
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymethylene with cage silsesquioxane: densely grafted structure prevents side-chain crystallization†
Yu
Tomioka
a,
Tomoki
Yasui
a,
Kensuke
Naka
 ab and
Hiroaki
Imoto
*abc
ab and
Hiroaki
Imoto
*abc
aFaculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, Goshokaido-cho, Matsugasaki, Sakyo-ku, Kyoto 606-0962, Japan. E-mail: himoto@kit.ac.jp
bMaterials Innovation Lab, Kyoto Institute of Technology, Goshokaido-cho, Matsugasaki, Sakyo-ku, Kyoto 606-0962, Japan
cFOREST, JST, Honcho 4-1-8, Kawaguchi, Saitama 332-0012, Japan
First published on 29th January 2025
Abstract
Cage silsesquioxane (POSS, polyhedral oligomeric silsesquioxane) is a crucial building block in the development of organic–inorganic hybrid polymers. However, polymers incorporating POSS units in their side chains often exhibit poor film-forming properties because of the high crystallinity of POSS. In this study, polymethylene densely grafted with POSS units was synthesized. This POSS-polymethylene structure produced a homogeneous, transparent cast film, in contrast to the turbid film formed by POSS-polyacrylate. The significant difference in the film quality was attributed to the inhibition of POSS crystallization, facilitated by the dense tethering of POSS on the polymethylene backbone.
Introduction
The integration of inorganic elements into organic polymers is a significant strategy for achieving enhanced performance and functionality, which are often unattainable using commodity polymers.1,2 Siloxane, which is particularly valued for its stability, transparency, structural diversity, and modifiability, is a prime candidate for this purpose.3 A notable siloxane variant, polyhedral oligomeric silsesquioxane (POSS), is well-suited for creating polymerizable molecular structures.4 POSS has been incorporated into polymers at various points, including the main chain,5 side chain,6 cross-linking sites,7 and terminal ends.8 Among these, side-chain polymers are essential for achieving high-POSS-content polymers. The synthesis of side-chain monomers is relatively simple because mono-functional POSS derivatives can be easily obtained from commercially available sources.9 However, homopolymers containing POSS in each repeating unit are rare (Fig. 1a).10 The high crystallinity of POSS often leads to the aggregation of high-POSS content polymers, producing turbid and brittle films.10a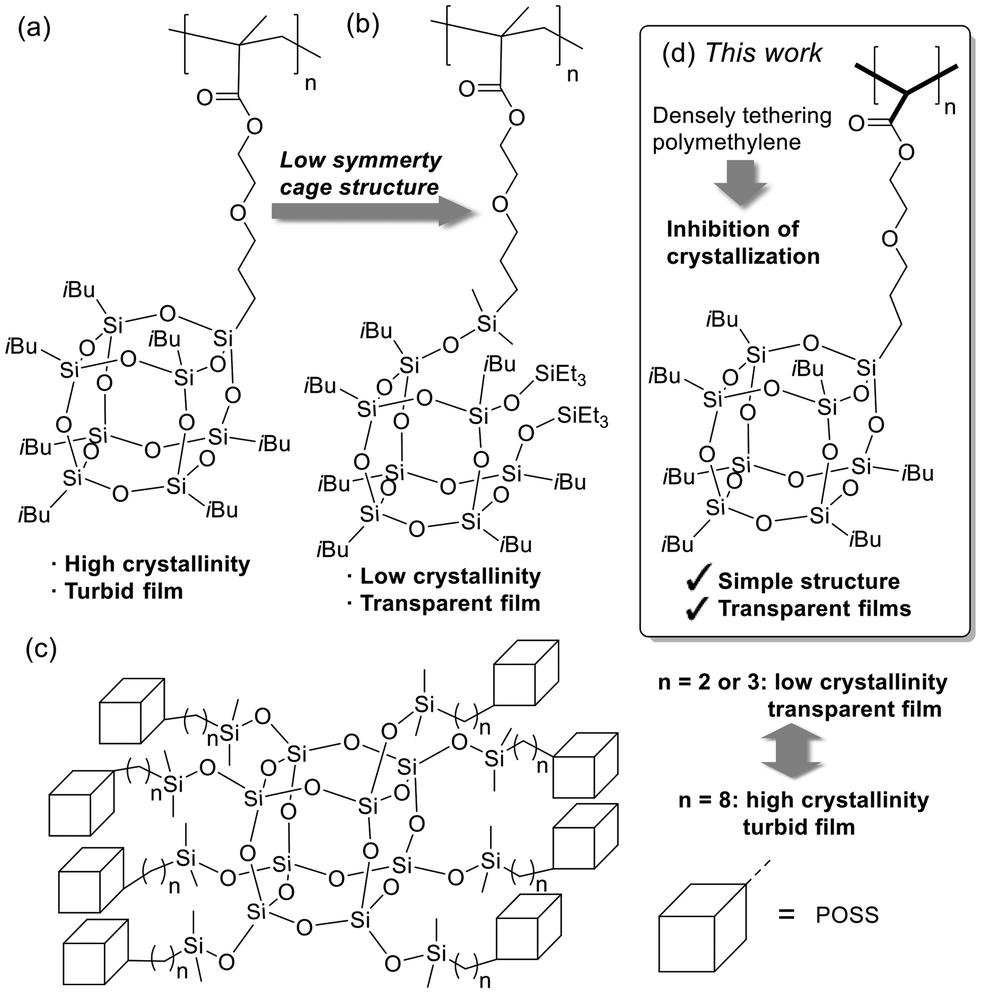 | ||
| Fig. 1 (a) Cubic and (b) open-cage POSS polymers. (c) POSS dendrimers on OS core. (d) POSS polymethylene (this work). | ||
To address the challenges posed by the crystallinity, we focused on the development of amorphous POSS homopolymers.10a,11 For instance, by incorporating partially hydrolyzed cage structures, the crystallinity of homopolymers can be reduced as the cages disrupt the symmetry of the POSS units, allowing for the formation of amorphous, optically transparent films (Fig. 1b).11a–c However, the selective synthesis of open-cage POSS monomers remains highly dependent on the substituents attached to POSS. Therefore, a versatile and simple strategy for inhibiting POSS crystallization is highly desired.
In a previous study, we investigated a dendritic POSS octamer in which eight POSS units were grafted onto an octasilicate (OS) core (Fig. 1c).12 Shorter linkers between the POSS units and the OS core produced transparent films, whereas longer linkers promoted POSS crystallization.12a This result suggests that densely substituted POSS units can prevent the formation of a crystalline structure, a concept that we extended to POSS-tethered polymers.
Polymethylene serves as an excellent platform for densely aligning functional units in polymer side chains.13 Unlike the POSS side chains in polymethacrylate, which tend to aggregate, the densely aligned structure of POSS in polymethylene is expected to inhibit crystallization. For polymethylene synthesis, Pd-catalyzed denitrogenative polymerization of diazoacetate monomers is highly effective.13 In this process, Pd–Ph species, formed via transmetalation from tetraphenylborate (BPh4−), initiate polymerization. During propagation, the Pd center reacts with the diazoacetate monomer, generating a Pd–carbene intermediate through denitrogenation, followed by migratory insertion of the carbene carbon into the Pd–Ph bond. Ihara et al. created polymethylenes with bulky side chains, such as cyclotriphosphazenes and poly(benzyl ether) dendrons, through the Pd-catalyzed denitrogenative polymerization of diazoacetate monomers.14 Importantly, compared to less sterically hindered side chains, bulky side chains can suppress undesirable side reactions, enabling well-controlled polymerization. This polymerization method is particularly advantageous for synthesizing POSS-tethered polymethylenes because of the bulkiness of the POSS units.
In this study, we design a novel POSS-diazoacetate monomer, where the Pd-catalyzed denitrogenative polymerization yields a well-controlled POSS-tethering polymethylene. As expected, the resulting polymer forms a transparent film, whereas the polyacrylate variant undergoes aggregation and produces a turbid film. X-ray diffraction (XRD) analysis provides insights into the assembly structures of the POSS units in polymethylene and polyacrylate.
Results and discussion
The syntheses of the POSS-diazoacetate and POSS-acrylate monomers (3 and 4, respectively) are outlined in Scheme 1. The procedures for synthesizing 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and 410a were adapted from previous reports. Heptaisobutyl-POSS alcohol 1 was prepared according to a previously described procedure.10a Condensation of 1 with bromoacetyl bromide yielded POSS-bromoacetate 2, which was subsequently reacted with N,N′-bis(p-toluenesulfonyl)hydrazine to produce POSS-diazoacetate monomer 3. Similarly, the condensation of 1 with acryloyl chloride afforded POSS-acrylate monomer 4. The structures and purities of the synthesized compounds were confirmed using a combination of NMR spectroscopy, MALDI-TOF MS, elemental analysis, Fourier transform infrared (FT-IR) spectroscopy, and size exclusion chromatography (SEC).
15 and 410a were adapted from previous reports. Heptaisobutyl-POSS alcohol 1 was prepared according to a previously described procedure.10a Condensation of 1 with bromoacetyl bromide yielded POSS-bromoacetate 2, which was subsequently reacted with N,N′-bis(p-toluenesulfonyl)hydrazine to produce POSS-diazoacetate monomer 3. Similarly, the condensation of 1 with acryloyl chloride afforded POSS-acrylate monomer 4. The structures and purities of the synthesized compounds were confirmed using a combination of NMR spectroscopy, MALDI-TOF MS, elemental analysis, Fourier transform infrared (FT-IR) spectroscopy, and size exclusion chromatography (SEC).
 | ||
| Scheme 1 Syntheses of POSS monomers 3 and 4. Cube indicates heptaisobutyl-substituted POSS unit. | ||
The reaction conditions for the synthesis of POSS-tethered polymethylene P1 were optimized (Table 1). The number-average molecular weight (Mn) and weight-average molecular weight (Mw) were estimated by SEC using polystyrene standards. Polymerization was initially conducted in tetrahydrofuran (THF) using allylpalladium(II) chloride (π-allylPdCl) as the catalyst, albeit with a low conversion of only 15% (entry 1). Inspired by the work of Ihara and Shimomoto,16 we introduced sodium tetraphenylborate (NaBPh4) as a co-catalyst (entry 2), which significantly improved both the molecular weight (Mn = 8100) and conversion (73%), with a low dispersity index (Mw/Mn = 1.11). This narrow dispersity suggests that the bulky side chains of POSS effectively suppress the side reactions.
| Entry | Catalyst | Temp. [°C] | [3]/[Pd]a | [3] [mM] | M n | M w/Mnb | Conversionb [%] |
|---|---|---|---|---|---|---|---|
| a Molar ratio of monomer 3 and Pd catalyst. b Estimated by SEC with polystyrene standards (THF, 1 mL min−1). | |||||||
| 1 | π-AllylPdCl | 25 | 50 | 200 | 5400 | 1.09 | 15 |
| 2 | π-AllylPdCl/NaBPh4 | 25 | 50 | 200 | 8100 | 1.11 | 73 |
| 3 | π-AllylPdCl/NaBPh4 | −15 | 50 | 200 | 7700 | 1.11 | 70 |
| 4 | π-AllylPdCl/NaBPh4 | 50 | 50 | 200 | 7500 | 1.10 | 66 |
| 5 | π-AllylPdCl/NaBPh4 | 25 | 30 | 200 | 6700 | 1.09 | 66 |
| 6 | π-AllylPdCl/NaBPh4 | 25 | 100 | 200 | 7100 | 1.14 | 52 |
| 7 | π-AllylPdCl/NaBPh4 | 25 | 200 | 200 | 7100 | 1.14 | 42 |
| 8 | π-AllylPdCl/NaBPh4 | 25 | 50 | 500 | 7500 | 1.11 | 70 |
Further variations in the reaction temperature, such as lowering it to −15 °C or raising it to 50 °C (entries 3 and 4), resulted in slight decreases in the molecular weight and conversion. Adjusting the ratio of the monomer to the Pd catalyst ([3]/[Pd]) (entries 5–7) and changing the monomer concentration (entry 8) did not significantly enhance the Mn (up to 7500) or conversion (up to 70%). The lack of significant improvement with increased [3]/[Pd] ratios might be attributed to chain transfer reactions, as suggested by Ihara and Shimomoto in similar polymerization systems.17 Therefore, the optimal conditions were identified, as described in entry 2.
Polymer P1 was synthesized under these optimized conditions and purified by preparative HPLC to remove the low-molecular-weight components (Scheme 2a). During purification, the dimer D1 was isolated as a mixture of cis and trans isomers (cis/trans = 21/79), as identified by NMR spectroscopy and MALDI-TOF MS. This dimer likely formed through C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation between diazo-bearing carbons of the monomers during polymerization, as previously reported in similar systems.14b,17 Polymer P2, the POSS-tethered polyacrylate, was synthesized via free-radical polymerization using 2,2′-azobis(isobutyronitrile) (AIBN) as the initiator, following a reported procedure for POSS-tethered polymethacrylates (Scheme 2b).10a While monomer conversion was relatively high (81%), the isolated yield was reduced to 44% due to the purification process involving preparative HPLC (Fig. S27†). SEC analysis confirmed that the molecular weights of P1 (Mn = 8100) and P2 (Mn = 8400) were similar, although P2 exhibited higher dispersity (Mw/Mn = 1.45) than P1 (Mw/Mn = 1.08), likely because of the nature of free-radical polymerization in P2.
C bond formation between diazo-bearing carbons of the monomers during polymerization, as previously reported in similar systems.14b,17 Polymer P2, the POSS-tethered polyacrylate, was synthesized via free-radical polymerization using 2,2′-azobis(isobutyronitrile) (AIBN) as the initiator, following a reported procedure for POSS-tethered polymethacrylates (Scheme 2b).10a While monomer conversion was relatively high (81%), the isolated yield was reduced to 44% due to the purification process involving preparative HPLC (Fig. S27†). SEC analysis confirmed that the molecular weights of P1 (Mn = 8100) and P2 (Mn = 8400) were similar, although P2 exhibited higher dispersity (Mw/Mn = 1.45) than P1 (Mw/Mn = 1.08), likely because of the nature of free-radical polymerization in P2.
 | ||
| Scheme 2 Syntheses of (a) polymer P1 and dimer D1, and (b) polymer P2. | ||
In the 1H-NMR spectra, the signals corresponding to the isobutyl groups (δ = 1.86 (methyne), 0.95 (methyl), 0.59 (methylene) ppm) in P1 (Fig. S10†) were notably broader than those in P2 (Fig. S18†), indicating that the molecular mobility of the POSS units in P1 was more restricted. Additionally, the 1H-NMR spectrum of P1 exhibited a broad signal corresponding to the methylene units in the main chain around 3.4–2.8 ppm. The assignment of this signal was supported by the 1H–13C HSQC NMR spectrum (Fig. S13†). Ihara and Shimomoto previously reported that polymerization catalyzed by π-allylPdCl results in atactic polymethylene, with broad methylene signals around 3 ppm.18 Therefore, it was reasonable to conclude that P1 had an atactic structure. The weak and broad signal around 7.2 ppm in the 1H NMR spectrum, corresponding to the terminal group of P1, was insufficiently resolved to determine the Mn value from integral ratios.
Chloroform (CHCl3) solutions of polymers P1 and P2 (75 g L−1) were cast onto glass substrates and dried under ambient conditions (25 °C, 1 atm) for 24 h. The cast film of P1 was transparent, whereas that of P2 appeared turbid (Fig. 2a). Even after annealing at around the melting points for 24 h, the film appearances remained unchanged (Fig. S31†). This suggests that the dense tethering structure of the polymethylene backbone in P1 effectively inhibited the aggregation of the POSS units. In contrast, the polyacrylate backbone of P2 allowed for greater molecular mobility, promoting the crystallization of POSS. The scanning electron microscopy (SEM) images of P1 and P2 revealed no micrometer-scale surface roughness (which can cause light scattering), although cracks were present. This indicates that the aggregates in P2 were likely at the submicron scale, contributing to visible-light scattering and resulting in turbidity. The transmittance spectra (Fig. 2b) confirmed that P1 exhibited significantly higher transparency than P2 in the visible-light range (350–800 nm).
 | ||
| Fig. 2 (a) Photographs (left) and SEM images (right) of P1 and P2. (b) Transmittance spectra of P1 and P2. | ||
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were used to evaluate the thermal properties of P1, P2, and monomeric POSS 1 (Fig. 3 and Table 2). The TGA results showed that the decomposition temperatures at 5 wt% loss (Td5) were 299 °C (P1), 340 °C (P2), and 239 °C (1) (Fig. 3a). Polymerization enhanced the thermal stability of both P1 and P2 compared to that of monomeric POSS 1. The higher thermal stability of P2 compared to that of P1 is likely due to aggregation of the POSS units, which is more pronounced in polyacrylate-based structures. Generally, polyacrylates exhibit better thermal stability than polymethylenes.19
 | ||
| Fig. 3 (a) TGA thermograms and (b) DSC curves (second scan) of P1, P2, and 1 (under N2, 10 °C min−1). Arrows indicate the melting points. | ||
DSC measurements (Fig. 3b) revealed that the melting points (Tm) of P1, P2, and 1 were 122, 162, and 170 °C, respectively. The melting peaks of P1 and P2 were reproducible across three heating–cooling cycles. During the cooling process, a clear crystallization peak was observed for P2 (Fig. S30†). The data indicate that the crystalline domains in P2 were significantly larger than those in P1 and comparable to those in 1. The estimated melting enthalpy (ΔH) of P1 (1.2 J g−1) was much lower than that of P2 (17.9 J g−1), suggesting that the crystallinity of POSS in P1 was greatly reduced by the polymethylene backbone. Furthermore, the smaller estimated melting entropy (ΔS) of P1 (3.1 mJ K−1 g−1) compared that of P2 (40.9 mJ K−1 g−1) supports the postulate that the molecular mobility of the POSS units in P1 was more restricted. The ΔH and ΔS values for 1 (19.8 J g−1 and 44.6 mJ K−1 g−1, respectively) were similar to those of P2, indicating that the POSS units in P2 were able to crystallize, similarly to monomeric POSS, due to the higher molecular mobility afforded by the polyacrylate backbone.
XRD analysis was performed to further investigate the aggregation structures of the POSS units in P1, P2, and 1 (Fig. 4a). The peak positions in the XRD patterns of P1 and P2 corresponded well with those of 1, indicating that the signals are associated with aggregation of the POSS units. However, the XRD peak of P1 was much broader than that of P2, reflecting the lower crystallinity of the POSS units in P1. This difference explains why P1 formed a transparent film, whereas P2 produced a turbid film.
 | ||
| Fig. 4 (a) XRD patterns of P1, P2, and 1. (b) Schematic of plausible mechanism for differences in the crystallization behaviors of P1 and P2. | ||
A plausible mechanism for the inhibition of POSS crystallization in P1 is shown in Fig. 4b. The tightly packed structure of the POSS units along the polymethylene backbone in P1 prevents crystallization during film formation. Additionally, the densely tethered POSS units provide insufficient space for crystallization along the polymethylene chains. In contrast, the POSS units in P2, which have a polyacrylate backbone, have higher molecular mobility and sufficient space to form crystallized structures.
Conclusions
Polymethylene P1 was successfully synthesized to achieve dense alignment of the POSS units in the side chain. The Pd-catalyzed denitrogenative polymerization of POSS-diazoacetate monomer 3 was well controlled, with the bulky POSS units ensuring low dispersity index in P1. The cast film of P1 was transparent and homogeneous, in stark contrast to its polyacrylate counterpart, P2, which formed aggregates, resulting in a turbid film. DSC and XRD analyses revealed that the crystallization of the POSS units in P1 was effectively suppressed by the densely tethered polymethylene backbone. This study demonstrated a novel molecular design approach for the development of side-chain POSS polymers with excellent film-forming properties. Further studies on a range of POSS polymethylenes are ongoing, and the results will be reported in future publications.Experimental
Materials
Potassium carbonate (K2CO3), dichloromethane (CH2Cl2), sodium chloride (NaCl), 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), chloroform (CHCl3), ethyl acetate (EtOAc), triethylamine (Et3N), acetonitrile (MeCN), hydrochloric acid (35%), α,α′-azobisisobutyronitrile (AIBN), and tetrahydrofuran (THF, HPLC grade) were purchased from Nacalai Tesque (Kyoto, Japan). CH2Cl2 (super dehydrated grade), distilled water, anhydrous magnesium sulfate (MgSO4), sodium hydrogen carbonate (NaHCO3), n-hexane, toluene (super dehydrated grade), and CDCl3 (D = 99.8%) were purchased from FUJIFILM Wako Pure Chemical Co. (Osaka, Japan). Bromoacetyl bromide, acryloyl chloride, and sodium tetraphenylborate (NaBPh4) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Superdehydrated THF (super dehydrated grade) was purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). Allylpalladium(II) chloride dimer ([π-allylPdCl]2) was purchased from Sigma-Aldrich (Hattiesburg, MS). Heptaisobutyl-hydroxyethoxypropyl CC-POSS (1)10a and N,N′-ditosylhydrazine15 were prepared according to literature procedures. The starting material for 1 (heptaisobutyl trisilanol POSS) was purchased from Hybrid Plastics Inc. (Hattiesburs, MS).Measurements
1H (400 MHz), 13C (100 MHz), and 29Si (80 MHz) nuclear magnetic resonance (NMR) spectra were recorded at 25 °C on a Bruker AVANCE III 400 NMR spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) in CDCl3. The following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The NMR measurements were performed with a sample concentration of about 30 mg in 0.4 mL of solvent. The polymer's 13C-NMR spectra were collected following 12 hours of signal accumulation. The 2D NMR (HSQC and HMBC) spectra were recorded at 25 °C on a Bruker AVANCE III 500 NMR spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) in CDCl3. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) data were recorded on a JMS-S3000 instrument (JEOL Ltd, Tokyo, Japan) using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as the matrix (20 mg mL−1 in CHCl3) and sodium trifluoroacetate as a cationizing agent (1 mg mL−1 in THF). The molecular weights were determined by size exclusion chromatography (SEC) using an LC-6AD column (Shimadzu, Kyoto, Japan) equipped with a Shodex KF-805L column (Showa Denko, Tokyo, Japan) and an RI detector RID-20A (Shimadzu). Preparative high-performance liquid chromatography (HPLC) for purification was performed on an LC-6AD (Shimadzu) instrument with a Shodex KF-2003 (Showa Denko) tandem column system using CHCl3 as the eluent (4.0 mL min−1). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed by using Shimadzu DTG-60 and DSC-60 Plus (Shimadzu) instruments, respectively, under N2 atmosphere, at heating rate of 10 °C min−1. Transmittance spectra were recorded using a JASCO spectrophotometer (V-670 KNN, JASCO). X-ray diffractometry (XRD) studies were performed on a Rigaku MiniFlex600-C X-ray diffractometer with Cu-Kα radiation (λ = 1.5406 Å) in the 2θ/θ mode at room temperature. The 2θ scan data were collected at 0.01° intervals; the scan speed was 10° (2θ) min−1. Scanning electron microscopy (SEM) images of the films were obtained using a VE-8800 system (KEYENCE, Osaka, Japan). Elemental analyses were performed on a YANAKO CHN Corder MT-5. Fourier transform infrared (FT-IR) spectra were obtained using a JASCO FT/IR-4600 spectrometer (JASCO).Synthesis procedure and characterization data
:1) afforded 3 as a pale yellow powder (1.48 g, 1.50 mmol, 47%). 1H-NMR (in CDCl3, 400 MHz): δ (ppm): 4.79 (s, 1H), 4.32–4.29 (m, 2H), 3.64–3.61 (m, 2H), 3.43 (t, J = 7.0 Hz, 2H), 1.92–1.78 (m, 7H), 1.72–1.64 (m, 2H), 0.95 (d, J = 6.8 Hz, 42H), 0.62–0.57 (m, 16H). 13C-NMR (in CDCl3, 100 MHz): δ (ppm): 167.0, 73.6, 68.6, 64.2, 46.4, 25.8, 24.0, 22.9, 22.6, 8.3. 29Si-NMR (in CDCl3, 80 MHz): δ (ppm): −67.4, −67.7, −67.9. MALDI-TOF-MS (m/z. [M + Na]+): 1009.3141, (calc.); 1009.3028 (observed). Anal. calcd for C35H74N2O15Si8: C, 42.56; H, 7.55; N, 2.84. Found: C, 42.36; H, 7.74; N, 2.48.
Film fabrication
A CHCl3 solution (0.20 mL) of the polymer (15 mg) was prepared. The solution was cast onto a glass substrate and dried at 25 °C for 24 h.Author contributions
Y. Tomioka: synthesis, structural analysis, data curation, writing – original draft; T. Yasui: structural analysis, data curation, writing – original draft; K. Naka: conceptualization, investigation, writing – review and editing, supervision; H. Imoto: conceptualization, investigation, writing – original draft, writing – review and editing, funding acquisition project administration, supervision.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate Prof. Hiroaki Shimomoto (Ehime University) for the constructive discussion on the synthesis and structural analysis of polymethylene.References
- (a) Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jaekle, Wiley, 2018 Search PubMed; (b) New Polymeric Materials Based on Element-Blocks, ed. Y. Chujo, Springer, Singapore, 2019 Search PubMed.
- (a) J. Bedard and S. Chitnis, Chem. Mater., 2023, 35, 8338–8352 CrossRef CAS; (b) F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed; (c) M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 109–126 CrossRef CAS; (d) M. Gon, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2017, 90, 463–474 CrossRef CAS.
- Silicones and Silicone-Modified Materials, ed. S. J. Clarson, J. J. Fitzgerald, M. J. Owen and S. D. Smith, ACS Symposium Series, 2000, vol. 729 Search PubMed.
- For reviews, see: (a) F. Chen, F. Lin, Q. Zhang, R. Cai, Y. Wu and X. Ma, Macromol. Rapid Commun., 2019, 40, 1900101 CrossRef PubMed; (b) H. Shi, J. Yang, M. You, Z. Li and C. He, ACS Mater. Lett., 2020, 2, 296–316 CrossRef CAS; (c) C. Wang, L. Zhou, Q. Du, T. Shan, K. Zheng, J. He, H. He, S. Chen and X. Wang, Polym. Int., 2022, 71, 379–392 CrossRef CAS; (d) Y. Liu, X.-Y. Yan, Q.-Y. Guo, H. Lei, X. Liu, X. Li, Y. Wu, W. Zhang, G. X. Liu and S. Z. D. Cheng, Macromol. Chem. Phys. Commun., 2023, 224, 2200357 CrossRef CAS; (e) L. Miao, L. Zhan, S. Liao, Y. Li, T. He, S. Yin, L. Wu and H. Qiu, Macromol. Rapid Commun., 2024, 45, 2300601 CrossRef CAS PubMed; (f) L. Li, H. Wang and S. Zheng, J. Polym. Sci., 2024, 64, 583–613 CrossRef.
- Recent papers, see: (a) H. Wang, L. Li, T. Zhang and S. Zheng, Eur. Polym. J., 2024, 202, 112652 CrossRef CAS; (b) H. Wang, G. Hang, J. Hu, Y. Gao, L. Li and S. Zheng, ACS Appl. Polym. Mater., 2023, 5, 4274–4287 CrossRef CAS; (c) T. Kamitani, M. Nakamura, S. Watase, T. Suzuki, H. Imoto and K. Naka, ACS Appl. Polym. Mater., 2023, 5, 5169–5176 CrossRef CAS; (d) H. Chi, M. Wang, J. Li, H. Tian, Y. T. Chong, S. H. Lim, Y. Wang and F. Wang, ACS Appl. Polym. Mater., 2022, 4, 5882–5890 CrossRef; (e) J. Guan, Z. Zhang and R. M. Laine, Macromolecules, 2022, 55, 5403–5411 CrossRef CAS.
- Recent papers, see: (a) F. Feng, S. Zhang, W. Wang, C. Hong, M. Zhang, F. Yang, Y. Peng, F. Jia and H. Liu, Giant, 2024, 19, 100304 CrossRef CAS; (b) A. R. Uribe, Polymer, 2023, 275, 125933 CrossRef; (c) S. Iwao, N. Kurono, W. Higashiguchi, T. Hayakawa, N. Ohta, K. Kamitani, S. Fujii, Y. Nakamura and T. Hirai, Chem. Lett., 2022, 51, 781–783 CrossRef CAS; (d) A. Takahashi, T. Okada, K. Nakano, Y. Ishida and A. Kameyama, Polym. J., 2021, 53, 1213–1222 CrossRef CAS; (e) Z. Li, Z. Li, J. Hu, X. Feng, M. Zhang, G. Duan, R. Zhang and Y. Li, ACS Macro Lett., 2021, 10, 1563–1569 CrossRef CAS PubMed; (f) N. Hasan, K. Busse, A. Ullah, H. Hussain and J. Kressler, Langmuir, 2021, 37, 13399–13408 CrossRef CAS PubMed; (g) J.-F. Yin, H. Xiao, P. Xu, J. Yang, Z. Fan, Y. Ke, X. Ouyang, G. X. Liu, T. L. Sun, L. Tang, S. Z. D. Cheng and P. Yin, Angew. Chem., Int. Ed., 2021, 60, 22212–22218 CrossRef CAS PubMed.
- Recent papers, see: (a) S. Samanta, S. Sarkar and N. K. Singha, ACS Appl. Mater. Interfaces, 2023, 15, 24812–24826 CrossRef CAS PubMed; (b) J. Yang, Y. Zhang, M. Hao, J. Zhi and X. Qian, Polymer, 2023, 268, 125719 CrossRef CAS; (c) Q. Liu, M. Ye, G. Yu and A. Han, J. Appl. Polym. Sci., 2023, 140, e53429 CrossRef CAS; (d) X. Xu, J. D. Skelly and J. Song, ACS Appl. Mater. Interfaces, 2023, 15, 2693–2704 CrossRef CAS PubMed; (e) S. Ida, T. Hikida, A. Kawai, T. Matsuda, S. Suzuki, H. Imoto, K. Naka and S. Kanaoka, Polym. Chem., 2023, 14, 2771–2778 RSC; (f) K. A. Stewart, D. P. DeLellis, J. J. Lessard, J. F. Rynk and B. S. Sumerlin, ACS Appl. Mater. Interfaces, 2023, 15, 25212–25223 CrossRef CAS PubMed.
- Recent papers, see: (a) J. Dong, L. Li, P. Qiu, Y. Pan, Y. Niu, L. Sun, Z. Pan, Y. Liu, L. Tan, X. Xu, C. Xu, G. Luo, Q. Wang and H. Wang, Adv. Mater., 2023, 35, 2211487 CrossRef CAS PubMed; (b) Y. Shao, B. Hou, W. Li, X. Yan, X. Wang, Y. Xu, Q. Dong, W. Li, J. He and W.-B. Zhang, Macromolecules, 2023, 56, 1562–1571 CrossRef CAS; (c) X. Liu, L. Tang, Y. Chen, M. Fu, Z.-H. Guo, W. Tang and K. Yue, Macromol. Rapid Commun., 2023, 44, 2200292 CrossRef CAS PubMed; (d) C. M. MacInnis, G. R. Younes and M. Marić, J. Appl. Polym. Sci., 2022, 139, e53225 CrossRef CAS.
- (a) J. F. Brown and L. H. Vogt, J. Am. Chem. Soc., 1965, 87, 4313–4317 CrossRef CAS; (b) F. J. Feher, S. H. Phillips and J. W. Ziller, Chem. Commun., 1997, 33, 829–830 RSC; (c) F. J. Feher, Chem. Commun., 1998, 34, 399–400 RSC.
- (a) Y. Sato, H. Imoto and K. Naka, J. Polym. Sci., 2020, 58, 1456–1462 CrossRef CAS; (b) Y. Tomioka, R. Tanaka, T. T. Pham, S. Yusa, H. Imoto and K. Naka, Polym. J., 2024, 56, 653–660 CrossRef CAS; (c) S. Tajbakhsh and M. Marić, J. Polym. Sci., 2020, 58, 2741–2754 CrossRef CAS; (d) X.-H. Zheng, J.-F. Zhao, T.-P. Zhao, T. Yang, X.-K. Ren, C.-Y. Liu, S. Yang and E.-Q. Chen, Macromolecules, 2018, 51, 4484–4493 CrossRef CAS; (e) C.-G. Chae, Y.-G. Yu, H.-B. Seo, M.-J. Kim, M. Y. L. N. Kishore and J.-S. Lee, Polym. Chem., 2018, 9, 5179–5189 RSC.
- (a) A. Igarashi, H. Imoto and K. Naka, Polym. Chem., 2022, 13, 1228–1235 RSC; (b) A. Igarashi, Y. Ueda, R. Katoh, H. Imoto and K. Naka, J. Polym. Sci., 2021, 59, 131–138 CrossRef CAS; (c) T. Nakano, K. Okamoto, H. Imoto and K. Naka, Polym. J., 2023, 55, 193–201 CrossRef CAS; (d) K. Okamoto, A. Igarashi, H. Imoto and K. Naka, J. Polym. Sci., 2022, 60, 214–220 CrossRef CAS.
- (a) Y. Yasumoto, T. Yamanaka, S. Sakurai, H. Imoto and K. Naka, Polym. J., 2016, 48, 281–287 CrossRef CAS; (b) H. Araki and K. Naka, Polym. J., 2012, 44, 340–346 CrossRef CAS; (c) R. Suzuki, L. Li, H. Imoto, H. Takagi, S. Sakurai and K. Naka, Polym. J., 2022, 54, 1179–1190 CrossRef CAS.
- (a) E. Ihara, Polym. J., 2025, 57, 1–23 CrossRef CAS; (b) H. Shimomoto, Polym. J., 2020, 52, 269–277 CrossRef CAS; (c) E. Ihara, Poly(substituted Methylene) Synthesis: Construction of C–C Main Chain from One Carbon Unit, in Polymer Materials. Advances in Polymer Science, ed. K. S. Lee and S. Kobayashi, Springer, 2010, vol. 231 Search PubMed.
- (a) F. Kato, A. Chandra, M. Tokita, H. Asano, H. Shimomoto, E. Ihara and T. Hayakawa, ACS Macro Lett., 2018, 7, 37–41 CrossRef CAS PubMed; (b) H. Shimomoto, R. Hohsaki, D. Hiramatsu, T. Itoh and E. Ihara, Macromolecules, 2020, 53, 6369–6379 CrossRef CAS.
- T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195–3197 CrossRef CAS PubMed.
- H. Shimomoto, H. Asano, T. Itoh and E. Ihara, Polym. Chem., 2015, 6, 4709–4714 RSC.
- E. Ihara, M. Akazawa, T. Itoh, M. Fujii, K. Yamashita, K. Inoue, T. Itoh and H. Shimomoto, Macromolecules, 2012, 45, 6869–6877 CrossRef CAS.
- (a) H. Shimomoto, J. Kawamata, H. Murakami, K. Yamashita, T. Itoh and E. Ihara, Polym. Chem., 2017, 8, 4030–4037 RSC; (b) H. Shimomoto, Y. Miyano, K. Kinoshita, T. Itoh and E. Ihara, Polym. Chem., 2023, 14, 1007–1018 RSC.
- M. Milovanović, R. Bošković, T. Tošić, L. Katsikas and I. G. Popović, Polym. Degrad. Stab., 2006, 91, 3221–3229 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, MALDI-TOFMS, and SEC. See DOI: https://doi.org/10.1039/d4py01222d |
| This journal is © The Royal Society of Chemistry 2025 |