
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the divergent reactivity of allenylstannanes generated from the O-directed free radical hydrostannation reaction of (±)-trans-3-(2-phenylcyclopropyl)prop-2-yn-1-ol. EPR evidence for the reversible addition of Ph3Sn radicals to vinyl triphenyltins†‡
K. Lawrence E.
Hale
a,
Alistair J.
Fielding
 *b and
Karl J.
Hale§
*a
*b and
Karl J.
Hale§
*a
aThe School of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, UK
bThe School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Byrom Street, Liverpool L3 3AF, UK. E-mail: A.J.Fielding@ljmu.ac.uk
First published on 28th March 2025
Abstract
(±)-trans-3-(2-Phenylcyclopropyl)prop-2-yn-1-ol (5) undergoes O-directed rt free radical hydrostannation with 2 equiv. of Bu3SnH or Ph3SnH in PhMe to produce the α-cyclopropyl-β-stannylvinyl radicals 26 and 27, which rapidly ring-open to give the benzylic stannylhomoallenyl radicals 28 and 29. These, in turn, react with the excess stannane that is present to provide 21 and 23 as primary reaction products. The triphenylstannylallene 23 also undergoes a competitive Ph3Sn˙ addition to its central allene carbon. This affords the allylically-stabilised radical 31c, which itself reacts with the stannane to produce (Z)-6-phenyl-2,3-bis(triphenylstannyl)hex-3-en-1-ol (24). EPR studies of the reaction of 5 with Ph3SnH (1 equiv.) and cat. Et3B/O2 in PhMe at 250 K have failed to identify the radicals 27 and 29 in the reaction mixtures. Rather, a sharp dd is always observed whose multiplicity is consistent with it being the tris-Ph3Sn-stabilised free radical 33. The latter is suggested to arise from a reversible O-directed Ph3Sn˙ addition to 24. The radical 33 has 1Hβ values of 1.32 mT (13.2 G) and 0.57 mT (5.7 G) and a g of 2.0020.
Introduction
In the preceding paper,1 we studied the kinetics of ring-opening of the intermediary α-cyclopropyl-β-stannylvinyl radicals that are formed when cyclopropylacetylenic alcohols are subjected to O-directed free radical hydrostannation with stannanes and cat. Et3B/O2. It was found that the high log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A values of these ring-openings (13.27–14.95 s−1) only satisfactorily align with a unimolecular EH1 homolytic mechanism for cyclopropane ring-cleavage. As a consequence, an entirely free radical mechanism2 was reaffirmed for the O-directed hydrostannation of dialkyl acetylenes,3 not the ionic mechanism of the stannylvinyl cation theory.4
A values of these ring-openings (13.27–14.95 s−1) only satisfactorily align with a unimolecular EH1 homolytic mechanism for cyclopropane ring-cleavage. As a consequence, an entirely free radical mechanism2 was reaffirmed for the O-directed hydrostannation of dialkyl acetylenes,3 not the ionic mechanism of the stannylvinyl cation theory.4
In this follow-on paper, we now describe our studies on the O-directed free radical hydrostannation3 of the 3-(2-phenylcyclopropyl)-prop-2-yn-1-ol probe 5, under cat. Et3B/O2-initiated conditions. Specifically, we will show that in both PhMe and THF/H2O, the products that arise, originate from an entirely homolytic pathway, thus reinforcing the mechanistic conclusions of the earlier kinetic study.1
We will also detail here our EPR studies of the O-directed hydrostannation of 5 with Ph3SnH/cat. Et3B in PhMe, which have now provided good spectroscopic support for the formation of 1,2-bis-(Ph3Sn) radical adducts from the primary vinyltriphenylstannane products of these reactions at low temperature. Observations that now require the original Hale–Manaviazar 2005 mechanism for the O-directed free radical hydrostannation of disubstituted alkyl acetylenes with Ph3SnH/cat. Et3B/O2 to be restored in its entirety, but with further augmentation and refinement as outlined below.2a
Results and discussion
Although Baines5 and Stratakis6 have each independently demonstrated that (trans-2-phenylcyclopropyl)ethyne (4)5 and 3-(2-phenylcyclopropyl)prop-2-yn-1-ol (5)6 are both highly useful mechanistic probes for distinguishing between vinylic free radical and vinylic carbocation pathways in organic reactions,5,6 some have found the current pathways to 55–7 to be somewhat difficult to implement.8 We have therefore devised a completely new route to 4 and 5 (Scheme 1) which now allows both probes to be conveniently prepared on multi-gram scale in good yield (41–56%, over 4 steps), without recourse to harsh −78 °C organolithium-based reaction conditions.
 | ||
| Scheme 1 A new synthesis of the alkyne probes 4 and 5. | ||
Our new pathway to 4 and 5 (see Scheme 1) sets off from commercially available (±)-trans-2-phenylcyclopropane-1-carboxylic acid (1), and requires just four steps to reach 5: Weinreb amidation with EDCI.HCl, semi-reduction of 2 with DIBAL, Ohira–Bestmann alkynylation9 of the aldehyde 3, and alkyne hydroxymethylation under the mild Zn(OTf)2/TMEDA conditions of Hale and Manaviazar10 for base-sensitive acetylenes. Our new route to (±)-5 is presented in full in Scheme 1.
According to Baines,5 it is possible to generate the α-2-phenyl-cyclopropylvinyl cation 6 through protonation of the alkyne 4 with conc. H2SO4 in THF/H2O (4:1) at reflux (Scheme 2) and, once it is formed, 6 undergoes rapid cyclopropane ring-cleavage (kring-opening = 2 × 1012 s−1) to give the homoallenyl benzylic cation 9 alongside 6.
 | ||
| Scheme 2 Baines’ successful interception of the 2-phenylcyclopropylvinyl cation 6 with H2O.5 | ||
Significantly, both intermediates are capable of being successfully intercepted with the H2O that is present in the medium, with the benzylic alcohol 8 forming alongside the methyl ketone 7 in 10.7:1 ratio.
Likewise, Velegraki and Stratakis6 were able to successfully isolate the enone 10 exclusively in 82% yield from the Au-catalysed hydration of alkynol 5 (Scheme 3), which confirmed that this reaction was proceeding via a gold-stabilised vinyl cation that could efficiently be trapped with the H2O that was present. Importantly, the structure of 11 very closely resembled the generalised tin-stabilised vinyl cation 18 (Scheme 4) that has been postulated to be a key intermediate in the O-directed free radical hydrostannation of dialkylacetylenes by some contributors to the field.4
 | ||
| Scheme 3 Velegraki and Stratakis’ vinyl cation trappings with H2O.6 | ||
 | ||
| Scheme 4 Our proposed use of the probe 5 to test out the stannylvinyl cation mechanistic theory4 of alkyne hydrostannation. | ||
These outcomes of Baines5 and Stratakis6 suggested to us that it should be possible to use the aforementioned 4:1 THF:H2O conditions of Baines,5 to readily trap the tin/cyclopropyl-stabilised cation 18, and its so-derived benzylic stannyl homoallenyl cation 20, to obtain 14–17, if the doubly-stabilised ion pair 18 (R = Bu or Ph) was indeed a genuine reaction intermediate in the O-directed free radical hydrostannation of alkynol 5 with stannanes. This would, of course, be the position taken up by proponents of the stannylvinyl cation theory of alkyne hydrostannation (see Scheme 4).4
Accordingly, we initially set out to investigate the O-directed free radical hydrostannation of 5 under the standard rt experimental conditions2,4 of 2 equiv. of Bu3SnH and 0.1 equiv. of Et3B in PhMe, in the presence of O2, and found that the ring-opened stannylallene 21 formed exclusively in 43% yield (Scheme 5). It was produced alongside unreacted starting material. Likewise, when the very same reaction was performed with 5 and Bu3SnH in THF/H2O (4:1) at 72 °C for 2.5 h, the stannylallene 21 once again formed as the sole alkyne-derived product but, on this occasion, it was isolated in 31% yield alongside unreacted starting alkynol 5.
 | ||
| Scheme 5 Our O-directed hydrostannations2 with the probe 5. | ||
Significantly, neither the α-stannyl enone 14 (R = Bu) nor the benzylic alcohol 15 (R = Bu) (Scheme 4) were ever detected as reaction products in this aqueous mixture which, given Baines5 and Stratakis’6 earlier work of Schemes 2 and 3, most definitely ruled out the intermediacy of the tin-stabilized 2-phenylcyclopropyl-1-stannylvinyl cations 18open/18closed (R = Bu), and the benzylic stannylallenyl cation 20 (R = Bu) in such reactions. The fact that the stannylallene 21 (Scheme 5) was the only alkyne-derived product that formed in THF/H2O, only satisfactorily aligned with a reaction mechanism where the stannylvinyl radical 26 (Scheme 6) underwent fast EH1 eliminative ring-cleavage to give the benzylic radical 28, which then H-atom abstracted from the Bu3SnH. The stannylallene 21 could not possibly be forming through a Bu3SnH-mediated SN1-type cationic reduction of 20 (R = Bu), nor from a concerted SN2-type ionic reduction of the stannylvinyl cation 18 (R = Bu), otherwise 14 and 15 (R = Bu) (Scheme 4) would almost certainly have formed competitively. Their absence in THF/H2O only realistically pointed to an entirely free radical pathway3,11,12 being the true source of 21 (Scheme 6). Such a unimolecular EH1 mechanism would be in accord with the high logA values that were recorded for the related cyclopropane ring-cleavages examined in the previous paper.1
 | ||
| Scheme 6 The mechanism by which 21 and 23 arise from 5. | ||
Other evidence that strongly argued against the intermediacy of a stannylvinyl cation4 in such hydrostannation reactions came from the rt reaction of 5 with Ph3SnH (2 equiv.) and Et3B (0.1 equiv.)/O2 in PhMe at 0.1 M substrate concentration over 3.25 h (Scheme 5). Apart from the stannylallene 23 being formed, the (±)-bis-triphenylstannylated adduct 2413 was also co-created as part of a 1.39:1 mixture that favoured 23. Following SiO2 flash chromatography, the unseparated mixture was isolated in 67% yield. Separation of 23 and 24 did, however, prove possible by multi-elution SiO2 preparative TLC using 20:1 petrol:EtOAc as eluent. This allowed their structures to be securely determined.
Our structural assignment of 24 is based upon detailed 2D and DEPT NMR analysis, which confirmed the presence of 35 aromatic and 9 non-aromatic protons in the 600.13 MHz 1H spectrum of 24 in CDCl3. As well as this, 48 carbons were detected in the 13C NMR spectrum of 24. The residency of two Ph3Sn groups was deduced from there being only 8 aromatic carbon signals at δ 139.2, 138.9, 137.4, 137.1, 128.9, 128.8, 128.6 and 128.5 ppm, which revealed NMR equivalency for all the Ph groups that were present in the two Ph3Sn residues. In the 1H spectrum of 24, there was a scalar 3J coupling between the olefinic H(4) resonance at δ 6.52 ppm and the two adjacent H(5) atoms that appeared as part of 4-proton multiplet centred around δ 2.28 ppm. That multiplet also contained the protons for H(6) which were coupled to the H(5) protons. As for H(2), it resonated as much a less shielded dd at δ 3.27 ppm, and it showed 3J couplings of 6.6 and 6.0 Hz with its neighbouring diastereotopic H(1) protons, which appeared as ddd signals at δ 4.03 and 3.87 ppm. Its attached C(2) itself resonated at δ 43.5 ppm in the 13C NMR spectrum in CDCl3, and importantly, it showed the expected 2J119/117Sn–13C coupling of 36.2 Hz with its neighbouring SnPh3 at C(3). The latter alkenic resonance appeared at δ 141.1 ppm, and the fact that it was a quaternary carbon was proven by DEPT spectroscopy. The other olefinic carbon at C(4) appeared at δ 145.3 ppm and, as one would anticipate, it showed the requisite HSQC correlation with the H(4) signal at δ 6.52 ppm, which overlapped with the resonances for two Ph protons. Importantly, H(4) showed a large 3J1H–119/117Sn coupling of 166.8 Hz with the Ph3Sn group resident at C(3) which confirmed the (Z)-olefin geometry for 24 and the fact that vicinal SnPh3 groups must be present at C(2) and C(3). Collectively, these observations unambiguously defined the structure of 24, and thus lent considerable weight to the EPR interpretation made herein.
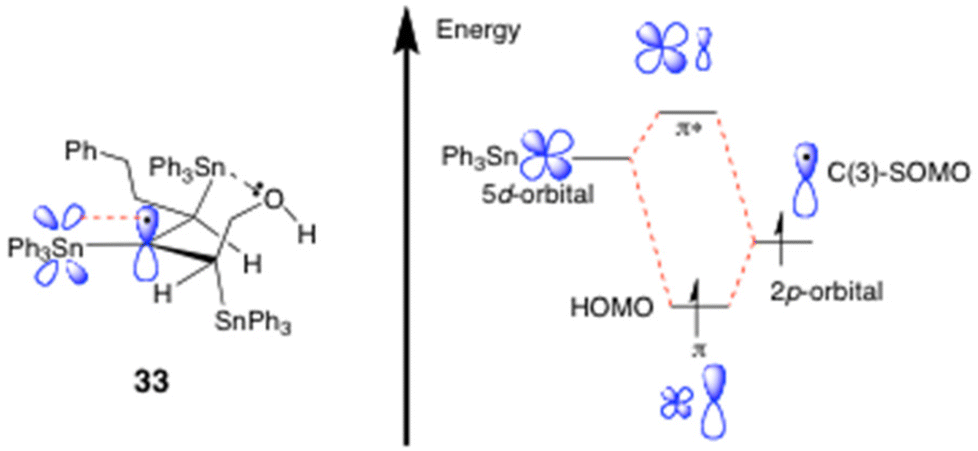
The 2,3-bis(triphenylstannyl)hex-3-en-1-ol 24 that was co-formed in the hydrostannation of 5 seemingly arises from the radical 30 by a reversible O-directed addition3,11,12 to the central C(3)-carbon of the allene (Scheme 7). The resulting tertiary allylic radical 31a then mesomerically isomerises and equilibrates with accompanying fast bond rotation, to give the most stable radical 31c prior to this abstracting a H-atom to give 24. The O-directed Ph3Sn˙ radical addition to 30 would be highly favourable due to the radical 31c being tertiary, allylic, and doubly hyperconjugatively stabilised14 by its α- and β-Ph3Sn groups. Unfortunately, our extensive EPR examination15 of the reaction of 5 with Ph3SnH (1 equiv.) and cat. Et3B/O2 in PhMe at 230–250 K (Scheme 7 and Fig. 1) failed to identify either 27, the benzylic stannyl-homoallenyl radical 29 or the 2,3-bis(triphenylstannyl)hex-3-en-2-yl radical 31c in any of the reaction mixtures that were generated. Presumably this is because all three radicals rapidly transit into the products 23 and 24. Invariably, the main species that was always seen accumulating over the course of 10 min to 1 h was consistent with it being the 2,3,4-tris(triphenylstannyl)hex-3-yl radical 33 (Scheme 7 and Fig. 1).
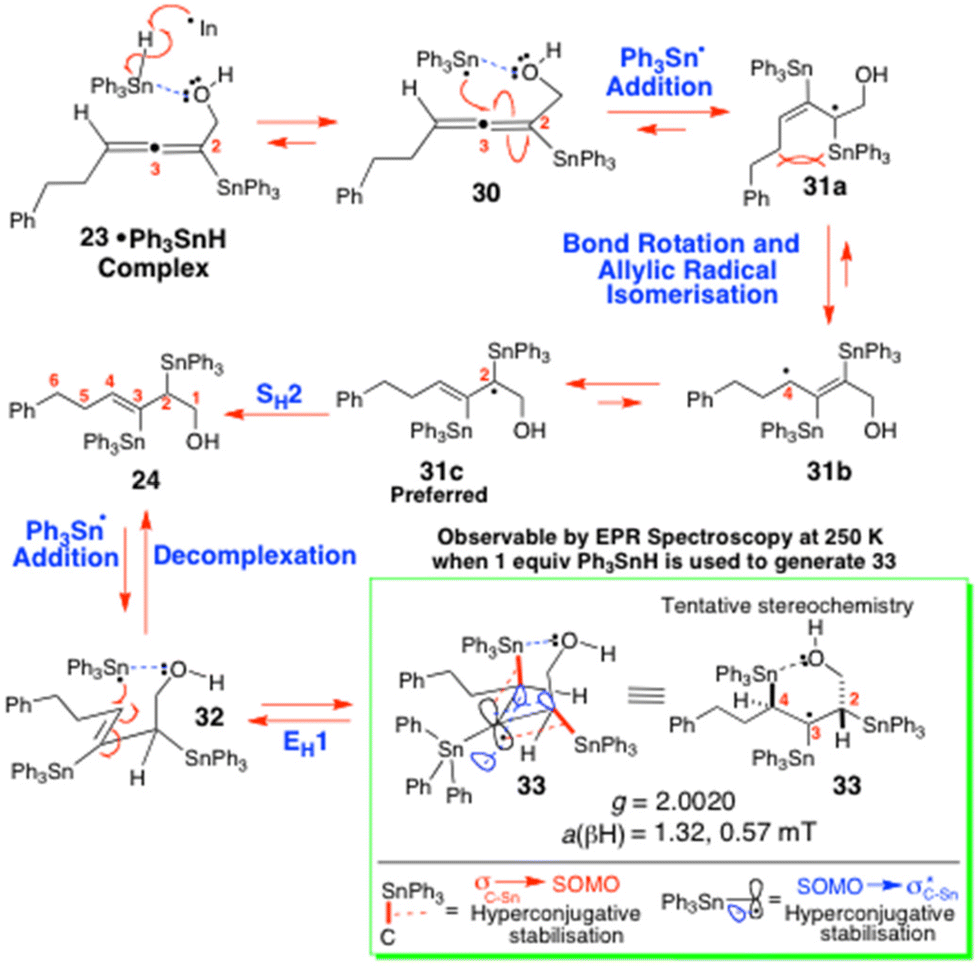 | ||
| Scheme 7 The mechanism by which 24 might be arising and being converted into the 2,3,4-tris(triphenylstannyl)hexyl radical 33. | ||
 | ||
| Fig. 1 The EPR spectrum of the 2,3,4-tris(triphenylstannyl)hex-3-yl radical 33 in PhMe at 250 K with EasySpin simulation16 overlayed. | ||
Presumably, radical 33 forms readily from 24 as a result of the new C(3)-radical centre being triply hyperconjugatively stabilised,12,14,15 it being tertiary, and it also being extraordinarily sterically shielded by the three proximal Ph3Sn groups, which each collectively help to prevent it from undergoing fast H-atom abstraction from the Ph3SnH. The end-result is a radical of fairly high longevity, sufficient for 33 to be readily observed in PhMe solution at −23 °C by EPR spectroscopy.15
Now with regard to the dd observed for radical 33, its multiplicity is fully consistent with the unpaired electron coupling with its two non-equivalent H-atom neighbours at C(2) and C(4) (Scheme 7). EasySpin simulations16 have revealed 1Hβa values of 1.32 mT (13.2 G) and 0.57 mT (5.7 G) for these splittings and, most reassuringly, our g of 2.0020 for 33 matches up very well with the g values of 2.002017a and 2.0020517b reported for the Me3SnCH2CH2˙ radical, where substantial β-C–Sn bond hyperconjugation14 is suggested17 to occur.
The different magnitudes of the two 1Hβ hyperfine values can be attributed to variations in the dihedral angle between the β-H-atoms at C(2) and (C4) and the SOMO, as described by the Heller–McConnell equation,18 as well as differences in spin density resulting from the varying degrees of hyperconjugation between the Ph3Sn substituents and the C(3) radical, as it dynamically oscillates between 33 and 32.
We strongly suspect that the O–Sn coordinated radical 32 is responsible for the broad singlet that is also present in this EPR spectrum, but clearly, such an unsupported assignment can only be considered tentative at best. For a further discussion of the EPR spectrum of 32 and 33 and the various EPR experiments that were performed, see the ESI.‡
As for the mechanism of this proposed triple hyperconjugative stabilisation of radical 33, it is almost certainly complex. It potentially arises from the two vicinal β-Ph3Sn-C bonds at C(2) and C(4) both primarily engaging in strong hyperconjugation with the partially-filled radical SOMO (i.e. σC(2) and C(4)–Sn → SOMO electron transfer) and, concurrently, the C(3) radical itself hyperconjugatively delocalising into the empty vicinal C-SnPh3 antibonding orbitals at C(2) and C(4) (i.e. SOMO → σ*C(2)–Sn and C(4)–Sn electron transfer).1,14,15
Clearly there must be very different degrees of σSn–C → SOMO and SOMO → σ*Sn–C(Ph) hyperconjugation dynamically occurring within 33, due to the C(4)-SnPh3 bond repeatedly being broken and reforming as the radical switches between itself and 32, most especially given that the C(2)-SnPh3 bond always remains intact throughout these interconversions. While we are not in a position to accurately assess the precise extent of these differing hyperconjugative interactions at present, these primary effects are depicted in valence bond format in Scheme 7 and Fig. 2, to enable readers to readily visualise the exact electron-delocalising hyperconjugative movements potentially involved. One obstacle to gauging the true degree of hyperconjugation that is occurring will stem from the intermolecular nature of the O–Sn interaction that is involved. No doubt this O-atom will be stabilising2,12a the complexed Sn radical in 32.
 | ||
| Fig. 2 How the C–Sn σ-bonds and σ*-orbitals of the β-Ph3Sn groups might be hyperconjugatively stabilising the radical 33. The radical SOMO can behave both as an electron-acceptor and as a donor. | ||
As for the C(3)-α-Ph3Sn group, it is suggested that it will most likely stabilise the C(3) radical via the delocalisative mechanism shown in Fig. 3.19a Such a mode of stabilisation would involve the pz electron of the radical behaving as an electron donor and delocalising into the σ*antibonding orbitals of the three Sn–C σ bonds that connect the Ph groups to the C(3)–Sn.19,20 In other words it will be a SOMO → σ*Sn–C(Ph)-type radical stabilising interaction and, once more, a Valence Bond representation most readily allows one to easily see this.
 | ||
| Fig. 3 The α-Ph3Sn SOMO → σ*Sn–C(Ph) delocalising stabilisation of 33. | ||
Similar stabilising interactions have previously been proposed by Sekiguchi and coworkers,19 to explain the high stability of (t-Bu2MeSi)3M˙ radicals, where M is Sn, Ge or Si. In those instances, the Sn, Ge and Si radical SOMOs were suggested to beneficially interact with the σ*antibonding orbitals of the Si–C(t-Bu) bonds to bring about substantial radical delocalisation and stabilisation. In the case of the (t-Bu2MeSi)3Sn˙ radical,19a X-ray crystallography further revealed that it had shorter Si–Sn bonds than normal, which provided very good supportive evidence for the existence of such hyperconjugation. It is thus already well established19 how the σ*antibonding orbitals of group 14a α-metal bonds can readily engage in radical-stabilising hyperconjugative interactions with adjacent radicals. Similar hyperconjugative stabilisation has also recently been reported for α-triphenylstannyl phosphinocarbenes21 where the carbene lone pair likewise donates into the Sn–C(Ph) σ* orbitals of the Ph3Sn.
Now given that the C(4)-SnPh3 group of 33 regioselectively weakens and subsequently undergoes stereospecific EH1 elimination back into 32, to ultimately return (Z)-24, while its C(2)-SnPh3 counterpart remains totally undisturbed (Scheme 7), this observation provides very strong and convincing experimental support for the C(4)-SnPh3 being regioselectively involved in strong internal O–Sn coordination with the terminal hydroxyl of 33. Such an event would clearly lengthen and selectively weaken the C(4) C–Sn bond to guarantee that it preferentially breaks to bring about this eliminative outcome.
Clearly our present EPR study is significant for it has provided the first in situ spectroscopic evidence for O-coordinated Ph3Sn˙ radicals preferentially adding in 1,2-fashion reversibly to the least- hindered alkene carbon of the (Z)-trisubstituted vinyl triphenyltin products of these O-directed hydrostannations at low temperature (250 K/−23 °C).3,22–24 It has thus powerfully shown that these events can give doubly hyperconjugatively stabilised 1,2-bis-triphenylstannylalkyl tertiary radical adducts21 that can stereospecifically eliminate under O–Sn coordinative control,2,12e to return the original (Z)-configured vinylstannane exclusively, in the form of its O-complexed Ph3Sn˙ radical. The latter can then subsequently decomplex or re-add.
Our current work now very strongly suggests that one of the main reasons why (Z) → (E) isomerisation is NOT seriously detrimental in Et3B-initiated hydrostannations of this type, at room temperature or below, is because these competing Ph3Sn˙ radical additions to the (Z)-trisubstituted vinyltriphenyltin products, and the subsequent eliminations that return those (Z)-vinyltriphenyltins, both proceed under O–Sn coordinative control;3,12 which powerfully prevents full central C–C bond rotation from ever taking place within the bis-tin-1,2-radical adducts prior to the Ph3Sn˙ radical elimination occurring.
Our EPR work on 33 has thus provided remarkable new insights into the complex mechanistic course of the rt O-directed free radical hydrostannation reaction with Ph3SnH, and it has likely helped to explain why these radical reactions typically proceed with such excellent levels of stereo- and regio-control, and without significant competing (Z)/(E) product isomerisation under the room or lower reaction temperature circumstances we always perform these reactions.
By way of contrast, when such O-directed free radical hydrostannations are conducted at high reaction temperatures, for prolonged periods, under the AIBN-mode of initiation particularly,23,24 the normally unfavourable,2a geometrically-isomerising, 1,1-mode of Ph3Sn˙ radical addition/elimination20,21 gradually starts to repeatedly occur upon the product trisubstituted (Z)-vinyltins of these reactions, albeit it in a minor way.
Nonetheless, such a constantly-recurrring competitive mode of isomerising tin radical addition/elimination, proceeding alongside the much more favourable, non-isomerising, 1,2-mode of addition/elimination in the (Z)-trisubstituted vinyltin systems, will typically lead to a stereochemically adverse outcome over time. It appears that when the ordinarily unfavourable2a 1,1-mode of R3Sn˙ radical addition/elimination23,24 occurs, there is often no appropriate restraining element within the intermediary 1,1-adduct, to prevent central C–C-bond rotation from occurring before the normally fast EH1 elimination proceeds. Consequentially, prolonged high temperature alkyne free radical hydrostannations instigated by Bu3SnH/AIBN (in the main)24 will often encounter significant (Z)/(E)-isomerisation. Therefore, extended reaction times at high reaction temperature should be avoided, if excellent product stereocontrol is desired.
We trust that the present paper has now fully clarified how the O-directed free radical hydrostannation of propargylically-oxygenated dialkylacetylenes mechanistically proceeds with Ph3SnH/cat. Et3B at rt or below (see Scheme 8), and why competing Ph3Sn˙ radical-induced (Z) → (E) trisubstituted vinyl triphenyltin isomerisation is not usually problematical in such reactions.
 | ||
| Scheme 8 The mechanism2 of the rt O-directed hydrostannation of propargylically-oxygenated dialkylacetylenes Ph3SnH/cat. Et3B, and why the favourable rt 1,2-addition/elimination of Ph3Sn˙ radicals does not cause (E)/(Z) isomerisation, while the high temperature, disfavoured, minor 1,1-addition/elimination pathway frequently does over extended timeframes. | ||
While the 1,2-mode of Ph3Sn˙ radical addition and elimination does continuously occur upon the (Z)-vinyltriphenyltin products of these reactions, while active Ph3SnH is still present, such processes are generally inconsequential due to O–Sn coordinative control seemingly operating throughout, and this preventing full central C–C-bond rotation from taking place in the intermediary 1,2-di-tin adducts before stannyl radical elimination occurs. It is thus typically non-isomerising and benign, when it does occur, under our standard rt or below reaction conditions.
This contrasts very sharply with its sterically unfavourable, high temperature, 1,1-additive/eliminative counterpart (Scheme 8) which is gradually isomerising over time.23,24
Conclusions
Alkyne hydrometallation reactions25 are continuing to play a prominent role in the fields of complex natural product total synthesis and medicinal chemistry, and the highly α- and (Z)-selective O-directed free radical hydrostannation of propargylically-oxygenated dialkylacetylenes with Ph3SnH and cat. Et3B/O2 in PhMe3 remains one of the most expedient and reliable methods for positioning (Z)-trisubstituted alkenes within highly complex target structures,26 particularly alkene motifs that are flanked by allylic stereocentres. In this aspect, this protocol has proven particularly powerful when it has been allied with the Marshall chiral allenylzinc addition to aldehydes,26d,27,28 the Carreira asymmetric alkynylation,29 and the Hale–Manaviazar alkyne hydroxymethylation10 reactions.In full agreement with the work previously published by our two teams over the period 2005–2021,2,3,15 the results reported here, and the paper that precedes it,1 once more define an entirely free radical mechanism2,3,15,23 for the O-directed hydrostannation of dialkylacetylenes with stannanes under cat. Et3B/O2 initiation (Scheme 8), and they further argue against the recently hypothesised roles for stannylvinyl cation intermediates4 in these processes.
As a result of the new EPR work performed here on the probe 5 with Ph3SnH/cat. Et3B, a new triply hyperconjugatively stabilised O-coordinated radical 33 has had its structure securely determined at low temperature, and its O-coordinated Ph3Sn˙ radical precursor 32 has additionally been potentially characterised. The detection of these two key radical intermediates has now given unique mechanistic insights into why many room temperature Ph3Sn˙ radical additions to the (Z)-vinyl triphenyltin products of these reactions [i.e. (Z)-41] do not cause significant erosive (Z) → (E)-isomerisation.
This is likely because the room temperature or below Et3B/O2-mediated Ph3Sn˙ radical addition reactions follow a predominantly sterically-controlled 1,2-addition/elimination pathway that operates under strong internal O–Sn coordinative control. The existence of prolonged internal O–Sn coordination within these adducts would clearly prevent central C(1)–C(2) bond rotation from freely proceeding which, in turn, would powerfully halt the (Z) → (E)-isomerisation event.
While this benign 1,2-addition/elimination pathway will continue to dominate the high temperature alkyne hydrostannation process, the much less favourable 1,1-addition/elimination pathway will also gradually start to compete and have a presence at higher temperatures, even if in a very minor way, in comparative terms.
Nonetheless, the continued repeated occurrence of this process, over time, will eventually allow significant (Z) → (E)-product isomerisation to proceed, in vinyltin systems where central C(1)–C(2) bond rotation is rotationally possible, and the process cannot be easily restrained before stannyl radical elimination occurs (see Scheme 8 for a mechanistic depiction of this process with Ph3SnH, but similar arguments hold with other R3SnH reagents).
With the new experimental data that has been gathered here and in the previous paper,1 it is hoped that the longstanding debate about how the rt O-directed free radical hydrostannation of propargylically-oxygenated dialkyl acetylenes with Ph3SnH/cat. Et3B/O2 mechanistically proceeds will now finally be settled. What is demonstrably clear from all of the mechanistic work conducted to date1–3,11a,12,15,23 is that an entirely free radical, O-directed, mechanism operates both for this and the high temperature Bu3SnH variant of this reaction under both the cat. Et3B/O2 and AIBN initiated conditions.
The present paper has also spectroscopically demonstrated that stannyl radical 1,2-addition/elimination processes are occurring constantly and dynamically at low temperatures throughout the course of the alkyne hydrostannation process, until all of the tin hydride has been consumed, and it has shown that such competitive side-reactions are not stereochemically erosive in their nature, at least not in the room temperature variant of the Ph3SnH/cat. Et3B dialkylacetylene hydrostannation reaction.2,3,26,28
However, for analogous high temperature hydrostannation protocols, conducted over extended periods,16 such addition/elimination processes can have a very dramatic and quite profound effect on the final (Z)/(E)-selectivity attained,16 but in a time- and temperature-dependent manner, by allowing the normally unfavourable, stereochemically erosive, 1,1-addition/elimination process to gradually contribute to outcome in the manner shown in Scheme 8.16
Of course, because EPR spectroscopy is a highly sensitive technique for detecting the presence of reasonably long-lived free radicals, and even very tiny quantities of a particular radical can give rise to a quite reasonable signal, it is difficult to quantitatively assess to what degree 33 is being formed relative to 24 in terms of a providing a relative ratio between the two entities at any point in time, since 24 is EPR inactive. While it would indeed be very interesting to ascertain this, using paramagnetic reference standards, such work is far from trivial to conduct, and it can be fraught with errors.
Nonetheless, we might try to look into this in the very near future to give a much greater idea of the true extent of competitive Ph3Sn˙ radical addition that is typically going on.
Finally, we would point out that our low temperature EPR data from the Ph3SnH/cat. Et3B mediated hydrostannation of 5 and 24 mechanistically aligns with the sterically-controlled outcomes of past alkene free radical hydrostannation reactions (Scheme 9),30 which generally have the tin radical adding reversibly, predominantly at the least hindered alkenic carbon. In particular, our results are strongly consonant with the work of Sommer and Kuivila30a on the photochemical addition of Me3Sn˙ radicals to methylcyclohexene (45). They are also in agreement with the studies of Mitchell22a and Fish.30d
 | ||
| Scheme 9 Past free radical hydrostannations of alkenes.30 | ||
They are likewise concordant with the more recent hydrostannation observations of Hale and Manaviazar on 2-methylene propane-1,3-diol (47) in solution (Scheme 9),30b and Wuest beforehand.30c
Experimental details
New experimental procedures for the preparation of the (±)-trans-3(2-phenylcyclopropyl)-prop-2-yn-1-ol, mechanistic probe 5
To a well-stirred solution of (±)-trans-2-phenylcyclopropane carboxylic acid 1 (5.0 g, 30.83 mmol) and N,O-dimethylhydroxylamine hydrochloride (3.31 g, 33.91 mmol, 1.1 equiv.) in dry CH2Cl2 (49.7 mL) at rt under N2 was added Et3N (12.9 mL, 92.48 mmol, 3 equiv.) in a slow stream. The initial slightly cloudy solution gradually formed a thick white precipitate by the time the Et3N addition was complete. The reaction mixture was stirred for 5 min before EDCI (7.09 g, 36.99 mmol, 1.2 equiv.) was added in one portion. The thick slurry was stirred at rt for 24 h, maintaining the N2 atmosphere throughout. TLC analysis (4
 :1 petrol:EtOAc as eluent, PMA stain) showed the amide 2 as a dark black/blue spot; it was faster-moving than the starting acid 1, and was formed cleanly. The reaction mixture was diluted with CH2Cl2 (30 mL) and washed with H2O (20 mL). The aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were then washed with H2O (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by gradient-elution SiO2 flash chromatography with petrol–EtOAc (10:1 → 8:1 → 5:1) to give the title Weinreb amide 2 (5.03 g, 79%) as a runny oil. 1H NMR of 2 (600.13 MHz, CDCl3) δ: 7.30–7.26 (m, 2H, H7), 7.19 (tt, J = 7.8, 1.2 Hz, 1H, H8), 7.15–7.11 (m, 2H, H6), 3.69 (s, 3H, –OMe), 3.23 (s, 3H, –NMe), 2.50 (ddd, J = 9.0, 6.0, 4.2 Hz, 1H, H4), 2.41 (very br s, 1H, H2), 1.63 (ddd, J = 9.0, 5.4, 4.2 Hz, 1H, H3a), 1.30 (ddd, J = 8.4, 6.0, 4.2 Hz, 1H, H3b) ppm. 13C NMR of 2 (150.9 MHz, CDCl3) δ: 173.1 (C1), 140.8 (C5), 128.4 (C7), 126.23 (C8), 126.19 (C6), 61.6 (–OMe), 32.6 (–NMe), 25.9 (C4), 21.5 (C2), 16.4 (C3) ppm. Electrospray HRMS of 2: Calcd for C12H15NO2Na [M + Na]+: 228.1001. Found: 228.1004.
:1 petrol:EtOAc as eluent, PMA stain) showed the amide 2 as a dark black/blue spot; it was faster-moving than the starting acid 1, and was formed cleanly. The reaction mixture was diluted with CH2Cl2 (30 mL) and washed with H2O (20 mL). The aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were then washed with H2O (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by gradient-elution SiO2 flash chromatography with petrol–EtOAc (10:1 → 8:1 → 5:1) to give the title Weinreb amide 2 (5.03 g, 79%) as a runny oil. 1H NMR of 2 (600.13 MHz, CDCl3) δ: 7.30–7.26 (m, 2H, H7), 7.19 (tt, J = 7.8, 1.2 Hz, 1H, H8), 7.15–7.11 (m, 2H, H6), 3.69 (s, 3H, –OMe), 3.23 (s, 3H, –NMe), 2.50 (ddd, J = 9.0, 6.0, 4.2 Hz, 1H, H4), 2.41 (very br s, 1H, H2), 1.63 (ddd, J = 9.0, 5.4, 4.2 Hz, 1H, H3a), 1.30 (ddd, J = 8.4, 6.0, 4.2 Hz, 1H, H3b) ppm. 13C NMR of 2 (150.9 MHz, CDCl3) δ: 173.1 (C1), 140.8 (C5), 128.4 (C7), 126.23 (C8), 126.19 (C6), 61.6 (–OMe), 32.6 (–NMe), 25.9 (C4), 21.5 (C2), 16.4 (C3) ppm. Electrospray HRMS of 2: Calcd for C12H15NO2Na [M + Na]+: 228.1001. Found: 228.1004.
To a well-stirred −20 °C solution of the Weinreb amide 2 (5.03 g, 24.51 mmol) in dry PhMe (223 mL) under N2 was added i-Bu2AlH (1.0 M solution in hexanes, Aldrich, 24.51 mL, 24.51 mmol, 1 equiv.) dropwise over 18 min. Stirring was continued at −20 °C under N2 and the reaction was continuously monitored by TLC. After 3.5 h, the reaction was deemed to be essentially over by TLC analysis (4
 :1 petrol:EtOAc, eluent, PMA stain), with only a very tiny quantity of the starting amide 2 still seen to be remaining, and the faster-moving aldehyde 3 dominating the plate; it stained black in PMA stain. The mixture was quenched by the careful dropwise addition of MeOH. The reaction mixture was then diluted with Et2O (20 mL). Saturated aqueous Rochelle salt solution (10 mL) was then added cautiously via a pipette. More Et2O (80 mL) was added, followed by more saturated aqueous Rochelle salt solution (40 mL). The mixture was transferred to a separatory funnel, vigorously shaken, and when the layers had separated, the organic layer was removed. The aqueous layer was then extracted further with Et2O (2 × 20 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by SiO2 flash chromatography with petrol–EtOAc (15:1) to give the title aldehyde 3 (3.12 g, 87%) as a colourless oil. 1H NMR of 3 (600 MHz, CDCl3) δ: 9.32 (d, J = 4.8 Hz, 1H, –CHO), 7.29 (m, 2H, H7), 7.22 (tt, J = 7.2, 1.2 Hz, 1H, H8), 7.13–7.09 (complex m, 2H, H6), 2.62 (ddd, J = 9.6, 6.6, 4.2 Hz, 1H, H4), 2.165 (dddd, J = 8.4, 5.4, 4.8, 4.2 Hz, 1H, H2), 1.72 (dt, J = 9.6, 4.8 Hz, 1H, H3a), 1.52 (ddd, J = 8.4, 6.6, 4.8 Hz, 1H, H3b). 13C NMR of 3 (150.9 MHz, CDCl3) δ: 199.7 (C1), 138.9 (C5), 128.6 (C7), 126.8 (C8), 126.2 (C6), 33.7 (C2), 26.5 (C4), 16.4 (C3).
:1 petrol:EtOAc, eluent, PMA stain), with only a very tiny quantity of the starting amide 2 still seen to be remaining, and the faster-moving aldehyde 3 dominating the plate; it stained black in PMA stain. The mixture was quenched by the careful dropwise addition of MeOH. The reaction mixture was then diluted with Et2O (20 mL). Saturated aqueous Rochelle salt solution (10 mL) was then added cautiously via a pipette. More Et2O (80 mL) was added, followed by more saturated aqueous Rochelle salt solution (40 mL). The mixture was transferred to a separatory funnel, vigorously shaken, and when the layers had separated, the organic layer was removed. The aqueous layer was then extracted further with Et2O (2 × 20 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by SiO2 flash chromatography with petrol–EtOAc (15:1) to give the title aldehyde 3 (3.12 g, 87%) as a colourless oil. 1H NMR of 3 (600 MHz, CDCl3) δ: 9.32 (d, J = 4.8 Hz, 1H, –CHO), 7.29 (m, 2H, H7), 7.22 (tt, J = 7.2, 1.2 Hz, 1H, H8), 7.13–7.09 (complex m, 2H, H6), 2.62 (ddd, J = 9.6, 6.6, 4.2 Hz, 1H, H4), 2.165 (dddd, J = 8.4, 5.4, 4.8, 4.2 Hz, 1H, H2), 1.72 (dt, J = 9.6, 4.8 Hz, 1H, H3a), 1.52 (ddd, J = 8.4, 6.6, 4.8 Hz, 1H, H3b). 13C NMR of 3 (150.9 MHz, CDCl3) δ: 199.7 (C1), 138.9 (C5), 128.6 (C7), 126.8 (C8), 126.2 (C6), 33.7 (C2), 26.5 (C4), 16.4 (C3).
To a stirred rt solution of the aldehyde 3 (2.68 g, 18.33 mmol) in dry MeOH (160 mL) at rt under N2 was added K2CO3 (5.07 g, 36.67 mmol, 2 equiv.) in one portion. A solution of freshly prepared dimethyl-1-diazo-2-oxopropylphosphonate (5.63 g, 1.6 equiv.) (Ohira–Bestmann reagent)9 in dry MeOH (23.3 mL) was then added dropwise to the reaction mixture via cannula over 15 min, whilst maintaining the N2 atmosphere. The reactants were thereafter stirred at rt for 21 h, whereupon TLC analysis (neat petrol as eluent) showed a single faster-moving product 4 had formed cleanly; it stained black on a glass-backed TLC plate in PMA stain. The reaction mixture was diluted with CH2Cl2 (80 mL) and H2O (50 mL) and separated. The aqueous layer was then further extracted with CH2Cl2 (30 mL × 3) and the combined organic extracts were dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by SiO2 flash chromatography with neat petrol to give the title alkyne 4 (1.88 g, 72%), as a colourless oil. 1H NMR of 4 (600.13 MHz, CDCl3) δ: 7.27–7.23 (m, 2H, H8), 7.19–7.17 (m, 1H, H9), 7.07–7.05 (m, 2H, H7), 2.27 (ddd, J = 8.4, 6.0, 4.2 Hz, 1H, H5), 1.89 (d, J = 1.8 Hz, 1H, H1), 1.49 (m, 1H, H3), 1.31 (ddd, J = 9.0, 6.0, 4.8 Hz, 1H, H4a), 1.22 (ddd, J = 8.4, 6.0, 4.8 Hz, 1H, H4b) ppm. 13C NMR of 4 (150.9 MHz, CDCl3) δ: 140.4 (C6), 128.4 (C8), 126.3 (C9), 125.9 (C7), 86.2 (C2), 64.8 (C1), 26.0 (C5), 17.4 (C4), 10.8 (C3) ppm. A satisfactory mass spectral confirmation of identity could not be obtained for the alkyne 4 at the QUB mass spectrometry facility. Nonetheless, there was excellent 13C NMR spectral agreement between our version of 4 and the spectrum that had previously been reported in the literature; that data was as follows: 100 MHz 13C NMR data for 4 in CDCl3: δ: 140.4 (C6), 128.3 (–Ph), 126.2 (–Ph), 125.9 (–Ph), 86.1 (C2), 64.7 (C1), 26.0 (C5), 17.3 (C4), 10.8 (C3) ppm (see: ref. 7). Significantly, these workers did report a satisfactory HRMS for [M + H]+ = 143.0857. Calcd for C11H11 [M + H]+ = 143.086.
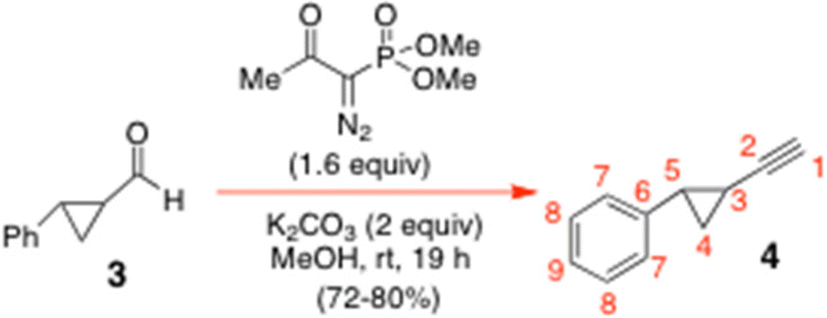
To a very vigorously stirred rt suspension of Zn(OTf)2 (9.45 g, 25.99 mmol, 2.2 equiv.) and TMEDA (3.9 mL, 25.99 mmol, 2.2 equiv.) in dry PhMe (40 mL) under N2 was added Et3N (3.62 mL, 25.99 mmol, 2.2 equiv.) in one portion. The reactants were stirred vigorously at rt for 2 h before a solution of the alkyne 4 (1.68 g, 11.81 mmol) in PhMe (9.2 mL) was introduced via cannula maintaining the N2 atmosphere throughout the addition. The reaction mixture was then heated at 60 °C under N2 for 15 min, whereupon solid paraformaldehyde (0.78 g, 25.99 mmol, 2.2 equiv.) was added in one portion, whilst maintaining the N2 atmosphere throughout the addition. The reactants were stirred under N2 at 60 °C for 25 h, whereafter, the reaction was judged complete by TLC analysis (7
 :1 petrol:EtOAc eluent, PMA stain). The reaction was then quenched by adding saturated aqueous NaHCO3 solution (20 mL) and Et2O (40 mL). The aqueous layer was extracted with Et2O (40 mL × 2) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude oily residue so obtained was purified by gradient-elution SiO2 flash chromatography with petrol–EtOAc (20:1 → 10:1 → 5:1) to give the title alkynol 5 (1.88 g (93%)) as a thick colourless oil that crystallised as a white solid upon storage. Our 1H NMR data for 5 (600.13 MHz, CDCl3) δ: 7.27–7.24 (m, 2H, 2 × H9), 7.19–7.16 (m, 1H, H10), 7.07–7.05 (m, 2H, 2 × H8), 4.25 (dd, J = 6.0, 1.8 Hz, 2H, H1ab), 2.25 (ddd, J = 8.4, 6.0, 4.8 Hz, 1H, H6), 1.85 (t, J = 6.0 Hz, 1H, –OH), 1.51 (m, 1H, H4), 1.30 (ddd, J = 9.0, 5.4, 4.8 Hz, 1H, H5a), 1.24 (ddd, J = 8.4, 6.0, 4.8 Hz, 1H, H5b). Our 13C NMR for 5 (150.9 MHz, CDCl3) δ: 140.5 (C7), 128.4 (C9), 126.2 (C10), 125.9 (C8), 88.1 (C3), 74.9 (C2), 51.3 (C1), 26.1 (C6), 17.5 (C5), 11.2 (C4). LR electrospray MS: Calcd for C24H26LiO2 [2M + 2H + Li]+: 353.20928. Found: 353.2003. Literature6 75 MHz 13C NMR data for 5 in CDCl3: δ: 140.4 (C7), 128.3 (–Ph), 126.1 (–Ph), 125.8 (–Ph), 87.8 (C3), 75.0 (C2), 51.0 (C1), 26.0 (C6), 17.4 (C5), 11.1 (C4) ppm. Lit. HRMS:6 Calcd for C12H13O [M + H]+: 173.0966. Found: 173.0960 (see: ref. 6).
:1 petrol:EtOAc eluent, PMA stain). The reaction was then quenched by adding saturated aqueous NaHCO3 solution (20 mL) and Et2O (40 mL). The aqueous layer was extracted with Et2O (40 mL × 2) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The crude oily residue so obtained was purified by gradient-elution SiO2 flash chromatography with petrol–EtOAc (20:1 → 10:1 → 5:1) to give the title alkynol 5 (1.88 g (93%)) as a thick colourless oil that crystallised as a white solid upon storage. Our 1H NMR data for 5 (600.13 MHz, CDCl3) δ: 7.27–7.24 (m, 2H, 2 × H9), 7.19–7.16 (m, 1H, H10), 7.07–7.05 (m, 2H, 2 × H8), 4.25 (dd, J = 6.0, 1.8 Hz, 2H, H1ab), 2.25 (ddd, J = 8.4, 6.0, 4.8 Hz, 1H, H6), 1.85 (t, J = 6.0 Hz, 1H, –OH), 1.51 (m, 1H, H4), 1.30 (ddd, J = 9.0, 5.4, 4.8 Hz, 1H, H5a), 1.24 (ddd, J = 8.4, 6.0, 4.8 Hz, 1H, H5b). Our 13C NMR for 5 (150.9 MHz, CDCl3) δ: 140.5 (C7), 128.4 (C9), 126.2 (C10), 125.9 (C8), 88.1 (C3), 74.9 (C2), 51.3 (C1), 26.1 (C6), 17.5 (C5), 11.2 (C4). LR electrospray MS: Calcd for C24H26LiO2 [2M + 2H + Li]+: 353.20928. Found: 353.2003. Literature6 75 MHz 13C NMR data for 5 in CDCl3: δ: 140.4 (C7), 128.3 (–Ph), 126.1 (–Ph), 125.8 (–Ph), 87.8 (C3), 75.0 (C2), 51.0 (C1), 26.0 (C6), 17.4 (C5), 11.1 (C4) ppm. Lit. HRMS:6 Calcd for C12H13O [M + H]+: 173.0966. Found: 173.0960 (see: ref. 6).
O-Directed free radical hydrostannation studies with the alkynol 5 and spectral data for 21, 23 and 24
To a round-bottomed flask containing a well-stirred solution of 5 (0.1 g, 0.58 mmol) in PhMe (5.81 mL) under N2 was added Bu3SnH (0.31 mL, 1.1613 mmol, 2 eq.) dropwise, followed by Et3B (0.06 mL, 1 M soln in hexanes, 0.05807 mmol, 0.1 equiv.). Air (5 mL) was then added via a syringe, and the reactants were left to stir under N2 for 24 h at rt, before being concentrated in vacuo. The crude residue was purified by gradient-elution SiO2 flash chromatography with petrol–EtOAc (50
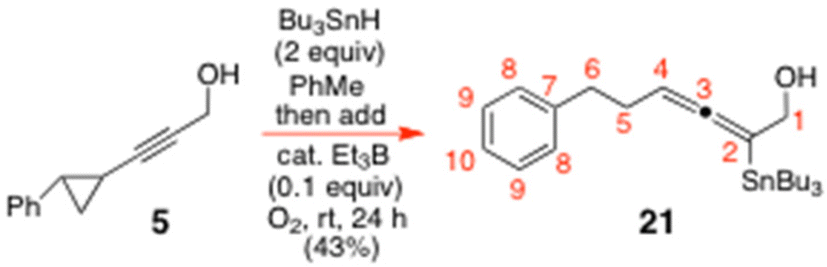 :1 → 25:1) to give the pure allenyltin 21 (130 mg, 43%) as an oil. 1H NMR of 21 (600.13 MHz, CDCl3) δ: 7.33–7.26 (m, 2H, Ph), 7.22–7.16 (m, 3H, Ph), 4.91 (m, 1H, H4), 4.05 (m, 2H, H1), 2.79–2.63 (m, 2H, H6), 2.32 (m, 2H, H5), 1.51 (m, 6H, –C
:1 → 25:1) to give the pure allenyltin 21 (130 mg, 43%) as an oil. 1H NMR of 21 (600.13 MHz, CDCl3) δ: 7.33–7.26 (m, 2H, Ph), 7.22–7.16 (m, 3H, Ph), 4.91 (m, 1H, H4), 4.05 (m, 2H, H1), 2.79–2.63 (m, 2H, H6), 2.32 (m, 2H, H5), 1.51 (m, 6H, –C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2– of SnBu3), 1.36 (t, J = 6.0 Hz, C1-OH), 1.31 (q, 6H, J = 7.2 Hz, 3 × –C2Me of –SnBu3), 0.95 (m, 6H, –C2Sn– of SnBu3), 0.90 (t, 9H, J = 7.2 Hz, 3 × Me of SnBu3). 13C NMR of 21 (150.9 MHz, CDCl3) δ: 199.6 (C3), 141.8 (C7), 128.5 (C9), 128.3 (C8), 125.9 (C10), 96.4 (C2, 1J119Sn–13C = 304.8 Hz, 1J117Sn–13C = 291.2 Hz), 85.5 (C4, 3J119/117Sn–13C = 39.2 Hz), 63.0 (C1, 2J119/117Sn–13C = 31.7 Hz), 35.9 (C6, 5J119/117Sn–13C = 9.1 Hz), 30.43 (C5, 4J119/117Sn–13C = 16.6 Hz), 29.0 (3 × –
2– of SnBu3), 1.36 (t, J = 6.0 Hz, C1-OH), 1.31 (q, 6H, J = 7.2 Hz, 3 × –C2Me of –SnBu3), 0.95 (m, 6H, –C2Sn– of SnBu3), 0.90 (t, 9H, J = 7.2 Hz, 3 × Me of SnBu3). 13C NMR of 21 (150.9 MHz, CDCl3) δ: 199.6 (C3), 141.8 (C7), 128.5 (C9), 128.3 (C8), 125.9 (C10), 96.4 (C2, 1J119Sn–13C = 304.8 Hz, 1J117Sn–13C = 291.2 Hz), 85.5 (C4, 3J119/117Sn–13C = 39.2 Hz), 63.0 (C1, 2J119/117Sn–13C = 31.7 Hz), 35.9 (C6, 5J119/117Sn–13C = 9.1 Hz), 30.43 (C5, 4J119/117Sn–13C = 16.6 Hz), 29.0 (3 × –![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2Me of SnBu3, 3J119/117Sn–13C = 21.1 Hz), 27.3 (3 × –H2CH2Me of SnBu3, 2J119Sn–13C = 57.3 Hz, 2J117Sn–13C = 54.3 Hz), 13.7 (3 ×
H2Me of SnBu3, 3J119/117Sn–13C = 21.1 Hz), 27.3 (3 × –H2CH2Me of SnBu3, 2J119Sn–13C = 57.3 Hz, 2J117Sn–13C = 54.3 Hz), 13.7 (3 × ![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) of SnBu3), 10.2 (3 × –H2Sn of SnBu3, 1J119Sn–13C = 339.5 Hz, 1J117Sn–13C = 324.4 Hz). LRMS Electrospray: Calcd for C24H42LiOSn [M + 2H + Li]+: 473.2418. Found: 473.2976.
of SnBu3), 10.2 (3 × –H2Sn of SnBu3, 1J119Sn–13C = 339.5 Hz, 1J117Sn–13C = 324.4 Hz). LRMS Electrospray: Calcd for C24H42LiOSn [M + 2H + Li]+: 473.2418. Found: 473.2976.
To a round-bottomed flask containing a well-stirred solution of 5 (0.1 g, 0.5807 mmol) in THF
 :H2O (4:1, 5.81 mL) under N2 was added Bu3SnH (0.31 mL, 1.1613 mmol, 2 equiv.) dropwise. The reactants were heated to between 72 °C, whereupon Et3B (0.06 mL, 1 M soln in hexanes, 0.05807 mmol, 0.1 equiv.) was added, followed by air (5 mL) added via a syringe. The reaction mixture was allowed to stir at 72 °C for 1 h, whereafter more Et3B (0.06 mL, 0.05807 mmol, 0.1 eq.) was again added. The reactants were then allowed to stir for 1.5 h at 72 °C. The reaction mixture was quenched by dilution with EtOAc (30 mL) and aqueous NaCl solution (30 mL). The organic layer was washed with H2O (50 mL), dried with MgSO4, filtered and concentrated in vacuo. The residue was purified by SiO2 flash chromatography with petrol–EtOAc (50:1) to give the pure allenyltin 21 (93 mg, 31%) as an oil.
:H2O (4:1, 5.81 mL) under N2 was added Bu3SnH (0.31 mL, 1.1613 mmol, 2 equiv.) dropwise. The reactants were heated to between 72 °C, whereupon Et3B (0.06 mL, 1 M soln in hexanes, 0.05807 mmol, 0.1 equiv.) was added, followed by air (5 mL) added via a syringe. The reaction mixture was allowed to stir at 72 °C for 1 h, whereafter more Et3B (0.06 mL, 0.05807 mmol, 0.1 eq.) was again added. The reactants were then allowed to stir for 1.5 h at 72 °C. The reaction mixture was quenched by dilution with EtOAc (30 mL) and aqueous NaCl solution (30 mL). The organic layer was washed with H2O (50 mL), dried with MgSO4, filtered and concentrated in vacuo. The residue was purified by SiO2 flash chromatography with petrol–EtOAc (50:1) to give the pure allenyltin 21 (93 mg, 31%) as an oil.
To a 50 mL pear-shaped flask inside a glove-bag filled with dry N2 gas was added Ph3SnH (0.57 g, 1.624 mmol). The flask was capped by a rubber septum, removed from the glove bag, and an N2-filled balloon connected to the septum via a wide-gauge needle. A small magnetic stirring bar was introduced into the flask against a counter-flow of N2, followed by the crystalline alkynol 5 (0.14 g, 0.812 mmol). Dry PhMe (8.12 mL) was added to the reaction vessel via syringe, and the contents were manually swirled to assist in dissolution. With vigorous stirring, Et3B (0.081 mL, 1 M soln in hexanes, 0.1 equiv.) was then added to the reaction flask dropwise at rt over 10 seconds. Air (5 mL) was then added to the reaction vessel via syringe, and the reactants were thereafter stirred at rt for 3 h 10 min. TLC analysis (10
 :1 petrol:EtOAc as eluent; anisaldehyde as TLC stain) thereupon indicated that a seemingly single faster-moving major product had formed, but it transpired that this single spot was actually a mixture of two main components 23 and 24 (formed in 1.39:1 ratio), when the reaction was examined by multi-elution TLC analysis. At this point, the reaction was judged to be complete, whereafter the crude reaction mixture was concentrated in vacuo on a rotary evaporator. The oil so obtained was purified by gradient-elution SiO2 flash chromatography initially using petrol:CH2Cl2 (3:1 → 2:1 → 1:1) to remove non-alkyne-derived tin-by-products, and then with petrol:EtOAc (25:1 → 20:1 → 10:1) to obtain the seemingly homogenous mixture of 23 and 24 (0.43 g, 67%). For structural analysis purposes only, however, a small portion of that crude unseparated mixture was dissolved in EtOAc and applied to two glass-backed TLC plates. These were then eluted and briefly air-dried on multiple occasions until the two main components were deemed to be separated. The respective compounds 23 and 24 were then scraped from the plate, and eluted from the TLC silica by suspension in EtOAc and filtration. The now separated individual components 23 and 24 were thereafter further purified individually by SiO2 flash chromatography with 10:1 petrol:EtOAc to give 23 and 24 which were both obtained as oils.
:1 petrol:EtOAc as eluent; anisaldehyde as TLC stain) thereupon indicated that a seemingly single faster-moving major product had formed, but it transpired that this single spot was actually a mixture of two main components 23 and 24 (formed in 1.39:1 ratio), when the reaction was examined by multi-elution TLC analysis. At this point, the reaction was judged to be complete, whereafter the crude reaction mixture was concentrated in vacuo on a rotary evaporator. The oil so obtained was purified by gradient-elution SiO2 flash chromatography initially using petrol:CH2Cl2 (3:1 → 2:1 → 1:1) to remove non-alkyne-derived tin-by-products, and then with petrol:EtOAc (25:1 → 20:1 → 10:1) to obtain the seemingly homogenous mixture of 23 and 24 (0.43 g, 67%). For structural analysis purposes only, however, a small portion of that crude unseparated mixture was dissolved in EtOAc and applied to two glass-backed TLC plates. These were then eluted and briefly air-dried on multiple occasions until the two main components were deemed to be separated. The respective compounds 23 and 24 were then scraped from the plate, and eluted from the TLC silica by suspension in EtOAc and filtration. The now separated individual components 23 and 24 were thereafter further purified individually by SiO2 flash chromatography with 10:1 petrol:EtOAc to give 23 and 24 which were both obtained as oils.
1H NMR of 23 (600.13 MHz, CDCl3) δ: 7.64–7.53 (complex m, 6H, 3J119Sn–1H = ca. 49.8 Hz, o-C, –SnPh3), 7.41–7.33 (complex m, 9H, m- and p-C, –SnPh3), 7.24 (d, 1H, J = 7.8 Hz, p-C, Ph), 7.16 (t, 2H, J = 7.8 Hz, m-C, Ph), 7.07 (d, 2H, J = 6.6 Hz, o-C, Ph), 4.99 (complex m, 1H, H4), 4.24 (m, 2H, 2 × H1), 2.57 (complex m, 1H, H6a), 2.49 (complex m, 1H, H6b), 2.33–2.15 (complex m, 2H, H5a,b), 1.46 (t, 1H, J = 6.0 Hz, –O) ppm.
13C NMR of 23 (150.9 MHz, CDCl3) δ: 202.7 (C3), 141.6 (quaternary C of Ph), 138.0 (3 × quaternary C of Ph3Sn), 137.1 (2J119/117Sn–13C = 37.7 Hz, 6 × o-H of Ph3Sn), 129.1 (4J119/117Sn–13C = 10.6 Hz, 3 × p-H of Ph3Sn), 128.6 (3J119/117Sn–13C = 51.3 Hz, 6 × m-CH of Ph3Sn), 128.5 (2 × o-H of Ph), 128.3 (2 × m-H of Ph), 125.9 (p-H of Ph), 96.1 (C2), 86.7 (C4), 63.4 (C1), 35.5 (C6, 5J117/119Sn–13C = 15.1 Hz), 30.2 (C5, 4J117/119Sn–13C = 21.1 Hz) ppm.
1H NMR of 24 (600.13 MHz, CDCl3) δ: 7.52–7.39 (2× complex m, each 6H [i.e. 12H in total] o-C, –SnPh3), 7.38–7.27 (complex m, 18H, m- and p-C, –SnPh3), 7.08–7.03 (complex m, 3 H, 2 × m-CH, and 1 p-CH, of Ph ring at C6), 6.52 (complex m, 3H, comprised of 2 multiplets for 2 × o-C of Ph ring at C6 superimposed upon H4 (t), 3J119Sn–H4 = 166.8 Hz), 4.03 (ddd, 1H, J = 10.2 Hz, 7.2 Hz, 6.0 Hz, H1a), 3.87 (ddd, 1H, J = 11.4 Hz, 6.0 Hz, 6.0 Hz, 3J119Sn–1H1 = 66 Hz, 3J117Sn–1H1 = ca. 62 Hz, H1b), 3.27 (dd, 1H, J = 6.6, Hz, 6.0 Hz, 1J119/117Sn–1H2 = ca. 64 Hz, H2), 2.33–2.23 (complex m, 4H, H5 and H6), 1.58 (t 1H, J = 5.4 Hz, –CH2O of C1) ppm.
13C NMR of 24 (150.9 MHz, CDCl3) δ: 145.3 (C4, 2J119/117Sn–13C(4) = 54.3 Hz), 141.14 (C3), 141.11 (quaternary C of Ph), 139.2 (3× quaternary C of Ph3Sn, 1J119Sn–13C = 479.9 Hz, 1J117Sn–13C = 458.7 Hz), 138.9 (3× quaternary C of Ph3Sn, 1J119Sn–13C = 504.0 Hz, 1J117Sn–13C = 482.9 Hz), 137.4 (2J119/117Sn–13C = 36.2 Hz, 6 × o-CH of Ph3Sn), 137.1 (6 × o-CH of Ph3Sn, 2J119/117Sn–13C = 36.2 Hz), 128.9 (3 × p-CH of Ph3Sn, 4J119/117Sn–13C = 10.6 Hz), 128.8 (3 × p-CH of Ph3Sn, 4J119/117Sn–13C = 10.6 Hz), 128.6 (6 × m-CH of Ph3Sn, 3J119/117Sn–13C = 48.3 Hz), 128.5 (6 × m-CH of Ph3Sn, 2J119/117Sn–13C = 48.3 Hz), 128.3 (2 × o-CH of Ph), 128.1 (2 × m-CH of Ph), 125.7 (p-CH of Ph), 65.7 (C1, 2J119/117Sn–13C1 = 15.1 Hz), 43.5 (C2, 2J117/119Sn–13C2 = 36.2 Hz), 38.0 (C5, 3J117/119Sn–13C5 = 42.3 Hz, 4J119/117Sn–13C5 = 9.1 Hz), 35.7 (C6, 4J117/119Sn–13C6 = 9.0 Hz) ppm.
NMR assignment of the structure of the triphenylstannylallene 23
The triphenylstannylallene 23 had its structural identity supported by high field NMR analysis. Specifically, the triphenylstannylallene 23 gave rise to the highly characteristic signal2c for the central C(3)-quaternary carbon of an allene at δ 202.7 ppm in its 150.9 MHz 13C NMR spectrum in CDCl3. As was the case with 21, where the C(2) carbon resonated at δ 96.4 ppm (1J119Sn–13C = 304.8 Hz, 1J117Sn–13C = 291.2 Hz), in 23, the other C(2)-quaternary carbon bearing the Ph3Sn substituent appeared at δ 96.1 ppm, it again being considerably more deshielded than the C(4)-allenyl carbon bearing the H-atom, which resonated some 10 ppm further upfield at δ 86.7 ppm. In the HSQC spectrum of 23, C(4) gave rise to the anticipated cross peak with the H(4)-allenyl proton multiplet at δ 4.99 ppm.As regards the 600 MHz 1H NMR spectrum of 23, numerical integration of the aromatic region confirmed that there were 20 aromatic H-atoms in the structure, which clearly pointed to only four phenyl rings being present. Noticeably, the –CH2OH hydroxyl resonance appeared as a triplet (J = 6.0 Hz) at the highly shielded position of δ 1.46 ppm. According to Willem and Gielen11 such a shift is highly indicative of such an OH being involved in a Sn–O coordinative interaction. However, hand-held molecular models of 23 suggested that such an O–Sn coordinative interaction could not be internal between the allylic –CH2OH and the allenyl SnPh3 due this OH being far too removed from the Sn atom. It is much more likely therefore that 23 is self-associating, forming a symmetrical dimer, in which the OH of one molecule of 23 coordinates intermolecularly to the Sn atom of another 23 molecule. Possibly this better explains the highly shielded resonance position of this hydroxyl.
The (Z)-6-phenyl-2,3-bis(triphenylstannyl)hex-3-en-1-ol (24) had its structure determined by extensive 600 MHz multi-dimensional 1H NMR spectroscopy in CDCl3, and DEPT NMR analysis. The latter revealed the presence of a deshielded hydroxymethyl carbon [C(1)] at δ 65.7 ppm. Importantly, this signal showed a 2J119/117Sn–13C(2) coupling of 15.1 Hz with the adjacent SnPh3 group at C(2). The fact that this 2J was much lower in magnitude than most typical 2J119/117Sn–13C couplings (which are generally ca. 17–38 Hz)31 was consistent with an electronegative OH group being stationed at C(1), and the C(1)–O bond being capable of taking up a near antiperiplanar orientation with the C(2)–Sn bond to maximise the O-atom's J-lowering effect.

As one might expect for a pair of allylic and benzylic carbons, the C(5) and C(6) –CH2– groups of 24 resonated at δ 38.0 (C5) and 35.7 (C6) ppm respectively, as verified by DEPT-135 spectroscopy. The latter showed the requisite negative peaks for these two carbons, which confirmed their methylenic (–CH2–) identity. An expansion of this region further revealed that the less shielded C(6) benzylic-carbon at δ 35.7 ppm exhibited an averaged long-range 4J119/117Sn–13C(6) coupling of ca. 9.0 Hz with the Sn atom at C(3). Although this averaged 4J119/117Sn–13C(6) coupling was fairly small, its existence did nevertheless allow this carbon to be confidently assigned to C(6), and it confirmed that a Ph3Sn group was resident at C(3). That same Sn-atom also showed a much larger averaged 3J119/117Sn–C(5) coupling of 42.3 Hz with the allylic C(5)–carbon which resonated at δ 38.0 ppm, which further reinforced this assignment. The allylic C(5) itself appeared to be involved in a long-range 4J coupling (9.1 Hz) with the SnPh3 resident at C(2).
As for the allylic carbon at C(2), it was assigned on the basis of its downfield chemical shift at δ 43.5 ppm, and its 2J119/117Sn–C(3) coupling of 36.2 Hz with the C(3)-SnPh3 substituent. Taken together, these three Sn–C J couplings provided very strong evidence for two SnPh3 groups being present on successive carbons at C(2) and C(3) within 24. Unfortunately, the 1J119/117Sn–13C couplings associated with C(2) and C(3) were essentially invisible. Undoubtedly this is due to the wide spectral width of these couplings and the low signal intensities that so arise from the low natural abundance of the 119/117Sn isotopes.
With respect to the olefinic C(3) quaternary carbon of 24, it resonated as a low intensity signal at δ 141.14 ppm in CDCl3. Its strongly downfield position unambiguously confirmed it was an alkenic-type carbon, and the fact that it was a quaternary carbon was verified by the absence of this signal from the DEPT-135 spectrum of 24. Importantly, C(3) also showed a long-range HMBC correlation with the H(2)-signal at δ 3.27 ppm. H(2) also showed strong HMBC correlations with C(4) at δ 145.3 ppm and C(1) at δ 65.7 ppm, which further confirmed their mutual proximity and skeletal connectivity.
Further proof that a vinyl triphenyltin was present within 24 came from the 600 MHz 1H NMR spectrum of 24 in CDCl3. Specifically, the olefinic signal for H(4) resonated as part of a highly complex multiplet centred at around δ 6.52 ppm, which quantitative NMR signal integration revealed contained 3H atoms in total, two of which were ultimately assignable to Ph protons. The chemical shift region around δ 6.5–6.6 ppm is typically where (Z)-trisubstituted vinyl triphenyltin olefinic protons resonate, and so our knowledge of this fact enabled us to make this assignment with confidence.3a Saying this, however, the presence of the stannylvinylic H(4) within this highly complicated 3H multiplet could only be unambiguously confirmed from its associated vicinal 3J1H–119/117Sn coupling of ca. 166.8 Hz between H(4) and the Sn atom at C(3). Its magnitude very clearly indicated that these two atoms were antiperiplanar to one another and, on this basis, we have assigned (Z)-geometry to the C(3)–C(4)-alkene present within 24.
The COSY spectrum of 24 subsequently pinpointed a strong vicinal coupling between H(4) and its two neighbouring allylic protons at H(5), which themselves resonated as part of a much more extensive and highly complex 4H-multiplet cluster positioned at around δ 2.28 ppm, which also contained the resonances for the two H(6) protons.
The quantitative signal integration to which we have just referred did ultimately reveal that 9 non-aromatic H-atoms were present alongside 35 aromatic H-atoms in 24, and so this careful quantification of the proton count did ultimately lead to great confidence in the structure that was ultimately assigned to 24.
Other findings that supported the assigned structure of 24 included the strong vicinal couplings of 6.6 and 6.0 Hz observed between H(2) and its diastereotopic H(1) neighbours which appeared as ddd signals at δ 4.03 and 3.87 ppm. Their multiplicities were attributable to couplings with the OH triplet (J = 5.4 Hz) at δ 1.58 ppm, the H(2) multiplet at δ 3.27 ppm, and each other. Those same 2J and 3J1H–1H couplings were subsequently ratified by appropriate cross peaks in the COSY spectrum of 24 in CDCl3. The very shielded resonance position for the OH triplet at δ 1.58 ppm was strongly suggestive of this C(1)-OH being involved in transient, but repeated, internal complexation events with the β-C(3)-Sn atom,11 but this is the main evidence for such a proposal.
Extra evidence for the presence of two Ph3Sn groups within the skeleton of 24 was provided by the 150.9 MHz 13C NMR spectrum of 24 in CDCl3, which contained 6 aryl carbon C–H signals at δ 137.4, 137.1, 128.9, 128.8, 128.6 and 128.5 ppm. There were also signals at δ 139.2 and 138.9 ppm for the quaternary carbons of those two Ph3Sn groups. The fact that only eight signals were observed for these two substituents confirmed that the three Ph groups present within each Ph3Sn subunit were magnetically equivalent and were each only producing four separate carbon signals.
To lend further support to our final structural assignment of 24, only four other aromatic carbon signals could be detected in the 13C NMR spectrum of 24. These appeared at δ 141.11 (quaternary C of Ph), 128.3 (2 × o-CH of Ph), 128.1 (2 × m-CH of Ph), and 125.7 (p-CH of Ph) ppm, and their magnetic equivalency and visibility clearly corroborated an additional Ph group being present at C(6).
Accordingly, the (Z)-6-phenyl-2,3-bis(triphenylstannyl)hex-3-en-1-ol structure (24) was eventually assigned to this co-product that was being formed alongside 23 in the hydrostannation reaction of 5 with Ph3SnH/cat. Et3B.
Data availability
All data supporting this article are included in the Experimental details section of this paper and the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank QUB, LJMU and Dr Joseph S. Vyle of QUB for generous gifts of chemicals. We also thank Mr Adam Brookfield for access to the EPSRC UK National EPR Facility and for technical support and chemical purchases under EPSRC grant EP/W014521/1.References
- H. A. Watson, K. L. E. Hale, J. M. Marron, S. Manaviazar, A. J. Fielding and K. J. Hale, Org. Biomol. Chem., 2024, 23 10.1039/d4ob01846j.
- (a) P. Dimopoulos, J. George, D. A. Tocher, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5377 CrossRef CAS PubMed; (b) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Tetrahedron, 2020, 76, 131061 CrossRef CAS; (c) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Chem. Commun., 2019, 55, 14454 RSC.
- (a) P. Dimopoulos, A. Athlan, S. Manaviazar, J. George, M. Walters, L. Lazarides, A. E. Aliev and K. J. Hale, Org. Lett., 2005, 7, 5369 CrossRef CAS PubMed; (b) Review: K. J. Hale, S. Manaviazar and H. A. Watson, Chem. Rec., 2019, 19, 238 CrossRef CAS PubMed.
- (a) M. S. Oderinde, R. D. J. Froese and M. G. Organ, Angew. Chem., Int. Ed., 2013, 52, 11334 CrossRef CAS PubMed; (b) M. S. Oderinde, R. D. J. Froese and M. G. Organ, Chem. – Eur. J., 2014, 20, 8579 CrossRef CAS PubMed; (c) M. S. Oderinde and M. G. Organ, Chem. – Eur. J., 2013, 19, 2615 CrossRef CAS PubMed; (d) M. S. Oderinde, H. N. Hunter, R. D. J. Froese and M. G. Organ, Chem. – Eur. J., 2012, 18, 10821 CrossRef CAS PubMed.
- S. E. Gottschling, T. N. Grant, K. K. Milnes, M. C. Jennings and K. M. Baines, J. Org. Chem., 2005, 70, 2686 CAS.
- G. Velegraki and M. Stratakis, J. Org. Chem., 2013, 78, 8880 CAS.
- A. B. Charette and A. Giroux, J. Org. Chem., 1996, 61, 8718 CrossRef CAS PubMed.
- M. Schmittel, A. A. Mahajan, G. Bucher and J. W. Bats, J. Org. Chem., 2007, 72, 2166 CrossRef CAS PubMed.
- (a) S. Ohira, Synth. Commun., 1989, 19, 561 CrossRef CAS; (b) S. Muller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 521 CrossRef; (c) Review: M. Dhameja and J. Pandey, Asian J. Org. Chem., 2018, 7, 1502 CrossRef CAS; (d) For a highly convenient method of synthesising Ohira–Bestmann reagent, see: J. Pietruszka and A. Witt, Synthesis, 2006, 4266 CrossRef CAS This is the protocol that was used to prepare the reagent we used here. It is an excellent protocol that gives high quality Ohira–Bestmann reagent that can be stored successfully for 2 years, if kept in a −20 °C freezer under N2.
- K. J. Hale, Z. Xiong, L. Wang, S. Manaviazar and R. Mackle, Org. Lett., 2015, 17, 198 CrossRef CAS PubMed.
- For other O-directed Ph3SnH hydrostannations: (a) R. Willem, A. Delmotte, I. De Borger, M. Biesemans, M. Gielen and F. Kayser, J. Organomet. Chem., 1994, 480, 255 CrossRef CAS; (b) F. Kayser, M. Biesemans, A. Delmotte, I. Verbruggen, I. De Borger, M. Gielen and R. Willem, Organometallics, 1994, 13, 4026 CrossRef CAS; (c) K. Micoine, P. Persich, J. Llaveria, M.-H. Lam, A. Merderna, F. Loganzo and A. Furstner, Chem. – Eur. J., 2013, 19, 7370 CrossRef CAS PubMed.
- (a) K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS PubMed; (b) N. P. Tsvetkov, E. Gonzales-Rodriguez, A. Hughes, G. dos Passos Gomes, F. D. White, F. Kuriakose and I. V. Alabugin, Angew. Chem., Int. Ed., 2018, 57, 3651 CrossRef CAS PubMed; (c) E. Gonzalez-Rodriguez, M. A. Abdo, G. dos Passos Gomes, S. Ayad, F. D. White, N. P. Tsvetkov, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2020, 142, 8352 CrossRef CAS PubMed; (d) C. Hu, L. Kuhn, F. D. Makurvet, E. S. Knorr, X. Lin, R. K. Kawade, F. Mentink-Vigier, K. Hamson and I. V. Alabugin, J. Am. Chem. Soc., 2024, 146, 4187 CrossRef CAS PubMed; (e) For a related RO→ Sn interaction facilitating an unusual alkoxy radical elimination, as opposed to a stannyl radical addition to an alkyne or a β-stannyl radical elimination, see: T. Harris, G. dos Passos Gomes, R. J. Clark and I. V. Alabugin, J. Org. Chem., 2016, 81, 6007 CrossRef CAS PubMed.
- For an extensive review on molecules that contain two Sn atoms, see: W. J. Kerr and P. L. Pauson, Chapter 4.15 Functions Containing Two Atoms of the Same Metallic Element, in Comprehensive Organic Functional Group Transformations, 1995, vol. 4, p. 667 Search PubMed.
- (a) I. V. Alabugin, Stereoelectronic Effects, John Wiley & Sons Ltd, 2016, ch. 3, 5 and 9 CrossRef; (b) I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed; (c) I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109 CAS; (d) C. Hu, J. Mena and I. V. Alabugin, Nat. Rev. Chem., 2023, 7, 405 CrossRef PubMed.
- For our own groundbreaking EPR spectroscopy work on the observation of β-triphenylstannylvinyl radicals that were generated in PhMe and THF via the O-directed free radical hydrostannation of dialkyl acetylenic alcohols with Ph3SnH/cat. Et3B and O2, see: H. A. Watson, A. J. Fielding and K. J. Hale, Chem. Commun., 2021, 57, 7449 RSC.
- EasySpin: (a) S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed; (b) S. Stoll and R. D. Britt, Phys. Chem. Chem. Phys., 2009, 11, 6614 RSC.
- (a) A. G. Davies, B. P. Roberts and M.-W. Tse, J. Chem. Soc., Perkin Trans. 2, 1978, 145 RSC; (b) T. Kawamura and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 648 CrossRef CAS; (c) P. J. Krusic and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 846 CrossRef CAS.
- C. Heller and H. M. McConnell, J. Chem. Phys., 1960, 32, 1535 CrossRef CAS.
- For a related α-stabilising delocalisative hyperconjugative interaction between the radical SOMO of a (t-Bu2MeSi)3Sn˙ radical and its proximal antibonding σ*(Si-Ct-Bu) orbitals, see: (a) A. Sekiguchi, T. Fukawa, V. Ya Lee and M. Nakamoto, J. Am. Chem. Soc., 2003, 125, 9250 CAS For similar stabilising interactions in other (t-Bu2MeSi)2M˙ radicals, see also: (b) S. Inoue, M. Ichinohe and A. Sekiguchi, Organometallics, 2008, 27, 1358 CrossRef CAS ; (for M = Si); (c) A. Sekiguchi, T. Fukawa, M. Nakamoto, V. Ya Lee and M. Ichinohe, J. Am. Chem. Soc., 2002, 124, 9865 CAS (for M = Si and Ge); (d) For a very informative review on such group 14 “heavy” atom stabilised radicals and their bonding interactions, see: V. Ya Lee and A. Sekiguchi, Acc. Chem. Res., 2007, 40, 410 CrossRef PubMed.
-
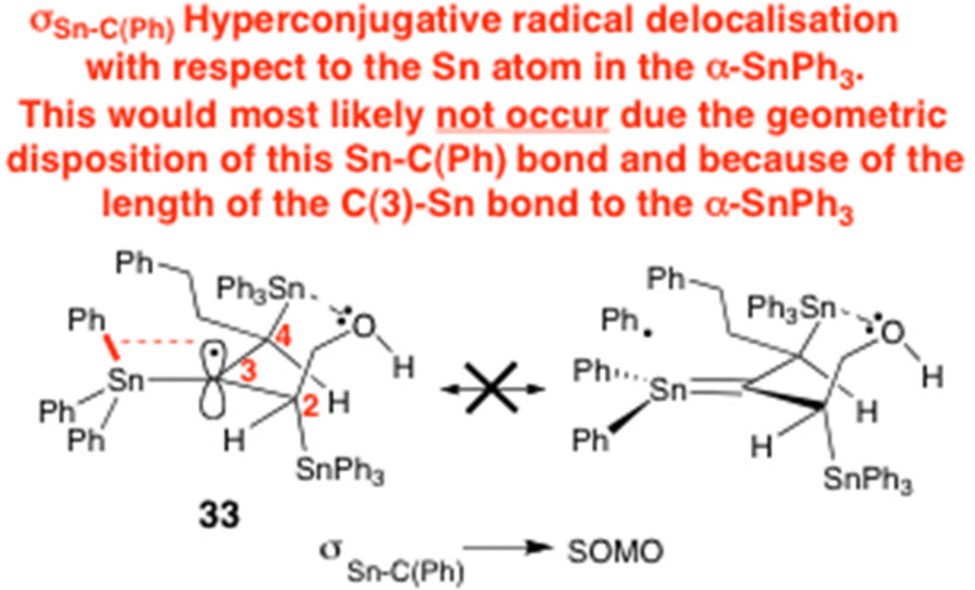
(a) A most helpful reviewer of our current paper has very kindly informed us that the long C(3)-SnPh3 bond length of 33 (ca. 2.14 Å) would almost certainly geometrically preclude the Sn–C(Ph) σ bond of 33 from ever being able to delocalisatively stabilise the C(3) radical in 33via a hyperconjugative mechanism:
Rather, this reviewer suggests that the main stabilising influence of the α-Ph3Sn group would emanate from its ability to engage in significant geminal hyperconjugative delocalisation of the radical into the σ* Sn–C(Ph) antibonding orbitals of the Ph3Sn group (i.e. via SOMO → σ*Sn–C(Ph) interactions) (see: Fig. 3 of our main manuscript). We are most grateful to this learned reviewer for their highly valuable intellectual input and insights here in rationalising the high stability of radical 33; (b) Whilst on the topic of the radical stabilisation of 33, it is perhaps pertinent to point out that the teams of Kochi (ref. 17b) and Mackey (see ref. 19b: J. H. Mackey and D. E. Wood, Mol. Phys., 1970, 18, 783 CrossRef CAS) have both suggested that 2p→ 5d radical delocalisation is a significant factor in helping to electronically-stabilise α-stannyl methyl radicals, on the basis of EPR g-factor and metal d-orbital odd-electron spin density measurements, as well as CNDO calculations. However, we would point out that the existence of pπ→ dπ* stabilisation in organotin compounds is still a subject of great controversy, and has been so for quite some time. See: (c) R. C. Poller, in The Chemistry of Organotin Compounds, Logos Press Limited, 1970, ch. 1, p. 5 Search PubMed. Readers should therefore bear this in mind when assessing the likely possible contribution of pπ → dπ bonding to the stability of 33 In this aspect, this same reviewer of our article, who commented on the long C(3)-SnPh3 bond length of 33 (ca. 2.14 Å) precluding Sn–C(Ph) σ bond hyperconjugative stabilisation of the radical in 33 is very much of the opinion that the Kochi and Mackey 2p → 5d radical stabilisation hypothesis would be equally unfeasible for similar reasons, due to the poor degree of orbital overlap that would result. We concur fully with the opinion of this learned reviewer. Nevertheless, we show below how such a pπ → dπ homoconjugative α-effect could potentially stabilise the radical in 33, if such stabilisation was operational:
. - R. Wei, X.-F. Wang, C. Hu and L. L. Liu, Chem. Commun., 2024, 60, 9793 RSC.
- Our observation of the radical adduct 33 takes on even greater significance if one considers that Mitchell and Lehnig and their two teams had never previously been able to observe such 1,2-bis-trimethyltin tertiary radical adducts of general structure Me3SnCH2C˙(R)SnMe3 by EPR spectroscopy, when this type of radical was generated by UV mediated Me3Sn˙ radical addition to the corresponding vinyltins. Presumably this is due to the normally very fast rate of β-scission of such radical adducts, once they are formed. See: (a) T. N. Mitchell, W. Reimann and C. Nettelbeck, Organometallics, 1985, 4, 1044 CrossRef CAS; (b) Until now, we too have been unable to observe a 1,2-bis-triphenyltin adduct forming from a trisubstituted vinyl triphenyltin by EPR spectroscopy until the vinyl triphenyltin 24 was exposed to proximally O-coordinated Ph3Sn radicals (see ref. 15 for our past EPR studies); (c) For similar negative EPR results with regard to the observation of β-trialkylstannylvinyl radicals and their adducts, see the important paper of: K. Suzuki, N. Sugihara, Y. Nishimoto and M. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202201883 CrossRef CAS PubMed.
- (a) C. Nativi and M. Taddei, J. Org. Chem., 1988, 58, 820 CrossRef; (b) D. P. Curran and T. McFadden, J. Am. Chem. Soc., 2016, 138, 7741 CrossRef CAS PubMed.
- M. Taniguchi, K. Nozaki, K. Miura, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1992, 65, 349 CrossRef CAS.
- (a) For a recent excellent review on alkyne hydrometallation reactions with group IV metal hydrides, see the following book chapter by: T. Wiesner and M. Haas, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2024, DOI:10.1016/B978-0-323-96025-0.00125-3 (b) For McLaughlan and Roberts’ recent highly regiocontrolled PtCl2/XPhos-catalysed hydrostannation of terminal aryl acetylenes and propargylic alcohols, see: D. D. Roberts and M. G. McLaughlin, Adv. Synth. Catal., 2023, 365, 1602 CrossRef CAS; (c) For McLaughlin's recent deployment of the PtCl2/XPhos/Et3SiH-catalyst system in the regioselective hydroboration of terminal alkyl, aryl and heteroaryl acetylenes with HBPin, see: K. L. E. Hale, D. D. Roberts and M. G. McLaughlin, Eur. J. Org. Chem., 2025, e202401355 CrossRef CAS.
- (a) (+)-Pumiliotoxin B formal total synthesis: S. Manaviazar, K. J. Hale and A. LeFranc, Tetrahedron Lett., 2011, 52, 2080 CrossRef CAS; (b) (−)-(3R)-Inthomycin C total synthesis: K. J. Hale, M. Grabski, S. Manaviazar and M. Maczka, Org. Lett., 2014, 16, 1164 CAS; (c) (−)-(3R)-Inthomycin revision of absolute stereochemistry: K. J. Hale, S. Hatakeyama, F. Urabe, J. Ishihara, S. Manaviazar, M. Grabski and M. Maczka, Org. Lett., 2014, 16, 3536 CAS; (d) Synthesis of the C(7)–C(22)-sector of (+)-acutiphycin by two-directional double O-directed free radical hydrostannation of a diyne, see: K. J. Hale, M. Maczka, A. Kaur, S. Manaviazar, M. Ostovar and M. Grabski, Org. Lett., 2014, 16, 1168 CAS; (e) For Furstner's elegant asymmetric total synthesis of (+)-isomigrastatin via an O-directed dialkylacetylene free radical hydrostannation with Ph3SnH/cat. AIBN, see ref. 11c.
- J. A. Marshall and S. Xie, J. Org. Chem., 1995, 60, 723 CrossRef.
- For our recent asymmetric total synthesis of the HDAC-inhibitory (+)-trichostatins A and C via the O-directed free radical hydrostannation of an alkyne derived from Marshall chiral allenylzinc addition to an aldehyde, see the paper that follows this, in this issue of OBC. See: K. Pan, S. Manaviazar and K. J. Hale, Org. Biomol. Chem., 2025, 23 10.1039/d4ob01848f.
- N. K. Anand and E. M. Carreira, J. Am. Chem. Soc., 2001, 123, 9687 CrossRef CAS PubMed.
-
(a) R. Sommer and H. Kuivila, J. Org. Chem., 1968, 33, 802 CrossRef CAS . It was in this paper that Sommer and Kuivila first postulated that fast reversible addition of tin radicals to acyclic alkenes could cause (Z)/(E) isomerisation;
(b) Dr Soraya Manaviazar of this laboratory has likewise successfully hydrostannated 2-methylene-propane-1,3-diol analogously to Wuest et al.,30c except in solution at room temperature, using our Ph3SnH/cat. Et3B/O2 method in PhMe:THF (9:1) over 24 h; she obtained the product 48 in 49% yield after SiO2 flash chromatography. This result was first published in Scheme 87, of ref. 3c, on page 310. Naturally, the latter outcome further confirms that such Ph3Sn˙ radical additions can readily take place upon alkenes in solution at rt and, under our O-directed hydrostannation conditions. In this specific instance, the application of our method even allowed a successful trapping of the tertiary radical adduct. For the earlier Wuest work, see: ;
(c) Y. Ducharme, S. Latour and J. D. Wuest, Organometallics, 1984, 3, 208 CrossRef CAS . Note how his procedure used neat Ph3SnH and cat. AIBN to thermally hydrostannylate 2-methylene-propane-1,3-diol at 64 °C, over 5 h, in 74% yield;
(d) R. H. Fish, J. Organomet. Chem., 1972, 42, 345 CrossRef CAS.
- (a) A. G. Davies, Organotin Chemistry, Wiley-VCH, 2nd edn, 2004, ch. 2 Search PubMed; (b) T. N. Mitchell and B. Kowall, Mag. Reson. Chem., 1995, 33, 325 CAS.
- M. Lehnig, H.-U. Buschhaus, W. P. Neumann and T. H. Apoussidis, Bull. Soc. Chim. Belg., 1980, 89, 90 Search PubMed (refer to material that can be found in the ESI‡).
- T. Berclaz and M. Geoffroy, Chem. Phys. Lett., 1977, 52, 606 CAS (refer to material that can be found in the ESI‡).
- M. Lehnig and K. Dören, J. Organomet. Chem., 1981, 210, 331 CAS (refer to material that can be found in the ESI‡).
- P. J. Krusic and J. K. Kochi, J. Am. Chem. Soc., 1968, 90, 7155 CAS (refer to material that can be found in the ESI‡).
- M. Castaing, M. Pereyre, M. Ratier, P. M. Blum and A. G. Davies, J. Chem. Soc., Perkin Trans. 2, 1979, 589 CAS (refer to material that can be found in the ESI‡).
Footnotes |
| † Dedicated to the memory of Professor Alwyn G. Davies FRS of UCL, whose numerous profound mechanistic contributions to the field of organometallic free radical chemistry will have enduring impact. Sadly, he died on 1st September 2023, aged 97 years, but his fine work, always done correctly, with great thoroughness and thought, will forever guide and inspire future generations. |
| ‡ Electronic supplementary information (ESI) available: EPR experimental procedures, additional discussion of the EPR results, and copies of the NMR and mass spectra for the compounds.32–36 See DOI: https://doi.org/10.1039/d4ob01847h |
| § Current address: Halazar Pharma Ltd, Edgware, Middlesex, HA8 7RB, UK. E-mail: k.hale120@btinternet.com. |
| This journal is © The Royal Society of Chemistry 2025 |