
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biobased oleyl glycidyl ether: copolymerization with ethylene oxide, postmodification, thermal properties, and micellization behavior†
Gregor M.
Linden
,
Sandra
Schüttner
,
Nora
Fribiczer
,
Sebastian
Seiffert
 and
Holger
Frey
*
and
Holger
Frey
*
Department of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
First published on 21st April 2025
Abstract
Oleyl glycidyl ether (OlGE) is a highly hydrophobic monomer synthesized from a biobased fatty alcohol and epichlorohydrin. When combined with hydrophilic monomethoxy poly(ethylene glycol) (mPEG) macroinitiators, well-defined, highly amphiphilic AB block copolymers are obtained via anionic ring-opening polymerization (Đ ≤ 1.08). Surprisingly, an investigation of the copolymerization kinetics of OlGE and ethylene oxide revealed an almost ideally random copolymerization (rEO = 1.27, rOlGE = 0.78) despite the significant structural differences. Both statistical and block copolymers were investigated regarding their behavior in aqueous solution. The block copolymers of the type mPEG-b-POlGE featured two distinct melting temperatures (Tms). Besides a melting transition of mPEG, a second Tm is attributed to the crystallization of the cis-alkenyl side chains of the OlGE units. Varying degrees of side chain hydrogenation of the POlGE homopolymer using potassium azodicarboxylate (PADA) allowed for tailoring of the Tm. The thiol–ene click reaction permitted subsequent functionalization. This work does not merely highlight the prospect of novel polyether surfactants, it also suggests the potential of biobased long-chain polyethers for the development of drug delivery systems featuring temperature-controlled release.
Introduction
Surfactants play a key role in numerous everyday applications, in which the combination of components with different polarity is required. They typically alter the surface properties of water. These applications include surfactants for cosmetics, coatings, paints, biotechnology, water purification, and many others.1,2 Polymer surfactants provide a means to tune and finely control these behaviors in manifold ways, due to their vast structural options. Mostly, when discussing polymer surfactants, reference is made to block copolymers. However, the term ‘surfactant’ also includes polysoaps, which are random copolymers of hydrophilic and hydrophobic monomers, among many other architectural configurations. The domain of polymer surfactants is extensive, and comprehensive reviews are available.1,3 The hydrophilic component of surfactants can either be charged, as seen in examples such as poly(acrylic acid),4 quaternized arylamines,5 and sulfonates,6 or uncharged, as in saccharides,7 polyoxazolines,8 and poly(ethylene glycol) (PEG).9 The industry provides numerous series of ethoxylated compounds as surfactants, such as e.g. Tween®, Myrj®, Span®, Triton X®, and Brij®.10Fats and oils represent renewable feedstocks for the chemical industry. Their substantial production volume (208.1 Mt in 2019) enables the cost-efficient supply of a diverse range of linear alkyl and alkenyl alcohols from triglycerides.11–15 The side product glycerol can be converted to epichlorohydrin and is equally commercially available as Epicerol® in a green process from Solvay. Glycerol is chlorinated twofold with HCl and after one elimination reaction, epichlorohydrin is formed.16–18 Epichlorohydrin can be utilized to introduce epoxide moieties in molecules.19 With a growing emphasis on the development of biobased chemicals that do not affect the food chain,20,21 it is crucial to underscore the effective utilization of triglycerides derived from non-edible oil plants as an alternative to using food crops.22 Moreover, converting waste into chemicals proves to be significantly more economical when compared to biofuel or electricity production.23 Ethylene oxide, the monomer used to produce PEG can be generated from biobased sources using two primary methods: dehydration of bioethanol to produce ethylene, followed by oxidation24,25 or electrosynthesis from bioethanol.26 Another viable approach involves production from CO2 and water.27
The field of drug solubilization via implementation of multifunctional polymer micelles is highly active in research, focusing on the purposeful treatment of diseases. These systems effectively handle the limitations of free hydrophobic drugs such as low solubility, dose-limiting toxicity, and inadequate biodistribution.28,29 The trigger to release drugs from external stimuli-responsive polymers can be changes in pH, temperature, magnetic fields, light, engineered sensitivities to enzymes, or radiofrequency.30–32 The lower critical solution temperature of poly(N-isopropyl acrylamide) can be employed to trigger drug release during local hyperthermia.33 Magnetically responsive nanoparticles (Fe3O4 or Fe2O3) can be incorporated into micelles or liposomes loaded with drugs while the drug release can be induced by an alternating external field.34 Brazel et al. employed poly(EG)-b-poly(ε-caprolactone) micelles in combination with iron oxide nanoparticles. The release of doxorubicin was demonstrated to occur by melting of the crystalline core when subjected to external magnetic field-induced heating.35 Block copolymers of PEG and glycidyl ethers with a hydrophobic chain have been synthesized and investigated for their use as cosurfactants,36 and show low cytotoxicity for splenic immune cells.37 ABA-type triblock copolymers of these structures can also be employed to prepare thermoresponsive hydrogels, capable of taking up Nile red as a model for a hydrophobic drug. By variation of the alkyl side chain length, the Tm of the hydrophobic block was altered to 49 °C in pure water.38
Here we introduce the monomer oleyl glycidyl ether (OlGE), prepared from oleyl alcohol and epichlorohydrin. This monomer was utilized in anionic ring-opening polymerization (AROP), affording homo- and block copolymers as well as statistical copolymers with ethylene oxide. Long-chain glycidyl ethers permit to modify the properties of highly water-soluble PEG, thereby allowing the fine-tuning of the physical characteristics of aqueous solutions.9,39 Furthermore, mPEG-b-poly(alkyl glycidyl ether) surfactants were synthesized and the ‘efficiency-boosting effect’ was investigated. The polymers increased the efficiency of medium- and longer-chain surfactants to stabilize water/oil interfaces.36 Analogously, terpenoid-derived systems that cannot crystallize due to their branched structure were explored and showed comparable results.40,41 We demonstrate that OlGE is a potentially biorenewable and versatile platform for a variety of surfactants.
Experimental section
Information regarding the reagents used, detailed monomer and polymer synthesis, as well as the analysis techniques employed, is available in the ESI.†Results and discussion
Monomer synthesis
The precursor oleyl alcohol is typically obtained through the hydrogenation of methyl oleate in industrial settings. Conventional industrial methods employ hydrogen gas and catalysts for the hydrogenation of methyl oleate. However, we opted for a more classical reduction method that employs lithium aluminum hydride (LiAlH4, Scheme 1).42 This choice was made to avoid the need for a high-pressure hydrogen gas apparatus. However, when considering large-scale synthesis, hydrogen gas would be the reducing agent of choice. Methyl oleate is a byproduct of transesterification, a process that involves triglycerides commonly sourced from large quantities of bio-derived oils and fats.12 Additionally, epichlorohydrin can be obtained from both petroleum-based resources and glycerol, which is a byproduct of biodiesel production and is available in abundance from fats and oils.16,17 The combination of oleyl alcohol and green epichlorohydrin obtained by this pathway results in a completely bio-based monomer. | ||
| Scheme 1 Two-step reaction for the synthesis of the oleyl glycidyl ether monomer. | ||
Oleyl glycidyl ether (OlGE) features a long hydrophobic side chain with a cis double bond at the C9 carbon atom. This prevents the side chain from crystallizing at room temperature and can serve as an anchoring point for post-polymerization modification if incorporated along the polymer backbone. The two-step reaction involves the reduction of methyl oleate followed by an etherification of the oleyl alcohol with epichlorohydrin (ECH) via phase transfer catalysis, as illustrated in Scheme 1. Hence, oleyl alcohol and epichlorohydrin underwent a two-phase reaction in the presence of a phase transfer catalyst under highly alkaline conditions, resulting in the formation of OlGE with yields of up to 71%. However, a known side reaction occurred during this process: the deprotonation of ECH with subsequent ring-opening led to 3-chloroallylalcohol. This alcohol is capable of reacting with ECH, leading to the formation of 3-chloroallyl glycidyl ether.19 The byproduct was effectively removed using Kugelrohr distillation, facilitated by the significant difference in boiling points between the byproduct and OlGE.
All experimental details as well as the characterization are given in the ESI.†
Synthesis of oleyl glycidyl ether-based polymers
All polymers were synthesized using standard AROP techniques. Experimental details along with characterization are provided in the ESI.† For block copolymers, the monofunctional initiator monomethoxy poly(ethylene glycol) (mPEG) with molar masses of 2000 g mol−1 (45 ethylene glycol units) and 5000 g mol−1 (114 ethylene glycol units) was utilized. Prior to polymerization, mPEG was deprotonated to an extent of 90% using KOtBu with the aid of [18]crown-6 and subsequently, the dried monomer OlGE was added. After the polymerization, the active chain ends were terminated using acidified methanol, and the resulting polymer was purified by precipitation. POlGE homopolymers were synthesized in the same way, but potassium(2-benzyloxy)ethanolate was used as the initiator. Since POlGE homopolymers are not the primary focus of this study, they have been excluded in Scheme 2. In the case of statistical copolymers, CsOH·H2O was employed as the deprotonating agent. This choice was made to maintain consistency in the experimental procedures between the copolymerization kinetics measurements (as described below) and the actual polymer synthesis. The procedure involved dissolving the cesium(2-benzyloxy)ethanolate initiator salt in dry DMSO and THF, after which dried OlGE was introduced into the reaction vessel. Subsequently, ethylene oxide was condensed to the custom-made anionic flask. Statistical copolymers with an overall number of both repeating units of 120 were targeted. Slight deviations stem from difficult measuring of liquefied EO at −80 °C, as small temperature increases led to a volume expansion of EO in the graduated ampule. | ||
| Scheme 2 Synthesis of block and statistical copolymers containing oleyl glycidyl ether by employing AROP techniques. | ||
Table 1 presents a summary of the synthesized polymers, including their characterization data and the intended composition compared to the calculated composition. Notably, all polymers based on oleyl glycidyl ether exhibited narrow dispersities ranging from 1.06 to 1.11. The shift in the SEC traces towards higher molar masses supports the block copolymer formation (Fig. S5 and S6†). Statistical copolymers showed slightly larger dispersities in the range of 1.11 to 1.15, although they still maintained a monomodal distribution, as illustrated in Fig. S9.† SEC analysis revealed an increase in molar mass for all polymers with the incorporation of higher amounts of OlGE. Variations are anticipated due to differences in the hydrodynamic volume behavior compared to the PEG calibration, which is particularly notable for POlGE homopolymers with their long side chains, leading to a bottlebrush-like structure (Fig. S12†). We would like to emphasize that achieving higher molar masses of the POlGE homopolymer was not tested, as this was beyond the scope of our study. It is essential to recognize that the molar mass calculated from SEC should be considered a rough estimate given the relative nature of the method employed. The NMR spectra can be found in the ESI (Fig. S3, S4, S7 and S8†). Differences observed between the theoretical and achieved degree of polymerization can be attributed to contamination by small amounts of OlGE homopolymer, initiated by residual traces of water. These polymers were subsequently removed during the work-up process.
| Polymer compositiona | n(OlGE)th | n(EO)th | M n(SEC)b (g mol−1) | M n(NMR)a (g mol−1) | Đ |
|---|---|---|---|---|---|
| a Determined via1H NMR analysis. b Eluent THF, PEG calibration, RI detector, ththeoretical. | |||||
| mPEG45-b-POlGE2.4 | 3 | — | 3000 | 2800 | 1.06 |
| mPEG45-b-POlGE5.0 | 6 | — | 4300 | 3600 | 1.06 |
| mPEG45-b-POlGE7.5 | 9 | — | 4500 | 4400 | 1.06 |
| mPEG45-b-POlGE11.3 | 12 | — | 4800 | 5700 | 1.08 |
| mPEG114-b-POlGE2.9 | 3 | — | 6400 | 5900 | 1.08 |
| mPEG114-b-POlGE5.8 | 6 | — | 7700 | 6900 | 1.07 |
| mPEG114-b-POlGE7.3 | 9 | — | 8200 | 7400 | 1.06 |
| mPEG114-b-POlGE9.9 | 12 | — | 9100 | 8200 | 1.07 |
| P(EG123-co-OlGE7) | 6 | 114 | 7500 | 7800 | 1.14 |
| P(EG94-co-OlGE13) | 12 | 108 | 7800 | 8500 | 1.15 |
| P(EG103-co-OlGE35) | 30 | 90 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
16000 |
1.12 |
| POlGE13 | 10 | — | 3400 | 4400 | 1.07 |
| POlGE25 | 25 | — | 5800 | 8300 | 1.11 |
Reactivity ratios of OlGE/EO by in situ1H NMR copolymerization kinetics
Knowledge of microstructure is crucial to understand polymer characteristics. Depending on the type of monomers and polymerization techniques employed, various microstructures can typically be achieved, ranging from random copolymers to nearly blocklike, tapered structures. These microstructures have a significant impact on various polymer properties such as thermal and mechanical properties, micellization, and solubility among others.43,44 In our study, we investigated the copolymerization behavior via in situ1H NMR analysis. To this end, EO and OlGE were copolymerized at 40 °C in an NMR tube equipped with a Teflon stopcock, using a mixture of DMSO-d6 and THF-d8 at a 1:5 (V/V) ratio. Cesium 2-(benzyloxy)ethanolate was used as the initiator for the polymerization. Cesium was chosen as a counterion for all statistical copolymers, as it enhances the propagation rate compared to potassium and reduces the time required for the kinetic investigation experiment.45 Experimental details are given in the ESI.† Monomer consumption was tracked by observing the monomer resonances at 2.68 ppm for OlGE and 2.58 ppm for EO during the copolymerization. Fig. 1 displays a selection of stacked spectra.
 | ||
| Fig. 1 Stacked selection of 1H NMR spectra of the copolymerization of OlGE and EO. Zoom-in shows the decrease of the OlGE (orange) and EO monomer signals (blue). Polymerization temperature 40 °C, solvent: DMSO-d6 and THF-d8 1:5 (V/V), 400 MHz. As spectra were collected every 2 min for 55 h, only every 200th spectrum is displayed. | ||
Both monomers exhibited nearly complete conversion after 55 hours. The gradual decrease of the signals is illustrated in Fig. 2, which shows the amount of unreacted monomer versus total conversion. Clearly, EO is incorporated more rapidly, while OlGE is consumed at a slightly lower rate. This already suggests that copolymerization is not ideally random.
 | ||
| Fig. 2 Unreacted monomer versus total conversion of the in situ1H NMR copolymerization kinetic study of EO with OlGE. Solvent: DMSO-d6/THF-d8 1:5 (V/V), 40 °C. | ||
To determine reactivity ratios r1,2 the data extracted from the kinetic experiment were fitted according to the Jaacks plot.46–48 For this, the following eqn (1) was used.
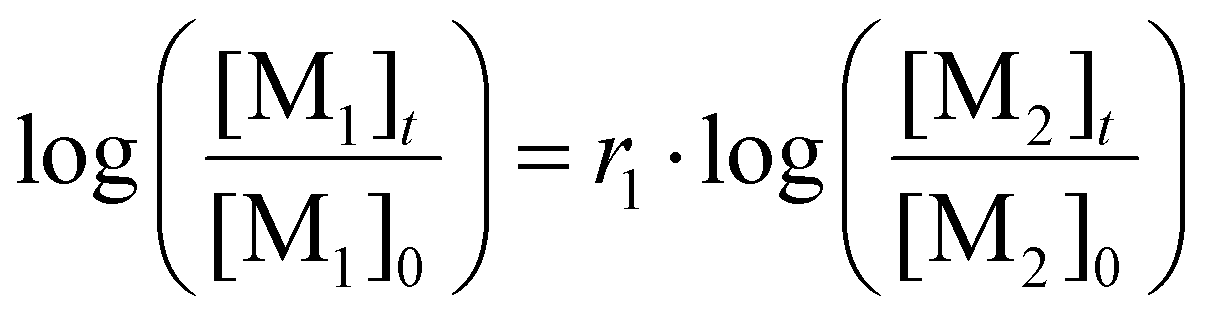 | (1) |
In an ideal copolymerization, the following correlation is defined: r1·r2 = 1. Both reactivity ratios were determined with the results rEO = 1.27 and rOlGE = 0.78 (Fig. S39†). As the coefficient of determination R2 was 0.99, an ideal or chain-end independent copolymerization can be concluded. The use of more complex terminal models such as the Meyer–Lowry method49 should be avoided as long as the more simple, non-terminal model explains the data well. This principle was coined by Sir William Hamilton as “Ockham's Razor”,50 and its application to copolymerization kinetics was already described.51 The fit is displayed in Fig. S40,† and the results are summarized in Table 2.
| Method | Model | r EO | r OlGE | |
|---|---|---|---|---|
| Jaacks | Non-terminal, ideal | 1.27 ± 0.01 | 0.78 ± 0.01 | R 2 = 0.99 |
| Meyer–Lowry | Terminal, non-ideal | 1.32 ± 0.12 | 0.80 ± 0.14 | NormRes = 0.38 |
The obtained reactivity ratios enable the simulation of the comonomer composition in the course of the copolymerization (Fig. 3), which directly translates to the monomer gradient in the copolymer chains formed. As previously indicated in Fig. 2, EO is initially incorporated with a slight preference during copolymerization. With increasing conversion, approaching full monomer conversion, more OlGE units are incorporated at the chain end. This subtle variation in reactivity ratios results in the enrichment of monomer units towards either the beginning or the terminus of the polymer chains, respectively. We would like to emphasize that this gradient is not pronounced, compared to other systems like the carbanionic copolymerization of isoprene and styrene.48 This result is remarkable in view of the large steric bulk of the OlGE monomer in comparison to EO.
 | ||
| Fig. 3 Simulated composition versus total conversion of the EO (blue)/OlGE (orange) comonomer pair for a hypothetical equimolar ratio. | ||
Even though glycidyl ethers (GEs) exhibit a substituted epoxide functionality, usually the correlation rEO ≈ rGE ≈ 1 is valid.37,52,53 This observation is counterintuitive, especially since propylene oxide copolymerizes with a much stronger gradient with EO, which is assigned to the methyl group substitution.54 Due to their ability to chelate the counterion and thereby activate the epoxide, glycidyl ethers show a transient “crown ether-effect”.55 Since the side chain contains additional oxygen atoms, glycidyl ethers react faster than the unsubstituted EO.56–58 Bulky and/or inflexible side chains, in contrast, prevent sufficient chelation and also lead to hindered nucleophilic attack of an active chain end at the epoxide functionality, resulting in less favored incorporation.40,58–60 This finding is confirmed by our study, as the bulky, apolar monomer is less prone to nucleophilic attack, translating to slower incorporation in the growing polymer chain.
Amphiphilic nature and CMC determination of OlGE copolymers
The determination of the CMC is commonly achieved by employing pyrene as a fluorescent probe.2,61 When pyrene transitions from a polar to an apolar environment, the emission spectrum experiences a pronounced shift towards higher values in the ratio of the I3 and I1 bands, correlated to the alteration in the polarity of the surrounding molecules. The experimental procedure used in this work is based on the method described by Zhu et al.62 In short, pyrene was mixed with the respective polymer solution in a serial dilution. Then the fluorescence spectra of pyrene were measured at 23 °C, and I3/I1 was plotted against the logarithmic polymer concentration. A Boltzmann sigmoidal fit (eqn (S1)†) was performed and the CMC was determined from the inflection point of the fit.40,61 The graphs are shown in the ESI (Fig. S18–S24).†The hydrophilic–lipophilic balance (HLB) is an empirical scale ranging from 1 to 20, employed to assess the utility of surfactants in various applications. Higher HLB values are indicative of surfactants with a greater affinity for polar environments, making them suitable for tasks such as solubilization and oil-in-water emulsification. Conversely, lower HLB values denote surfactants with a stronger preference for nonpolar environments, rendering them more appropriate for purposes such as water-in-oil emulsification and foam reduction. The HLB scale serves as a valuable tool for understanding the contrasting characteristics of surfactants.63
Table 3 gives an overview of the synthesized OlGE-containing polymers with their HLB values and the CMC determined by fluorometry. The sole water-soluble compound within the series of statistical copolymers was P(EG123-co-OlGE7), which exhibited a CMC of 24.5 mg L−1. Of the four block copolymers obtained from mPEG45, only two displayed water solubility and had a CMC threshold of 23.8 mg L−1. This observation is attributed to a significant increase in the weight percentage of the hydrophobic block due to the incorporation of additional 2.6 OlGE units, each with a high molar mass of 324 g mol−1, in contrast to ethylene glycol units with a mass of only 44 g mol−1 each. A comparable outcome was noted in the case of block copolymers based on mPEG114. The CMCs exhibited a notable reduction from 73.4 to 25.4 mg L−1, with just a slight 2.1 mg L−1 difference between mPEG114-b-POlGE7.3 and mPEG114-b-POlGE9.9. As reported by Lodge et al., the CMC decreases exponentially, but is weakly dependent on the hydrophobic block length for longer blocks and is notably less dependent on the hydrophilic block length.1,64 Thereby, the difference in CMC between polymers with a small hydrophobic block (mPEG114-b-POlGE2.9 and mPEG114-b-POlGE5.8) is more pronounced compared to block polymers with larger hydrophobic blocks (mPEG114-b-POlGE7.3 and mPEG114-b-POlGE9.9). Noteworthy, all three varieties of copolymers exhibited a CMC threshold of approximately 25 mg L−1. When contrasting our system with existing literature, the CMC of Brij® 98, a twentyfold ethoxylated oleyl alcohol, is reported to fall within the range of 7–29 mg L−1.65–68 Variations in CMC values for the same compound may arise from differences in measurement methods, among other factors.69 Block copolymers of mPEG114-b-poly(farnesyl glycidyl ether)m (m = 5 and 9) showed slightly lower CMCs of 53 and 15 mg L−1, respectively.40
| Polymer | mol% (OlGE) | w% (OlGE) | HLB | CMC (mg L−1) |
|---|---|---|---|---|
| a Values could not be determined due to insufficient solubility in water. | ||||
| P(EG123-co-OlGE7) | 5.4 | 30 | 14.1 | 24.5 |
| P(EG94-co-OlGE13) | 12.1 | 50 | 9.9 | n.d.a |
| P(EG103-co-OlGE35) | 25.4 | 71 | 5.7 | n.d.a |
| mPEG45-b-POlGE2.4 | 5.1 | 28 | 14.4 | 41.9 |
| mPEG45-b-POlGE5.0 | 10.0 | 45 | 11.0 | 23.8 |
| mPEG45-b-POlGE7.5 | 14.3 | 55 | 9.0 | n.d.a |
| mPEG45-b-POlGE11.3 | 20.1 | 65 | 7.1 | n.d.a |
| mPEG114-b-POlGE2.9 | 2.5 | 16 | 16.8 | 73.4 |
| mPEG114-b-POlGE5.8 | 4.8 | 27 | 14.5 | 44.2 |
| mPEG114-b-POlGE7.3 | 6.0 | 32 | 13.6 | 27.5 |
| mPEG114-b-POlGE9.9 | 8.0 | 39 | 12.2 | 25.4 |
Dynamic light scattering of OlGE copolymers
Polymer solutions of the water-soluble copolymers were investigated by dynamic light scattering (DLS) to determine the hydrodynamic radius of the respective micelles and larger aggregates. Additionally, the results were compared to TEM observation of the aggregates. The polymer concentration was significantly above the CMC determined via fluorometry. The amplitude autocorrelation function was subjected to a biexponential fit (represented by eqn (S4)†), given that TEM images hinted at the presence of multiple types of aggregates for all copolymers. The diffusion coefficient of each measured angle was received from the relation D = (τR·q2)−1. This diffusion coefficient was subsequently plotted versus q2 to calculate the z-average diffusion coefficient for each specific aggregate. Additional details on the calculation of diffusion coefficients and hydrodynamic radii are described in the ESI.† A summary of the results obtained from DLS analysis is provided in Table 4. Among the statistical copolymers examined, P(EG123-co-OlGE7) was the only sample that could be analyzed, as the others were insoluble. The block copolymer mPEG114-b-POlGE9.9 was not analyzed by DLS, as a turbid solution was obtained. P(EG123-co-OlGE7) exhibited aggregates with an RH,1 of 5.31 nm, corresponding to a single chain (micelle), as the comparable mPEG114 unimer exhibited a hydrodynamic radius of 2 nm.40 In contrast, the larger aggregates displayed a significantly greater RH,2 of 107 nm. This discrepancy is attributed to chain folding, resulting in the formation of multicompartment micelles. This phenomenon has been discussed in previous studies.70–72 The corresponding TEM micrograph equally indicates small circular structures as well as larger aggregates but with a smaller radius (Fig. S25†). While they are separated in the dry TEM micrographs, they probably stick to each other in solution, due to hydrophobic interactions between OlGE units in the corona-forming segments of different micelles. This leads to the formation and observation of aggregates with a greater RH,2. Potemkin et al. described this “stickiness” in a recent study.73| Polymer | D 1 (10−7 cm2 s−1) | D 2 (10−7 cm2 s−1) | R H,1 (nm) | R H,2 (nm) |
|---|---|---|---|---|
| P(EG123-co-OlGE7) | 4.64 ± 0.03 | 0.23 ± 0.01 | 5.3 ± 0.1 | 107 ± 5 |
| mPEG45-b-POlGE2.4 | 2.54 ± 0.02 | 0.51 ± 0.05 | 9.7 ± 0.1 | 48 ± 4 |
| mPEG45-b-POlGE5.0 | 0.51 ± 0.02 | 0.12 ± 0.01 | 48 ± 2 | 206 ± 5 |
| mPEG114-b-POlGE2.9 | 1.41 ± 0.02 | 0.35 ± 0.01 | 17.5 ± 0.2 | 70 ± 1 |
| mPEG114-b-POlGE5.8 | 0.55 ± 0.03 | 0.10 ± 0.01 | 45 ± 2 | 240 ± 20 |
| mPEG114-b-POlGE7.3 | 1.12 ± 0.01 | 0.21 ± 0.01 | 21.9 ± 0.2 | 117 ± 7 |
Block copolymers composed of mPEG45 generally displayed larger RH,1 values, despite having a significantly greater number of repeating units in comparison to the statistical copolymer. This observation implies the aggregation of multiple chains into micelles. When the size of the POlGE block is doubled, there is a notable four- to fivefold increase in the hydrodynamic radius of both aggregates. This finding aligns with the structures observed in the TEM micrographs (Fig. 4A and B).
 | ||
| Fig. 4 TEM micrograph of mPEG-b-POlGEn block copolymers in aqueous solution (0.1 g L−1). A: mPEG45-b-POlGE2.4, B: mPEG45-b-POlGE5.0, the circular structure in the upper left corner is caused by irradiation damage. C: mPEG114-b-POlGE2.9, D: mPEG114-b-POlGE5.8, E: mPEG114-b-POlGE7.3, F: mPEG114-b-POlGE9.9. Samples were treated with 2% uranyl acetate solution as a negative stain. Original micrographs obtained by the software can be found in the ESI (Fig. S26 and 27†). | ||
The behavior of block copolymers containing mPEG114 demonstrates subtle variations. While the hydrodynamic radius (RH,1,2) for the POlGEn block (with n = 2.9 and 7.3, C and E) only experiences a slight increase as n increases, the block copolymer with n = 5.8 (D) exhibits a larger RH,1,2 compared to the one with 7.3 OlGE monomer units. This outcome may seem unexpected, but it can be attributed to the fact that due to the larger hydrophobic block (n = 7.3), there are fewer chains present within the respective micelles compared to mPEG114-b-POlGE5.8, resulting in a reduced aggregation number. Consequently, the hydrodynamic radius does not increase as one might anticipate. The polymer mPEG114-b-POlGE2.9 reveals only small structures in the TEM micrograph, a finding that contrasts with the DLS analysis results indicating clustering in solution. The TEM micrograph of the block copolymer with the largest POlGE part, mPEG114-b-POlGE9.9 (F), displays well-resolved, fibrillar structures. Nevertheless, further analysis has been deferred to a later study. Original TEM micrographs can be found in the ESI (Fig. S25–S27†).
Thermal properties of polymers containing OlGE
PEG plays an important role as ointment base and tailoring the melting point slightly above human body temperature enables softening into a semi-solid during application and even spreading while lasting at the applied area.9,74,75 The synthesized OlGE (co)polymers were investigated by differential scanning calorimetry (DSC) (Table 5). The thermograms are displayed in the ESI (Fig. S13–S17†). Detection of glass transition temperatures (Tgs) was performed with the second heating curve with a heating rate of 10 K min−1. As OlGE has a long, flexible side chain, the Tg is shifted towards lower temperatures compared to PEG (<−60 °C).9 Melting temperatures (Tms) and melting enthalpies were determined with a 1 K min−1 heating rate. Reducing the heating/cooling rate to 1 K min−1 allowed for ordering of the side chains and prevention of recrystallization. Otherwise, a pronounced recrystallization was detected during the melting event in the measurements with a heating/cooling rate of 10 K min−1 (Fig. S13†). Substituted polyethers are typically amorphous materials. However, due to the elongated and linear structure of the side chain, POlGE displays a distinct Tm, despite the racemic monomer. This indicates a propensity for side chain crystallization. The microphase separation driven by crystallization was previously described in literature with comparable side chains, albeit for polyacrylates or polyesters.70,76,77| Entry | Polymer | OlGE, mol% |
T
ga/°C |
T
mb/°C |
ΔHmb/J g−1 |
|---|---|---|---|---|---|
| a Heating rate 10 °C min−1, second heating curve. b Heating rate 1 °C min−1, second heating curve. In the case of block copolymers, the first Tm and ΔHm denote the POlGE block crystallization, whereas the second denote the PEG block crystallization. | |||||
| 1 | POlGE13 | 100 | n.d. | −27.8 | 32.2 |
| 2 | POlGE25 | 100 | n.d. | −23.2 | 63.4 |
| 3 | P(EG123-co-OlGE7) | 5 | n.d. | 35.7 | 64.9 |
| 4 | P(EG94-co-OlGE13) | 12 | −81 | 12 | 46.5 |
| 5 | P(EG103-co-OlGE35) | 25 | −80 | -41.7 | 30.3 |
| 6 | mPEG45 | 0 | −60 | —/52.3 | —/164.7 |
| 7 | mPEG45-b-POlGE2.4 | 3 | −75 | n.d./48.1 | n.d./104.0 |
| 8 | mPEG45-b-POlGE5.0 | 10 | n.d. | −39.5/42.7 | 9.0/67.1 |
| 9 | mPEG45-b-POlGE7.5 | 14 | −78 | −46.8/37.2 | 6.6/48.2 |
| 10 | mPEG45-b-POlGE11.3 | 20 | −79 | −32.1/35.0 | 21.8/45.4 |
| 11 | mPEG114 | 0 | −60 | —/61.9 | —/190.1 |
| 12 | mPEG114-b-POlGE2.9 | 2 | −79 | n.d./57.4 | n.d./133.8 |
| 13 | mPEG114-b-POlGE5.8 | 5 | −80 | −50.5/53.7 | 4.2/108.5 |
| 14 | mPEG114-b-POlGE7.3 | 6 | −76 | −58.0/51.9 | 0.8/89.8 |
| 15 | mPEG114-b-POlGE9.9 | 8 | n.d. | −41.3/53.3 | 10.2/88.9 |
Statistical P(EG-co-OlGE) copolymers
The random incorporation of 5 mol% OlGE in P(EG-co-OlGE) copolymers (Table 5, entry 3) results in a notable reduction in crystallinity, as evidenced by the decrease of the melting enthalpy (ΔHm) of PEG. For comparison, mPEG114 (Table 5, entry 11), which closely resembles the polymer under investigation, was utilized. 12 mol% OlGE (Table 5, entry 4) further inhibited crystallization of the polyether backbone, and the material showed the PEG Tm already below room temperature. When 25 mol% OlGE was incorporated (Table 5, entry 5), the Tm (−41.7 °C) was solely dominated by the OlGE side chain.mPEG-b-POlGE block copolymers
Block copolymers initiated by mPEG45 showed decreasing ΔHm with increasing POlGE block (Table 5, entries 6–10), again indicating partial miscibility of the two different blocks. Comparing entries 8 and 9, an unusual behavior appears: with an increasing amount of POlGE (entry 9), the Tm of both copolymer blocks decreased. This indicated a transition zone where both blocks were too small to crystallize. By further increase of the POlGE content, the Tm of the POlGE block increased again (entry 10), whereas the thermal properties of the PEG block remained unchanged. Very similar thermal behavior was observed for mPEG114-b-POlGE block copolymers as well: both Tms decreased with increasing POlGE content (entries 13 and 14), but with higher POlGE content the Tms elevated again. This indicated that a more effective phase separation between the two polymers contributed to the rise in the Tms (entry 15).Post-polymerization modification
 | ||
| Fig. 5 1H NMR spectra of POlGE25 and the respective hydrogenated polymers (400 MHz, CDCl3). The value of H denotes the degree of hydrogenation. The intensity was locally normalized to the initiator benzyl group (gray). | ||
To confirm the anticipated effect of hydrogenation concerning side chain crystallization, the thermal properties after hydrogenation were investigated by DSC (Fig. S48†). With an increasing degree of hydrogenation, the Tm of POlGE25 rises above room temperature and reaches its peak at 51.9 °C when fully hydrogenated. Up to 53% of hydrogenation results in a significant increase of Tm, whereas the difference in Tm decreases with higher degrees of hydrogenation (entries 3 and 4). The melting enthalpy increases only slightly, going from 63.4 to 75.2 J g−1. This relatively small change occurs even though the hydrogenated side chains were expected to be hindered from crystallization due to the assumed miscibility of the saturated and unsaturated side chains. Here, the melting enthalpy is almost independent of the degree of hydrogenation, albeit the melting point increases by 75.1 °C in total. In summary, the hydrogenation experiments show that POlGE can be fully hydrogenated in a post-polymerization modification. With a Tm in the physiological range, copolymers containing OlGE repeating units possess potential for applications in drug delivery systems. Tailoring can be conveniently performed, as no copolymerization of high and low Tm monomers is necessary. Instead, one starting material is sufficient (Table 6).
Not only does the double bond content provide the opportunity to customize thermal properties, but it also allows for the modification of the structure to meet specific requirements. It enables the incorporation of hydroxyl groups or other functional groups, which enables the tuning of hydrophobicity and facilitates further coupling with various other groups.84–86
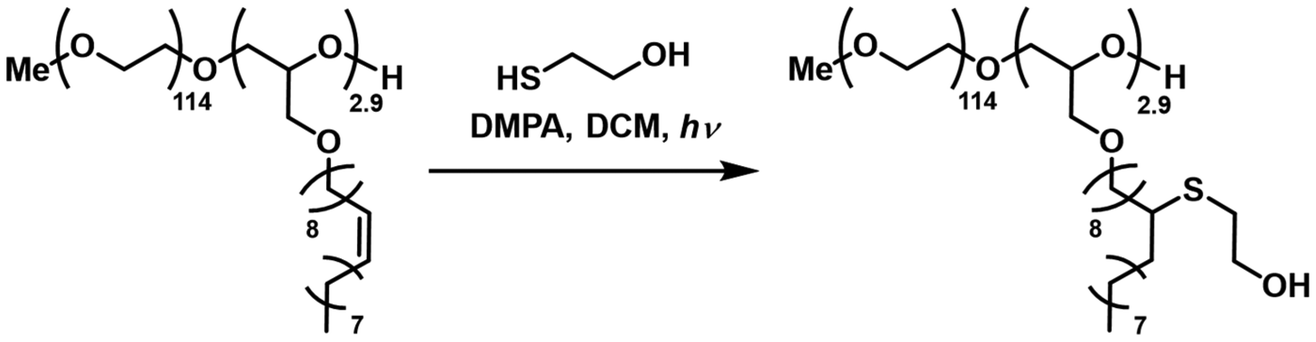 | ||
| Scheme 3 Thiol–ene click reaction of mPEG114-b-POlGE2.9 with thioglycol. Note that two possible addition products can be present. | ||
Complete conversion was confirmed by 1H NMR by the disappearance of the double bond and allylic proton signals. The appearance of the methine and methylene group of the respective thioether further confirmed the successful modification (Fig. S42†). SEC revealed a shift towards higher molar masses, again keeping the dispersity constant (Fig. S43†). DOSY NMR confirmed no residual thioglycol precursor after work-up by dialysis (Fig. S44†). MALDI-ToF MS analysis showed the new repeating unit with a molar mass of 402 g mol−1 (Fig. S45†).
Conclusion
Oleyl glycidyl ether (OlGE) has been introduced as a biorenewable, highly apolar epoxide building block for the AROP copolymerization with EO. Investigation of the reactivity ratios revealed a small deviation from ideal statistical copolymerization of OlGE (rOlGE = 0.78) with EO (rEO = 1.27), leading to an almost ideally random copolymer structure. This is a remarkable observation, considering the large steric bulk of the OlGE monomer in comparison to EO. In addition to statistical copolymers, using different mPEG macroinitiators yielded block copolymers. Capitalizing on block copolymerization of POlGE with mPEG, amphiphilic polymers were successfully produced. The CMCs of mPEG-based block copolymers show a limit in the same range as the established surfactant Brij® 98. The water-soluble statistical copolymer also showed micelle formation, owing to the long hydrophobic side chains. TEM and DLS revealed spherical and elongated micellar aggregates, which can be explained by the arrangement of the sterically demanding, linear side chains. The utilization of OlGE allows the synthesis of biobased surfactants from readily accessible starting materials. The bulk properties of the polymers showed two distinguishable Tms. The cis-alkenyl side chain was successfully modified by a thiol–ene click reaction, demonstrating the accessibility and emphasizing the versatility of the hidden functionality. This enables coupling of a wide range of thiol-containing compounds to the polymers. Partial hydrogenation of the double bond with potassium azodicarboxylate (PADA) enables adjusting the melting point of the materials, eradicating the mixing of monomers with high and low side chain Tm, respectively. The achieved range of melting points is suitable for developing potential thermoresponsive drug delivery systems, as it matches the human body temperature. In future drug delivery applications, active pharmaceutical ingredients could be released from micellar solutions, if the Tm of the precisely adjusted side chain is reached.In brief, our study shows that the biobased novel monomer OlGE and its copolymerization can be employed to generate a wide range of structurally varied polymeric surfactants. This approach can pave the way for replacing traditional petroleum-based surfactants and also unlocks the potential for generating diverse and tailored polymer architectures. “Green” and conveniently modifiable polymers with respect to the requirements of a user, particularly with respect to melting temperatures or hydrophobicity, can offer a future platform for customized drug-delivery systems.
Data availability
The data supporting this article have been included as part of the extended ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Monika Schmelzer for SEC measurements, Elena Berger-Nicoletti for MALDI-ToF MS measurements. Frank Depoix and Elisabeth Sehn are acknowledged for introduction to TEM measurements and Lisanne Lörke for experimental support. Rebecca Matthes is thanked for thorough correction of the manuscript.References
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- Amphiphilic block copolymers: Self-assembly and applications, ed. P. Alexandridis and B. Lindman, Elsevier, Amsterdam, 1st edn, 2000 Search PubMed.
- I. R. Schmolka, J. Am. Chem. Soc., 1977, 54, 110–116 CAS.
- G. Laruelle, J. François and L. Billon, Macromol. Rapid Commun., 2004, 25, 1839–1844 CrossRef CAS.
- Z. Gao, S. K. Varshney, S. Wong and A. Eisenberg, Macromolecules, 1994, 27, 7923–7927 CrossRef CAS.
- K. Lienkamp, C. Ruthard, G. Lieser, R. Berger, F. Groehn and G. Wegner, Macromol. Chem. Phys., 2006, 207, 2050–2065 CrossRef CAS.
- M. Koóš, in Encyclopedia of biophysics, ed. G. C. K. Roberts, Springer, Berlin, 1st edn, 2013, p. 237 Search PubMed.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- Biobased surfactants and detergents: Synthesis, properties, and applications, ed. D. G. Hayes, Academic Press, London, 2019 Search PubMed.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS PubMed.
- K. Noweck and W. Grafahrend, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- H. Baumann, M. Bühler, H. Fochem, F. Hirsinger, H. Zoebelein and J. Falbe, Angew. Chem., Int. Ed. Engl., 1988, 27, 41–62 CrossRef.
- ISTA Mielke GmbH, Oil World Annual 2020, Hamburg, 2020.
- Solvay, Solvay Epicerol® Earns Roundtable on Sustainable Biomaterials Certification, available at: https://www.solvay.com/sites/g/files/srpend221/files/tridion/documents/17_RSB%20Certificate_Final.pdf, accessed September 9, 2024.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean, 2008, 36, 657–661 CAS.
- Y. Meng, F. Taddeo, A. F. Aguilera, X. Cai, V. Russo, P. Tolvanen and S. Leveneur, Catalysts, 2021, 11, 765 CrossRef CAS.
- X.-P. Gu, I. Ikeda and M. Okahara, Bull. Chem. Soc. Jpn., 1987, 60, 667–672 CrossRef CAS.
- J. Hill, E. Nelson, D. Tilman, S. Polasky and D. Tiffany, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11206–11210 CrossRef CAS PubMed.
- A. García-Franco, P. Godoy, J. de La Torre, E. Duque and J. L. Ramos, Microb. Biotechnol., 2021, 14, 1871–1877 CrossRef PubMed.
- B. Thangaraj and P. R. Solomon, Clean Energy, 2020, 4, 89–106 CrossRef.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- BASF, Bioethanol. New catalyst geometry poised to re-shape the ethanol-to-ethylene conversion process, available at: https://chemicals.basf.com/global/en/Catalysts/hydrogenation-specialty/products-we-offer/alumina/Ethanol-to-Ethylene-E2E.html.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- C. Lucky, T. Wang and M. Schreier, ACS Energy Lett., 2022, 7, 1316–1321 CrossRef CAS.
- Y. Li, A. Ozden, W. R. Leow, P. Ou, J. E. Huang, Y. Wang, K. Bertens, Y. Xu, Y. Liu, C. Roy, H. Jiang, D. Sinton, C. Li and E. H. Sargent, Nat. Catal., 2022, 5, 185–192 CrossRef CAS.
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 77 Search PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- D. A. LaVan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21, 1184–1191 CrossRef CAS PubMed.
- X. Guo and F. C. Szoka, Acc. Chem. Res., 2003, 36, 335–341 CrossRef CAS PubMed.
- V. Torchilin, Eur. J. Pharm. Biopharm., 2009, 71, 431–444 CrossRef CAS PubMed.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- A. L. Glover, J. B. Bennett, J. S. Pritchett, S. M. Nikles, D. E. Nikles, J. A. Nikles and C. S. Brazel, IEEE Trans. Magn., 2013, 49, 231–235 Search PubMed.
- K. Schneider, P. Verkoyen, M. Krappel, C. Gardiner, R. Schweins, H. Frey and T. Sottmann, Langmuir, 2020, 36, 9849–9866 CrossRef CAS PubMed.
- P. Verkoyen, P. Dreier, M. Bros, C. Hils, H. Schmalz, S. Seiffert and H. Frey, Biomacromolecules, 2020, 21, 3152–3162 CrossRef CAS PubMed.
- P. Verkoyen, T. Johann, J. Blankenburg, C. Czysch and H. Frey, Polym. Chem., 2018, 9, 5327–5338 RSC.
- P. Verkoyen and H. Frey, Macromol. Rapid Commun., 2020, 41, 2000225 CrossRef CAS PubMed.
- S. Schüttner, M. Krappel, M. Koziol, L. Marquart, I. Schneider, T. Sottmann and H. Frey, Macromolecules, 2023, 56, 6928–6940 CrossRef.
- P. Verkoyen, P. Dreier, M. Bros, C. Hils, H. Schmalz, S. Seiffert and H. Frey, Biomacromolecules, 2020, 21, 3152–3162 CrossRef CAS PubMed.
- H. G. O. Becker, Organikum. Organisch-chemisches Grundpraktikum, Wiley-VCH, Weinheim, 21st edn, 2001 Search PubMed.
- K. Matyjaszewski, Y. Gnanou and L. Leibler, Macromolecular Engineering. Precise Synthesis, Materials Properties, Applications, Wiley, Weinheim, 2007 Search PubMed.
- B. Tieke, Makromolekulare Chemie. Eine Einführung, Wiley-VCH, Weinheim, 3rd edn, 2014 Search PubMed.
- S. Boileau, A. Deffieux, D. Lassalle, F. Menezes and B. Vidal, Tetrahedron Lett., 1978, 19, 1767–1770 CrossRef.
- V. Jaacks, Angew. Chem., 1967, 79, 419 CrossRef.
- V. Jaacks, Makromol. Chem., 1972, 161, 161–172 CrossRef CAS.
- M. Steube, T. Johann, M. Plank, S. Tjaberings, A. H. Gröschel, M. Gallei, H. Frey and A. H. E. Müller, Macromolecules, 2019, 52, 9299–9310 CrossRef CAS.
- V. E. Meyer and G. G. Lowry, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2843–2851 CrossRef CAS.
- R. Hoffman, B. K. Carpenter and V. I. Minkin, HYLE, 1997, 3, 3–28 Search PubMed.
- J. Blankenburg, E. Kersten, K. Maciol, M. Wagner, S. Zarbakhsh and H. Frey, Polym. Chem., 2019, 10, 2863–2871 RSC.
- C. Mangold, F. Wurm, B. Obermeier and H. Frey, Macromolecules, 2010, 43, 8511–8518 CrossRef CAS.
- C. Tonhauser, A. Alkan, M. Schömer, C. Dingels, S. Ritz, V. Mailänder, H. Frey and F. R. Wurm, Macromolecules, 2013, 46, 647–655 CrossRef CAS.
- Y. Deng, J. Ding, G. Yu, R. H. Mobbs, F. Heatley, C. Price and C. Booth, Polymer, 1992, 33, 1959–1962 CrossRef CAS.
- P. Dreier, R. Matthes, R. D. Barent, S. Schüttner, A. H. E. Müller and H. Frey, Macromol. Chem. Phys., 2023, 224, 2200209 CrossRef CAS.
- A. Lee, P. Lundberg, D. Klinger, B. F. Lee, C. J. Hawker and N. A. Lynd, Polym. Chem., 2013, 4, 5735–5742 RSC.
- J. Herzberger, K. Fischer, D. Leibig, M. Bros, R. Thiermann and H. Frey, J. Am. Chem. Soc., 2016, 138, 9212–9223 CrossRef CAS PubMed.
- J. Blankenburg, K. Maciol, C. Hahn and H. Frey, Macromolecules, 2019, 52, 1785–1793 CrossRef CAS.
- J. Herzberger, D. Leibig, J. C. Liermann and H. Frey, ACS Macro Lett., 2016, 5, 1206–1211 CrossRef CAS PubMed.
- K. Niederer, C. Schüll, D. Leibig, T. Johann and H. Frey, Macromolecules, 2016, 49, 1655–1665 CrossRef CAS.
- J. Aguiar, P. Carpena, J. A. Molina-Bolívar and C. Carnero Ruiz, J. Colloid Interface Sci., 2003, 258, 116–122 CrossRef CAS.
- H. Li, D. Hu, F. Liang, X. Huang and Q. Zhu, R. Soc. Open Sci., 2020, 7, 192092 CrossRef CAS PubMed.
- W. C. Griffin, J. Soc. Cosmet. Chem., 1949, 1, 311–326 Search PubMed.
- M. M. Mok, R. Thiagarajan, M. Flores, D. C. Morse and T. P. Lodge, Macromolecules, 2012, 45, 4818–4829 CrossRef CAS.
- J. Qiu, T. Pintauer, S. G. Gaynor, K. Matyjaszewski, B. Charleux and J.-P. Vairon, Macromolecules, 2000, 33, 7310–7320 CrossRef CAS.
- A. C. Caritá, R. R. M. Cavalcanti, M. S. S. Oliveira and K. A. Riske, Chem. Phys. Lipids, 2023, 255, 105327 CrossRef PubMed.
- M. E. N. P. Ribeiro, C. L. de Moura, M. G. S. Vieira, N. V. Gramosa, C. Chaibundit, M. C. de Mattos, D. Attwood, S. G. Yeates, S. K. Nixon and N. M. P. S. Ricardo, Int. J. Pharm., 2012, 436, 631–635 CrossRef CAS PubMed.
- C. Klammt, D. Schwarz, K. Fendler, W. Haase, V. Dötsch and F. Bernhard, FEBS J., 2005, 272, 6024–6038 CrossRef CAS PubMed.
- D. R. Perinelli, M. Cespi, N. Lorusso, G. F. Palmieri, G. Bonacucina and P. Blasi, Langmuir, 2020, 36, 5745–5753 CrossRef CAS PubMed.
- T. Terashima, J. Oleo Sci., 2020, 69, 529–538 CrossRef CAS PubMed.
- M. Hibino, K. Tanaka, M. Ouchi and T. Terashima, Macromolecules, 2022, 55, 178–189 CrossRef CAS.
- T. L. Nguyen, K. Ishihara and S.-I. Yusa, Polymers, 2022, 14, 577 CrossRef CAS PubMed.
- V. S. Kravchenko, V. Abetz and I. I. Potemkin, Polymer, 2021, 235, 124288 CrossRef CAS.
- PharmaCentral, Polyethylene Glycol | Uses, Suppliers, and Specifications, available at: https://pharmacentral.com/product/polyethylene-glycol-pharmaceutical-excipient/.
- The Dow Chemical Company, Formulating CARBOWAX™ SENTRY™ PEGs in ointment applications, available at: https://www.cphi-online.com/46/resourcefile/14/35/18/file.pdf.
- G. Hattori, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2018, 140, 8376–8379 CrossRef CAS PubMed.
- S. Imai, Y. Ommura, Y. Watanabe, H. Ogawa, M. Takenaka, M. Ouchi and T. Terashima, Polym. Chem., 2021, 12, 1439–1447 RSC.
- Merck, Safety data sheet., Oleyl alcohol, available at: https://www.sigmaaldrich.com/DE/de/sds/aldrich/369314?userType=anonymous, accessed 8 December 2024.
- Merck, Safety data sheet., Stearyl alcohol, available at: https://www.sigmaaldrich.com/DE/de/sds/sial/phr1115?userType=anonymous, accessed 8 December 2024.
- J. W. Hamersma and E. I. Snyder, J. Org. Chem., 1965, 30, 3985–3988 CrossRef CAS.
- J. T. Groves and K. W. Ma, J. Am. Chem. Soc., 1977, 99, 4076–4082 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- S. Schüttner, G. M. Linden, E. C. Hoffmann, P. Holzmüller and H. Frey, Polym. Chem., 2025, 16, 374–385 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, 1H NMR, 13C NMR, DOSY, evaluation data for 1H NMR copolymerization kinetic, additional characterization data (SEC, MALDI-ToF, DSC, TEM, DLS and fluorescence spectroscopy). See DOI: https://doi.org/10.1039/d5py00159e |
| This journal is © The Royal Society of Chemistry 2025 |