
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStandard purification methods are not sufficient to remove micellular lipophilic dye from polymer nanoparticle solution†
Eric H.
Sterin
 a,
Laura A.
Weinstein
a,
Chitran
Roy Chowdhury
a,
Emma C.
Guzzetti
a and
Emily S.
Day
*abc
a,
Laura A.
Weinstein
a,
Chitran
Roy Chowdhury
a,
Emma C.
Guzzetti
a and
Emily S.
Day
*abc
aDepartment of Biomedical Engineering, University of Delaware, Newark, DE 19713, USA. E-mail: emilyday@udel.edu
bDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA
cCawley Center for Translational Cancer Research, Helen F. Graham Cancer Center and Research Institute, Newark, DE 19713, USA
First published on 6th March 2025
Abstract
Tracking nanoparticles’ location is imperative for understanding cellular interactions, pharmacokinetics, and biodistribution. DiD is a lipophilic dye commonly used to label nanoparticles for trafficking studies. Herein, we show that DiD micelles form in polymer NP solutions during synthesis and can lead to false positive results in downstream assays. Potential methods to remove these micelles are also described.
Introduction
Nanoparticles (NPs) have been increasingly studied in medical applications over the past century due to their ability to carry therapeutic cargo and deposit it at desired sites of action while minimizing delivery to off-target locations.1–3 Understanding the biodistribution, pharmacokinetics, and in vivo fate of NPs is critical for their successful implementation. One of the most common methods to track NPs in vitro and in vivo is by labeling them with fluorescent markers and then monitoring their location with fluorescence imaging techniques (such as IVIS, intravital microscopy, confocal microscopy, and more).4 Labeling of NPs for such studies may involve encapsulation of a fluorescent molecule inside the NP, incorporating a fluorescent molecule into the bulk NP structure via steric interactions or hydrogen bonding, and/or conjugation of a fluorescent molecule to the inside or outside of the NP.5 Alternatively, the NPs may be loaded with nucleic acids that encode for a fluorescent protein (such as green or red fluorescent protein (GFP/RFP)), which allows for analysis of cargo localization as well as bioactivity.6,7 One consideration when using fluorescent labeling techniques for cellular uptake or biodistribution studies is that it is imperative to remove any unbound/non-encapsulated molecules from the NP solution—otherwise, the analysis of cargo delivery based on fluorescent signal may be flawed.8 To emphasize the importance of this challenge, in this work we compare the ability of different purification methods to remove unbound, lipophilic fluorophores from poly(ethylene glycol)–poly(lactic-co-glycolic acid) (PEG–PLGA) NPs as a model delivery system.In this study, DiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, a far-red fluorescent lipophilic dye) is encapsulated in PEG–PLGA NPs as a representative fluorescent label. DiD and derivatives are commonly used to label cell membranes,9 extracellular vesicles,10 and other lipophilic NPs,11,12 but these carbocyanine dyes can form “dye-only particles” in aqueous solution.13 We show that these may appear in non-negligible quantities during the high energy solvent evaporation synthesis method used to produce PEG–PLGA NPs (or other forms of polymeric NPs) (Scheme 1). While the true conformation of these dye-only particles is unknown, they are likely either unstructured aggregates or structured micelles, and we refer to them as micelles in the text for simplicity. Unfortunately, these micelles have similar size, charge, and, likely, electron densities as the NPs themselves, which makes it difficult to separately characterize or distinguish dye-only micelles from dye-labeled NPs by conventional techniques (for example, dynamic light scattering, nanoparticle tracking analysis, zeta potential, and electron microscopy). We further demonstrate that standard NP purification methods, including filtering and pelleting by centrifugation, do not significantly remove these micelles (Scheme 1). While typical NP characterization methods could not detect these micelles, their presence could be confirmed using density gradient centrifugation. Moreover, we show that failure to remove these micelles has the potential to lead to false positive results when evaluating cell uptake of DiD-labeled NPs (Scheme 1). Given the ubiquity of such lipophilic dyes as labeling tools in NP research, it is critical that researchers be aware of this issue and take measures to mitigate the risk of skewing cell uptake and/or biodistribution study results by properly eliminating particle-free dye molecules from dye-labeled NPs.
 | ||
| Scheme 1 Scheme depicting the synthesis of PEG–PLGA NPs encapsulating DiD (DiD NPs) for fluorescence-based trafficking studies. DiD micelles that form in the solution along with the DiD NPs are challenging to remove with conventional purification techniques and can lead to false positive signal. Created with https://BioRender.com. | ||
Materials and methods
NP and micelle synthesis
DiD-loaded PEG–PLGA NPs (DiD NPs) were synthesized through solvent evaporation (Fig. S1†). PLGA (39.5 kDa, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 L:G ratio) was dissolved in acetone at 2 mg mL−1. Separately, methoxy–PEG (5 kDa)–PLGA (30 kDa) was dissolved in dichloromethane (DCM) at 2 mg mL−1. These solutions were mixed at a 3:1 PLGA:PEG–PLGA ratio for a final volume of 1 mL, to which 50 μL of 0.4 mg mL−1 DiD in dimethyl sulfoxide (DMSO) was added. This polymer/DiD solution was added dropwise to 3 mL of 0.1% poly(vinyl alcohol) (PVA) in water and probe sonicated. The solvent was evaporated by stirring at 800 rpm overnight at room temperature. As a control, DiD micelles (without NPs) were separately formed using the same procedure but without including either polymer. Cy5-conjugated NPs (Cy5 NPs) (used for comparison in cell uptake studies) were synthesized by replacing 20% of the PLGA with Cy5-capped PLGA (30–50 kDa, 50:50 L:G ratio) and removing the DiD.
:50 L:G ratio) was dissolved in acetone at 2 mg mL−1. Separately, methoxy–PEG (5 kDa)–PLGA (30 kDa) was dissolved in dichloromethane (DCM) at 2 mg mL−1. These solutions were mixed at a 3:1 PLGA:PEG–PLGA ratio for a final volume of 1 mL, to which 50 μL of 0.4 mg mL−1 DiD in dimethyl sulfoxide (DMSO) was added. This polymer/DiD solution was added dropwise to 3 mL of 0.1% poly(vinyl alcohol) (PVA) in water and probe sonicated. The solvent was evaporated by stirring at 800 rpm overnight at room temperature. As a control, DiD micelles (without NPs) were separately formed using the same procedure but without including either polymer. Cy5-conjugated NPs (Cy5 NPs) (used for comparison in cell uptake studies) were synthesized by replacing 20% of the PLGA with Cy5-capped PLGA (30–50 kDa, 50:50 L:G ratio) and removing the DiD.
NP purification by centrifugal filtration or pelleting
DiD NP and DiD micelle solutions were purified either by filtering (Fig. S2A†) or pelleting (Fig. S2B†) in the absence or presence of Triton X-100 (TX) to disrupt the micelle structure, resulting in the following study groups: DiD micelles, DiD micelles + TX, DiD NPs, and DiD NPs + TX. Filtered samples (Fig. S2A†) were washed thrice in 50 kDa molecular weight cutoff (MWCO) filter tubes by centrifuging for 20 minutes (3200 rcf, 4 °C), and resuspending the samples with 1 mL water between each centrifugation step. For TX groups, 2.5% TX was used for the first wash, and water was used for the second two. Pelleted samples (Fig. S2B†) were purified in a similar manner, with each centrifugation lasting 20 minutes (20000 rcf, 4 °C). After the first centrifugation, the samples were suspended in either water or 2.5% TX, and after the second and third centrifugation steps, they were suspended in water.
NP characterization
Nanoparticle tracking analysis (NTA) was used to measure the hydrodynamic diameter of the NPs and micelles after purification. Samples were diluted in a 1:2000 sample-to-water ratio and then analyzed with a Nanosight NS300 (Malvern) using the appropriate camera level and gain for each NP or micelle type. The zeta potential was measured via electrophoretic light scattering on a LiteSizer500 (Anton Paar) using a 1:50 sample-to-water dilution. Transmission electron microscopy (TEM) was employed to characterize the morphology of the nanoparticles. TEM samples were prepared by placing equal concentrations of small volumes on hydrophilic carbon support films with copper grids at 400 mesh, staining with 2% uranyl acetate, and drying. Electron micrographs were captured using a Talos L120C transmission electron microscope at the Delaware Biotechnology Institute. Lastly, the remaining fluorescent signal after purification of each sample was measured. Since DiD is only weakly fluorescent in water, samples containing DiD were diluted in a 1:9 sample-to-DMSO ratio, while Cy5-containing samples were diluted in water at the same ratio. All measurements were made on a Synergy H1M plate reader (BioTek) using a 640/670 excitation/emission pair alongside a standard curve of the same molecule.
Density gradient centrifugation
Density gradient centrifugation (DGC) was used to separate species of varying densities within the same sample. A discontinuous iodixanol gradient of 60% (bottom), 45% (middle), and 35% (top) iodixanol were layered on top of each other (1 mL each) and the sample was added to the top (<500 μL). Samples were centrifuged for 16 hours (100000 rcf, 4 °C). Photographs were acquired of the separated samples. To collect relevant bands, the top layers were aspirated, and the desired layers were recovered and diluted 4× in water, placed in 50 kDa MWCO centrifugal filter tubes and centrifuged for 15 minutes (3200 rcf, 4° C) to remove residual iodixanol. Finally, the samples were washed twice more with 1 mL of water.
Cell uptake analysis
Cell uptake studies utilized THP-1 cells (American Type Culture Collection; cultured in suspension in a 37 °C, 5% CO2 humidified incubator in RPMI-1640 Medium containing 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic (growth medium) per the manufacturer's recommendation) that were differentiated into M0 macrophages using standard methods as follows. THP-1 cells were plated in the desired container for 24 hours in growth medium containing 150 nM phorbol 12-myristate 13-acetate (PMA). For flow cytometry, THP-1 cells were plated at 20000 cells per well in a 96-well plate, and for confocal microscopy, they were plated at 80000 cells per well in an 8-well chamber plate. After the 24 h period, the differentiation medium was removed, and the M0 macrophages were incubated with equal total fluorescence of DiD NPs, DiD NPs collected after DGC (DGC DiD NPs), free DiD, or Cy5 NPs in growth medium. After 1 hour the cells were washed as follows and analyzed by flow cytometry or confocal microscopy. For flow cytometry, the cell media was replaced with phosphate-buffered saline (PBS), then the cells were trypsinized for 2 minutes, pelleted by centrifugation (5 minutes, 300 rcf, 4 °C) and suspended in PBS (three times), then analyzed on a NovoCyte flow cytometer (Agilent). Events were gated on cells using forward and side scatter, then on singlets using forward scatter height versus area. To calculate median fluorescence intensity, all fluorophores were excited using the 640 nm laser and detected in the 675/30 channel. For confocal microscopy, cells were washed by replacing the media with PBS, then fixed with 4% formaldehyde for 20 minutes at room temperature. The membranes were labeled by incubating the fixed cells with a wheat germ agglutinin-AlexaFluor488 conjugate (WGA) diluted in PBS to 5 μg mL−1. After 10 min at room temperature, the solution was removed, the cells were washed with PBS, the chamber sides were removed, and the samples were sealed with mounting media containing DAPI to label nuclei. Images were taken on an Andor Dragonfly Spinning Disc microscope equipped with 405 nm, 488 nm, 561 nm, 638 nm, and 735 nm laser lines using standard DAPI, FITC, and Cy5 emission channels.
Results and discussion
The appearance of DiD micelles within the DiD NP solution was first qualitatively observed by the blue color in the retentate at various steps during purification by filtering. There is a distinct lack of blue color in the filtrate of both DiD micelles and DiD NPs without TX (Fig. 1A), indicating that unencapsulated DiD formed clusters during the synthesis procedure that are larger than the 50 kDa MWCO pores of the centrifugal filter and could not be removed using this purification technique. These micelles are visualized in TEM images of the retentate, where DiD micelles, DiD NPs, and DiD NPs + TX all exhibit similar morphology (Fig. 1A). Adding TX to the DiD micelle and DiD NP solutions during the first step of the filtration process leads to the emergence of a blue color in the filtrate due to the disruption of the micelles (Fig. 1A). TEM images of DiD micelles + TX were typically absent of any type of morphology, confirming micellular disruption (Fig. 1A shows a representative region with absence of spherical structures), although rare aggregates were also observed (Fig. S3A†). These findings are confirmed by size, charge, and fluorescence measurements of DiD micelles and NPs purified by centrifugal filtration with and without TX (Fig. 1B). With the addition of TX to DiD micelles during purification, a drop in total fluorescence as well as a more neutral zeta potential is measured, indicating DiD removal and less micellular stability. While the drop in fluorescence (Fig. 1B) correlates with the dye removal observed visually and in TEM images (Fig. 1A), some of the measured reduction in fluorescence could be attributed to TX-induced fluorescence quenching. However, as filtering removes a significant amount of TX from the NP solution, the impact should be negligible in the measurement. Although there is an increase in the apparent size of DiD micelles with the addition of TX during filtering (Fig. 1B), it is likely due to aggregates that adhered to the filter itself being recollected, as is the nature of MWCO filters.14 These aggregates are insignificant, as they were rarely observed in TEM images (Fig. S3A†). It can also be seen that adding TX during filtering of DiD NPs does not change the size or charge but does reduce the total fluorescence signal by a comparable level to the DiD micelles – meaning that adding TX seems to remove free dye from the DiD NP solution during filtering.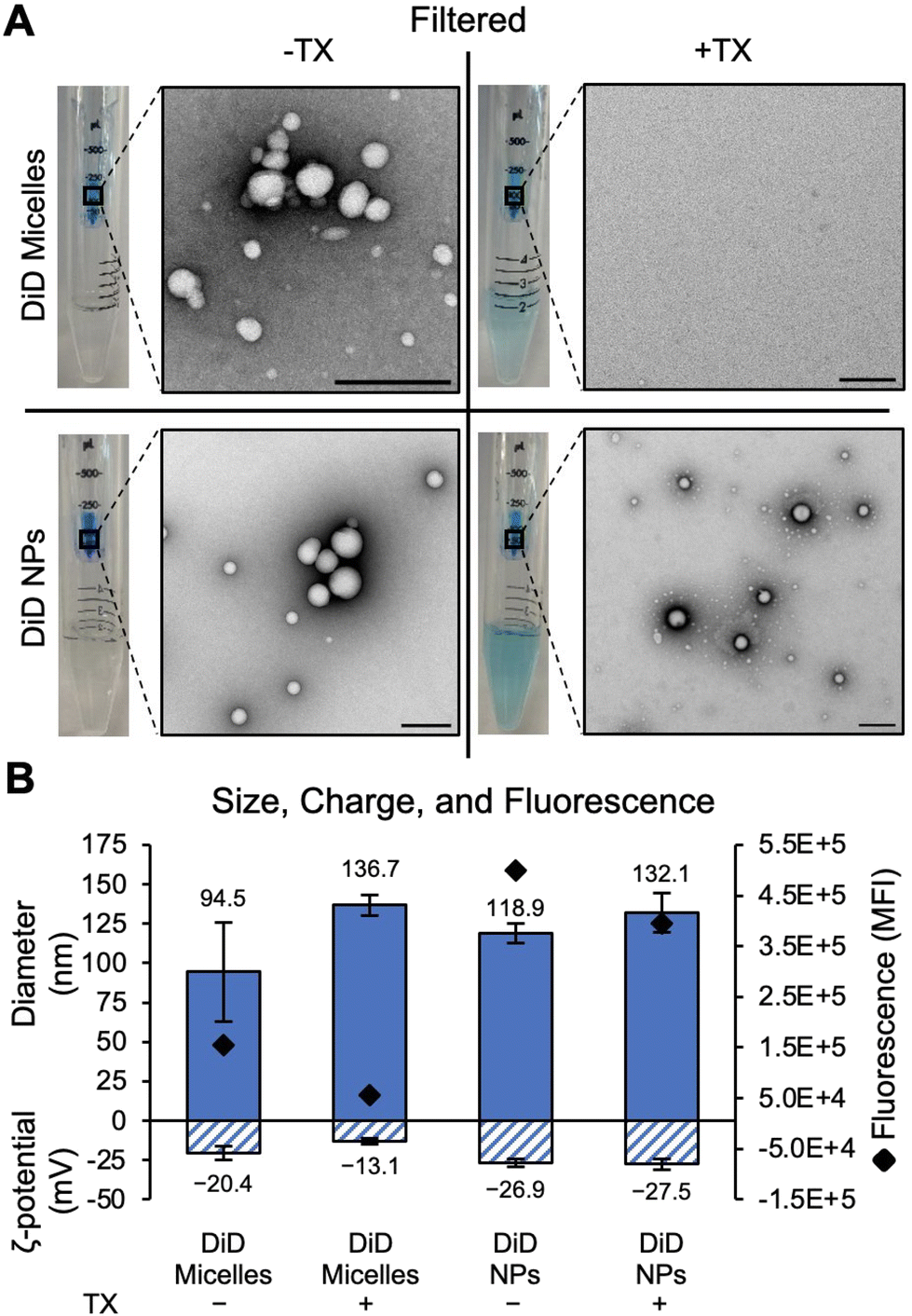 | ||
| Fig. 1 Characterization of DiD Micelles and DiD NPs purified by filtering with and without the addition of Triton X-100 (TX). (A) Photographs of filter tubes and representative TEM images (scale bars = 200 nm). (B) Hydrodynamic size, zeta potential, and total fluorescence of each sample. | ||
Adding TX during pelleting (Fig. 2) seems to be even more successful at removing free dye-only micelles. DiD micelles, DiD NPs, and DiD NPs + TX all retain a noticeable blue pellet while DiD micelles + TX do not; this is likely due to the disruption of the dye-only micelles which is visualized by the associated TEM images (Fig. 2A). While TEM images of DiD Micelles + TX purified by pelleting were largely void of any features, rare aggregates/particulates were observed (Fig. S3B†), similar to the finding for DiD Micelles + TX purified by filtering (Fig. S3A†). Quantitatively, adding TX to DiD micelles purified by pelleting nearly eliminates the fluorescent signal compared to DiD micelles without TX and reduces the hydrodynamic diameter (Fig. 2B) (the measured size may be due to the rare aggregates present in the samples). Likewise, adding TX to DiD NPs removes some, but not all, of the fluorescent signal and reduces the size as measured by NTA (Fig. 2B). The reduced fluorescence and diameter in the DiD NPs + TX group is likely due to the disruption of DiD micelles in the solution as well as the elimination of surface adsorbed dye, with the retained fluorescence being attributed to dye that is encapsulated in the NPs. Given that pelleting is a less gentle method of purification, it is not surprising that extra dye was removed from the solution by this technique as compared to purifying by filtration. There was no noticeable shift in zeta potential for DiD NPs pelleted in the absence or presence of TX (Fig. 2B), and the samples had similar TEM morphology, indicating the DiD NPs remained intact after pelleting with TX to remove DiD micelles.
 | ||
| Fig. 2 Characterization of DiD Micelles and DiD NPs purified by pelleting with and without the addition of TX. (A) Photographs of centrifuge tubes and representative TEM images (scale bars = 200 nm). (B) Hydrodynamic diameter, zeta potential, and total fluorescence of each sample. | ||
The results of the pelleting and filtering studies show that size and charge measurements alone provide minimal indication of the presence or absence of micelles in NP solutions before or after purification. Additional techniques (TEM, fluorescence measurement) and relevant controls were needed to identify the presence of multiple species within each solution. After demonstrating that DiD micelles could be reduced in NPs solutions by purifying in the presence of TX, we planned to study how this altered in vitro analysis of cellular uptake of the NPs. However, because TX could not be completely removed through centrifugal filtering or pelleting, cells exposed to these solutions were permeabilized, leading to subsequent cell death (Fig. S4†). Additionally, as the dye-only micelles are indistinguishable from the polymer NPs by NTA and zeta potential, we were unable to definitively prove that TX-washing removed all of the free DiD from the NP solution. To enable micelle elimination without inducing cell toxicity, a gentler/more cytocompatible detergent could be explored in the future.
Given the challenge of completely eliminating TX and free DiD from NP solutions by filtering/pelleting, we next investigated density gradient centrifugation (DGC) as a technique to separate DiD-loaded NPs from DiD micelles (Fig. 3). We hypothesized that the dye-only micelles would have a lower density than the polymer NPs containing DiD. To confirm this, we layered three discontinuous iodixanol phases (60%, 45%, and 35%) on top of each other and then added an aqueous phase containing either free DiD, empty NPs (prepared without DiD), DiD Micelles, DiD Micelles + TX, DiD NPs, or DiD NPs + TX (Fig. 3 and 4A). After ultracentrifugation, the components separate based on density, with lower density species stacked on top of higher density species. Free DiD settles underneath the aqueous phase at the top of the tube but above the layer of 35% iodixanol, as do DiD micelles (Fig. 3). DiD micelles + TX, due to disruption of their structure, become soluble molecules and remain in the uppermost aqueous phase. Empty NPs (prepared without DiD) exhibit a range of densities that span across the gradients we used, with bands appearing at both the 60%–45% interface and the 45%–35% interface. DiD NPs have both of those bands, plus a band at the same location as free DiD/DiD micelles. When TX is added to the DiD NPs, this band is solubilized and moves to the aqueous phase (Fig. 3). This proves that DiD NPs synthesized through solvent evaporation contain an extra species that is likely DiD micelles given the location of the band. Interestingly, with the addition of the TX, the NP band at the 45%–35% interface disappears. One potential explanation is that, as has been reported previously,15 adding TX to PLGA NPs acts as a plasticizer allowing the PLGA chains to form into a more energetically stable conformation. Another possible explanation is that TX is known to form micelles/sphere-like complexes with PEG.16 In this case, TX could interact with individual polymer chains on the exterior of the NPs causing new micelles to form and the NPs to change density. More research would be needed to fully understand these interactions and determine if this is a contributing factor. Finally, adding TX to the DiD NPs could be removing some of the encapsulated DiD from the NPs, leading to slight constriction of the NPs. In each of these cases, the NPs from the band at the 45%–35% interface could either move down to the 60%–45% interface or disassemble and move up to the aqueous phase.
 | ||
| Fig. 3 Representative images of different groups after density gradient centrifugation. | ||
 | ||
| Fig. 4 Method and characterization of DiD NPs purified by DGC. (A) Scheme of purification process. Created with https://BioRender.com. (B) TEM of DiD NPs after DGC. (C) Hydrodynamic diameter, zeta potential, and fluorescence of filtered DiD NPs before versus after DGC. | ||
Since DGC could successfully separate DiD NPs from DiD micelles, we collected and washed the bottom DiD NP band after DGC by centrifugal filtration to remove excess iodixanol (Fig. 4A). After DGC and washing, the DiD NPs retained their spherical morphology (Fig. 4B) and had similar size and charge to DiD NPs that had not undergone DGC (Fig. 4C) but with reduced total fluorescent signal (Fig. 4C) (attributed to the removal of the DiD micelles from the solution).
After characterizing the DiD NPs that had been purified by DGC, we next studied their cellular interactions as compared to non-purified DiD NPs and NPs synthesized with Cy5-tagged PLGA (i.e., Cy5 NPs). Flow cytometry of M0 macrophages (differentiated from THP-1 cells) that were treated with each sample showed there was ∼27.5% less signal in cells (based on median fluorescence intensity) exposed to DGC-purified DiD NPs as compared to unpurified DiD NPs (Fig. 5A). This reduced signal can be attributed to the removal of the DiD micelles from the NP solutions by DGC, as depicted in Fig. 4. Congruent with this result, confocal images of macrophages stained with DiD show labeling of the cell surface, as evidenced by the red DiD signal predominantly co-locating with the WGA-labeled membranes at the periphery of the cells (Fig. 5B, Movie S1†). In contrast, the DiD NPs purified by DGC to remove DiD micelles, as well as Cy5 NPs, appear primarily as puncta within the cell interior (Fig. 5B, Movies S2 and S3†). The unpurified DiD NPs, which contain both NP and micellular structures, appear both within the cell as puncta and co-located with WGA-labeled cell membrane (Fig. 5B, Movie S4†). This indicates that unencapsulated DiD in the DiD NP samples can cause errant fluorescent signal by “tagging” cells rather than revealing NP location. Therefore, studies that did not, or do not, remove excess dye are likely to have false positive signal that makes it challenging to accurately interpret the results of NP cellular uptake and trafficking studies. While not explored here, one can surmise that administering NP solutions that still contain lipophilic micelles to rodents could also impact the analysis of NP biodistribution as the micelles may tag any cell they come in contact with after intravenous delivery.
 | ||
| Fig. 5 Cell uptake of free dye, unpurified DiD NPs, DGC-purified DiD NPs, and Cy5 NPs measured by flow cytometry and confocal microscopy. (A) Representative flow cytometry histogram (top) and median fluorescence intensity (bottom) for cells treated with unpurified or DGC-purified DiD NPs. NT = untreated cells. n = 3. (B) Confocal microscopy images of M0 macrophages (differentiated from THP-1 cells) after exposure to free DiD, unpurified DiD NPs, DGC-purified DiD NPs, or Cy5 NPs. WGA = green, DAPI = blue, DiD or Cy5 = Red. Scale bar = 5 μm. Yellow arrows highlight DiD signal associated with cell membranes, while white arrows highlight DiD or Cy5 signal within the cell interior. | ||
Conclusions
This study shows that lipophilic dyes like DiD can form micelles or aggregates during the nanoprecipitation synthesis process used to produce PEG–PLGA NPs (Fig. 1–4). These micelles cannot be detected using common characterization methods such as dynamic light scattering, zeta potential, and TEM, and may lead to false positive signal in cellular uptake studies (Fig. 5). The removal of these micelles is crucial for elucidating the true position of the NPs in cellular uptake and biodistribution studies. While TX can be used to disrupt and remove the micelles, it is incompletely eliminated after centrifugal filtration or pelleting, which results in undesired cellular toxicity (Fig. S4†). Alternatively, DGC can be used to separate DiD-labeled NPs from DiD micelles without this effect (Fig. 3–5).These findings emphasize the importance of detailed characterization of fluorophore-labeled NPs and process optimization to ensure the removal of unbound fluorophores. The absolute purity of labeled NPs must be confirmed before they are used to make conclusions about cellular uptake, intracellular trafficking, biodistribution, or in vivo fate. While we used DiD-labeled PEG–PLGA as a model platform in this study, different combinations of fluorophores and NP compositions will need to be studied in future work since the presence and quantity of dye-only aggregates/micelles will likely vary based on the material composition and synthesis method. However, dyes containing lipophilic regions, such as DiD and other close derivatives (DiO, DiR, etc.), will generally form micelles during common synthesis methods. As such, researchers should use caution when using these dyes outside of their intended purpose of membrane-labeling. Alternatively, researchers can utilize dyes directly conjugated to the polymer used to form the NPs (such as Cy5-labeled PLGA), which can more easily be separated from produced NPs than micelle-forming agents. Following these principles will allow for more accurate characterization and analysis of NP systems which will yield more efficient translation.
Author contributions
ES: Conceptualization, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, writing – review & editing. LW: Investigation, visualization, writing – original draft, writing – review & editing. EG: Investigation. ED: Funding acquisition, project administration, resources, supervision, writing – original draft, writing – review & editing.Data availability
The data supporting this article are available in the manuscript or its ESI† or from the authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr John Sperduto for his input and expertise as well as the Delaware Biotechnology Institute for their support. This work was supported by the National Institutes of Health under award numbers R35GM149292 and R35GM119659 and by the University of Delaware Summer Scholars Program. E. H. S. acknowledges support from the National Science Foundation Graduate Research Fellowship Program under award number 1940700.References
- A. M. Vargason, A. C. Anselmo and S. Mitragotri, The evolution of commercial drug delivery technologies, Nat. Biomed. Eng., 2021, 5(9), 951–967 CrossRef PubMed . Available from: https://www.nature.com/articles/s41551-021-00698-w.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Engineering precision nanoparticles for drug delivery, Nat. Rev. Drug Discovery, 2020, 20(2), 101–124 CrossRef PubMed . Available from: https://www.nature.com/articles/s41573-020-0090-8.
- A. C. Anselmo and S. Mitragotri, Nanoparticles in the clinic: An update, Bioeng. Transl. Med., 2019, 4(3), e10143 CrossRef PubMed . Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC6764803/.
- J. S. Ni, Y. Li, W. Yue, B. Liu and K. Li, Nanoparticle-based Cell Trackers for Biomedical Applications, Theranostics, 2020, 10(4), 1923 CrossRef CAS PubMed . Available from: https:///pmc/articles/PMC6993224/.
- A. Vollrath, S. Schubert and U. S. Schubert, Fluorescence imaging of cancer tissue based on metal-free polymeric nanoparticles – a review, J. Mater. Chem. B, 2013, 1(15), 1994–2007 RSC . Available from: https://pubs.rsc.org/en/content/articlehtml/2013/tb/c3tb20089b.
- M. E. Karim, S. T. Haque, H. Al-Busaidi, A. Bakhtiar, K. K. Tha and M. M. B. Holl, et al., Scope and challenges of nanoparticle-based mRNA delivery in cancer treatment, Arch. Pharmacal Res., 2022, 45(12), 865–893 CrossRef CAS PubMed . Available from: https://link.springer.com/article/10.1007/s12272-022-01418-x.
- M. A. Islam, E. K. G. Reesor, Y. Xu, H. R. Zope, B. R. Zetter and J. Shi, Biomaterials for mRNA delivery, Biomater. Sci., 2015, 3(12), 1519–1533 RSC . Available from: https://pubs.rsc.org/en/content/articlehtml/2015/bm/c5bm00198f.
- T. Thomsen, A. B. Ayoub, D. Psaltis and H. A. Klok, Fluorescence-Based and Fluorescent Label-Free Characterization of Polymer Nanoparticle Decorated T Cells, Biomacromolecules, 2021, 22(1), 190–200 CrossRef CAS PubMed . Available from: https://pubs.acs.org/doi/full/10.1021/acs.biomac.0c00969.
- W. Zhang, P. Wei, L. Liu, T. Ding, Y. Yang and P. Jin, et al., AIE-enabled transfection-free identification and isolation of viable cell subpopulations differing in the level of autophagy, Autophagy, 2023, 19(12), 3062–3078 CrossRef CAS PubMed . Available from: https://www.tandfonline.com/doi/abs/10.1080/15548627.2023.2235197.
- T. T. Tang, B. Wang, M. Wu, Z. L. Li, Y. Feng and J. Y. Cao, et al., Extracellular vesicle–encapsulated IL-10 as novel nanotherapeutics against ischemic AKI, Sci. Adv., 2020, 6(33), 748–760 Search PubMed . Available from: https://www.science.org/doi/10.1126/sciadv.aaz0748.
- M. H. Chao, Y. T. Lin, N. Dhenadhayalan, H. L. Lee, H. Y. Lee and K. C. Lin, et al., 3D Probed Lipid Dynamics in Small Unilamellar Vesicles, Small, 2017, 13(13), 1603408 CrossRef PubMed . Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/smll.201603408.
- S. Osella, F. Di Meo, N. A. Murugan, G. Fabre, M. Ameloot and P. Trouillas, et al., Combining (Non)linear Optical and Fluorescence Analysis of DiD to Enhance Lipid Phase Recognition, J. Chem. Theory Comput., 2018, 14(10), 5350–5359 CrossRef CAS PubMed . Available from: https://pubs.acs.org/doi/full/10.1021/acs.jctc.8b00553.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Cyanines during the 1990s: a review, Chem. Rev., 2000, 100(6), 1973–2011 CrossRef CAS PubMed . Available from: https://pubs.acs.org/doi/full/10.1021/cr990402t.
- R. V. Levy and M. W. Jornitz, Types of Filtration, Adv. Biochem. Eng./Biotechnol., 2006, 98, 1–26 CAS . Available from: https://link.springer.com/chapter/10.1007/b104242.
- M. Hoda, S. A. Sufi, B. Cavuturu and R. Rajagopalan, Stabilizers Influence Drug–Polymer Interactions and Physicochemical Properties of Disulfiram-Loaded Poly-Lactide-Co-Glycolide Nanoparticles, Future Sci. OA, 2018, 4(2), FSO263 CrossRef PubMed , Available from: https://www.tandfonline.com/doi/abs/10.4155/fsoa-2017-0091.
- L. Ge, X. Zhang and R. Guo, Microstructure of Triton X-100/poly (ethylene glycol) complex investigated by fluorescence resonance energy transfer, Polymer, 2007, 48(9), 2681–2691 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5pm00013k |
| This journal is © The Royal Society of Chemistry 2025 |