
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSometimes less is more: avidity-dependent transport of targeted polymersomes across the blood–brain-barrier†
Ana
Alves
abc,
Peter
Pfeifer
d,
Andreia
Marinho
 e,
Claúdia
Nunes
e,
Salette
Reis
e,
Domingos
Ferreira
ac,
Marta
Correia-da-Silva
bf,
Paulo C.
Costa
ac,
Giuseppe
Battaglia
dg,
Íris L.
Batalha
*dh and
Cátia D. F.
Lopes
*d
e,
Claúdia
Nunes
e,
Salette
Reis
e,
Domingos
Ferreira
ac,
Marta
Correia-da-Silva
bf,
Paulo C.
Costa
ac,
Giuseppe
Battaglia
dg,
Íris L.
Batalha
*dh and
Cátia D. F.
Lopes
*d
aUCIBIO – Applied Molecular Biosciences Unit, MedTech-Laboratory of Pharmaceutical Technology, Faculty of Pharmacy, University of Porto, Rua Viterbo Ferreira, 228, 4050-313 Porto, Portugal
bLaboratório de Química Orgânica e Farmacêutica, Departamento de Ciências Químicas, Faculdade de farmácia da Universidade do Porto, Rua Viterbo Ferreira, 228, 4050-313 Porto, Portugal
cAssociate Laboratory i4HB – Institute for Health and Bioeconomy, Faculty of Pharmacy, University of Porto, Rua Viterbo Ferreira, 228, 4050-313 Porto, Portugal
dInstitute for Bioengineering of Catalonia (IBEC), Barcelona Institute of Science and Technology (BIST), Carrer Baldiri Reixac, 10-12, 08028 Barcelona, Spain. E-mail: clopes@ibecbarcelona.eu
eLAQV/REQUIMTE, Departamento de Ciências Químicas, Faculdade de farmácia da Universidade do Porto, Rua Viterbo Ferreira, 228, 4050-313 Porto, Portugal
fCIIMAR – Interdisciplinary Centre of Marine and Environmental Research, Marine Natural Products and Medicinal Chemistry Group, Avenida General Norton de Matos Matosinhos 4450-208, Matosinhos, Portugal
gCatalan Institution for Research and Advanced Studies, Passeig de Lluís Companys, 23, 08010, Barcelona, Spain
hDepartment of Life Sciences, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: icdlb20@bath.ac.uk
First published on 9th April 2025
Abstract
Over the past decade, roughly 10% of new FDA-approved drugs targeted central nervous system (CNS) disorders, while it has been estimated that 98% of small-molecule drugs and nearly all large-molecule therapeutics are unable to cross the blood–brain barrier (BBB). There is a clear need for novel therapeutic modalities that promote receptor-mediated transcytosis modulation and efficiently deliver drugs to the brain. Here, we show that poly(ethylene glycol)-b-poly(lactic acid) (PEG-b-PLA) polymersomes functionalised with a transferrin receptor (TfR)-targeted peptide can effectively deliver a glioblastoma small drug therapeutic (3,6-bis(2,3,4,6-tetra-O-acetyl-β-glucopyranosyl)xanthone; XGAc) through a two-dimensional model of the BBB and that the transport is dependent on the avidity of the nanoformulation. By adjusting the density of targeting peptides on polymersomes, we present a novel strategy to enhance the efficiency of BBB receptor-mediated transcytosis. These findings highlight the promise of precision-tuned polymersomes in overcoming the BBB and advancing treatments for glioblastoma and other brain diseases.
Introduction
Glioblastoma multiforme (GBM) has been classified by the World Health Organisation (WHO) as a grade IV brain tumour and is one of the most lethal human cancers.1 It is characterised by fast growth, high recurrence rate, and ability to spread rapidly within the brain, with a median survival of <2 years and a 5-year survival rate of only 5.8%.2 Treatment is extremely expensive (approx. USD 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per patient) and typically involves a combination of surgery, radiation therapy, and chemotherapy. Nevertheless, the prognosis remains poor due to several factors, including the tumour's highly infiltrative nature and genetic heterogeneity, the immunosuppressive microenvironment, and protection by the blood–brain barrier (BBB).3,4 Despite being partially disrupted in GBM, the BBB remains intact in the peritumoral region, harbouring invasive cells associated with drug resistance and recurrence.5
000 per patient) and typically involves a combination of surgery, radiation therapy, and chemotherapy. Nevertheless, the prognosis remains poor due to several factors, including the tumour's highly infiltrative nature and genetic heterogeneity, the immunosuppressive microenvironment, and protection by the blood–brain barrier (BBB).3,4 Despite being partially disrupted in GBM, the BBB remains intact in the peritumoral region, harbouring invasive cells associated with drug resistance and recurrence.5
Substantial efforts are underway to improve drug delivery to the brain, including strategies to disrupt the BBB using ultrasound and heat and the development of nanomedicines with the ability to permeate the BBB.6–8 One strategy for overcoming the BBB, which is already being explored in clinical trials,9 involves using transferrin receptor (TfR)-mediated transport. Transferrin (Tf) is a ca. 80 kDa glycoprotein involved in iron homeostasis known to be required for normal neuronal function.10 The TfR is enriched in brain capillary endothelial cells as opposed to endothelial cells in other tissues,11 enabling the targeted delivery of therapeutic agents to the brain. In addition to its role in the BBB, the TfR is also highly expressed in glioblastoma cells (reportedly up to 100-fold higher than healthy cells),12 further enhancing its relevance as a dual-targeting moiety capable of traversing the BBB and precisely targeting glioblastoma tumour cells.
Nevertheless, the role of transcytosis in TfR-mediated drug delivery has been the subject of extensive debate, with recent studies showing that both affinity and valency of antibodies targeting the TfR play a key role.13,14 In this sense, nanoparticle functionalisation with ligands targeting the TfR offers the much-needed versatility to enable precise control over ligand composition and density.
In previous work, we pioneered the development of a new synthetic xanthone, 3,6-bis(2,3,4,6-tetra-O-acetyl-β-glucopyranosyl)xanthone (XGAc) (Fig. 1), that showed potent anti-growth activity (GI50 < 1 μM) in several human glioblastoma cell lines (U251, U373, U87-MG)15 and exhibited antitumor efficacy against triple-negative breast cancer (TNBC), ovarian cancer, and pancreatic ductal adenocarcinoma (PDAC) cells.16 To overcome issues related to the drug's poor solubility and rapid hydrolysis by esterases, we further formulated XGAc both in egg phosphatidylcholine liposomes containing cholesterol15 and poly(ethylene glycol)-ε-caprolactone (PEG-PCL) polymersomes.17
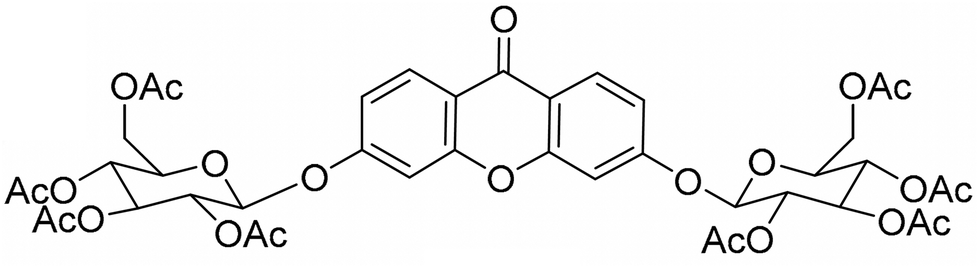 | ||
| Fig. 1 Chemical structure of 3,6-bis(2,3,4,6-tetra-O-acetyl-β-glucopyranosyl)xanthone (XGAc). | ||
Liposomes were the first clinically approved nanocarriers and, therefore, remain the most explored for drug delivery, with several reported clinical trials for the treatment of gliomas.18,19 However, polymeric nanoparticles, particularly polymersomes, represent viable alternatives due to their improved physicochemical properties, such as higher stability, extended circulation time, more controlled drug release, and ease of functionalisation.20
While nanoparticle functionalisation with Tf may seem an evident approach to achieve targeted delivery,21 endogenous Tf in the bloodstream can effectively compete for binding to TfR, rendering treatments largely ineffective.22
Here, we report on the development of a new therapeutic nanoparticle modality composed of poly(ethylene glycol)-b-poly(lactic acid) (PEG-b-PLA) diblock copolymer polymersomes functionalised with the T7 heptapeptide (His-Ala-Ile-Tyr-Pro-Arg-His) for the targeted delivery of XGAc both to the brain and glioblastoma tumours.
Results and discussion
T7 is a peptide discovered by phage display and exhibits high affinity to human TfR. Most importantly, T7 did not compete with Tf for receptor binding, suggesting the peptide binds to a different sequence of the TfR.23 Here, we have explored T7 as a targeting moiety to promote BBB crossing and potentially target glioblastoma cancer cells through binding to the TfR. As illustrated in Fig. 2, this peptide was conjugated to PEG20-b-PLA106 diblock copolymer by copper-catalysed alkyne–azide cycloaddition (CuAAC) with a conjugation efficiency of 68 ± 9 (mol/mol)%.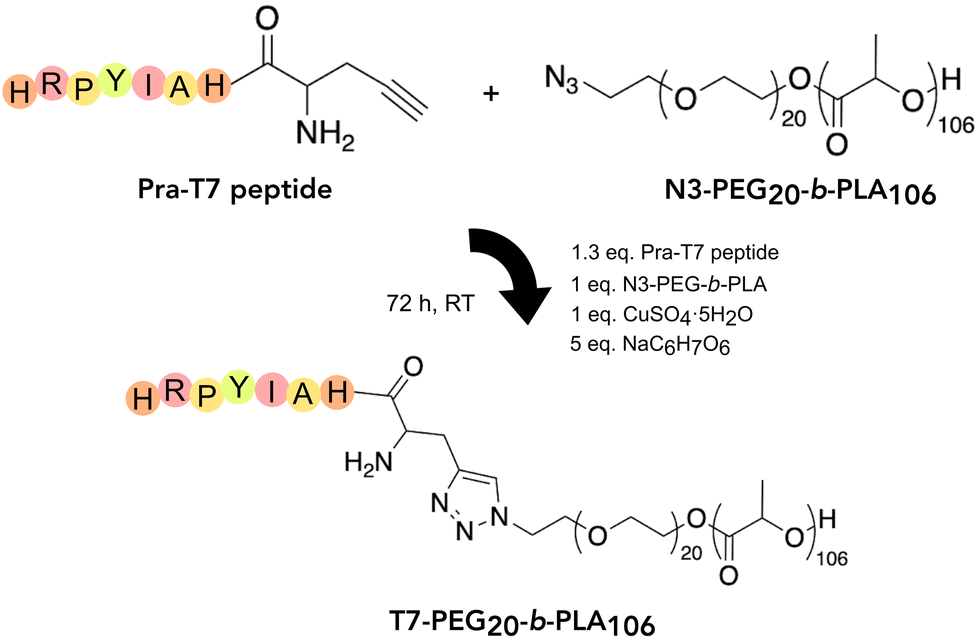 | ||
| Fig. 2 T7 conjugation to N3-PEG20-b-PLA106 diblock copolymer by copper-catalysed alkyne–azide cycloaddition (CuAAC). | ||
To investigate the BBB targeting and crossing capabilities, we prepared polymersomes with low (0.25 molar percentage, mol%) and high (0.50 mol%) T7 valencies. Within this range, the polymersomes exhibited similar size distributions to pristine (i.e., non-functionalised) polymersomes, as confirmed by dynamic light scattering (DLS) measurements (Fig. 3A–C). The mean particle size of the low and high T7 valency polymersomes was 101.7 ± 20.4 nm and 95.9 ± 18.8 nm, respectively, confirming their suitability to approach cell surface receptors and facilitate intracellular transport. Transmission electron microscopy (TEM) images (Fig. 3D–F) showed a spherical morphology of the pristine and T7-functionalised polymersomes.
 | ||
| Fig. 3 Characterisation of polymersomes in terms of size and morphology. Dynamic light scattering (DLS) autocorrelation function data (A) and size distribution by number (B) of pristine and T7-decorated PEG-b-PLA polymersomes. Summary table of size distribution results (C) showing a similar peak diameter for both nanoparticle types (n = 3). SD, standard deviation. Representative transmission electron micrographs (TEM) of pristine polymersomes (D), low T7 valency (0.25 mol%) (E), and high T7 valency (0.50 mol%) polymersomes (F). Scale bar: 100 nm. | ||
The similar physicochemical characteristics of pristine and T7-polymersomes provided a robust platform to evaluate the specific effects of T7 functionalisation on BBB binding and permeability. To assess the role of T7 valency in modulating cell interaction, polymersomes with low and high T7 valencies were incubated with bEnd.3 cells, a well-established in vitro model of mouse brain endothelial cells. Quantitative analysis of cell binding and internalisation were performed at specific time intervals at 37 °C to elucidate uptake dynamics. Our results revealed that T7-functionalised polymersomes exhibited increased binding and/or internalisation by bEnd.3 cells over time for both formulations (Fig. 4).
 | ||
| Fig. 4 Brain endothelial cell targeting and association of T7-PEG-PLA polymersomes. The graph shows the number of bound and/or internalised polymersomes per cell (after subtracting non-specific binding of pristine polymersomes). Data are presented as the mean ± standard deviation (n = 6). | ||
Interestingly, despite comparable uptake levels, low T7 valency polymersomes demonstrated superior BBB permeability compared to high T7 valency formulations and the pristine (non-functionalised) control (Fig. 5). This finding underscores the complexity of T7-functionalised polymersome interactions with the BBB and suggests that factors beyond binding affinity and cellular uptake govern effective transcytosis. Specifically, the reduced permeability of high T7 valency polymersomes may be attributed to steric hindrance or polymersome rigidity caused by excessive ligand density, which could reduce the T7-TfR complex endocytosis efficiency required for transcytosis. Higher avidity of 0.50 mol% T7 valency may also prevent polymersomes’ release from cell surfaces and impair exocytosis.24 Additionally, high T7 valency may affect the intracellular sorting mechanism of TfR, reducing transcytosis efficiency. To gain a deeper understanding of these mechanisms, further investigations are necessary.
 | ||
| Fig. 5 Blood–brain barrier transcytosis. Apparent permeability coefficients (Papp) for low (0.25 mol%) and high (0.50 mol%) T7 valency polymersomes (A). Truncated violin plots depict the data distribution for each group. The non-specific TfR transport was removed by subtracting the Papp values of pristine polymersomes (n = 3). Cumulative mass crossing of polymersomes across the in vitro BBB model over a 90-minute period (B). Data is normalised to pristine polymersomes to isolate the effect of the T7 peptide on BBB permeability (n = 3). | ||
Based on these findings, the low T7 valency polymersome emerged as the most promising candidate for drug delivery across the BBB. Therefore, XGAc was successfully encapsulated during the polymersome self-assembly process, with an efficiency of 76.00 ± 1.09 (mol/mol)% (Fig. 6A). Importantly, the encapsulation process did not significantly alter the physicochemical characteristics of the polymersomes, including size distribution or spherical morphology, as confirmed through DLS and TEM analyses (Fig. 6A–C).
 | ||
| Fig. 6 Characterisation of XGAc-loaded polymersomes. Summary table of size distribution, polydispersity index (PDI) and efficiency of XGAc encapsulation (EE) in pristine and low T7 valency polymersomes (A) (n = 3). SD, standard deviation. Representative transmission electron micrographs of pristine (B) and low T7 valency (0.25 mol%) (C) polymersomes. Scale bars represent 50 nm. Metabolic activity (MTT assay) of bEnd.3 cells after treatment with pristine polymersomes (D) (n = 3) or free XGAc, XGAc-loaded pristine and XGAc-loaded 0.25 mol% T7 polymersomes (E) (n = 3). | ||
The metabolic activity of bEnd.3 cells following treatment with XGAc-loaded low T7 valency polymersomes was assessed using the MTT assay (Fig. 6E). PEG-b-PLA polymersomes without XGAc did not significantly affect the metabolic activity of bEnd.3 cells, with viability levels comparable to untreated cells across all tested concentrations (Fig. 6D). This indicates that the polymersomes are inert and highly biocompatible, a critical feature for any drug delivery platform targeting sensitive tissues such as the brain. In contrast, XGAc-loaded polymersomes (Fig. 6E) exhibited a dose-dependent reduction in metabolic activity, reflecting the cytotoxic or therapeutic action of XGAc. Most importantly, our findings demonstrated a considerable improvement in cell metabolic activity when XGAc was encapsulated within the polymersomes compared to its free drug form, underscoring the protective role of polymersomes in modulating drug delivery. Specifically, cells exposed to the free XGAc exhibited a dose-dependent decline in metabolic activity, with viability dropping below 50% at higher concentrations, while cells treated with encapsulated XGAc retained over 70% metabolic activity at XGAc doses ≤ 10 μM. This difference shows that low T7 valency polymersomes attenuate the cytotoxic effects of free XGAc at the level of the BBB. Overall, our data suggests that carefully designed targeted polymersome systems may enable the safe and effective delivery of therapies across challenging biological barriers, paving the way for in vivo translation.
Conclusions
This study demonstrates the potential of the proposed therapeutic nanoparticle modality for overcoming the BBB and treating GBM. By leveraging polymersomes functionalised with the T7 peptide targeting the TfR, we achieved enhanced BBB penetration while significantly reducing the cytotoxicity of XGAc in brain endothelial cells compared to the free drug. These findings illustrate the prospect of our nanoparticle modality to impact glioblastoma treatment by combining precision targeting with therapeutic protection.Our ongoing research on refining T7 valency promises to unlock even greater BBB crossing efficiency and therapeutic efficacy. This work holds significant promise not only for glioblastoma treatment but also for expanding the application of targeted nanomedicine for other brain-related diseases, contributing to the advancement of BBB-crossing therapies in neuro-oncology and beyond.
Materials and methods
Nano-drug delivery system preparation
PEG45-b-PLA106 and N3-PEG20-b-PLA106 copolymers were synthesised via ring-opening polymerisation (ROP) and characterised by gel permeation chromatography and nuclear magnetic resonance spectroscopy. T7 was conjugated to N3-PEG20-b-PLA106 by CuAAC (ESI for details†).T7-functionalized polymersomes were self-assembled using the solvent displacement method. A mixture of pristine PEG45-b-PLA106, 10 mol% cyanine 5 (Cy5)-PEG20-b-PLA106, and 0.25 or 0.5 mol% T7-PEG20-b-PLA106 was dissolved in dimethylformamide (final concentration 20 mg mL−1 polymer). Pristine polymersomes, i.e., lacking the T7 peptide, were prepared using 90 and 10 mol% of pristine and Cy5-conjugated diblock copolymers, respectively. For drug-loaded formulations, 2 mg of the glioblastoma therapeutic XGAc was co-dissolved with the polymer mixture prior to self-assembly, allowing for encapsulation during polymersome formation. The polymer mixture (30% v/v) was injected into Milli-Q water (70% v/v) at a flow rate of 100 μL min−1 under magnetic stirring at 500 rpm using a syringe pump (New Era Pump Systems, Inc.), resulting in the spontaneous self-assembly of both drug-free and XGAc-loaded polymersomes. The resulting polymersome solution was dialysed against Milli-Q water for 1 hour using a 3.5 kDa dialysis membrane (CelluSep®, France) and then against PBS overnight. Finally, the solution was centrifuged at 1000g for 10 minutes to remove any precipitate. Polymersome and XGAc concentrations were determined by high-performance liquid chromatography using a Jupiter® C18 300 Å (150 × 4.6 mm) LC column with a particle size of 5 μm (Phenomenex, USA). The mobile phase consisted of a mixture of water and acetonitrile, with a gradient elution from 50:50 to 100% acetonitrile over 20 minutes, followed by a 5-minute wash with 100% acetonitrile, and a 5-minute re-equilibration to 50:50 water:acetonitrile. A 100 μL sample was injected at a flow rate of 1.0 mL min−1, with the column at room temperature. Polymer and XGAc detection were performed at 220 nm and 265 nm, respectively. A calibration curve was generated using known concentrations of the polymer and XGAc standards to determine their respective concentrations in the samples. XGAc encapsulation efficiency (EE%) was calculated as the ratio of the encapsulated drug to the total drug added during assembly. The polymersome morphology was examined via transmission electron microscopy (JEOL JEM 1010 80 kV, Japan). Diameter distribution was assessed by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, UK).
Cell culture
Mouse brain endothelial cells (bEnd.3) were purchased from the American Type Culture Collection (ATCC) and used between passages 11 and 18. Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 in complete media consisting of Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) Penicillin–Streptomycin (Gibco, ThermoFisher Scientific). Whenever stated, culture vessels were pre-coated overnight at 4 °C with 0.1 mg mL−1 Collagen I (Sigma-Aldrich) diluted in phosphate buffered saline (PBS), pH 7.4. For biocompatibility and binding studies, bEnd.3 cells were seeded in 96-well plates at a density of 31250 cells per cm2 and incubated for 24 hours previous polymersome treatment. For the BBB model, cells were seeded on the apical compartment of coated transwell membranes (0.4 μm pore size, 6.5 mm diameter, Corning) at 50000 cells per cm2 density. The culture medium was replaced on the third day in culture, and serum was removed from the basolateral compartment. On the sixth day in culture, before polymersome treatment, the integrity of the cell monolayer was confirmed by transendothelial electrical resistance (TEER) measurements with EVOM3™ (World Precision Instruments), following the manufacturer's instruction.
Biocompatibility assay
Cell metabolic activity of polymersome-treated cells was measured by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay to assess the biocompatibility of polymersomes. bEnd.3 cells were exposed to serial concentrations of free XGAc, unloaded polymersomes, and XGAc-loaded polymersomes for 24 hours. Cells cultured in fresh medium served as untreated controls. Following incubation, the culture medium was removed, and 0.1 mL of MTT solution (0.5 mg mL−1 in PBS) was added to each well and incubated for 3 hours. The MTT solution was discarded, and 0.1 mL of dimethyl sulfoxide (DMSO, Sigma-Aldrich) was used to dissolve the formazan crystals. The absorbance of each well was measured at 570 nm, with 630 nm used as a reference wavelength to correct for background interference. Cell metabolic activity was calculated as a percentage of the untreated control and expressed as mean ± standard deviation (SD).Brain endothelial cells targeting
To assess the binding and uptake of T7-functionalised polymersomes by brain endothelial cells, bEnd.3 cells were exposed to 5 μM polymersomes diluted in complete medium. After 10- and 60-minute incubation at 37 °C, the supernatant (containing unbound polymersomes) was transferred to a black 96-well plate. Fluorescence intensity was measured using a Tecan microplate reader with excitation and emission wavelengths of 633 and 677 nm, respectively. Similarly, the same volume of initial polymersome solution was measured to determine the initial dose. The amount of polymersomes was estimated using a calibration curve of known polymersome concentrations. The number of polymersomes associated with cells (i.e., bound or uptaken) at each time point was determined by subtracting the number of polymersomes remaining in the supernatant from the initial polymersome dose. Pristine polymersomes were used as a control to assess non-specific binding, thus serving as a baseline for specific binding. Therefore, the specific binding of T7-functionalised polymersomes was determined by subtracting the average binding of the control at each time point.The number of bound and/or internalised T7-polymersomes was further normalised to the cell number to account for any variations in cell seeding density or cell division. Cell number per well was determined after staining cell nuclei with Hoechst 33342 (0.1 μg mL−1 in PBS, Invitrogen) for 10 minutes at room temperature.
BBB transcytosis
To determine the BBB crossing ability of T7-polymersomes, confluent bEnd.3 cell monolayers on the apical transwell compartment were exposed to 3 μM of polymersomes diluted in complete medium and incubated at 37 °C. After 15, 30, 45, 60, and 90-minute incubation, the basolateral medium was sampled, and the removed medium was replaced with an equal volume of polymersome-free medium. Fluorescence intensity was measured as previously described. Apparent permeability (Papp) was calculated as elsewhere25 using the following equation:where dQ/dt is the rate of mass transfer across the monolayer, V is the volume in the receptor compartment in mL, A is the surface area of the monolayer in cm2, and C is the initial polymersome concentration in the donor compartment in mg mL−1. The Papp of the pristine polymersomes (without the T7 ligand) served as a baseline measurement for passive paracellular diffusion. The Papp of T7-polymersomes was normalised to that of the pristine polymersomes by subtracting the average Papp values of the pristine.

Author contributions
C. D. F. L., I. L. B., G. B., P. C., and M. C. D. S., contributed to the project conceptualisation. A. A., P. F., and A. M. performed the experiments. A. A., C. D. F. L., and I. L. B. analysed the data. G. B. and D. F. were responsible for funding acquisition and provision of resources. C. D. F. L., S. R., and C. N. were involved in methodology design and development. C. D. F. L., I. L. B., and M. C. D. S. supervised the project. The original draft was written by I. L. B. and C. D. F. L. and revised by all the authors.Data availability
The data supporting this article have been included in the ESI.† This includes a detailed description of the synthetic procedures and all the characterisation data for the synthesised polymers.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
I. L. B. would like to acknowledge the support of a fellowship from the La Caixa Foundation (ID 100010434) and the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 847648. The fellowship code is LCF/BQ/PI21/11830004. C. D. F. L. thanks funding from the European Union's Horizon Europe research and innovation program under the Marie Skłodowska-Curie grant agreement No. 101066836. A. A. would like to acknowledge Fundação para a Ciência e Tecnologia, I. P. (FCT) for the PhD scholarship (grant number SFRH/BD/144607/2019). C. N. thanks FCT for funding through the Individual Call to Scientific Employment Stimulus (grant number 10.54499/2022.05608.CEECIND/CP1724/CT0002). A. M. acknowledges the fellowship funding from REQUIMTE (REQUIMTE 2023-22). This research was partially funded by FCT in the scope of the strategic funding UIDB/04423/2020 and UIDP/04423/2020 (CIIMAR–Marine Natural Products and Medicinal Chemistry group) and UIDP/04378/2020 and UIDB/04378/2020 (UCIBIO-Research Unit on Applied Molecular Biosciences), and the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy-i4HB.References
- WHO Classification of Tumours Editorial Board, in World Health Organization Classification of Tumours of the Central Nervous System, International Agency for Research on Cancer, Lyon, 5th edn, 2021, ISBN-13: 978-92-832-4508-7 Search PubMed.
- A. C. Tan, D. M. Ashley, G. Y. López, M. Malinzak, H. S. Friedman and M. Khasraw, CA Cancer J. Clin., 2020, 70, 299–312 CrossRef PubMed.
- A. Bikfalvi, C. A. da Costa, T. Avril, J. V. Barnier, L. Bauchet, L. Brisson, P. F. Cartron, H. Castel, E. Chevet, H. Chneiweiss, A. Clavreul, B. Constantin, V. Coronas, T. Daubon, M. Dontenwill, F. Ducray, N. Enz-Werle, D. Figarella-Branger, I. Fournier, J. S. Frenel, M. Gabut, T. Galli, J. Gavard, G. Huberfeld, J. P. Hugnot, A. Idbaih, M. P. Junier, T. Mathivet, P. Menei, D. Meyronet, C. Mirjolet, F. Morin, J. Mosser, E. C. J. Moyal, V. Rousseau, M. Salzet, M. Sanson, G. Seano, E. Tabouret, A. Tchoghandjian, L. Turchi, F. M. Vallette, S. Vats, M. Verreault and T. Virolle, Trends Cancer, 2023, 9, 9–27 CrossRef CAS PubMed.
- T. I. Janjua, P. Rewatkar, A. Ahmed-Cox, I. Saeed, F. M. Mansfeld, R. Kulshreshtha, T. Kumeria, D. S. Ziegler, M. Kavallaris, R. Mazzieri and A. Popat, Adv. Drug Delivery Rev., 2021, 171, 108–138 CrossRef CAS PubMed.
- J. N. Sarkaria, L. S. Hu, I. F. Parney, D. H. Pafundi, D. H. Brinkmann, N. N. Laack, C. Giannini, T. C. Burns, S. H. Kizilbash, J. K. Laramy, K. R. Swanson, T. J. Kaufmann, P. D. Brown, N. Y. R. Agar, E. Galanis, J. C. Buckner and W. F. Elmquist, Neuro-Oncology, 2018, 20, 184–191 CrossRef CAS PubMed.
- M. Nowak, M. E. Helgeson and S. Mitragotri, Adv. Ther., 2020, 3, 1900073 CrossRef.
- A. E. Sloan, M. S. Ahluwalia, J. Valerio-Pascua, S. Manjila, M. G. Torchia, S. E. Jones, J. L. Sunshine, M. Phillips, M. A. Griswold, M. Clampitt, C. Brewer, J. Jochum, M. V. McGraw, D. Diorio, G. Ditz and G. H. Barnett, J. Neurosurg., 2013, 118, 1202–1219 Search PubMed.
- A. Idbaih, M. Canney, L. Belin, C. Desseaux, A. Vignot, G. Bouchoux, N. Asquier, B. Law-Ye, D. Leclercq, A. Bissery, Y. De Rycke, C. Trosch, L. Capelle, M. Sanson, K. Hoang-Xuan, C. Dehais, C. Houillier, F. Laigle-Donadey, B. Mathon, A. André, C. Lafon, J.-Y. Chapelon, J.-Y. Delattre and A. Carpentier, Clin. Cancer Res., 2019, 25, 3793–3801 CrossRef CAS PubMed.
- T. Okuyama, Y. Eto, N. Sakai, K. Nakamura, T. Yamamoto, M. Yamaoka, T. Ikeda, S. So, K. Tanizawa, H. Sonoda and Y. Sato, Mol. Ther., 2021, 29, 671–679 CrossRef CAS PubMed.
- D. F. Leitner and J. R. Connor, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 393–402 CrossRef CAS PubMed.
- W. Zhang, Q. Y. Liu, A. S. Haqqani, S. Leclerc, Z. Liu, F. Fauteux, E. Baumann, C. E. Delaney, D. Ly, A. T. Star, E. Brunette, C. Sodja, M. Hewitt, J. K. Sandhu and D. B. Stanimirovic, Fluids Barriers CNS, 2020, 17, 1–17 CrossRef PubMed.
- H. Yukawa, R. Tsukamoto, A. Kano, Y. Okamoto, M. Tokeshi, T. Ishikawa, M. Mizuno and Y. Baba, J. Cell Sci. Ther., 2013, 4, 1–7 Search PubMed.
- J. Niewoehner, B. Bohrmann, L. Collin, E. Urich, H. Sade, P. Maier, P. Rueger, J. O. Stracke, W. Lau, A. C. Tissot, H. Loetscher, A. Ghosh and P. O. Freskgård, Neuron, 2014, 81, 49–60 CrossRef CAS PubMed.
- N. Bien-Ly, Y. J. Yu, D. Bumbaca, J. Elstrott, C. A. Boswell, Y. Zhang, W. Luk, Y. Lu, M. S. Dennis, R. M. Weimer, I. Chung and R. J. Watts, J. Exp. Med., 2014, 211, 233–244 CrossRef CAS PubMed.
- A. Alves, M. Correia-da-Silva, C. Nunes, J. Campos, E. Sousa, P. M. A. Silva, H. Bousbaa, F. Rodrigues, D. Ferreira, P. C. Costa and M. Pinto, Molecules, 2019, 24, 409 CrossRef PubMed.
- J. Calheiros, L. Raimundo, J. Morais, A. C. Matos, S. A. Minuzzo, S. Indraccolo, E. Sousa, M. C. da Silva and L. Saraiva, Cancers, 2023, 15, 5718 CrossRef CAS PubMed.
- A. Alves, A. M. Silva, C. Nunes, S. Cravo, S. Reis, M. Pinto, E. Sousa, F. Rodrigues, D. Ferreira, P. C. Costa and M. Correia-da-Silva, Life, 2024, 14, 132 CrossRef CAS PubMed.
- D. Brandsma, V. Dieras, S. Altintas, C. Anders, M. Arnedos, H. Gelderblom, P. Soetekouw, A. Jager, M. van Linde and P. Aftimos, Neuro-Oncology, 2014, 16, ii50 CrossRef.
- N. A. Butowski, S. Han, J. W. Taylor, M. K. Aghi, M. Prados, S. M. Chang, J. L. Clarke, K. Bankiewicz, D. C. Drummond and J. Fitzgerald, J. Clin. Oncol., 2015, 33, TPS2081–TPS2081 Search PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667 CrossRef CAS PubMed.
- J. Lichota, T. Skjørringe, L. B. Thomsen and T. Moos, J. Neurochem., 2010, 113, 1–13 CrossRef CAS PubMed.
- J. H. Lee, J. A. Engler, J. F. Collawn and B. A. Moore, Eur. J. Biochem., 2001, 268, 2004–2012 CrossRef CAS PubMed.
- H. Deng, P. Dutta and J. Liu, Nanoscale, 2019, 11, 11227–11235 RSC.
- D. M. Leite, M. Seifi, L. Ruiz-Perez, F. Nguemo, M. Plomann, J. D. Swinny and G. Battaglia, Brain Commun., 2022, 4, fcac039 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00338a |
| This journal is © The Royal Society of Chemistry 2025 |