
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation of phenols by the excited state of an osmium(VI) nitrido complex†
Yu-Zhong
Lu
 a,
Li-Xin
Wang
a,
Rui-Yue
Qi
ab,
Jing
Xiang
*a,
Ji-Yan
Liu
*a and
Tai-Chu
Lau
*ac
a,
Li-Xin
Wang
a,
Rui-Yue
Qi
ab,
Jing
Xiang
*a,
Ji-Yan
Liu
*a and
Tai-Chu
Lau
*ac
aKey Laboratory of Optoelectronic Chemical Materials and Devices (Ministry of Education), School of Optoelectronic Materials and Technology, Jianghan University, Wuhan, 430056, China. E-mail: xiangjing35991@sohu.com
bHebei Key Laboratory of Heterocyclic Compounds, Handan University, Handan 056005, Hebei Province, China
cDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong, P. R. China. E-mail: bhtclau@cityu.edu.hk
First published on 26th February 2025
Abstract
The photoreaction of an osmium(VI) nitrido complex, [OsVI(N)(L)(CN)3]− (OsN), with various phenols has been investigated. Upon irradiation of OsN with visible light, the excited state (OsN*) is generated which reacts readily with a variety of phenols. OsN* reacts with mono- and di-substituted phenols, including 2,6-dimethylphenol, 2,6-dichlorophenol and 4-methylphenol to afford the corresponding osmium(II) benzoquinone monoimine and osmium(IV) benzoquinone monoiminato complexes. On the other hand, in the reactions of OsN* with bulky tri-substituted phenol such as 2,4,6-tri-tert-butylphenol, C–C bond cleavage occurred and [OsIV(L)(CN)3(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) tBu2Ph(−2H)O)]− was formed as the major product. The electronic effects of various para-substituents (X) on the oxidation of phenols were investigated by the method of initial rates (Rx). A Hammett plot of log(Rx/RH) versus σp is linear with a ρ value of −0.54. A linear correlation of log(Rx) with the oxidation potentials (E) of phenols was also found with a slope of −0.80. On the other hand, no correlations were found between log(Rx) and O–H bond dissociation energy (BDE), as well as the pKa of phenols. The oxidation of phenols by OsN* exhibits a negligible kinetic isotope effect (KIE), k(C6H5OH)/k(C6D5OD) ∼1. These results are consistent with a mechanism that involves an initial 1e− oxidation of the phenol followed by rapid proton transfer (ET-PT) to generate a phenoxy radical, this is followed by a N-rebound step to give the osmium products.
tBu2Ph(−2H)O)]− was formed as the major product. The electronic effects of various para-substituents (X) on the oxidation of phenols were investigated by the method of initial rates (Rx). A Hammett plot of log(Rx/RH) versus σp is linear with a ρ value of −0.54. A linear correlation of log(Rx) with the oxidation potentials (E) of phenols was also found with a slope of −0.80. On the other hand, no correlations were found between log(Rx) and O–H bond dissociation energy (BDE), as well as the pKa of phenols. The oxidation of phenols by OsN* exhibits a negligible kinetic isotope effect (KIE), k(C6H5OH)/k(C6D5OD) ∼1. These results are consistent with a mechanism that involves an initial 1e− oxidation of the phenol followed by rapid proton transfer (ET-PT) to generate a phenoxy radical, this is followed by a N-rebound step to give the osmium products.
Introduction
Transition metal nitrido (M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) complexes are intermediates in N2 fixation and potentially useful reagents for the nitrogenation of organic substrates.1,2 Compared to metal oxo (MO) species, there are relatively few nitrido complexes that are reactive towards common organic substrates. Examples include osmium(VI) nitrido complexes containing various N-based ligands such as 2,2′-bipyridine (bpy), 2,2′:6′,2′′-terpyridine (tpy), and tris(1-pyrazolyl)methane (tpm); they are highly electrophilic and readily undergo N-atom transfer to a variety of organic substrates.3 Moreover, Ru(VI) nitrido complexes bearing salen type ligands, such as [RuVI(N)(salchda)(CH3OH)]+ (RuN, salchda = N,N′-bis(salicylidene)-o-cyclohexyldiamine dianion),4 and several iron(V/IV)5 and Mn(V/VI)6 nitrido complexes are also highly reactive species that are capable of oxidizing various organic substrates.
N) complexes are intermediates in N2 fixation and potentially useful reagents for the nitrogenation of organic substrates.1,2 Compared to metal oxo (MO) species, there are relatively few nitrido complexes that are reactive towards common organic substrates. Examples include osmium(VI) nitrido complexes containing various N-based ligands such as 2,2′-bipyridine (bpy), 2,2′:6′,2′′-terpyridine (tpy), and tris(1-pyrazolyl)methane (tpm); they are highly electrophilic and readily undergo N-atom transfer to a variety of organic substrates.3 Moreover, Ru(VI) nitrido complexes bearing salen type ligands, such as [RuVI(N)(salchda)(CH3OH)]+ (RuN, salchda = N,N′-bis(salicylidene)-o-cyclohexyldiamine dianion),4 and several iron(V/IV)5 and Mn(V/VI)6 nitrido complexes are also highly reactive species that are capable of oxidizing various organic substrates.
The oxidation of phenols to quinones by various oxidants has been extensively studied, mainly because such reactions are relevant to many biological processes.7–13 Phenols can be oxidized by various pathways (Fig. 1), the most common one is initial 1e− oxidation resulting in the formation phenoxy radicals, which is followed by rapid loss of the phenolic proton (consecutive electron transfer-proton transfer, ET-PT). Further rapid loss of 1e− + 1H+ results in the formation of quinones.14,15 On the other hand, oxidation of phenols by some metal oxo species may involve an H-atom transfer mechanism (or concerted ET-PT).16–22 Oxidation via an initial electrophilic attack on the aromatic ring of phenol has also been reported for a ruthenium(IV) species.23,24
 | ||
| Fig. 1 Oxidation pathways of phenols by oxidants. | ||
Prior to this work, there was only one example on oxidation of phenols by a metal nitrido species. RuN readily reacts with phenols in the presence of pyridine to afford (salchda)ruthenium(II) p-benzoquinone imine complexes.25 The reactions were proposed to proceed via an initial electrophilic attack by RuN at the aromatic ring of phenols.
In an attempt to design a highly active metal nitrido complex, we turned our attention to the excited state chemistry of MN. We reported recently the synthesis of a highly luminescent Os(VI) nitrido complex, [OsVI(N)(L)(CN)3]− (OsN, HL = 2-(2-hydroxy-5-nitrophenyl)benzoxazole);26–33OsN is highly luminescent in both solid state and fluid solution (solid state: λem = 591 nm, ϕ = 11.7%, τ = 1.90 μs; in degassed CH2Cl2: λem = 594 nm, ϕ = 3.0%, τ = 0.48 μs). The CV of OsN shows an irreversible oxidation wave at Epa = 1.88 V and an irreversible reduction wave at Epa = −0.99 V vs. Saturated Calomel Electrode (SCE), which are tentatively assigned as the metal-centered OsVII/VI and OsVI/V process, respectively. The excited state (OsN*) of this complex is readily generated by visible light irradiation (λ > 460 nm). OsN* was found to be highly oxidizing and it reacts readily with various organic substrates, including alkanes, arenes, amines, alcohols, and dihydroxybenzenes.26–33
Herein, we reported the oxidation of phenols by OsN*. These reactions have a number of unusual/novel features. In contrast to the oxidation of phenols by RuN, which occurs via an electrophilic ring attack mechanism, oxidation by OsN* occurs by an ET-PT followed by an N-rebound mechanism. Similar to oxidation by RuN, oxidation of various mono- and di-substituted phenols by OsN* produced the corresponding osmium(II) benzoquinone imine complexes. However, because OsN* is a powerful oxidant, further oxidation of the osmium(II) products by OsN* occur to afford novel osmium(IV) iminato complexes as a second product. Moreover, oxidation of tri-substituted phenol such as 2,4,6-tBu3C6H2OH results in C–C bond cleavage of the substrate.
Results and discussion
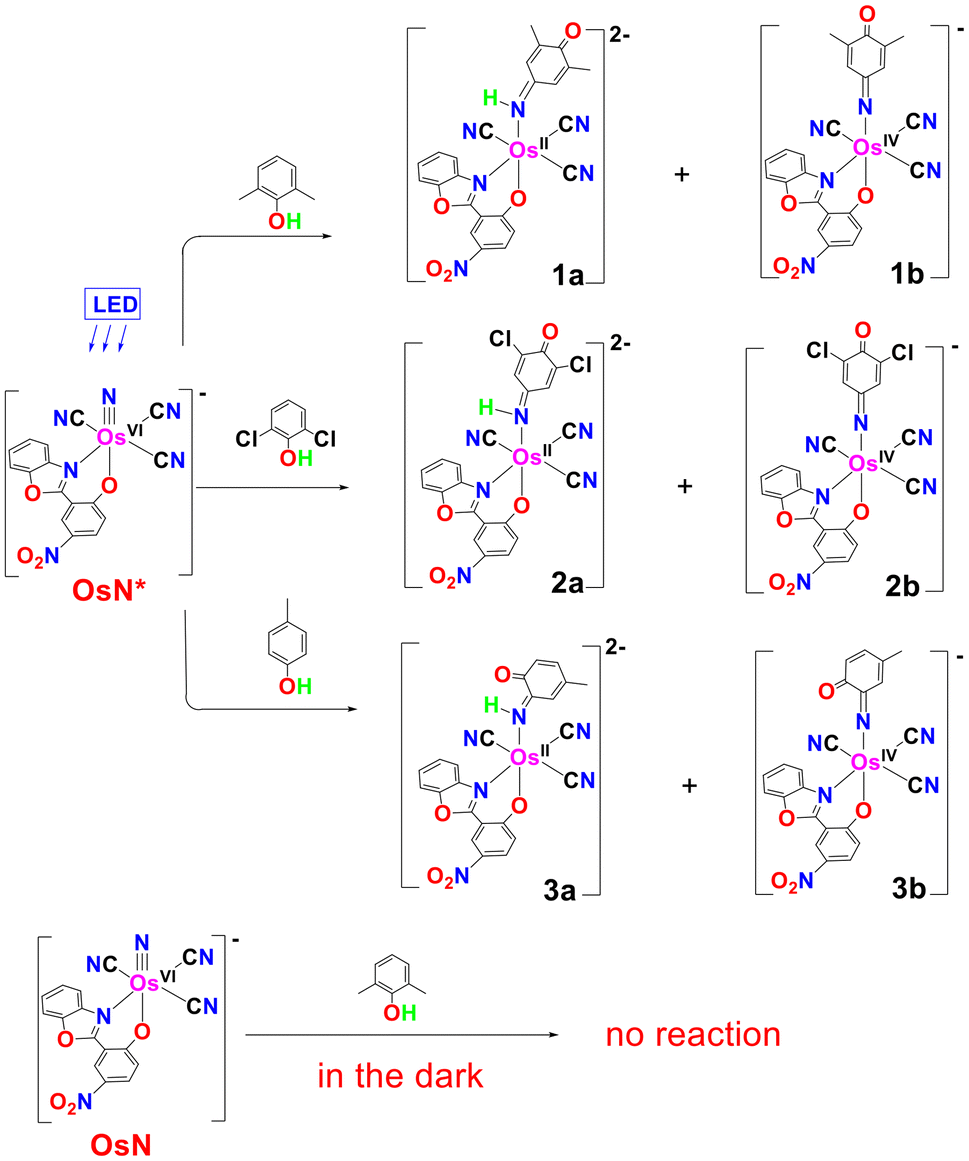
Upon irradiation with blue LED (λ > 460 nm) for 24 h, the light-yellow CH2Cl2 solution of OsN and 10 equiv. of 2,4,6-Me3C6H2OH rapidly turned dark red (Fig. 2). A mixture of the osmium(II) p-benzoquinone monoimine complex [OsII(L)(CN)3(NHMe2Ph(−2H)O)]2− (1a) and osmium(IV) p-benzoquinone monoiminato complex [OsIV(L)(CN)3(NMe2Ph(−2H)O)]− (1b), were isolated as PPh4+ salts with 20% and 26% yields, respectively. Similarly, the photoreaction of OsN with 2,6-Cl2C6H3OH afforded a mixture of (PPh4)2[OsII(L)(CN)3(NHCl2Ph(−2H)O)] [(PPh4)22a] and (PPh4)[OsIV(L)(CN)3(NCl2Ph(−2H)O)] [(PPh4)2b] with 32% and 25% yields, respectively. In addition, (PPh4)[OsII(L)(CN)3(NH3)] (OsNH3)28 was isolated with ∼10% yield in reactions with these phenols. Controlled experiments showed that no reaction between OsN and phenols was observed in the dark.
 | ||
| Fig. 2 Reactions of OsN with various phenols in the excited state and ground state. | ||
On the other hand, in the photoreaction with 4-methylphenol, OsN undergoes electrophilic attack at the ortho position to afford an [PPh4]2[OsII(L)(CN)3(o-NHMePh(−2H)O)] [(PPh4)23a] and [PPh4][OsIV(L)(CN)3(o-NMePh(−2H)O)] [(PPh4)3b], with yields of 37% and 32%, respectively.
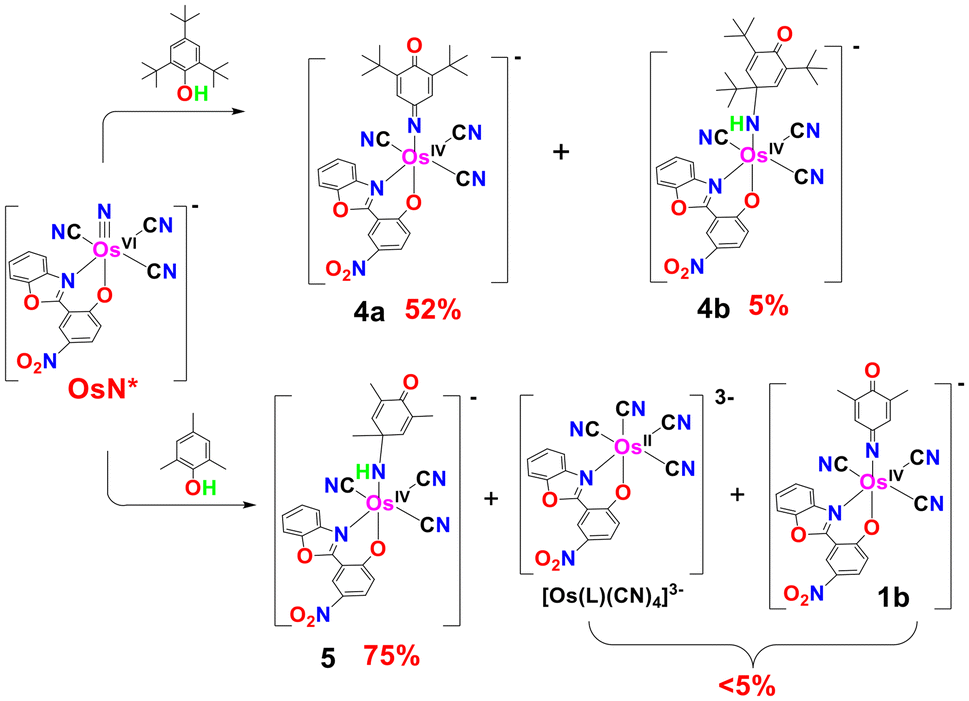
The photoreactions of OsN with 2,4,6-tri-tert-butylphenol and 2,4,6-tri-methylphenol were also investigated (Fig. 3); for these substrates with three alkyl substituents, direct electrophilic attack on the aromatic ring by OsN* may be inhibited. Reaction of OsN* with 2,4,6-tBu3C6H2OH afforded [OsIV(L)(CN)3(p-NtBu2Ph(−2H)O)]− (4a), isolated as PPh4+ salt with ∼52% yield. The structure of 4a (see characterization below) reveals formal C–C bond cleavage of 2,4,6-tBu3C6H2OH; GC/MS and GC show that the 2-methylpropene was formed with ∼50% yield. 1H NMR spectrum of (PPh4)4a shows that the remaining two tBu groups are symmetry-related, consistent with the para attack of 2,4,6-tBu3C6H2OH by OsN*. ESI/MS (−ve mode) of the photoreaction solution of OsN and 2,4,6-tBu3C6H2OH shows a predominant peak at m/z 743 (Fig. 4), attributed to [OsIV(L)(CN)3(p-NtBu2Ph(−2H)O)]− (4a). There is also a very minor peak at m/z 801, (estimated to be 5%), which is tentatively assigned to [OsIV(L)(CN)3(NH-tBu3Ph(−2H)O)]− (4b), arising from an electrophilic attack of OsN at the para position of the phenol.
 | ||
| Fig. 3 Photoreaction of OsN with 2,4,6-tBu3C6H2OH and 2,4,6-Me3 C6H2OH. | ||
 | ||
| Fig. 4 ESI/MS of photoreaction of OsN with 10 equiv. 2,4,6-tBu3C6H2OH for 4 h showing a predominant peak at m/z 743 and a small peak at m/z 801. | ||
In the case of 2,4,6-Me3C6H2OH with less bulky methyl substituents, electrophilic ring attack by OsN* occurred predominantly at the para position, and the amido complex, [OsIV(L)(CN)3{NH-(Me3Ph(−2H)O)}]− (5), was isolated as PPh4+ salt with ∼75% yield. The ESI/MS (−ve mode) of the product solution shows a predominant peak at m/z = 675 due to complex 5. In addition, there are two minor peaks at m/z 659.1 and 275.6, which are tentatively assigned to 1b and [OsII(L)(CN)4]3−, respectively. These two species should arise from C–C bond cleavage of 2,4,6-Me3C6H2OH; however, the total yields of these two products are estimated to be <5%.
All products have been characterized by IR, UV/vis, CV, ESI/MS, and 1H NMR (Fig. S1–S12†). All the Os(II) and Os(IV) compounds are diamagnetic, as evidenced by the sharp resonances found in the normal range in their 1H NMR spectra. The diamagnetism of these compounds is consistent with the low spin d6 and d4 electronic configurations for Os(II) and Os(IV), respectively. The IR spectra of (PPh4)21a and (PPh4)22a show v(CN) stretches at 2096, 2070 cm−1 and 2108, 2082 cm−1, and v(CO) stretches at 1609 cm−1 and 1610 cm−1, respectively. The IR spectra of (PPh4)1b and (PPh4)2b show v(CN) stretches at 2145, 2134 cm−1 and 2153, 2142 cm−1, and v(CO) stretches at 1629 cm−1 and 1639 cm−1, respectively which are at higher wavenumbers as compared with (PPh4)21a and (PPh4)22a. Similar v(CN) and v(CO) stretches are also found in (PPh4)23a and (PPh4)3b.
As shown in Fig. S6,† the UV/vis spectra of these compounds show ligand-centered π–π* transitions in the UV regions. In the osmium(II) products (PPh4)21a, (PPh4)22a, and (PPh4)23a, there are strong absorption bands at 500–650 nm with molar extinction coefficients (ε) of the order of 104 M−1 cm−1, which are assigned to Os(II) to p-benzoquinone monoimine charge transfer (MLCT) transitions. Notably, there is an intense absorption band with ε > 6 × 104 M−1 cm−1 in (PPh4)22a. In the osmium(IV) products (PPh4)1b, (PPh4)2b and (PPh4)3b, the broad absorption bands at ∼450 nm are probably due to LMCT transitions.
The cyclic voltammograms (CVs) of the osmium complexes (PPh4)21a, (PPh4)22a, and (PPh4)23a in CH3CN exhibit a reversible/quasi-reversible OsIII/II couples at −0.67 V, −0.38 V and −0.78 V vs. Fc+/0, respectively (Fig. 5). There is also an irreversible wave at Epc = −1.46 V, −1.35 V and −1.72 V, respectively which are tentatively assigned to the reduction of the benzoquinone imine ligands. The oxidation waves at E1/2 = 1.25 V, Epc = 1.46 V and 1.23 V, respectively, are assigned to OsIV/III. The CVs of the osmium(IV) products 1b, 2b and 3b exhibit irreversible OsIV/III waves at Ep = −0.65 V, −0.39 V and −0.66 V, respectively (Fig. 6); while the waves at Ep = −1.61 V, −1.35 V and −1.49 V, respectively are tentatively assigned to the reduction of benzoquinone imine ligands.
 | ||
| Fig. 5 CVs for 1a, 2a, and 3a in MeCN solutions containing 0.1 M [nBu4N](PF6) with a scan rate = 0.1 V s−1. | ||
 | ||
| Fig. 6 CVs for 1b, 2b, and 3b in MeCN solutions containing 0.1 M [nBu4N](PF6) with a scan rate = 0.1 V s−1. | ||
The molecular structures of (PPh4)1b, (PPh4)22a, (PPh4)3b, and (PPh4)4a have been determined by X-ray crystallography (Fig. 7). Selected bond parameters are summarized in Table 1. The mer-configuration of OsN is retained in these complexes. There are two PPh4+ in 2a and only one PPh4+ in 1b and 3b. The Os–N4 bond length in 2a is 1.927(3) Å, which is significantly shorter than related Os–N bond lengths in [OsIII{N(H)C(NH2)}(L1)(CN)3]− and [OsIII{N(H)CN}(L1)(CN)3]2− (HL1 = 2-(2-hydroxyphenyl)benzoxazole),32,33 indicating the presence of strong π back-bonding interaction between OsII and the benzoquinone monoimine ligand. On the other hand, the Os–N4 bond lengths are 1.781(4) and 1.768(4) Å, respectively for 1b and 3b, which are much shorter than that in 2a, indicating Os–N double bond character in these Os(IV) complexes. This is also evidenced by the more linear Os1–N4–C4 bond angles of 167.2(4)° and 167.9(5)° than that in 2a (136.6(3)°). The bond parameters of 3b and 4a are essentially identical to those in 1b, as the metal centers have the same oxidation state.
 | ||
| Fig. 7 The structures of (a) 1b, (b) 2a, (c) 3b and (d) 4a. | ||
| 1b | 2a | 3b | 4a | |
|---|---|---|---|---|
| Os1–C1 | 2.074 (5) | 2.053 (4) | 2.082 (5) | 2.081(5) |
| Os1–C2 | 2.077 (5) | 2.070 (4) | 2.081 (5) | 2.029(4) |
| Os1–C3 | 2.025 (5) | 2.009 (4) | 2.034 (6) | 2.066(5) |
| Os1–N4 | 1.781 (4) | 1.927 (3) | 1.768 (4) | 1.779(4) |
| Os1–N5 | 2.117 (4) | 2.129 (3) | 2.118 (4) | 2.115(3) |
| Os1–O1 | 2.010 (3) | 2.066 (3) | 2.028 (3) | — |
| Os1–O2 | — | — | — | 2.022(3) |
| Os1–N4–C4 | 167.2 (4) | 136.6 (3) | 167.9 (5) | 163.8(3) |
Substituent effects
The photoreactions of OsN with various 4-substituted phenols (4-X-C6H4OH; X = MeO, Me, H, F, Cl) were also investigated by UV/vis spectroscopy (Fig. S10†). The initial rates (Rx) of these reactions were obtained at 450 nm (Fig. 8a). Rx was found to increase with increasing electron donating properties of the substituents: MeO > Me > H > F > Cl. A linear correlation was obtained in the Hammett plot of log(Rx/RH) versus the substituent constant (σ), with a ρ value of −0.54 ± 0.01 (Fig. 8b), indicating that the phenol center is more positive in the transition state. log(Rx) also correlates well with the oxidation potentials (Eox) of these phenols with a slope of −0.80 (Fig. 8c).34 The results indicate that the reaction may proceed via an initial rate-limiting 1e− transfer (ET) from phenols to OsN*. No clear correlation between log(Rx) and pKa of phenols, suggesting that proton transfer (PT) is not involved in the rate-limiting step. There is also no clear relationship between log(Rx) and the O–H bond dissociation energies (BDEs) of the phenols (Fig. 8d),35 which does not support a hydrogen atom transfer (HAT) mechanism for the oxidation of phenols by OsN*. | ||
| Fig. 8 (a) UV/vis spectral changes for the photoreaction of OsN (2.5 × 10−5 M) with phenol (1.8 × 10−3 M) in C2H4Cl2. Inset shows the time-trace absorbance at 450 nm. (b) Hammett plot for the photoreaction of OsN with 4-substituted phenols in C2H4Cl2. Slope = −0.54. Intercept = −0.00217. (c) Plot of log(Rx) vs. Eox. (d) Plot of log(Rx) vs. BDE. | ||
Kinetic isotope effects (KIE)
The KIE for the reaction of OsN* with phenol was determined by ESI/MS, using an equimolar mixture of phenol (C6H5OH) and d6-phenol (C6D5OD) as substrate. As shown in Fig. 9a, the KIE value was estimated to be ∼(1 ± 0.05) from the ratios of the most intense peaks for two Os(IV) benzoquinone monoiminato products, (m/z = 631 and 635 for [OsIV(L)(CN)3(NC6H4O)]− and [OsIV(L)(CN)3(NC6D4O)]− respectively), assuming that the spraying and ionization efficiencies of the two ions are similar. The isotopic distribution pattern obtained is also in agreement with the simulated one. The KIE is also determined by UV/vis spectroscopy. As shown in Fig. S11,† the UV/vis spectral changes were obtained from the photoreaction of OsN with phenol (C6H5OH) and d6-phenol (C6D5OD) under the same conditions. The reactions were followed by the change in absorbance at 367 nm, which gives a KIE of RH/RD = 0.97 ± 0.01 (Fig. 9b). Based on the above results, the photoreaction of OsN with phenol exhibits negligible KIE, indicating that C–H bond cleavage is not involved in the rate-limiting step, in line with the conclusion obtained from investigation of substituent effects.
 | ||
Fig. 9 (a) ESI/MS for the photoreaction of OsN with equimolar of phenol and d6-phenol showing a predominant peak around m/z 631. Inserts show the expanded isotopic distribution of peak m/z 631, which agrees with the simulated isotopic distribution of m/z 631 and 635 with a mole ratio of 0.505![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.495 (KIE ∼1). (b) Time trace for absorbance at 367 nm of OsN (2.5 × 10−5 M) with 100 equiv. of phenol (red line) and d6-phenol (blue line) in C2H4Cl2. The linear fitting gives initial rates of RH = (−2.13 ± 0.06) × 10−3, r2 = 0.98 and RD = (−2.40 ± 0.10) × 10−3, r2 = 0.99; RH:RD = 0.97. :0.495 (KIE ∼1). (b) Time trace for absorbance at 367 nm of OsN (2.5 × 10−5 M) with 100 equiv. of phenol (red line) and d6-phenol (blue line) in C2H4Cl2. The linear fitting gives initial rates of RH = (−2.13 ± 0.06) × 10−3, r2 = 0.98 and RD = (−2.40 ± 0.10) × 10−3, r2 = 0.99; RH:RD = 0.97. | ||
Proposed mechanism
Based on the above experiments, the proposed mechanism for the photoreaction of OsN with phenols is illustrated in Fig. 10 (using 2,6-Me2C6H3OH as an example). The initial step is rate-limiting 1e− oxidation of 2,6-Me2C6H3OH (ET), followed by rapid proton transfer (PT) to give the OsVNH and phenoxy radical, which is supported by a linear correlation between log(Rx) and E of the phenols with a slope of −0.80. The proposed mechanism is also supported by the linear Hammett correlation with ρ value of −0.54 and a negligible KIE effect (k(C6H5OH)/k(C6D5OD) ∼1.03). Moreover, the initial rate (R) for phenolate is 60 times faster than that of phenol, further validating the initial ET mechanism (Fig. S12†). This is followed by tautomerism and N-rebound to give an OsIV amido species. An internal 2e− redox results in the OsII hydroquinone monoimine product. The other product, Os(IV) hydroquinone monoiminato complex, was formed by further 2e− oxidation by OsN* (2/3OsN* + 1a → 1b + 2/3OsNH3). This last step is supported by the isolation of OsNH3 in 10% yield. | ||
| Fig. 10 The proposed reaction mechanism for the reaction of OsN* with 2,6-Me2C6H3OH. | ||
For the photoreaction of OsN and 2,4,6-tBu3C6H2OH, the first four steps are similar (Fig. 11), i.e. 1e− oxidation of 2,4,6-tBu3C6H2OH (ET), followed by rapid proton transfer (PT) to give the OsVNH and phenoxy radical; followed by tautomerism and N-rebound to give an OsIV amido species. Further 2e− oxidation of this OsIV amido species by OsN* leads to C–C bond cleavage with the formation of 4a, 2-methylpropene, and OsNH3.
 | ||
| Fig. 11 The proposed mechanism for the C–C bond cleavage of 2,4,6-tBu3C6H2OH by OsN*. | ||
Conclusion
In conclusion, we have demonstrated novel reactivity of the excited state of an osmium(VI) nitrido complex towards phenols. The photoreactions of OsN with the parent phenol, as well as mono- and di-substituted phenols afforded Os(II) p-benzoquinone monoimine and osmium(IV) p-benzoquinone monoiminato complexes. On the other hand, oxidation of bulky tri-substituted phenol such as 2,4,6-tBu3C6H2OH resulted in C–C bond cleavage of the substrate. Mechanistic studies indicate that the photoreactions proceed via an initial 1e− oxidation followed by a rapid proton transfer (ET-PT) to generate phenoxy radicals, this is followed by a N-rebound step to give the osmium products. We believe that our work is a significant contribution to MN excited state as well as phenol oxidation chemistry.
Experimental
(PPh4)2[OsII(L)(CN)3(NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) Me2PhOH(−2H))] [(PPh4)21a] and (PPh4)[OsIV(L)(CN)3(NMe2PhOH(−2H))] [(PPh4)1b]
Me2PhOH(−2H))] [(PPh4)21a] and (PPh4)[OsIV(L)(CN)3(NMe2PhOH(−2H))] [(PPh4)1b]
10 tubes each containing OsN (5 mg, 5.7 μmol) and 2,6-Me2C6H3OH (73 mg, 0.6 mmol) in CH2Cl2 under argon were irradiated with blue LED light for 24 h, whereby the light-yellow solutions turned dark red. The solutions were combined, and the solvent was removed under reduced pressure. The residue was washed with diethyl ether (100 ml) to remove the unreacted 2,6-Me2C6H3OH. The residue was dissolved in a minimum amount of CH2Cl2 and then loaded onto a silica gel column. The first yellow band (PPh4)[OsIV(L)(CN)3(NMe2Ph(−2H)O)] [(PPh4)1b] was eluted by CH2Cl2/acetone (v:v, 4:1). Yellow needle crystals were obtained from slow diffusion of diethyl ether into a MeCN solution of (PPh4)1b. The second blue band was eluted by CH2Cl2/acetone/MeOH (v:v:v, 8:2:1). The solvent was removed under reduced pressure. The solid obtained was dissolved in H2O (10 ml) and excess PPh4Cl (30 mg) was added to give the blue precipitate. The crude product was further purified by slow diffusion of diethyl ether into a CH2Cl2 solution of (PPh4)21a. Yield for (PPh4)21a: 15 mg, 20% (based on the OsN consumed). Selected IR (KBr disc, cm−1): v(N–H) 3248; v(CN) 2096 and 2070; v(CO) 1645; v(CN) 1609; v(NO) 1303. 1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H, Ar–H), 7.92–7.78 (m, 8H, Ar–H), 7.77–7.64 (m, 18H, Ar–H), 7.62–7.48 (m, 18H, Ar–H), 7.38 (d, J = 8.0 Hz, 1H, Ar–H), 7.29 (m, 1H, Ar–H), 7.19 (t, J = 8.0 Hz,1H, Ar–H), 6.67 (s, 1H, Ar–H), 1.69 (s, 6H). ESI/MS (−ve mode) in MeOH: at m/z 329.7 ([M]2−); Anal. Calcd for C72H56N6O5P2Os: C, 64.66; H, 4.22; N, 6.28. Found: C, 64.80; H, 4.25; N, 6.32%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 269 (22472), 276 (21630), 287 (16020), 344 (14050), 377sh (11870), 432 (11270), 541 (17340), 586 (17670). Yield for (PPh4)1b: 15 mg, 26% (based on the OsN consumed). Selected IR (KBr disc, cm−1): v(CN) 2145 and 2135; v(CO) 1630; v(CN) 1611. 1H NMR (400 MHz, CDCl3): δ 9.04 (s, 1H, Ar–H), 8.19 (s, 2H, Ar–H), 8.07 (d, J = 9.5 Hz, 1H, Ar–H), 7.96–7.88 (m, 4H, Ar–H), 7.84–7.74 (m, 9H, Ar–H), 7.72 (d, J = 7.4 Hz, 1H, Ar–H), 7.69–7.61 (m, 8H, Ar–H), 7.53 (t, J = 8.3 Hz, 2H, Ar–H), 6.92 (d, J = 9.2 Hz, 1H, Ar–H), 3.15 (s, 6H, –CH3). ESI-MS (−ve mode) in MeOH: m/z 659 (M−); Anal. Calcd for C48H35N6O5OsP: C, 57.82; H, 3.54; N, 8.43. Found: C, 57.93; H, 3.60; N, 8.31%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 269 (23710), 277 (25070), 288 (23480), 330 (22610), 432 (33300).
(PPh4)2[OsII(L)(CN)3(NHCl2PhOH(−2H))] [(PPh4)22a] and (PPh4)[OsIV(L)(CN)3(NCl2PhOH(−2H))] [(PPh4)2b]
The synthesis and isolation of (PPh4)22a and (PPh4)2b are similar to that of (PPh4)21a and (PPh4)1b except that 2,6-Cl2C6H3OH was used instead of 2,6-dimethylphenol. Yield for (PPh4)22a: (25 mg, 32%, based on the OsN consumed). Selected IR (KBr disc, cm−1): v(N–H) 3235; v(CN) 2108 and 2082; v(CO) 1638; v(CN) 1609; v(NO) 1307; 1H NMR (400 MHz, CDCl3): δ 8.96 (d, J = 2.9 Hz, 1H), 7.90–7.82 (m, 8H), 7.80 (d, J = 2.9 Hz, 1H), 7.77–7.68 (m, 16 H), 7.65–7.56 (m, 16H), 7.55–7.48 (m, 2H), 7.37–7.29 (m, 3H), 7.23 (s, 1H); 6.78 (d, J = 9.4 Hz, 1H). ESI-MS (−ve mode) in MeOH: m/z 350.2 [M]2−. Anal. Calcd for C70H50Cl2N6O5OsP2: C, 61.00; H, 3.66; N, 6.10. Found: C, 61.10; H, 3.71; N, 6.18%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 262sh (18090), 269 (19810), 276 (18740), 285sh (12760), 338 (12110), 363sh (9360), 417sh (7070), 593 (60850). Yield for (PPh4)2b: 15 mg, 25% (based on the OsN consumed). Selected IR (KBr disc, cm−1): v(CN) 2153 and 2141; v(CO) 1639; v(CN) 1614. 1H NMR (400 MHz, CDCl3): δ 9.08 (s, 1H, Ar–H), 8.50 (s, 2H, Ar–H), 8.21 (d, J = 8.9 Hz, 1H, Ar–H), 7.97–7.89 (m, 4H, Ar–H), 7.84–7.75 (m, 9H, Ar–H), 7.72–7.58 (m, 11H, Ar–H), 7.13 (d, J = 9.2 Hz, 1H, Ar–H). ESI-MS (−ve mode) in MeOH: m/z 699 (M−); Anal. Calcd for C46H29Cl2N6O5OsP: C, 53.23; H, 2.82; N, 8.10. Found: C, 53.29; H, 2.76; N, 8.21%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 270 (40050), 277 (44140), 288sh (42330), 331sh (37880), 447 (60490).
(PPh4)2[OsII(L)(CN)3(NHMePhOH(−2H))] [(PPh4)23a] and (PPh4)[OsIV(L)(CN)3(NMePhOH(−2H))] [(PPh4)3b]
The synthesis and isolation of (PPh4)23a and (PPh4)3b are similar to that of (PPh4)21a and (PPh4)1b except that 4-MeC6H4OH was used instead of 2,6-Me2C6H3OH. Yield for (PPh4)23a, 28 mg, 37%. Selected IR (KBr disc, cm−1) for (PPh4)23a: v(N–H) 3253; v(CN) 2125 and 2086; v(CO) 1645; v(CN) 1612. 1H NMR (400 MHz, CDCl3): δ 9.03 (d, J = 2.9 Hz, 1H, Ar–H), 8.01 (s, 1H), 7.99 (s, 1H), 7.80–7.86 (m, 8H), 7.77–7.68 (m, 16 H), 7.65–7.56 (m, 16H), 7.18–7.10 (t, J = 8.0 Hz, 1H), 7.08 (d, J = 10.2 Hz, 1H), 6.72 (m, 2H), 6.38 (m, 2H), 5.32 (s, 1H) 1.27 (s, 3H), 3.50 (s, 3H, Me). ESI-MS (−ve mode) in MeOH: m/z 323.3 [M]2−. Anal. Calcd for C71H54N6O5OsP2: C, 64.44; H, 4.11; N, 6.35. Found: C, 64.10; H, 4.21; N, 6.18%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 262sh (18090), 269 (19810), 276 (18740), 285sh (12760), 338 (12110), 363sh (9360), 417sh (7070), 593 (60850). Yield for (PPh4)3b, 18 mg, 32%. Selected IR (KBr disc, cm−1): v(CN) 2139 and 2130; v(CO) 1649; v(CN) 1614. 1H NMR (400 MHz, CDCl3): δ 9.07 (d, J = 2.8 Hz, 1H), 8.65 (dd, J = 6.3, 2.8 Hz, 1H) 8.13 (dd, J = 9.3, 2.9 Hz, 1H), 7.90–7.82 (m, 4H), 7.77–7.84 (m, 8H), 7.65–7.73 (m, 10H), 7.60–7.53 (m, 2H), 7.21 (dd, J = 9.8, 2.2 Hz, 1H), 6.99 (d, J = 9.3 Hz, 1H); 5.77 (d, J = 9.8 Hz, 1H) 3.58 (s, 3H). ESI-MS (−ve mode) in MeOH: m/z 645 [M]−. Anal. Calcd for C47H33N6O5OsP: C, 57.43; H, 3.38; N, 8.55. Found: C, 57.10; H, 3.71; N, 8.18%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 262sh (18090), 269 (19810), 276 (18740), 285sh (12760), 338 (12110), 363sh (9360), 417sh (7070), 593 (60850).
(PPh4)2[OsIV(L)(CN)3(NtBu2Ph(−2H)O)] [(PPh4)4a] and (PPh4)[OsIV(L)(CN)3(NH-tBu3PhOH(−2H))] [(PPh4)4b]
The synthetic route of (PPh4)4a and (PPh4)4b is similar to that of (PPh4)21a and (PPh4)1b, except that the 2,4,6-tBu3C6H2OH is used instead of 2,6-Me2C6H3OH. The first yellow band (PPh4)4a was eluted by CH2Cl2/acetone (v:v, 10:1). Yellow needle crystals were obtained from the slow diffusion of diethyl ether into an acetone solution of (PPh4)4a. The solvent was removed under reduced pressure.
Yield for (PPh4)4a: 32 mg, 52%. Selected IR (KBr disc, cm−1): v(CN) 2142 and 2128, v(CO) 1638; v(CN) 1615; v(NO) 1307. 1H NMR (400 MHz, CDCl3): δ 9.03 (d, J = 2.8 Hz, 1H, Ar–H), 8.12 (s, 2H, Ph–H), 8.05 (dd, J = 9.3, 2.9 Hz, 1H, Ar–H), 7.92 (dd, J = 8.0, 6.0 Hz, 4H, PPh4-H), 7.83–7.76 (m, 9H, Ar–H and PPh4-H), 7.73 (d, J = 7.2 Hz, 1H, Ar–H), 7.68–7.62 (m, 8H, PPh4-H), 7.54 (dtd, J = 13.9, 7.8, 6.2 Hz, 2H, Ar–H), 6.89 (d, J = 9.4 Hz, 1H, Ar–H), 1.28 (s, 18H, CH3). ESI-MS (−ve mode) in MeOH: m/z 743 [M]2−. Anal. Calcd for C54H47N6O5OsP: C, 59.99; H, 4.38; N, 7.77. Found: C, 60.10; H, 4.31; N, 7.78%. UV/Vis (CH2Cl2): λmax [nm] (ε [mol−1 dm3 cm−1]): 230 (27380), 277 (15620), 288 (14640), 330 (13030), 428 (20930).
(PPh4)[OsIV(L)(CN)3(NH-Me3PhOH(–H))] [(PPh4)5]
The synthetic route of (PPh4)5 is similar to that of (PPh4)21a and (PPh4)1b, except that the 2,4,6-Me3C6H2OH is used instead of 2,6-Me2C6H3OH. (PPh4)5 was purified by silica gel CH2Cl2/acetone (5:3) as the eluent. Yield: 43 mg, 75% (based on the OsN consumed). UV/Vis (CH2Cl2) for (PPh4)5: λmax [nm] (ε/M−1 cm−1): 233 (42430), 270 (18030), 277 (19430), 293 (20360), 353 (20460), 457 (6420). Selected IR (KBr disc, cm−1) for 5: v(CN) 2135 and 2127; v(N–H) 3247; v(CN) 1610. ESI-MS (−ve mode) in MeOH: m/z 675 [M]−. Anal. Calcd for C49H39N6O5OsP: C, 58.09; H, 3.88; N, 8.30. Found: C, 58.10; H, 3.81; N, 8.28%. 1H NMR (400 MHz, CDCl3): δ 8.93 (d, J = 2.8 Hz, 1H, Ar–H), 7.96 (d, J = 6.6 Hz, 1H, Ar–H), 7.89 (t, J = 7.2 Hz, 4H), 7.75 (td, J = 7.6, 3.7 Hz, 8H), 7.65–7.57 (m, 10H, Ar–H and PPh4-H), 7.43 (t, J = 7.1 Hz, 1H, Ar–H), 7.20 (t, J = 8.4 Hz, 1H, Ar–H), 6.78 (s, 2H, Ph–H), 6.57 (d, J = 9.4 Hz, 1H, Ar–H), 2.23 (s, 3H, CH3), 1.27 (s, 6H, CH3).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The authors will supply the relevant data in response to reasonable requests.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22371092), the Excellent Discipline Cultivation Project by JHUN (2023XKZ038), the Project of Hebei Key Laboratory of Heterocyclic Compounds (no. KF202402), and the Graduate Scientific Research Foundation of Jianghan University (KYCXJJ202426).References
- (a) J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, Nitridomanganese(V) Complexes: Design, Preparation, and Use as Nitrogen Atom-Transfer Reagents, Acc. Chem. Res., 1997, 30, 364–372 CrossRef CAS; (b) J. T. Groves and T. Takahashi, Activation and transfer of nitrogen from a nitridomanganese(V) porphyrin complex. Aza analog of epoxidation, J. Am. Chem. Soc., 1983, 105, 2073–2074 CrossRef CAS; (c) R. A. Eikey and M. M. Abu-Omar, Nitrido and imido transition metal complexes of Groups 6–8, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS.
- (a) J. F. Berry, Terminal Nitrido and Imido Complexes of the Late Transition Metals, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS; (b) J. M. Smith, Reactive Transition Metal Nitride Complexes, Prog. Inorg. Chem., 2014, 58, 417–470 CAS; (c) M. N. Cosio and D. C. Powers, Prospects and challenges for nitrogen-atom transfer catalysis, Nat. Rev. Chem., 2023, 7, 424–438 CrossRef CAS PubMed; (d) S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Nitrogen Fixation via Splitting into Nitrido Complexes, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- (a) T. J. Meyer and M. H. V. Huynh, The Remarkable Reactivity of High Oxidation State Ruthenium and Osmium Polypyridyl Complexes, Inorg. Chem., 2003, 42, 8140–8160 CrossRef CAS PubMed; (b) T. J. Crevier and J. M. Mayer, Direct Attack of Phenyl Anion at an Electrophilic Osmium−Nitrido Ligand, J. Am. Chem. Soc., 1998, 120, 5595–5596 CrossRef CAS; (c) S. N. Brown, Insertion of a Metal Nitride into Carbon−Carbon Double Bonds, J. Am. Chem. Soc., 1999, 121, 9752–9753 CrossRef CAS.
- (a) W.-L. Man, W. W. Y. Lam and T.-C. Lau, Reactivity of Nitrido Complexes of Ruthenium(VI), Osmium(VI), and Manganese(V) Bearing Schiff Base and Simple Anionic Ligands, Acc. Chem. Res., 2014, 47, 427–439 CrossRef CAS PubMed; (b) W.-L. Man, W. W. Y. Lam, S.-M. Yiu, T.-C. Lau and S.-M. Peng, Direct Aziridination of Alkenes by a Cationic (Salen)ruthenium(VI) Nitrido Complex, J. Am. Chem. Soc., 2004, 126, 15336–15337 CrossRef CAS PubMed; (c) W.-L. Man, W. W. Y. Lam, H.-K. Kwong, S.-M. Yiu and T.-C. Lau, Ligand-Accelerated Activation of Strong C-H Bonds of Alkanes by a (Salen)ruthenium(VI)–Nitrido Complex, Angew. Chem., Int. Ed., 2012, 51, 9101–9104 CrossRef CAS PubMed.
- (a) J. J. Scepaniak, C. S. Vogel, M. M. Khusniyarov, F. W. Heinemann, K. Meyer and J. M. Smith, Synthesis, Structure, and Reactivity of an Iron(V) Nitride, Science, 2011, 331, 1049–1052 CrossRef CAS PubMed; (b) S. B. Muñoz III, W.-T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Styrene Aziridination by Iron(IV) Nitrides, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 CrossRef PubMed.
- (a) H. Shi, H. K. Lee, Y. Pan, K.-C. Lau, S.-M. Yiu, W. W. Y. Lam, W.-L. Man and T.-C. Lau, Structure and reactivity of a manganese(VI) nitrido complex bearing a tetraamido macrocyclic ligand, J. Am. Chem. Soc., 2021, 143, 15863–15872 CrossRef CAS PubMed; (b) H. Shi, R. Liang, D. L. Phillips, H. K. Lee, W.-L. Man, K.-C. Lau, S.-M. Yiu and T.-C. Lau, Structure and Reactivity of One- and Two-Electron Oxidized Manganese(V) Nitrido Complexes Bearing a Bulky Corrole Ligand, J. Am. Chem. Soc., 2022, 144, 7588–7593 CrossRef CAS PubMed.
- (a) G. W. Burton and K. U. Ingold, Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function, Acc. Chem. Res., 1986, 19, 194 CrossRef CAS; (b) L. Doyle, A. Magherusan, S. Xu, K. Murphy, E. R. Farquhar, F. Molton, C. Duboc, L. Que Jr. and A. R. McDonald, Class Ib Ribonucleotide Reductases: Activation of a Peroxido-MnIIMnIII to Generate a Reactive Oxo-MnIIIMnIV Oxidant, Inorg. Chem., 2024, 63, 2194–2203 CrossRef CAS PubMed; (c) K. J. Fisher, M. L. Feuer, H. M. C. Lant, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, Concerted proton-electron transfer oxidation of phenols and hydrocarbons by a high-valent nickel complex, Chem. Sci., 2020, 11, 1683–1690 RSC.
- C. Tommos and G. T. Babcock, Proton and hydrogen currents in photosynthetic water oxidation, Biochim. Biophys. Acta, Bioenerg., 2000, 1458, 199 CrossRef CAS PubMed.
- (a) S. Kundu, E. Miceli and E. R. Farquhar, Mechanism of phenol oxidation by heterodinuclear Ni Cu bis (μ-oxo) complexes involving nucleophilic oxo groups, Dalton Trans., 2014, 43, 4264–4267 RSC; (b) D. Dhar, G. M. Yee, T. F. Markle, J. M. Mayer and W. B. Tolman, Reactivity of the copper(III)-hydroxide unit with phenols, Chem. Sci., 2017, 8, 1075–1085 RSC; (c) L. Yang, R. Ito, H. Sugimoto, Y. Morimoto and S. Itoh, Oxidation mechanism of phenols by copper(II)–halide complexes, Chem. Commun., 2024, 60, 7586–7589 RSC; (d) J. Y. Lee, R. L. Peterson, K. Ohkubo, I. Garcia-Bosch, R. A. Himes, J. Woertink, C. D. Moore, E. I. Solomon, S. Fukuzumi and K. D. Karlin, Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex, J. Am. Chem. Soc., 2014, 136, 9925–9937 CrossRef CAS PubMed.
- (a) T. J. Meyer, M. H. V. Huynh and H. H. Thorp, The possible role of proton–coupled electron transfer (PCET) in water oxidation by photosystem II, Angew. Chem., Int. Ed., 2007, 46, 5284 CrossRef CAS PubMed; (b) P. Mondal and A. R. McDonald, Phenol Oxidation by a Nickel(III)–Fluoride Complex: Exploring the Influence of the Proton Accepting Ligand in PCET Oxidation, Chem. – Eur. J., 2020, 26, 10083–10089 CrossRef CAS PubMed.
- J. A. Stubbe, D. G. Nocera, C. S. Yee and M. C. Y. Chang, Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer?, Chem. Rev., 2003, 103, 2167 CrossRef CAS PubMed.
- R. D. Webster, New insights into the oxidative electrochemistry of vitamin E, Acc. Chem. Res., 2007, 40, 251 CrossRef CAS PubMed.
- M. H. V. Huynh and T. J. Meyer, Proton-coupled electron transfer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed.
- T. F. Markle, I. J. Rhile, A. G. DiPasquale and J. M. Mayer, Probing concerted proton–electron transfer in phenol–imidazoles, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8185 CrossRef CAS PubMed.
- N. Song and D. M. Stanbury, Oxidation of Phenol by Tris (1, 10-phenanthroline) osmium(III), Inorg. Chem., 2012, 51, 4909 CrossRef CAS PubMed.
- J. Bonin, C. Costentin, C. Louault, M. Robert, M. Routier and J.-M. Savéant, Intrinsic reactivity and driving force dependence in concerted proton–electron transfers to water illustrated by phenol oxidation, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3367 CrossRef CAS PubMed.
- A. Al-Ajlouni, A. Bakac and J. H. Espenson, Kinetics and mechanism of the oxidation of phenols by the oxochromium(IV) ion, Inorg. Chem., 1993, 32, 5792 CrossRef CAS.
- D. T. Y. Yiu, M. F. W. Lee, W. W. Y. Lam and T. C. Lau, Kinetics and mechanisms of the oxidation of phenols by a trans-dioxoruthenium(VI) complex, Inorg. Chem., 2003, 42, 1225 CrossRef CAS PubMed.
- T. Osako, K. Ohkubo and M. Taki, Oxidation mechanism of phenols by dicopper−dioxygen (Cu2/O2) complexes, J. Am. Chem. Soc., 2003, 125, 11027–11033 CrossRef CAS PubMed.
- D. E. Lansky and D. P. Goldberg, Hydrogen Atom Abstraction by a High-Valent Manganese(V)−Oxo Corrolazine, Inorg. Chem., 2006, 45, 5119–5125 CrossRef CAS PubMed.
- M. L. Neidig, A. Decker, O. W. Choroba, F. Huang, M. Kavana and G. R. Moran, Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12966–12973 CrossRef CAS PubMed.
- A. Gunay and K. H. Theopold, C−H Bond Activations by Metal Oxo Compounds, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- W. K. Seok and T. J. Meyer, Multiple electron oxidation of phenols by an oxo complex of ruthenium(IV), J. Am. Chem. Soc., 1988, 110, 7358 CrossRef CAS.
- W. K. Seok, J. C. Dobson and T. J. Meyer, Mechanisms of oxidation of phenol and cyclohexene by an oxo complex of ruthenium(IV), Inorg. Chem., 1988, 27, 3–5 CrossRef CAS.
- J. H. Xie, W. L. Man, C. Y. Wong, X. Y. Chang, C. M. Che and T. C. Lau, Four-electron oxidation of phenols to p-benzoquinone imines by a (salen) ruthenium(VI) nitrido complex, J. Am. Chem. Soc., 2016, 138, 5817–5820 CrossRef CAS PubMed.
- (a) J. Xiang, X. X. Jin, Q. Q. Su, S. C. Cheng, C. C. Ko, W. L. Man, M. Y. Xue, L. L. Wu, C. M. Che and T. C. Lau, Photochemical nitrogenation of alkanes and arenes by a strongly luminescent osmium(VI) nitrido complex, Commun. Chem., 2019, 2, 40 CrossRef; (b) J. Xiang, H.-T. Shi, W.-L. Man and T.-C. Lau, Design of Highly Electrophilic and Stable Metal Nitrido Complexes, Acc. Chem. Res., 2024, 57, 2700–2716 CrossRef CAS PubMed.
- L. J. Luo, Q.-Q. Su, S. C. Cheng, J. Xiang, W. L. Man, W. M. Shu, M. H. Zeng, S. M. Yiu, C. C. Ko and T. C. Lau, Tunable luminescent properties of tricyanoosmium nitrido complexes bearing a chelating O^N ligand, Inorg. Chem., 2020, 59, 4406–4413 CrossRef CAS PubMed.
- (a) J. Xiang, Y. Pan, L.-L. Liu, L.-X. Wang, H. Yang, S.-C. Cheng, S.-M. Yiu, C.-F. Leung, C.-C. Ko, K.-C. Lau and T.-C. Lau, Visible Light-Induced Oxidation of Alcohols by a Luminescent Osmium(VI) Nitrido Complex: Evidence for the Generation of PhIO+ as a Highly Active Oxidant in the Presence of PhIO, J. Am. Chem. Soc., 2023, 145, 9129–9135 CrossRef CAS PubMed; (b) J. Xiang, M. Peng, Y. Pan, L. J. Luo, S. C. Cheng, X. X. Jin, S. M. Yiu, W. L. Man, C. C. Ko, K. C. Lau and T. C. Lau, Visible light-induced oxidative N-dealkylation of alkylamines by a luminescent osmium(VI) nitrido complex, Chem. Sci., 2021, 12, 14494–14498 RSC; (c) J. Xiang, J. Zhu, M.-M. Zhou, L.-L. Liu, L.-X. Wang, M. Peng, B.-S. Hou, S.-M. Yiu, W.-P. To, C.-M. Che, K.-C. Lau and T.-C. Lau, Oxidative C–O bond cleavage of dihydroxybenzenes and conversion of coordinated cyanide to carbon monoxide using a luminescent Os(VI) cyanonitrido complex, Chem. Commun., 2022, 58, 7988–7991 RSC.
- (a) Q. Q. Su, K. Fan, X. D. Huang, J. Xiang, S. C. Cheng, C. C. Ko, L. M. Zheng, M. Kurmood and T. C. Lau, Field-induced slow magnetic relaxation in low-spin S= 1/2 mononuclear osmium(V) complexes, Dalton Trans., 2020, 49, 4084–4092 RSC; (b) L. L. Liu, L. X. Wang, M. Peng, J. Xiang, H. Yang, S. M. Yiu and T. C. Lau, Ring nitrogenation of aromatic amines by the excited state of an osmium(VI) nitrido complex, Inorg. Chem., 2023, 62, 1447–1454 CrossRef CAS PubMed.
- J. Xiang, W. L. Man, S. M. Yiu, S. M. Peng and T. C. Lau, Reaction of an osmium(VI) nitrido complex with cyanide: formation and reactivity of an osmium(III) hydrogen cyanamide complex, Chem. – Eur. J., 2011, 17, 13044–13051 CrossRef CAS PubMed.
- J. Xiang, Q. Wang, S. M. Yiu, W. L. Man, H. K. Kwong and T. C. Lau, Aerobic oxidation of an osmium(III) N-hydroxyguanidine complex to give nitric oxide, Inorg. Chem., 2016, 55, 5056–5061 CrossRef CAS PubMed.
- J. Xiang, Q. Wang, S. M. Yiu and T. C. Lau, Dual pathways in the oxidation of an osmium(III) guanidine complex. formation of osmium(VI) nitrido and osmium nitrosyl complex, Inorg. Chem., 2017, 56, 2022–2028 CrossRef CAS PubMed.
- J. Xiang, Q.-Q. Su, L.-J. Luo and T.-C. Lau, Synthesis and reactivity of an osmium(III) aminoguanidine complex, Dalton Trans., 2019, 48, 11404–11410 RSC.
- C. Li and M. Z. Hoffman, One-electron redox potentials of phenols in aqueous solution, J. Phys. Chem. B, 1999, 103, 6653–6656 CrossRef CAS.
- J. H. Xie, L. Ma, W. W. Y. Lam, K. C. Lau and T. C. Lau, Hydrogen atom transfer reactions of ferrate(VI) with phenols and hydroquinone. Correlation of rate constants with bond strengths and application of the Marcus cross relation, Dalton Trans., 2016, 45, 70–73 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2406139–2406142. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi03144j |
| This journal is © the Partner Organisations 2025 |