 (sh), 966 (s), 938 (s) [ν(MoO)], 852 (sh), 822 (s) [ν(As–O)], 748 (s), 656 (w), 612 (w), 549 (m), 494 (s), 462 (s) [δ(Mo–O(Mo))].
2.6 Synthesis of Na0.7(NH4)1.3[C6H5PMoV6O15(OH)3{AsO2(CH3)2}3]·9H2O (NaNH4-C6H5PMo6)
The polyanion C6H5PMo6 was synthesized using a method analogous to PMo6, replacing sodium phosphate with phenyl phosphonic acid (C6H5PO3H2) (0.040 g, 0.25 mmol). The pH of the solution is adjusted to 6.8 with 6M NaOH, followed by stirring and heating at 80 °C for 1 hour in a closed vial. The reaction mixture is later kept for cooling at room temperature in the closed vial for two days to procure good quality red block crystals. After isolation of the initial product, the dark red solution is kept for crystallization in open air for 2 weeks. The combined isolated yield after two weeks is 0.160 g (39% based on P). IR was used to characterize the products separated at different time intervals. Elemental analysis (%): calcd (found) for NaNH4-C6H5PMo6: Na, 1.01 (1.15); Mo, 36.3 (36.8); As, 14.2 (14.4); P, 2.0 (2.0); C, 9.08 (9.01); H, 3.12 (3.37); N, 1.15 (1.19). FT-IR data (KBr pellet) in cm−1: 3416 (s), 3218 (s) [ν(O–H)], 3017 (w), 2928 (w) [ν(C–H)], 2071 (w), 1623 (m) [δ(O–H)], 1441 (sh) [ν(CC), ν(P–C)], 1404 (m) [δ(C–H)], 1275 (w), 1255 (sh) [ν(As–C)], 1142 (m), 1079 (sh), 1058 (m) [ν(P–O)], 965 (s), 940 (m) [ν(MoO)], 854 (sh), 821 (s) [ν(As–O)], 748 (s), 655 (w), 611(w), 578 (w), 567 (sh), 557 (m), 493 (s), 461 (s) [δ(Mo–O(Mo))].
2.7 Synthesis of (4-fluorophenyl) phosphonic acid (LPF)
The reported synthetic procedure13 was slightly modified and optimized as follows: 1-bromo-4-fluoro benzene (9.0 mmol), triethyl phosphite (1.89 mL, 10.8 mmol), and nickel chloride catalyst (0.117 g, 0.90 mmol) were accurately weighed and transferred into a standard microwave reaction vessel equipped with a pressure controller. The assembly was placed in a CEM Discover MW reactor, and the reaction mixture was irradiated at 140 °C for 5 hours under continuous stirring at a power setting of 200 W. After the reaction, the crude product was purified by vacuum distillation to obtain pure diethyl (4-fluorophenyl) phosphonate. 1H NMR (CDCl3, 400 MHz, ppm): δ 1.18 (t, 6H, CH3), 3.99 (m, 4H, CH2CH3), 7.01 (m, 2H, Ar), 7.68 (m, 2H, Ar); 19F NMR (CDCl3, 376.5 MHz, ppm): δ −106; 31P{1H} NMR (CDCl3, 161.83 MHz, ppm): δ 18.3; 13C{1H} NMR (CDCl3, 100 MHz, ppm): δ 16.21, 62.12, 115.5, 124.4 (d, 1JCP = 189.4 Hz), 134.4, 165.5 (d, 1JCF = 252.1 Hz).The phosphonic ester was then refluxed at 110 °C for 48 hours in 20 mL of concentrated hydrochloric acid (12 M). The resulting mixture was fully evaporated using a rotavapor, and the resulting white solid was washed with n-hexane and dried, yielding (4-fluorophenyl) phosphonic acid (LPF) as a fine white powder. 1H NMR (D2O, 400 MHz, ppm): δ 7.04 (m, 2H, Ar), 7.57 (m, 2H, Ar); 19F NMR (D2O, 376.5 MHz, ppm): δ −107.9; 31P{1H} NMR (D2O, 161.83 MHz, ppm): δ 15.4; 13C{1H} NMR (D2O, 100 MHz, ppm): δ 115.7, 127.2 (d, 1JCP = 183.5 Hz), 132.8, 164.4 (d, 1JCF = 248.4 Hz).
2.8 Synthesis of NaNH4[4-FC6H4PMoV6O15(OH)3{AsO2(CH3)2}3]·8H2O (NaNH4-FC6H4PMo6)
(NH4)6Mo7O24·4H2O (0.053 g, 0.04 mmol), N2H4·2HCl (0.011 g, 0.1 mmol) and (4-fluorophenyl) phosphonic acid (LPF), FC6H4PO3H2 (0.009 g, 0.05 mmol) is added to 3 mL 0.5 M sodium dimethylarsinate buffer (pH 7) and the resulting mixture is stirred at room temperature for 1 hour. Dark-red block-shaped crystals start forming after one to two days. Total isolated yield after one week is 0.035 g (42% based on P). Elemental analysis (%): calcd (found) for NaNH4-FC6H4PMo6: Na, 1.45 (1.63); Mo, 36.2 (38.4); As, 14.2 (14.7); P, 1.95 (1.82); C, 9.07 (8.14); H, 2.85 (2.95); N, 0.88 (1.10). FT-IR data (KBr pellet) in cm−1: 3406 (s) [ν(O–H)], 3027 (w), 2929 (w) [ν(C–H)], 2079 (w), 1631 (m) [δ(O–H)], 1499 (w) [ν(C–F)], 1441 (sh) [ν(CC), ν(P–C)], 1401 (m) [δ(C–H)], 1281 (w), 1223 (w) [ν(As–C)], 1166 (sh), 1137 (w), 1062 (m) [ν(P–O)], 965(s), 937(sh) [ν(MoO)], 827 (s) [ν(As–O)], 747 (s), 661 (w), 609 (w), 574 (w), 528 (sh), 488 (s), 466 (s) [δ(Mo–O(Mo))].
2.9 Synthesis of (4- trifluoromethoxyphenyl) phosphonic acid (LPOCF3)
The reported synthetic procedure13 was modified with the reaction mixture containing 1-bromo-4-(trifluoromethoxy) benzene (1.34 mL, 9.0 mmol), triethyl phosphite (1.89 mL, 10.8 mmol), and NiCl2 (0.117 g, 0.90 mmol) being heated up to 130 °C for 5 hours with a power input of 150 W in a standard microwave reaction pressure vial. This reaction was repeated 5 times to obtain approximately 15 mL of crude product which was further purified through vacuum distillation yielding the desired product diethyl (4-trifluoromethoxyphenyl) phosphonate ester. 1H NMR (CDCl3, 400 MHz, ppm): δ 1.28 (t, 6H, CH3), 4.07 (m, 4H, CH2CH3), 7.24 (m, 2H, Ar), 7.82 (m, 2H, Ar); 19F NMR (CDCl3, 376.5 MHz, ppm): δ −57.7; 31P{1H} NMR (CDCl3, 161.83 MHz, ppm): δ 17.6; 13C{1H} NMR (CDCl3, 100 MHz, ppm): δ 16.3, 62.3, 120.3 (q, 1JCF = 256.7 Hz), 127.2 (d, 1JCP = 188.9 Hz), 120.5 (d, 2JCP = 15.4 Hz), 133.8 (d, 3JCP = 10.6 Hz), 152.2.(4-Trifluoromethoxyphenyl) phosphonic acid (LPOCF3) was isolated as a fine white powder after refluxing the phosphonic ester in a similar procedure used for LPF. 1H NMR (D2O, 400 MHz, ppm): δ 7.39 (d, 2H, Ar), 7.74 (m, 2H, Ar); 19F NMR (D2O, 376.5 MHz, ppm): δ −56.6; 31P{1H} NMR (D2O, 161.83 MHz, ppm): δ 11.4; 13C{1H} NMR (D2O, 100 MHz, ppm): δ 120.5 (q, 1JCF = 255 Hz), 121.1 (d, 2JCP = 13.4 Hz), 133.4 (d, 3JCP = 9.9 Hz), 133.9 (d, 1JCP = 173.8 Hz), 150.6.
2.10 Synthesis of Na0.5(NH4)1.5[4-F3COC6H4PMoV6O15(OH)3{AsO2(CH3)2}3]·0.3NaO2As(CH3)2·9H2O (NaNH4-F3COC6H4PMo6)
The polyanion F3COC6H4PMo6 was synthesized in a similar manner to FC6H4PMo6 by replacing (4-fluorophenyl) phosphonic acid with (4-trifluoromethoxyphenyl) phosphonic acid (LPOCF3), F3COC6H4PO3H2, (0.012 g, 0.05 mmol). The reaction mixture (pH 7) was continuously heated and stirred at 80 °C for 1 hour and dark-red block-shaped crystals started forming within a day after cooling to room temperature. The total isolated yield of the compound after one week is 0.030 g (38% based on P). Elemental analysis (%): calcd (found) for NaNH4-F3COC6H4PMo6: Na, 1.07 (1.08); Mo, 33.5 (35.0); As, 14.4 (15.2); P, 1.80 (1.32); C, 9.50 (8.66); H, 2.98 (3.20); N, 1.22 (1.36). FT-IR data (KBr pellet) in cm−1: 3398 (s), 3247 (s) [ν(O–H)], 3022 (w), 2931 (w) [ν(C–H)], 2077 (w), 1628 (m) [δ(O–H)], 1496 (w) [ν(C–F)], 1456 (sh) [ν(CC), ν(P–C)], 1401 (m) [δ(C–H)], 1261 (m), 1226 (w) [ν(As–C)], 1166 (w), 1140 (sh), 1068(m) [ν(P–O)], 966 (s), 939 (sh) [ν(MoO)], 829 (s) [ν(As–O)], 751 (s), 657 (w), 618 (sh), 580 (m), 495 (s), 465 (s) [δ(Mo–O(Mo))].
2.11 Synthesis of Na[HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]·NH4O2As(CH3)2·11H2O (Na-Mo7)
The mixed-valent polyanion Mo7 was synthesized by dissolving (NH4)6Mo7O24·4H2O (0.265 g, 0.215 mmol) in 5 mL 0.5 M sodium cacodylate buffer (pH 7) followed by addition of N2H4·2HCl (0.52 g, 0.5 mmol) with continuous stirring. pH of the dark red solution is adjusted to 6.9–7.0 with 6 M NaOH followed by heating at 50 °C for 1 hour with continuous stirring. The isolated crystal yield after 2 weeks is around 0.040 g (11% based on Mo). Elemental analysis (%): calcd (found) for Na-Mo7: Na, 1.30 (1.69); Mo, 38.0 (37.7); As, 17.1 (18.3); C, 5.44 (5.12); H, 3.08 (3.20); N, 0.79 (0.83). FT-IR data (KBr pellet) in cm−1: 3403 (s) [ν(O–H)], 3010 (w), 2922 (w) [ν(C–H)], 2054 (w), 1633 (m) [δ(O–H)], 1401 (m) [δ(C–H)], 1274 (w), 1255 (sh) [ν(As–C)], 969 (s) [ν(MoO)], 849 (sh), 820 (s) [ν(As–O)], 747 (s), 653 (w), 613 (w), 555 (sh), 524 (sh), 495 (s), 455 (s) [δ(Mo–O(Mo))].
3. Results and discussion
The polyanions [HPMoV6O15(OH)3{AsO2(CH3)2}3]2− (HPMo6),[HOPMoV6O15(OH)3{AsO2(CH3)2}3]2− (PMo6), [CH3PMoV6O15(OH)3{AsO2(CH3)2}3]2− (CH3PMo6), [HO2CCH2PMoV6O15(OH)3{AsO2(CH3)2}3]2− (HO2CCH2PMo6), [HO2CC2H4PMoV6O15(OH)3{AsO2(CH3)2}3]2− (HO2CC2H4PMo6), [C6H5PMoV6O15(OH)3{AsO2(CH3)2}3]2− (C6H5PMo6), [FC6H4PMoV6O15(OH)3{AsO2(CH3)2}3]2− (FC6H4PMo6), [F3COC6H4PMoV6O15(OH)3{AsO2(CH3)2}3]2− (F3COC6H4PMo6) and [MoVIMoV6O15(OH)4{AsO2(CH3)2}3]− (Mo7), were synthesized in a one-pot aqueous reaction by stirring a solution containing ammonium heptamolybdate/hydrazine hydrochloride/heterogroup/sodium cacodylate buffer in the molar ratio of 1/2/1/20. The molecular structures of the polyanions [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4) and [HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]− feature three edge-shared {MoV2O10} fragments interconnected via edges by hydroxo bridges (confirmed by bond valence sum (BVS) calculations, Tables S2–S10†) forming a six-membered molybdenum(V)-oxo ring surrounding the RPO3 or MoO4 hetero group, and being capped by three peripheral dimethylarsinate ligands, creating a flowerpot-like structure (Fig. 1a and b). BVS calculations for the crystallographically independent molybdenum atoms resulted in values close to 5 (Tables S2–S10†), further confirming the MoV oxidation state. The reduced MoV2 pairs exhibit a severely distorted octahedral coordination geometry, and the coordination of each molybdenum center is mediated by two μ2-oxo, one μ2-hydroxo, one terminal oxo group, one oxo-donor from the terminal dimethylarsinate and one oxo-donor from the central heterogroup.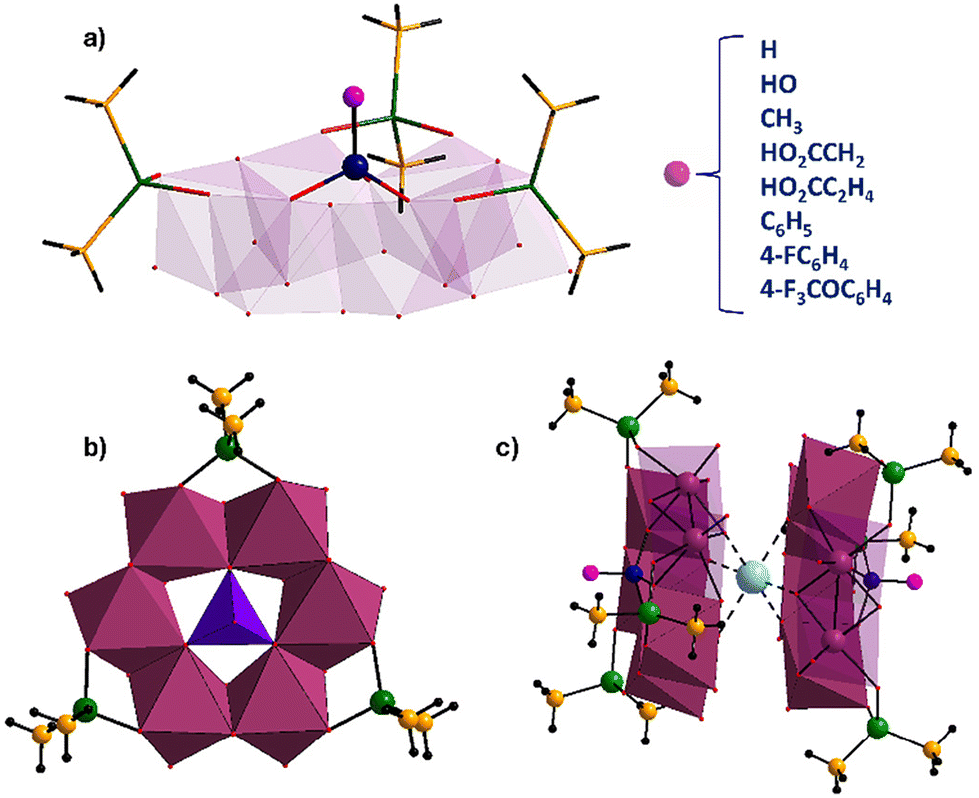 | ||
| Fig. 1 (a) Combined polyhedral/ball-and-stick representation of polyanion RPMo6 [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4) and (b) the mixed-valent heptamolybdate [HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]−. (c) In the solid state, all nine novel polyanions form sodium ion-mediated dimers. Color code: MoO6, purple octahedra; MoO4, violet tetrahedron; As, green; C, yellow; P, navy blue; O, red; H, black; Na, light turquoise. | ||
As a result, the cyclic hexanuclear {Mo6O24} core comprises exclusively edge-shared octahedra with alternating short Mo–Mo bonded contacts (ca. 2.6 Å) and longer Mo⋯Mo nonbonding contacts (ca. 3.6 Å).
In the solid state, the individual polyanions form Na+ counter cation-mediated dimers via six Na⋯O interactions (ca. 2.3 Å) with oxo groups from the bonded MoV2 pairs opposite to the side where the dimethylarsinates are bound (Fig. 1c). It appears that the formation of the Na+-mediated polyanion sandwich in the solid state is pivotal for the crystallization of the polyanions, as we could never isolate any polyanion without such motif.9a,b
The novel polyanions can be obtained in the pH range 6.6–8.2 at room temperature as well as 50 or 80 °C, with optimal yields at pH 6.8–6.9. In all cases, the solutions turned dark red due to the reduction of the molybdenum centres from 6+ to 5+, with good quality crystals being formed within a week in decent yields. However, if the solutions are left standing open to the air during crystallization for a long time such as a month then [MoVI18MoV12O84{AsO2(CH3)2}18]18− {Mo30} crystals started forming alongside the target compounds, probably due to a drop in pH and partial air-oxidation of the MoV centers (Fig. 2).8 This reemphasizes the fact that in POM chemistry pH plays a vital role in the reaction outcomes.
 | ||
| Fig. 2 Schematic representation of the pH importance in the synthesis of polyoxomolybdates in aqueous solution; (bottom and right) the novel polyanions described here. (Left) The Mo30 wheel [MoVI18MoV12O84{AsO2(CH3)2}18]18− and (top) the neutral [MoV4O8{AsO2(CH3)2}4]. Color code: MoVO6, purple octahedra; MoVIO6, blue octahedra; MoO4, violet tetrahedron; P, navy blue; As, green; C, yellow; O, red; H, black. | ||
When using sodium molybdate (Na2MoO4·2H2O) instead of ammonium heptamolydate ((NH4)6Mo7O24·4H2O) as a reagent the target compounds could be obtained, but we observed extended crystallization periods and lower yields. Above pH 6.5, the same reaction mixture without addition of the phosphorous-containing heterogroups produced the heptanuclear mixed-valent polyoxomolybdate [MoVIMoV6O15(OH)4{AsO2(CH3)2}3]1− (Mo7) with a central tetrahedral molybdate ion surrounded by a reduced MoV6-oxo ring. Varying the amount of reductant or the order of adding the reactants did not alter the outcome, suggesting that the suitable pH range and counter cations facilitated the crystal formation. However, the use of hydrazine hemisulphate (N2H4·0.5 H2SO4) as reducing agent for the Mo7 reaction resulted in a disordered Mo atom in the center of the structure, likely due to its partial substitution by sulphate.
The central hetero group and the three dimethylarsinate ligands are all grafted on the same side of the polyanions. Within each polyanion, the central hetero elements are slightly elevated above the plane of the six molybdenum atoms and are bound via three μ3-oxo groups resulting in a distorted tetrahedral coordination geometry. The central heterogroup can be systematically substituted by incorporation of phosphate (HOPO32−), phosphite (HPO32−), methyl phosphonate (CH3PO32−), phosphonoacetate (HO2CCH2PO32−), phosphonopropionate (HO2CC2H4PO32−), phenyl phosphonate (C6H5PO32−), (4-fluorophenyl)phosphonate (4-FC6H4PO32−) and (4-trifluoromethoxyphenyl)phosphonate (4-F3COC6H4PO32−), wherein the overall charge of the polyanion mirrors with that of the heterogroup.6 The mixed-valent heptamolybdate [MoVIMoV6O15(OH)4{AsO2(CH3)2}3]1− (Mo7) comprises molybdenum atoms in both tetrahedral and octahedral coordination modes. The diphosphonate derivative [Mo7O16(O3PCH2PO3)3]8− also contains one MoVI and six MoV centers, but the latter do not form an edge-shared six-membered ring, as the three MoV2 units are bridged by a diphosphonate group.12
The eight novel phosphomolybdates(V) [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4) and the mixed-valent heptamolybdate [HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]− can be structurally described as follows: (a) terminal MoOterm bonds of 1.675–1.708 Å; (b) Mo–O(As) bonds of 2.066–2.104 Å and Mo–O(Mo) bonds of 2.107–2.128 Å; and (c) Mo–O(P/Mo) bonds of 2.274–2.355 Å. The dimethylarsinate ((CH3)2AsO2) capping groups act as bidentate ligands and the As center is tetrahedrally coordinated with average As–O(Mo) distances from 1.68–1.70 Å.4d The terminal P–O bond (1.52 Å) in PMo6 and the terminal MoVI–O bond (1.82 Å) of the central tetrahedral molybdenum atom in Mo7 are both monoprotonated corresponding to a hydroxo group, as confirmed by BVS calculations (Tables S2 and S10†).
The crystalline nature and phase purity of the synthesized polyanions were assessed through powder-XRD (PXRD) measurements on crystals directly taken from the mother liquor, see Fig. S18.† The diffractograms showed a good agreement between the experimental patterns and simulated patterns derived from the respective single-crystal X-ray diffraction analyses. Differences in diffraction peak intensities are attributed to various crystal orientations. The PXRD patterns of all compounds complement each other confirming their isostructural nature.
Upon closer examination of the diffraction patterns for the phosphite derivative Na-HPMo6 and the phosphate derivative NaNH4-PMo6 (Fig. S19†), an additional peak was observed at low 2θ angles 7.5–10°. Further studies revealed that polyanions with short groups on the central P hetero atom lose their crystal waters rapidly upon exposure to atmospheric air, causing the clear red crystals to transition into orange powder within one hour.14 Different degrees of hydration for Na-HPMo6 result in modifications of the packing arrangements in the crystalline lattice, thereby influencing the positions and intensities of the diffraction peaks observed in the air-dried sample. The rehydration of Na-HPMo6 in the presence of water vapor at room temperature overnight brings back the compound to the freshly synthesized states as shown by PXRD and TGA, indicating a reversible dehydration-rehydration process (Fig. 3). Conversely, compounds with longer groups attached to the hetero atom such as NaNH4-HO2CC2H4PMo6 maintained their structural integrity and crystalline nature even after extended drying periods (Fig. S19†).
 | ||
| Fig. 3 (left) Powder XRD patterns for freshly prepared, air-dried and rehydrated Na-HPMo6 and (right) thermograms for air dried and rehydrated Na-HPMo6 from room temperature to 400 °C under N2 atmosphere. | ||
3.1 NMR spectroscopy
To investigate the solution properties of the P-containing polyanions [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4), we conducted multinuclear NMR spectra (31P, 1H, 19F, 13C{1H}) on the salts dissolved in water. The 13C{1H} NMR spectra required overnight collection due to the low solubility of the polyanions. The 1H solution NMR spectra in H2O/D2O of the reference compounds cacodylic acid (H-Cac) (pH 4.1) and sodium cacodylate buffer (Na-Cac) (H-Cac adjusted to pH 7.0 with NaOH pellets) exhibit a distinct peak at 1.8 and 1.7 ppm, respectively, alongside the solvent water peak at 4.7 ppm. Their corresponding 13C{1H} NMR spectra show a sharp singlet at 17.0 and 17.5 ppm, respectively, consistent with the expected signal from the two equivalent methyl groups on the AsV atom.7aThe 31P{1H} NMR spectrum of NaNH4-PMo6 displays three singlets at 2.80, 2.65 and 2.50 ppm, with an intensity ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1. Notably, the peak at 2.80 ppm could be attributed to PMo6, whereas the signals at 2.65 and 2.50 ppm likely correspond to protonated phosphate species (Fig. 4a). The 1H NMR spectrum of NaNH4-PMo6 dissolved in H2O/D2O at room temperature exhibits two distinct singlets at δ = 2.3 and 2.0 ppm (intensity ratio 1:1) (Fig. 4b), whereas the 13C{1H} NMR spectrum shows two sharp resonances at 19.0 and 18.0 ppm, clearly reflecting the inequivalent nature of the two methyl groups on each As atom (Fig. 4c). The 1H NMR peak at 1.7 ppm is likely from a minute amount of cocrystallized or decomposed free cacodylate, whereas the minor peaks at 2.2 and 1.9 ppm could not be assigned. The solutions were monitored by NMR over a period of 1 week, during which the spectra remained unchanged, indicating no significant decomposition of PMo6 (Fig. S20†). This observation suggests that in PMo6 and the other derivatives, the cacodylate groups are strongly bound to the polyanion, which is supported by Mo–O(As) bond lengths of 2.076–2.099 Å. For comparison, the Mo–O(P) bonds in Haushalter's [(H3O)2NaMo6P4O24(OH)7]2− {P4MoV6} are 2.1073–2.1075 Å,9b and for both compounds are much shorter than the Mo–O(C) bonds (2.260–2.322 Å) in Kortz's amino acid-decorated [SeIVMo6O21(O2CCH2NH3)3]2− (which is one of 18 derivatives with different amino acids bound, including chiral ones).6e
:5:1. Notably, the peak at 2.80 ppm could be attributed to PMo6, whereas the signals at 2.65 and 2.50 ppm likely correspond to protonated phosphate species (Fig. 4a). The 1H NMR spectrum of NaNH4-PMo6 dissolved in H2O/D2O at room temperature exhibits two distinct singlets at δ = 2.3 and 2.0 ppm (intensity ratio 1:1) (Fig. 4b), whereas the 13C{1H} NMR spectrum shows two sharp resonances at 19.0 and 18.0 ppm, clearly reflecting the inequivalent nature of the two methyl groups on each As atom (Fig. 4c). The 1H NMR peak at 1.7 ppm is likely from a minute amount of cocrystallized or decomposed free cacodylate, whereas the minor peaks at 2.2 and 1.9 ppm could not be assigned. The solutions were monitored by NMR over a period of 1 week, during which the spectra remained unchanged, indicating no significant decomposition of PMo6 (Fig. S20†). This observation suggests that in PMo6 and the other derivatives, the cacodylate groups are strongly bound to the polyanion, which is supported by Mo–O(As) bond lengths of 2.076–2.099 Å. For comparison, the Mo–O(P) bonds in Haushalter's [(H3O)2NaMo6P4O24(OH)7]2− {P4MoV6} are 2.1073–2.1075 Å,9b and for both compounds are much shorter than the Mo–O(C) bonds (2.260–2.322 Å) in Kortz's amino acid-decorated [SeIVMo6O21(O2CCH2NH3)3]2− (which is one of 18 derivatives with different amino acids bound, including chiral ones).6e
 | ||
| Fig. 4 Multinuclear NMR study ((a) 31P{1H}, (b) 1H, (c) 13C{1H}) of NaNH4-PMo6 (pink spectra) dissolved in H2O/D2O at room temperature (pH 7). The spectra of the reference compounds are shown in blue and black. | ||
Considering that the novel polyanions are formed easily in a one-pot procedure we performed in situ31P{1H} NMR studies, in order to optimize the synthetic procedures. For the synthesis of PMo6, a reaction mixture containing ammonium heptamolybdate, sodium phosphate, and hydrazine hydrochloride in sodium cacodylate buffer (pH 7) with a molar ratio of Mo:P at 36:1 was stirred and monitored at two different temperatures. This six-fold molybdenum excess was initially used to ensure optimal product yield. During the synthesis, we observed that at room temperature PMo6 did not form during the first hour (Fig. S21a†), whereas at 80 °C a gradual conversion from Na3PO4 at pH 7 (δ = 2.5 ppm) to PMo6 (δ = 3.0 ppm) occurred over the course of one hour, ultimately resulting in a clean singlet at 3.0 ppm, indicative of complete conversion (Fig. S21b†). To further understand the effect of molar ratios, reaction progress was monitored at 80 °C across various Mo:P ratios. Notably, with a stoichiometric Mo:P ratio of 6:1, only 35% conversion to the target product was observed after one hour (Fig. S21†), underscoring the importance of molybdenum excess in achieving higher conversion rates.
Regarding the NMR characteristics of dissolved crystalline material for the other compounds, the phosphite-analogue HPMo6 displayed the expected NMR resonances in H2O/D2O with prominent sharp singlets at δ = 2.4 and 1.9 ppm for 1H NMR (Fig. S22a†) as well as δ = 19.4 and 17.9 ppm in 13C{1H} NMR (Fig. S22b†) attributed to the cacodylate methyl groups. The 31P{1H} spectrum showed a significant downfield shift at 12.9 ppm, compared to the reference shift at 2.90 ppm for Na2HPO3 (adjusted from pH 9.2 to pH 7 with 4M HCl). Additionally, the 31P NMR spectra displayed doublet peaks at 15.12 and 10.84 ppm (1JP–H = 693 Hz) due to the strong coupling of the phosphorus atom with the directly attached proton (Fig. S23†).6d
The 1H NMR spectrum of the methylphosphonate analogue CH3PMo6 exhibits two sharp equal-intensity resonances at 2.4 and 2.0 ppm (for the inequivalent methyl protons of the coordinated cacodylates), a peak at 1.8 ppm for free cacodylate (possibly some minor polyanion decomposition due to heating at 40 °C to increase solubility in H2O) and a doublet centered at 0.015 ppm (2JP–H = 18 Hz) due to coupling of the central methylphosphonate protons with the phosphorous nucleus, significantly upfield shifted from the doublet at 1.06 ppm (2JP–H = 16.4 Hz) for the reference CH3PO3H2 dissolved in water (adjusted to pH 6 with 6M NaOH) (Fig. S24a†). The 13C{1H} NMR of CH3PMo6 shows the anticipated two peaks at 19.6 and 17.5 ppm for the two inequivalent methyl groups of each bound cacodylate, along with a sharp peak at 16.9 ppm (corresponding to the free cacodylates) and a doublet at 13.85 ppm (1JP–C = 150.8 Hz) (originating from the methyl group on the P heteroatom), shifted slightly downfield from the doublet of the reference centered at 13.3 ppm (1JP–C = 140.7 Hz) (Fig. S24b†). The 31P{1H} NMR of CH3PMo6 shows a sharp singlet at 37.2 ppm, shifted downfield as compared to the 24.0 ppm signal for the reference methylphosphonate (Fig. S25a†), whereas the non-decoupled 31P NMR spectrum of CH3PMo6 depicts a clear quartet centered at δ = 37.2 ppm (2JP–H = 18.5 Hz), and more upfield at δ = 24 ppm (2JP–H = 16.2 Hz) the quartet for the reference (Fig. S25b†).6d
The salts of the polyanions HO2CCH2PMo6, HO2CC2H4PMo6 and C6H5PMo6 exhibited low solubility in H2O and so we used d6-DMSO as solvent to acquire clear and reliable NMR spectra. In 31P{1H} NMR the polyanion HO2CCH2PMo6 displays a major peak δ = 20.6 ppm, as compared to 15.9 ppm for the reference and a minor peak at 16.8 ppm, which could not be assigned (Fig. S26a†).6a It was difficult to obtain a good 13C{1H} NMR spectrum due to the low solubility of the compound. On the other hand, the 31P{1H} spectrum of HO2CC2H4PMo6 in d6-DMSO exhibits a sharp signal at 31.5 ppm, whereas the reference signal is at 25.5 ppm (Fig. S26b†).6a The 13C{1H} NMR of HO2CC2H4PMo6 redissolved in d6-DMSO (Fig. S27†) displays two signals at 18.2 and 20.1 ppm (for the inequivalent methyl carbons of coordinated cacodylates), along with signals at δ = 24.4 (23.4), 27.9 (28.2), 174.4 (174.4) ppm from the 2-carboxyethyl phosphonate hetero group (values in parentheses are the chemical shifts for the corresponding C atoms of free 2-carboxyethyl phosphonic acid dissolved in d6-DMSO). The peak at 24.4 ppm is a doublet (1JP–C = 150.8 Hz), arising from C–P coupling.
The 13C{1H} NMR spectrum of C6H5PMo6 in d6-DMSO shows resonances at δ = 126.7 (128.6), 129.2 (131.5), 131.4 (131.0) and 137.0 (134.5) ppm for the phenyl group (values in parentheses are for the corresponding C atoms of free phenylphosphonic acid dissolved in d6-DMSO). Two further signals at 20.0 and 17.8 ppm were observed originating from the inequivalent methyl carbons of the cacodylate groups (Fig. 5). The 13C{1H} peaks in the aromatic region at 137.0 ppm (1JP–C = 191 Hz) and 126.7 ppm (2JP–C = 20.1 Hz) could be ascribed to the ipso and ortho carbon atoms of the phenyl ring, whereas the peak at 131.4 ppm (3JP–C = 10.1 Hz) is due to the meta carbon atom. The 31P{1H} spectrum of C6H5PMo6 exhibited a major peak at 20.0 for the hetero element, as compared to a signal at 13.6 ppm for the neat phenylphosphonate. The minor peak at 19.6 ppm we cannot assign with confidence (Fig. S28a†).
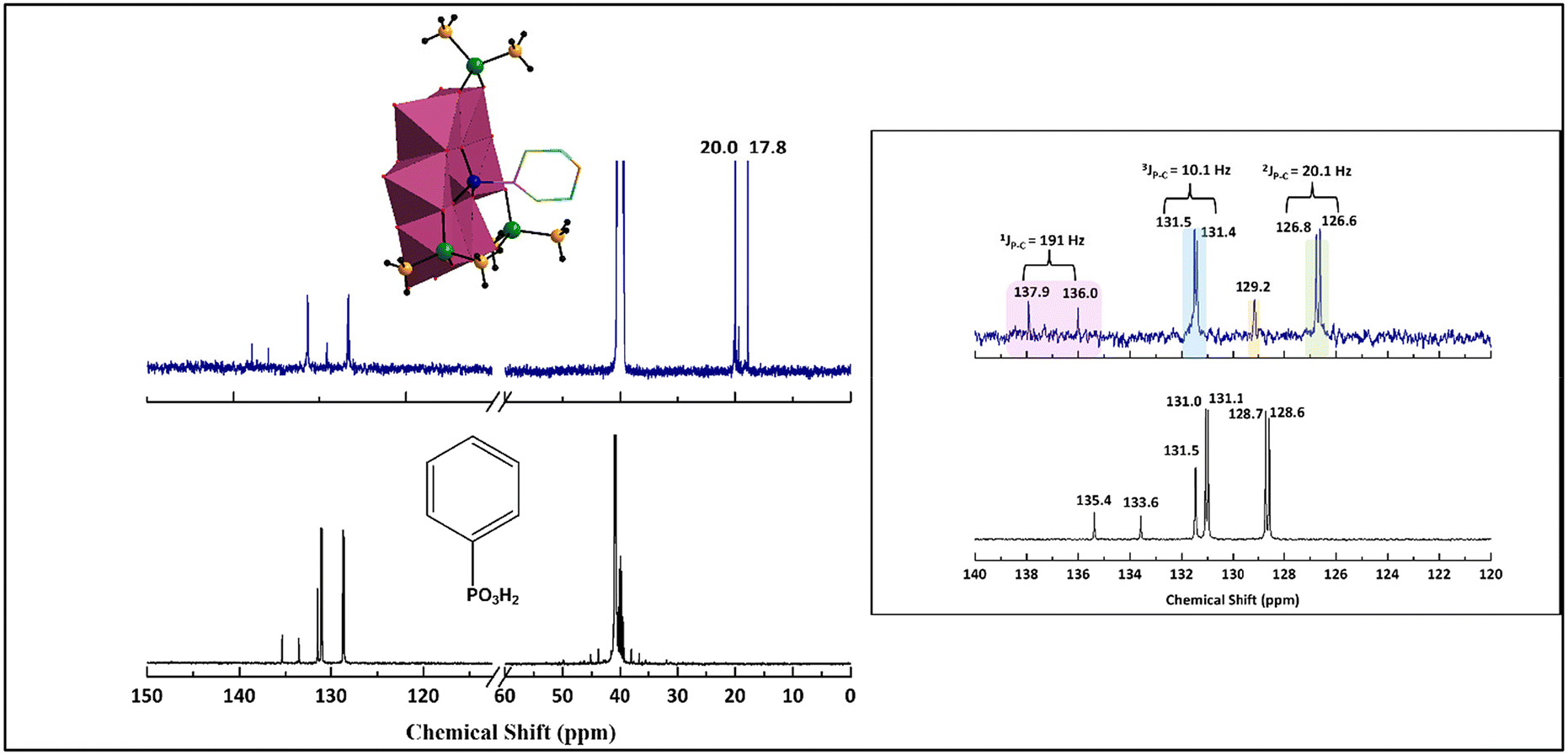 | ||
| Fig. 5 The 13C{1H} NMR spectra of NaNH4-C6H5PMo6 (upper left in blue) and the reference C6H5PO3H2 (lower left in black) dissolved separately in d6-DMSO at room temperature. The right panel showcases the enhanced spectra for the aromatic region only for NaNH4-C6H5PMo6 (blue) and C6H5PO3H2 (black). The peaks corresponding to structurally and hence magnetically inequivalent carbon atoms are labelled accordingly. | ||
The 31P{1H} NMR spectra for the two fluorinated polyanions 4-FC6H4PMo6 and 4-F3COC6H4PMo6 were recorded in H2O/D2O at pH 6 and displayed singlets at 22.8 ppm (Fig. S28b†) and 22.0 ppm (Fig. S28c†), respectively, indicating structural integrity. The chemical shifts of the free hetero groups (4-fluorophenyl) phosphonic acid (LPF) and (4-trifluoromethoxyphenyl) phosphonic acid (LPOCF3) in the same solvent at the same pH appear at 12.1 and 11.8 ppm, respectively (Fig. S28†). The 19F NMR spectra for 4-FC6H4PMo6 and 4-F3COC6H4PMo6 in H2O/D2O appear at −108.8 and −57.7 ppm, respectively (Fig. S29†).
Overall, it can be concluded that all our compounds which were easily water-soluble (HPMo6, 4-FC6H4PMo6 and 4-F3COC6H4PMo6) were shown to be solution-stable by multinuclear NMR. On the other hand, those compounds which needed heating and/or sonication to accomplish dissolution in water, showed some extra peaks due to possible minor transformation/decomposition of the polyanion (PMo6 and CH3PMo6). Those compounds (HO2CCH2PMo6, HO2CC2H4PMo6 and C6H5PMo6) which were less soluble in water could be dissolved in DMSO and exhibited clean NMR spectra. It is to be noted that, the mixed-valent heptamolybdate [MoVIMoV6O15(OH)4{AsO2(CH3)2}3]1− (Mo7) lacks sufficient solubilty in all tested solvents to obtain clear spectra required for detailed analysis.
3.2 ESI-mass spectrometry
ESI mass spectra of the polyanions were acquired from aqueous solution in both positive and negative ion mode. In positive ion mode only Mo7 resulted in a high-quality spectrum. In the negative ion mode all polyanions analyzed showed high-quality ESI-MS spectra with systematic patterns (Fig. S30–S36†). All compounds showed two strong cluster of ions, a first between m/z 680–820 and a second between m/z 1320 and 1410 depending on the central moiety. For a detailed discussion we selected as two representative examples C6H5PMo6 and Mo7. All mass spectrometry data are summarized in Table S11.†The polyanion C6H5PMo6 showed a first cluster of signals at m/z 692.54 and a second cluster at m/z 1406.07 (see Fig. 6). The first cluster was assigned to a doubly-charged negative ion with an elemental composition of [Mo6PAs3C12H27O24]2−. The observed isotope pattern is in full agreement with the structure as demonstrated by comparison to a simulation of the spectrum (see Fig. 6).
 | ||
| Fig. 6 Negative mode ESI mass spectra of polyanion C6H5PMo6. (A) Full scan full range view with two main signal clusters; (B) expanded region around m/z 690 with experimental spectra in upper panel and simulated isotope pattern in bottom panel; (C) expanded region around m/z 1406 with experimental spectra in upper panel (please note signals of dimeric species at 0.5 Da mass difference at lower intensity in between main signals) and simulated isotope pattern in bottom panel. | ||
The second signal cluster at m/z 1406.07 was assigned to originate from two individual species in the gas phase. Firstly, a monomeric, singly-charged species with an elemental composition of [Mo6PAs3C12H27O24Na]− with a mass difference of 1 Da between isotope peaks. Secondly, in between these signals a second species with isotope peak distances of 0.5 Da was observed, which was assigned to a dimeric species with an elemental composition of Na[Mo6PAs3C12H27O24Na]22−. Again, simulated spectra are in full agreement with experimental spectra. As demonstrated from single-crystal XRD data, the polyanion C6H5PMo6 dimerizes also in the gas phase mediated by a central Na+ ion, reminiscent of a 15-crown-5 crown ether interacting with a sodium cation. From the relative ion intensities of the two monomeric species and the single dimeric species a ratio of dimer/monomer of 0.57 in the gas phase was determined.
The polyanion Mo7 showed a first cluster of signals at m/z 683.54 and a second cluster at m/z 1390.08 (see Fig. 7). The first cluster was assigned to a doubly-charged negative ion with an elemental composition of [Mo7As3C6H18O24]2−. The observed isotope pattern is in full agreement with the structure as demonstrated by comparison to a simulation of the spectrum (see Fig. 7). The second cluster at m/z 1390.08 was assigned to originate from two individual species in the gas phase. Firstly, a monomeric, singly-charged species with an elemental composition of [Mo7As3C6H21O25]− with a mass difference of 1 Da between isotope peaks. Secondly in between these signals a second species with isotope differences of 0.5 Da was observed, which was assigned to a dimeric species with an elemental composition of Na[Mo7As3C6H22O25]22−. Again, simulated spectra are in full agreement with experimental spectra. In this case the ratio of dimer to monomer in the gas phase was determined to be 0.24.
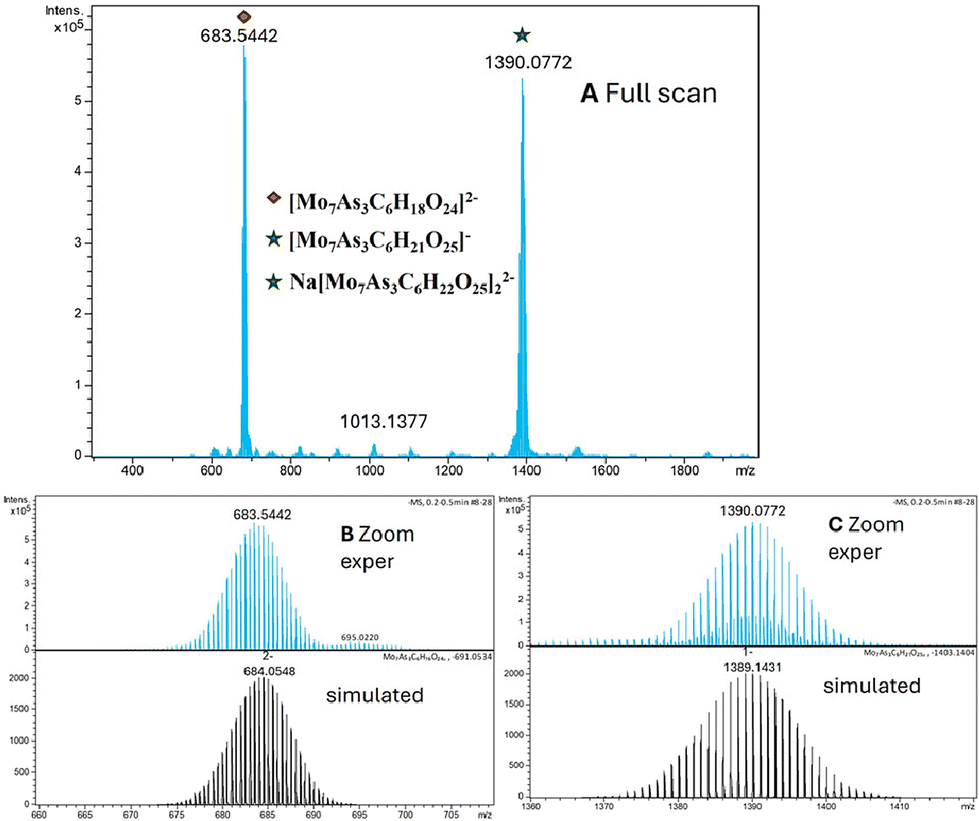 | ||
| Fig. 7 Negative mode ESI mass spectra of polyanion Mo7. (A) Full scan full range view with two main signal clusters; (B) expanded region around m/z 680 with experimental spectra in upper panel and simulated isotope pattern in bottom panel; (C) expanded region around m/z 1390 with experimental spectra in upper panel (please note: signals of dimeric species at lower intensity in between main signals) and simulated isotope pattern in bottom panel. | ||
Additionally, the polyanion Mo7 allowed for acquisition of a spectrum in the positive ion mode with two clusters observed at m/z 760.57 for a doubly positive ion and a second cluster centered around m/z 1498.17 corresponding to a singly positively charged ion (see Fig. S37†). It is worth mentioning that for the polyanion HO2CC2H4PMo6 exclusively the signals at higher m/z values at 1403.22 were observed with the monomeric dianion signals being absent (see Fig. S34†).
3.3 Ion mobility mass spectrometry
With a view to obtain further evidence for the assignment of the higher m/z signal envelope as overlapping monomeric and dimeric species we decided to attempt separation of the ions using ion mobility techniques. Ion mobility separation of POMs has a precedent in early work by Cronin's group on Keplerates.15a Further ion mobility separation of complex inorganic species have been achieved by Clever's group on Pd cages,15b and summarised in a recent review by Geue et al.15cThe two polyanions Mo7 and CH3PMo6 were selected for investigations with the latter described in detail and the former shown in the ESI (see Fig. S38†). For CH3PMo6 quadrupole selection isolated the peaks of the isotope envelope centred at m/z 1346 with an isolation width of 35 Da (see Fig. 8). The multiple isotopes were subjected to ion mobility separation in a cyclic ion mobility mass spectrometer. Extracted ion mobilograms were obtained at m/z 1346.7 corresponding to the dimeric doubly-charged species and m/z 1346.2 corresponding to a mixture of monomeric and dimeric species. For the dimeric species a single species was observed in the mobilogram, whereas for the ions at m/z 1346.2 two species were detected at distinct drift times of 18.4 ms and 24.9 ms. ESI spectra were recorded separately for both species confirming the identity of a dimeric [2M − 2H]2− ion at 18.4 ms and a monomeric [M − H]− ion at 24.9 ms. We believe this to be the first example of ion mobility separation of isobaric oligomeric POMs in the gas phase. The term isobaric refers to the fact that half of the monoisotopic peaks of the dimeric species are isobaric to the monoisotopic peaks of the monomeric species.
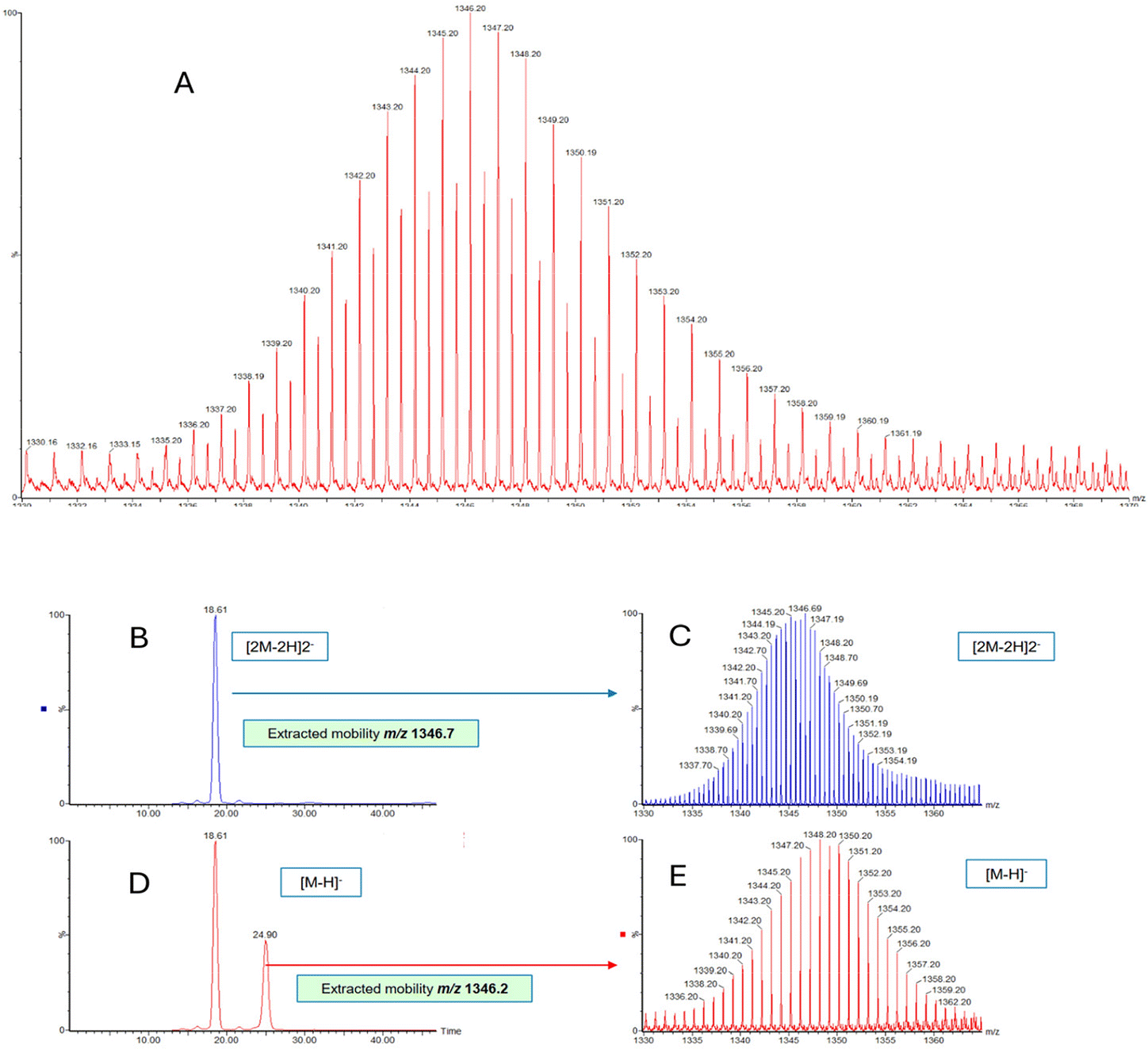 | ||
| Fig. 8 Negative mode ESI ion mobility mass spectra of compound CH3PMo6 and corresponding mobilograms. (A) Expanded view of ESI mass spectrum with two overlapping species; (B) extracted ion mobilogram of m/z 1346.7 showing single dimeric species [2M − 2H]2−. (C) ESI mass spectrum corresponding to dimeric species [2M − 2H]2−. (D) Extracted ion mobilogram of m/z 1346.2 showing single monomeric species [M − H]2−. (E) ESI mass spectrum corresponding to monomeric species [M − H]−. | ||
4. Conclusions
We have performed a comprehensive study on the synthesis and characterization of organically functionalized, water soluble and stable reduced polyoxomolybdates. As a result, we have isolated a family of eight dimethylarsinate-functionalized phosphomolybdates(V), [RPMoV6O15(OH)3{AsO2(CH3)2}3]2− (R = H, HO, CH3, HO2CCH2, HO2CC2H4, C6H5, 4-FC6H4, 4-F3COC6H4) and the mixed-valent heptamolybdate [HOMoVIMoV6O15(OH)3{AsO2(CH3)2}3]−. All nine polyanions were synthesized via facile one-pot open beaker conditions in aqueous solution, by careful adjustment of the reagent ratios, ionic strength, and most prominently pH. The incorporation of various organophosphonates including fluorinated derivatives as well as phosphate and phosphite within a stable hexanuclear molybdenum(V)-oxo ring provided a nice platform to investigate solubility and stability in aqueous medium at around pH 6–7. We performed extensive multinuclear (1H, 31P, 19F, 13C) NMR studies to investigate the solution properties of the novel polyanions. Due to the systematic modification of the organic function on the polyanions their interaction with biomolecules is of interest, which is currently in progress and will be reported separately. Furthermore, we are in the process of expanding the current family of compounds by making other derivatives. Interestingly, the polyanions crystallized as a sodium-ion mediated dimer, which appears to be in equilibrium with the free polyanion after dissolution of the solid salts in water. ESI-MS allowed to prove the coexistence of both monomer and dimer, and it was even possible to separate both entities and then obtain clean spectra for each by using ion mobility mass spectrometry, indicating the power of this technique when it comes to separating two components during the measurement.Data availability
• The data supporting this article have been included as part of the ESI.†• Electronic supplementary information (ESI) available: single-crystal XRD data, bond valence sum calculations, ESI-MS data and spectra, multinuclear NMR spectra, FT-IR spectra, thermograms, and powder XRD patterns.
• CCDC 2394921–2394926 and 2406188–2406190, respectively.
• Crystallographic data for nine compounds have been deposited at the CCDC under the accession numbers 2394921 (NaNH4-PMo6), 2394922 (Na-Mo7), 2394923 (Na-HPMo6), 2394924 (NaNH4-CH3PMo6), 2394925 (NaNH4-C6H5PMo6), 2394926 (HO2CC2H4PMo6·dmso), 2406188 (NaNH4-FC6H4PMo6), 2406189 (NaNH4-HO2CCH2PMo6) and 2406190 (NaNH4-F3COC6H4PMo6).
Author contributions
U. K. supervised the project and guided the research direction. V. S. synthesized the compounds, conducted material characterization, and drafted this paper. A. P. was responsible for synthesizing and characterizing the fluorinated organophosphonate groups and the corresponding polyanions. S. B. discovered the parent compound with initial studies performed by A. S. B. S. B. contributed to the single crystal – XRD measurements, data analysis and discussions. J. H. and D. S. performed the mass spectrometry experiments, with N. K. analysing the data and writing the mass spectrometry section. J. O., D. K., R. G., A. K. assisted with the reproduction and characterization of the compounds during their bachelor's thesis. All authors reviewed and contributed to the final version of the manuscript.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
U. K. thanks the German Research Council (DFG KO-2288/31-1) and Constructor University (formerly Jacobs University) for research support. The polyanion structures in Fig. 1, 2, 4 and 5 were generated with Diamond, version 3.2 (Crystal Impact GbR). We acknowledge ion mobility mass spectrometry measurements carried out by Dr Susan Slade and Dr Malcolm Anderson from Waters (Wilmslow, UK).References
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef; (b) M. T. Pope and A. Müller, Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef; (c) A. Proust, R. Thouvenot and P. Gouzerh, Functionalization of polyoxometalates: towards advanced applications in catalysis and materials science, Chem. Commun., 2008, 1837–1852 RSC; (d) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Hybrid Organic−Inorganic Polyoxometalate Compounds: From Structural Diversity to Applications, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed; (e) S.-S. Wang and G.-Y. Yang, Recent Advances in Polyoxometalate-Catalyzed Reactions, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed; (f) N. Lotfian, M. M. Heravi, M. Mirzaei and B. Heidari, Applications of inorganic–organic hybrid architectures based on polyoxometalates in catalyzed and photocatalyzed chemical transformations, Appl. Organomet. Chem., 2019, 33, e4808 CrossRef; (g) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Functionalization and post-functionalization: a step towards polyoxometalate-based materials, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC; (h) M.-P. Santoni, G. S. Hanan and B. Hasenknopf, Covalent multi-component systems of polyoxometalates and metal complexes: Toward multi-functional organic–inorganic hybrids in molecular and material sciences, Coord. Chem. Rev., 2014, 281, 64–85 CrossRef CAS; (i) J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Hybrid polyoxometalate materials for photo(electro-) chemical applications, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS; (j) K. Xia, K. Yamaguchi and K. Suzuki, Recent advances in hybrid materials of metal nanoparticles and polyoxometalates, Angew. Chem., Int. Ed., 2023, 62, e202214506 CrossRef CAS PubMed; (k) J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Polyoxometalates in medicine, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed; (l) M. B. Čolović, M. Lacković, J. Lalatović, A. S. Mougharbel, U. Kortz and D. Z. Krstić, Polyoxometalates in biomedicine: Update and overview, Curr. Med. Chem., 2020, 27, 362–379 CrossRef PubMed; (m) H. Soria-Carrera, E. Atrián-Blasco, R. Martín-Rapún and S. G. Mitchell, Polyoxometalate–peptide hybrid materials: from structure–property relationships to applications, Chem. Sci., 2023, 14, 10–28 RSC.
- (a) A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Hybrid polyoxometalates as post-functionalization platforms: from fundamentals to emerging applications, Chem. Soc. Rev., 2020, 49, 382–432 RSC; (b) J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Supramolecular assemblies of organo-functionalised hybrid polyoxometalates: from functional building blocks to hierarchical nanomaterials, Chem. Soc. Rev., 2022, 51, 293–328 RSC.
- (a) C. Rinfray, S. Renaudineau, G. Izzet and A. Proust, A covalent polyoxomolybdate-based hybrid with remarkable electron reservoir properties, Chem. Commun., 2014, 50, 8575–8577 RSC; (b) C. Li, N. Mizuno, K. Yamaguchi and K. Suzuki, Self-assembly of anionic polyoxometalate–organic architectures based on lacunary phosphomolybdates and pyridyl ligands, J. Am. Chem. Soc., 2019, 141, 7687–7692 CrossRef CAS PubMed; (c) C. Li, K. Yamaguchi and K. Suzuki, Ligand–Directed Approach in Polyoxometalate Synthesis: Formation of a New Divacant Lacunary Polyoxomolybdate [γ–PMo10O36]7−, Angew. Chem., Int. Ed., 2021, 60, 6960–6964 CrossRef CAS PubMed; (d) A. Jimbo, C. Li, K. Yonesato, T. Ushiyama, K. Yamaguchi and K. Suzuki, Molecular hybrids of trivacant lacunary polyoxomolybdate and multidentate organic ligands, Chem. Sci., 2023, 14, 10280–10284 RSC.
- (a) A. Rosenheim and R. Bilecki, Über Molybdänsäure–alkylarsinate. (Zur Kenntnis der Iso– Und Heteropolysalze. VIII. Mitteilung.), Ber. Dtsch. Chem. Ges., 1913, 46, 539–557 CrossRef CAS; (b) W. Kwak, L. M. Rajković, J. K. Stalick, M. T. Pope and C. O. Quicksall, Synthesis and Structure of Hexamolybdobis(organoarsonates), Inorg. Chem., 1976, 15, 2778–2783 CrossRef CAS; (c) K. M. Barkigia, L. M. Rajković, M. T. Pope and C. O. Quicksall, New type of heteropoly anion. Tetramolybdo complexes of dialkyl- and diarylarsinates, J. Am. Chem. Soc., 1975, 97, 4146–4147 CrossRef CAS; (d) K. M. Barkigia, L. M. Rajković-Blazer, M. T. Pope, E. Prince and C. O. Quicksall, Molybdoarsinate Heteropoly Complexes. Structure of the Hydrogen Tetramolybdodimethylarsinate(2-) Anion by X-Ray and Neutron Diffraction, Inorg. Chem., 1980, 19, 2531–2537 CrossRef CAS.
- (a) W. Kwak, M. T. Pope and T. F. Scully, Stable Organic Derivatives of Heteropoly Anions, Pentamolybdobisphosphonates, J. Am. Chem. Soc., 1975, 97, 5735–5738 CrossRef CAS; (b) J. K. Stalick and C. O. Quicksall, Two Heteropoly Anions Containing Organic Groups. Crystal and Molecular Structures of Ammonium Pentamolybdobis(methylphosphonate) Pentahydrate, (NH4)4[(CH3P)2Mo5O21]·5H2O, and Sodium Tetramethylammonium Pentamolybdobis(ethylammoniumphosphonate) Pentahydrate, Na[N(CH3)4][(NH3C2H4P)2Mo5O21]·5H2O, Inorg. Chem., 1976, 15, 1577–1584 CrossRef CAS; (c) T. Ozeki, H. Ichida, H. Miyamae and Y. Sasaki, Crystal Structure of K4[H2P2Mo5O21]·2H2O, Bull. Chem. Soc. Jpn., 1988, 61, 4455–4457 CrossRef CAS; (d) D. G. Lyxell and R. Strandberg, The structure of tetraguanidinium pentamolybdodiphenylphosphonate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1535–1538 CrossRef.
- (a) U. Kortz, C. Marquer, R. Thouvenot and M. Nierlich, Polyoxomolybdates Functionalized with Phosphonocarboxylates, Inorg. Chem., 2003, 42, 1158–1162 CrossRef CAS PubMed; (b) P. Manna, S. Bhattacharya and U. Kortz, Arylarsonate- and Phosphonate-Capped Polyoxomolybdates, [(RC6H4As)2Mo6O24]n− and [(R′C6H4P)2Mo5O21]n−, Inorg. Chem., 2021, 60, 7161–7167 CrossRef CAS PubMed; (c) M. Carraro, A. Sartorel, G. Scorrano, C. Maccato, M. H. Dickman, U. Kortz and M. Bonchio, Chiral Strandberg–Type Molybdates [(RPO3)2Mo5O15]2− as Molecular Gelators: Self–Assembled Fibrillar Nanostructures with Enhanced Optical Activity, Angew. Chem., Int. Ed., 2008, 47, 7275–7279 CrossRef CAS PubMed; (d) U. Kortz, J. Vaissermann, R. Thouvenot and P. Gouzerh, Heteropolymolybdates of Phosphate, Phosphonate, and Phosphite Functionalized by Glycine, Inorg. Chem., 2003, 42, 1135–1139 CrossRef CAS PubMed; (e) U. Kortz, M. G. Savelieff, F. Y. A. Ghali, L. M. Khalil, S. A. Maalouf and D. I. Sinno, Heteropolymolybdates of AsIII, SbIII, BiIII, SeIV, and TeIV Functionalized by Amino Acids, Angew. Chem., Int. Ed., 2002, 41, 4070–4073 CrossRef CAS.
- (a) S. Bhattacharya, X. Ma, A. S. Mougharbel, M. Haouas, P. Su, M. F. Espenship, D. H. Taffa, H. Jaensch, A.-J. Bons, T. Stuerzer, M. Wark, J. Laskin, E. Cadot and U. Kortz, Discovery of a Neutral 40-PdII-Oxo Molecular Disk, [Pd40O24(OH)16{(CH3)2AsO2}16]: Synthesis, Structural Characterization, and Catalytic Studies, Inorg. Chem., 2021, 60, 17339–17347 CrossRef CAS PubMed; (b) S. Bhattacharya, A. Barba-Bon, T. A. Zewdie, A. B. Müller, T. Nisar, A. Chmielnicka, I. A. Rutkowska, C. J. Schürmann, V. Wagner, N. Kuhnert, P. J. Kulesza, W. M. Nau and U. Kortz, Discrete, Cationic Palladium(II)–Oxo Clusters via f–Metal Ion Incorporation and their Macrocyclic Host–Guest Interactions with Sulfonatocalixarenes, Angew. Chem., Int. Ed., 2022, 61, e202203114 CrossRef CAS PubMed; (c) X. Ma, S. Bhattacharya, T. Nisar, A. B. Müller, V. Wagner, N. Kuhnert and U. Kortz, Mixed-valent palladium(IV/II)-oxoanion, [PdIVO6PdII6((CH3)2AsO2)6]2−, Chem. Commun., 2023, 59, 904–907 RSC; (d) J. Zhang, S. Bhattacharya, A. B. Müller, L. Kiss, C. Silvestru, N. Kuhnert and U. Kortz, Mixed Noble Metal–Oxo Clusters: Platinum(IV)–Gold(III) Oxoanion [PtIV2AuIII3O6((CH3)2AsO2)6]−, Chem. Commun., 2023, 59, 5918–5921 RSC; (e) J. Zhang, S. Bhattacharya, B. E. Khsara, T. Nisar, A. B. Müller, M. Besora, J. M. Poblet, V. Wagner, N. Kuhnert and U. Kortz, PtIV-Containing Hexaplatinate(II) [PtIVPtII6O6(AsO2(CH3)2)6]2− and Hexapalladate(II) [PtIVPdII6O6(AsO2(CH3)2)6]2−, Inorg. Chem., 2023, 62, 13184–13194 CrossRef CAS PubMed.
- A. Sundar, S. Bhattacharya, J. Oberstein, X. Ma, B. S. Bassil, T. Nisar, D. H. Taffa, M. Wark, V. Wagner and U. Kortz, Organically Functionalized Mixed-Valent Polyoxo-30-molybdate Wheel and a Neutral Tetramolybdenum(V) Oxo Cluster, Inorg. Chem., 2022, 61, 11524–11528 CrossRef CAS PubMed.
- (a) R. C. Haushalter and F. W. Lai, [Et4N]6[Na14Mo24P17O97(OH)31]·xH2O: A Hollow Cluster Filled with 12 Na⊕ Ions and a H3PO4 Molecule, Angew. Chem., Int. Ed. Engl., 1989, 28, 743–746 CrossRef; (b) R. C. Haushalter and F. W. Lai, Synthesis of a New One-Dimensional Sodium Molybdenum Phosphate Polymer: Structure of [(H3O)2NaMo6P4O24(OH)7]2−, Inorg. Chem., 1989, 28, 2904–2905 CrossRef CAS; (c) R. C. Haushalter and L. A. Mundi, Reduced Molybdenum Phosphates: Octahedral-Tetrahedral Framework Solids with Tunnels, Cages, and Micropores, Chem. Mater., 1992, 4, 31–48 CrossRef CAS.
- (a) G. Cao, R. C. Haushalter and K. G. Strohmaier, A Novel Polyoxo Molybdenum(V) Organophosphonate Anion Having a Sandwich Structure: Synthesis and Crystal Structure of [N(C2H5)4]2Na3(H3O)4{Na[Mo6O15(O3PC6H5)(HO3PC6H5)3]2}·14H2O, Inorg. Chem., 1993, 32, 127–128 CrossRef CAS; (b) M. I. Khan, Q. Chen and J. Zubieta, Hydrothermal synthesis and crystal structure of (NH4)5Na4{Na[Mo6O15− (HO3PC6H5)3(O3PC6H5)]2}·6H2O, Inorg. Chim. Acta, 1993, 206, 131–133 CrossRef CAS; (c) M. I. Khan, Q. Chen and J. Zubieta, Oxomolybdenum(V) polyanion clusters. Hydrothermal syntheses and structures of (NH4)5Na4{Na [Mo6O12(OH)3(O3PC6H5)4)]2}·6H2O and (C6H5CH2NMe3)4K4{K2[Mo6O12(OH)3(O3PC6H5)4]2}·10H2O and their relationship to the binuclear (Et4N)[Mo2O4Cl3(H2O)3]·5H2O, Inorg. Chim. Acta, 1995, 235, 135–145 CrossRef CAS; (d) Y. D. Chang and J. Zubieta, Investigations into the syntheses and structures of clusters of the Mo-O-REO32− systems (E = P and As), Inorg. Chim. Acta, 1996, 245, 177–198 CrossRef CAS; (e) M. J. Manos, A. D. Keramidas, J. D. Woollins, A. M. Z. Slawin and T. A. Kabanos, The first polyoxomolybdenum carbonate compound: Synthesis and crystal structure of (NH4)5[(Mo2VO4)3(μ6-CO3)(μ-CO3)3(μ-OH)3]·0.5CH3OH, J. Chem. Soc., Dalton Trans., 2001, 3419–3420 RSC; (f) M. J. Manos, J. D. Woollins, A. D. Keramidas, A. M. Z. Slawin and T. A. Kabanos, Polyoxomolybdenum(V) Sulfite Complexes: Synthesis, Structural, and Physical Studies, Angew. Chem., Int. Ed., 2002, 41, 2801–2805 CrossRef CAS.
- (a) E. Cadot, A. Dolbecq, B. Salignac and F. Sécheresse, Self-Condensation of [MoV2O2S2]2+ with Phosphate or Arsenate Ions by Acid-Base Processes in Aqueous Solution: Syntheses, Crystal Structures, and Reactivity of [(HXO4)4Mo6S6O6(OH)3]5−, X = P, As, Chem. – Eur. J., 1999, 5, 2396–2403 CrossRef CAS; (b) A. Dolbecq, E. Cadot, D. Eisner and F. Sécheresse, Regioselective S/O substitutions in heteropolyoxothioanions: 31P NMR study and X-ray crystal structure of the half-substituted anion [(HPO4)4Mo6S3O9(OH)3]5−, Inorg. Chim. Acta, 2000, 300–302, 151–157 CrossRef CAS.
- (a) E. Dumas, C. Sassoye, K. D. Smith and S. C. Sevov, Synthesis and Characterization of [Mo7O16(O3PCH2PO3)3]8−: A Mixed-Valent Polyoxomolybdenum Diphosphonate Anion with Octahedrally and Tetrahedrally Coordinated Molybdenum, Inorg. Chem., 2002, 41, 4029–4032 CrossRef CAS PubMed; (b) C. du Peloux, A. Dolbecq, P. Mialane, J. Marrot and F. Sécheresse, Template synthesis of {(MoV2O4)(O3PCH2PO3)}n clusters (n = 3, 4, 10): solid state and solution studies, Dalton Trans., 2004, 1259–1263 RSC.
- G. Keglevich, A. Grün, A. Bölcskei, L. Drahos, M. Kraszni and G. T. Balogh, Synthesis and Proton Dissociation Properties of Arylphosphonates: A Microwave-Assisted Catalytic Arbuzov Reaction with Aryl Bromides, Heteroat. Chem., 2012, 23, 574–582 CrossRef CAS.
- (a) D. Barats-Damatov, L. J. W. Shimon, Y. Feldman, T. Bendikov and R. Neumann, Solid-State Crystal-to-Crystal Phase Transitions and Reversible Structure−Temperature Behavior of Phosphovanadomolybdic Acid, H5PV2Mo10O40, Inorg. Chem., 2015, 54, 628–634 CrossRef CAS PubMed; (b) B. Herzog, W. Bensch, T. Ilkenhans, R. Schlögl and N. Deutsch, Single crystal and powder diffraction studies of the structure of heteropolymolybdophosphoric acid catalysts, Catal. Lett., 1993, 20, 203–219 CrossRef CAS.
- (a) P. J. Robbins, A. J. Surman, J. Thiel, D.-L. Long and L. Cronin, Use of ion-mobility mass spectrometry (IMS-MS) to map polyoxometalate Keplerate clusters and their supramolecular assemblies, Chem. Commun., 2013, 49, 1909–1911 RSC; (b) K. E. Ebbert, L. Schneider, A. Platzek, C. Drechsler, B. Chen, R. Rudolf and G. H. Clever, Resolution of minor size differences in a family of heteroleptic coordination cages by trapped ion mobility ESI-MS, Dalton Trans., 2019, 48, 11070–11075 RSC; (c) N. Geue, R. E. P. Winpenny and P. E. Barran, Ion Mobility Mass Spectrometry for Large Synthetic Molecules: Expanding the Analytical Toolbox, J. Am. Chem. Soc., 2024, 146, 8800–8819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2394921–2394926 and 2406188–2406190. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi03215b |
| This journal is © the Partner Organisations 2025 |