
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of the HDAC inhibitor (+)-(R)-trichostatin A via O-directed dialkylacetylene free radical hydrostannation with Ph3SnH/Et3B. The unusual inhibitory effect of a proximal α-OPv group on the course of a vinyl iodide Stille cross-coupling†‡
Ke
Pan
,
Soraya
Manaviazar§
and
Karl J.
Hale§
 *
*
The School of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast BT9 5AG, UK. E-mail: k.hale120@btinternet.com
First published on 24th March 2025
Abstract
In this paper, a new asymmetric total synthesis of optically pure (+)-trichostatin A (1a) is described via a route that utilises a Marshall chiral allenylzinc addition between 9 and 4-dimethylaminobenzaldehyde (10) and an O-depivaloylation at its early stages. O-Directed free radical hydrostannation of the resulting propargylic alcohol 15 with Ph3SnH/cat. Et3B/O2 in PhMe at rt thereafter provided the (Z)-α-triphenylstannylvinyltin 16 in 80–89% yield, with complete stereocontrol and very high α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β regioselectivity (25:1). A stereoretentive I–Sn exchange reaction between 16 and I2 (1.4 equiv.) in CH2Cl2 (−78 °C to rt, 1 h) subsequently secured the vinyl iodide 18 in 84–96% yield. The latter was transformed into the enal 4 by successive TPAP/NMO (Ley–Griffith) oxidation and a high yielding (80%) Stille reaction between the α-iodo enal 20 and Me4Sn, catalysed by Pd(PPh3)4 in DMF at 60 °C, under the Baldwin–Lee conditions, which use CsF and CuI as promoters. A Wittig reaction between 4 and Ph3P
:β regioselectivity (25:1). A stereoretentive I–Sn exchange reaction between 16 and I2 (1.4 equiv.) in CH2Cl2 (−78 °C to rt, 1 h) subsequently secured the vinyl iodide 18 in 84–96% yield. The latter was transformed into the enal 4 by successive TPAP/NMO (Ley–Griffith) oxidation and a high yielding (80%) Stille reaction between the α-iodo enal 20 and Me4Sn, catalysed by Pd(PPh3)4 in DMF at 60 °C, under the Baldwin–Lee conditions, which use CsF and CuI as promoters. A Wittig reaction between 4 and Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et (5), saponification, and DDQ oxidation next afforded (+)-trichostatic acid (22). Helquist's ethyl chloroformate mixed-anhydride/TBSONH2 coupling procedure (ref. 17e) thereafter secured (+)-trichostatin A (1a) in good yield. This new total synthesis of 1a is the first-ever successful application of the O-directed dialkylacetylene free radical hydrostannation with Ph3SnH/cat. Et3B/O2 in a dialkylaniline N-containing disubstituted alkynol system, and it now provides a convenient means of accessing many novel trichostatin analogues for future biological screening.
CHCO2Et (5), saponification, and DDQ oxidation next afforded (+)-trichostatic acid (22). Helquist's ethyl chloroformate mixed-anhydride/TBSONH2 coupling procedure (ref. 17e) thereafter secured (+)-trichostatin A (1a) in good yield. This new total synthesis of 1a is the first-ever successful application of the O-directed dialkylacetylene free radical hydrostannation with Ph3SnH/cat. Et3B/O2 in a dialkylaniline N-containing disubstituted alkynol system, and it now provides a convenient means of accessing many novel trichostatin analogues for future biological screening.
Introduction
For some time now, we have been studying the scope,1,2a mechanism,3,4 and utility1 of the O-directed free radical hydrostannation reaction of various propargylically-oxygenated dialkylacetylenes with stannanes,1–4 and the protocol that has consistently emerged best for most acyclic synthetic applications is the room temperature Ph3SnH/cat. Et3B variant of this reaction performed in PhMe (Scheme 1).2a | ||
| Scheme 1 The rt O-directed free radical hydrostannylative synthesis of (Z)-trisubstituted alkenes with Ph3SnH/cat. Et3B. | ||
But that is not to say that the thermally-mediated Bu3SnH/AIBN counterpart4 of this reaction does not have an equally important role to play in synthesis, particularly where tandem radical cyclisation is a cardinal requirement, as in Alabugin's traceless polyaromatic ring construction reactions,5 where his team's powerful contributions have been particularly elegant, innovative and synthetically impactful. Those same studies5 have also provided profound computational and experimental insights into the detailed mechanistic workings of the free radical hydrostannation reaction of propargylically-oxygenated disubstituted acetylenes, and affirmed the entirely free radical O-directed mechanism proposed for this process.3,4a–c
The O-directed Ph3SnH/cat. Et3B variant1,2 of this reaction has now shown its versatility in a wide range of complex settings that have included total syntheses of the natural products (+)-pumiliotoxin B6 and (−)-(3R)-inthomycin C.7 A noteworthy double O-directed free radical hydrostannation has also been introduced for the simultaneous installation of two structurally distinct trisubstituted alkenes within target structures. Its synthetic utility was powerfully demonstrated by the fully stereocontrolled route that was developed to the C(7)–C(22)-sector of (+)-acutiphycin.8 The Furstner team has likewise elegantly employed an O-directed free radical hydrostannation with Ph3SnH, cat. AIBN and PhMe in their total synthesis of (+)-isomigrastatin.9 In each of these synthetic applications, I–SnPh3 exchange has played a central role in final trisubstituted alkene elaboration10 and, for the synthesis of all-carbon branched alkene structures, the CuI/CsF Baldwin–Lee11 and Farina Ph3As variants12 of the Stille reaction have both proven themselves immensely valuable.6–8,10 Other Pd(0)-mediated cross-coupling methods (e.g. Suzuki, Negishi, and Cuprate) and carbonylations have shown themselves to be equally applicable in this sphere.10
While there have now been many impressive displays of the utility of the rt Ph3SnH/cat. Et3B O-directed hydrostannation in synthesis,6–8,10 there remain some classes of alkynes on which this reaction has yet to be successfully applied. Prominent amongst these substrates are propargyloxy dialkylacetylenes with an unprotected amine functionality. These generally form a complex with the Et3B initiator, to prevent the O2-mediated SH2 radical initiation event from ever taking place. Although a stoichiometric excess of Et3B can sometimes allow radical initiation to occur, frequently the outcomes of these processes are disappointing.
Despite all of the past difficulties, we recently decided to investigate whether weakly basic amines, such as anilines, might prove compatible with the Ph3SnH/cat. Et3B O-directed hydrostannation method. In this paper, we now report that the 4-dimethylanilino functionality is indeed well tolerated in this process, as demonstrated by our new asymmetric total synthesis of (+)-trichostatin A, where this reaction protocol was very effectively deployed alongside a Marshall chiral allenylzinc addition process.13
We also document here, for the first time ever, the profound inhibitory effect that an allylic OPv group can have on the course of a Pd-catalysed cross-coupling of a vinyl iodide. The latter observation was made quite by chance, whilst we were attempting to apply such a coupling to a trisubstituted vinyl iodide with such a functionality. Given this synthetic impasse, we thought it important to record this difficulty here, to prevent others from becoming similarly ensnared in the future.
(+)-Trichostatin A (1a) is a highly potent, naturally-occurring, histone deacetylase (HDAC) inhibitor that has elicited considerable medicinal interest as a possible antifungal,14a anticancer,14b,c immunosuppressive,14d and anti-Duchenne muscular dystrophy drug,15 but its clinical use has so far been precluded by its remarkably short plasma half-life (<6.3 min at 80 mg kg−1)14c and its pronounced genotoxicity. It is widely thought that if these undesirable properties could somehow be ablated by careful structural modification, as was recently found for glycosylated (+)-trichostatin C (1b),16 and HDAC specificity could also be improved, such trichostatin analogues might potentially become useful new treatments for a host of diseases. It was with such pharmaceutical and synthetic objectives in mind that we first embarked on the development of a new improved total synthesis17 of (+)-trichostatin A (1a) (R = OH), for the purpose of providing novel C(4)-analogues17e for drug screening and X-ray crystallographic/NMR studies. It was hoped that such studies might provide useful new insights into how individual HDAC isozymes work, as well as new therapeutic drugs.
Accordingly, we duly formulated the retrosynthetic plan of Scheme 2 for the synthetic acquisition17 of (+)-(R)-trichostatin A (1a). Underpinning our approach would be the O-directed free radical hydrostannation of alkyne 8 with Ph3SnH/Et3B, which would be allied with an I–Sn exchange and a Stille cross-coupling with Me4Sn. An O-depivaloylation and an alcohol oxidation would thereafter procure 4. The latter would then be chain extended with the ylide 5, the C(1)-carboxyl subsequently unmasked, and the C(7)-ketone thereafter elaborated, prior to C(1)-oxyamidation with TBSONH2. An O-desilylation would then complete the synthesis of 1a.17 Alkyne 8 would itself be created through a new Marshall chiral allenylzinc addition between the known chiral allenylzinc 913 and 4-dimethylaminobenzaldehyde (10) under Pd(0)-catalysis.
 | ||
| Scheme 2 Retrosynthetic planning for (+)-(6R)-trichostatin A (1a). | ||
Results and discussion
Our first objective was the preparation of the chiral allenylzinc 9 from the known propargyl O-mesylate 11 (Scheme 3)13 by the method of Marshall and Xie.13 Specifically, 11 was added dropwise to a solution of Pd(OAc)2 (0.05 equiv.) and Ph3P (0.05 equiv.) that had been pre-aged for 20 min in dry THF at −20 °C, under N2. The reactants were then stirred for a further 5 min post-addition, before 4-dimethylaminobenzaldehyde (10) was added dropwise, followed by Et2Zn (3 equiv.). Stirring was then continued at −20 °C for 48 h to give an essentially inseparable 1:1 mixture of two alcohol diastereomers at C(5). This was inconsequential for further synthetic progression, since the C(5)-alcohols of 12 would ultimately be oxidized to the C(7) ketone in 1a, at the penultimate step, and this same reaction had fully controlled the stereochemistry of the key C(4)-Me group in 12 (this would be C(6) in 1a). After O-silylation with TBSCl, the alkynyl O-pivaloate 8 was subjected to an O-directed free radical hydrostannation with Ph3SnH (1.5 equiv.) and Et3B (0.1 equiv.) in PhMe under our standard rt conditions. This reaction proceeded efficiently, affording 7 as two C(5)-diastereomeric products in 78% yield after 18 h. With 7 in hand, the key I–Sn exchange was investigated to obtain 6. It was found that when this reaction was performed with just 1.5 equiv. of NIS at 0 °C, it did not proceed to completion, even after 2 h. However, when an additional 4 equiv. of NIS was added at rt,7 and the reactants were stirred for a further 2 h, the reaction did deliver the slightly impure vinyl iodide 6, as a much slower-moving product spot on TLC analysis. The necessity for such a large excess of the NIS was indicative of strong internal O–Sn coordination occurring within 7, between the pivaloyloxy-carbonyl-O and the SnPh3 substituent, which preferentially enhanced the reactivity of the Ph groups.7 As a result, a highly polar vinyltin triiodide initially needed to form, which was then eventually replaced by the desired vinyl iodide after prolonged stirring at rt. Iodide 6 was next subjected to a Stille cross-coupling with Me4Sn under the normally successful Baldwin–Lee conditions.11 Surprisingly, this reaction performed very poorly; an outcome that we have attributed to an analogous strong internal coordinative effect from the nearby OPv group, which we believe displaces the iodide from the initially formed vinyl-palladium(II) iodide to give 13,18 whose high stability then halts iodide return, to prevent the key transmetallation step from ever occurring with the Me4Sn or hypervalent [Me4SnF]−.
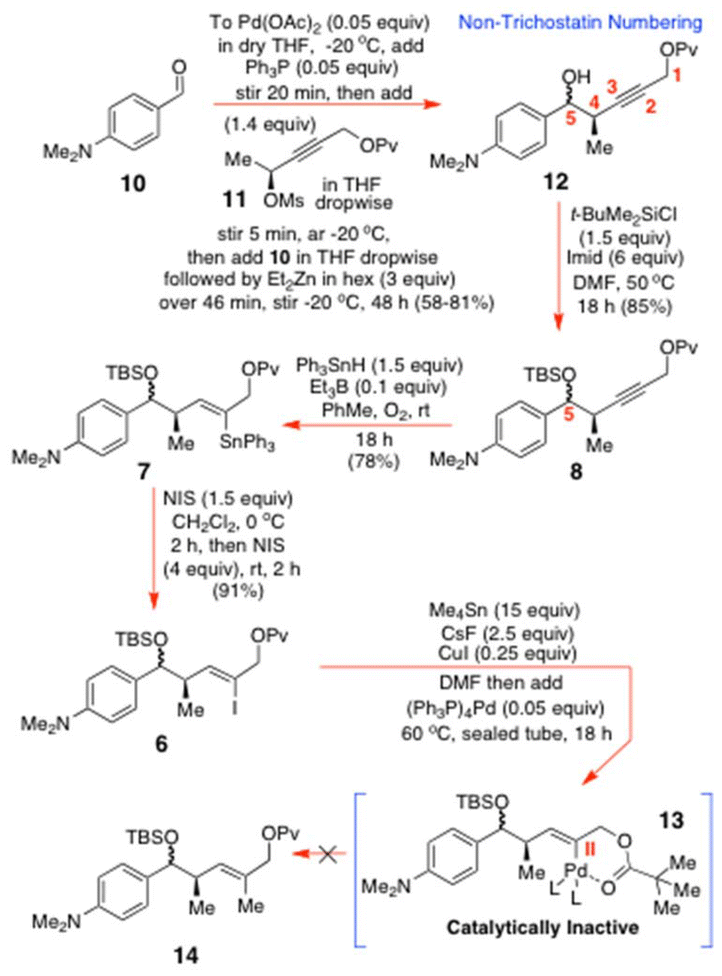 | ||
| Scheme 3 Initially investigated route to 7 and 14. | ||
In light of this setback, we decided to cleave the OPv group from 8 with i-Bu2AlH (2 equiv.) in CH2Cl2 at −78 °C. The reaction took 2 h to reach completion, affording the desired alkynol 15 in 83% yield after extractive work up and SiO2 flash chromatography. It was later found more convenient and higher yielding (96–98% yield) to use K2CO3 in MeOH to accomplish this transformation over 5 h at rt (Scheme 4). The resulting alkynol 15 was then subjected to O-directed hydrostannation with Ph3SnH (1.7 equiv.) and Et3B (0.1 equiv.) and O2 in PhMe (1 M in 15) for 19 h at rt. This afforded a 25:1 mixture of α:β vinyltriphenylstannanes 16 and 17, from which the two α-stannylated C(5)-diastereomers 16 could typically be purified with 80–89% yield after SiO2 flash chromatography.
 | ||
| Scheme 4 The O-directed hydrostannylative route to (+)-(R)-trichostatin A (1). | ||
Now that the Pv protecting group had been detached, the requisite I–Sn exchange reaction proceeded successfully with 16 to give 18 in >90% yield using only 1.4 equiv. of I2. This result provided good confirmatory evidence that the OPv group had been adversely involved in strong internal O-coordination to the metal centre in both 7 and 13, and our combined findings now pointed to a new strategic way forward.
The iodo-allylic alcohol 18 was next subjected to a Baldwin–Lee Stille cross-coupling with Me4Sn.11 It provided 19 efficiently in 80% yield after 22 h at rt. Importantly, this reaction had to be performed in complete darkness, inside a sealed vessel under N2, to prevent light-induced vinyl iodide decomposition. Notwithstanding this success, difficulties soon arose when attempts were made to oxidise alcohol 19 under Swern, PCC, or MnO2 oxidation conditions, due to the presence of the dimethylamino group. Only cat. TPAP/NMO19 afforded the desired enal 4, but in a disappointing 20% yield.
Even so, this positive outcome did prompt us to evaluate the Ley–Griffith TPAP (0.05 equiv.)/NMO (2 equiv.) oxidising system19 on the iodoallylic alcohol 18 in CH2Cl2 in the presence of 4 Å sieves. This reaction worked reasonably well at rt, providing the desired α-iodo-enal 20 in 54–64% yield after just 15 min. To our surprise, a much slower-moving by-product was also formed alongside 20; this is suspected to be the N-oxide based upon 1H and 13C NMR analysis. Fortunately, it could be separated from 20 by SiO2 flash chromatography. The unstable iodo-enal 20 was thereafter used quickly for a Baldwin–Lee Stille cross-coupling with Me4Sn (15 equiv.), CsF (2.5 equiv.) and CuI (0.25 equiv.) in DMF at 60 °C; a reaction that needed to be conducted in darkness. The success of this coupling (76–80% yield) was confirmed by the presence of two new allylic methyl doublets at δ 1.62 ppm (J = 1.2 Hz) and 1.61 ppm (J = 1.6 Hz) in CDCl3. Due to 20 and 4 both having near identical TLC plate mobilities after a single elution, it was necessary to follow the progress of this reaction by multi-elution TLC with 3:1 petrol/CH2Cl2 as the eluent. The product enal 4 moved as a slightly slower single spot, when compared with the iodo-enal 20; 4 also stained dark blue when the TLC plate was heated with the anisaldehyde/H2SO4 stain. The iodo-enal 20 stained brown. It is worth noting that performing this coupling at a temperature significantly below 60 °C led to a marked diminution in the yield of 4; so 60 °C is essential and optimal.
Surprisingly the Wittig condensation of aldehyde 4 with stabilised P-ylide 5 took 6–10 d to reach completion at 38–40 °C in PhMe/CH2Cl2 (1.25:1), and again, 4 and 3 had near identical TLC mobilities. When complete, the reaction furnished the dienoate 3 in 86–90% yield with total geometric control.
Unfortunately, the subsequent O-desilylation of 3 with n-Bu4NF in THF, or CsF in DMF, or HF/pyridine did not lead to the expected alcohol but, instead, a multitude of products. With n-Bu4NF, 4-dimethylaminobenzaldehyde (10) was observed amongst the products, confirming that a retro-vinylogous aldol cleavage occurred at C(6)/C(7). This unexpected result led to an immediate change in our synthetic planning.
Without separation, the two C(7)-diastereoisomers of ester 3 were saponified with 90–98% yield with LiOH (4 equiv.) in 1:1:1 THF/MeOH/H2O. The known acid 2117f was then oxidatively converted into (+)-trichostatic acid (22) by brief exposure to 1.5 equiv. of DDQ in 20:1 CH2Cl2/H2O at 0 °C for just 7 min. Such precise timing was essential to avoid extensive decomposition of the (+)-trichostatic acid 22 so formed. Although, in principle, the viability of this reaction had already been demonstrated by Hosokawa and Tatsuta in 2005,17f these workers never supplied an experimental procedure for this reaction, which meant that considerable experimentation was needed on our part to obtain a successful outcome. Nonetheless, with the new procedure reported here, the above reaction can now deliver 22 in 54–71% yield, with only a small amount of the starting material ever remaining at the reaction end.
With acid 22 in hand, it could be converted into the mixed anhydride 23 by treatment with 2 equiv. of ethyl chloroformate at 0 °C in THF.17b,e Exposure to 2 equiv. of TBSO-NH2 according to Helquist's procedure17e thereafter afforded 24, which was deprotected with CsF in MeOH to obtain (+)-(R)-trichostatin A (1) in 40–60% yield over 2 steps, without loss of chirality.17 The latter was proven by our synthetic conversion of 2220 into its naturally-occurring β-glucoside, (+)-trichostatin C (1b) (NMR data in the ESI‡), which also, simultaneously, unambiguously confirmed the assigned structure of that natural product.16,20
Conclusions
With this new enantioselective total synthesis of (+)-trichostatin A that has been developed,21 we have provided yet another powerful demonstration of the utility of the room temperature O-directed free radical hydrostannation of propargylically-oxygenated dialkylacetylenes with Ph3SnH and Et3B/O2 in complex molecule total synthesis,1–3,6–8,22 and we have exemplified how it can be successfully deployed in an alkyne system that bears the dialkylaniline N-functionality. We have also shown how vinyl iodides with a proximal –CH2OPv group can internally O-coordinate to a vinyl palladium(II) intermediate in a manner that prevents it from successfully engaging in transmetallation18 and reductive elimination.Experimental
Procedures for the total synthesis of (+)-(R)-trichostatin A (1)
For the synthesis of propargyl alcohol 15 see the accompanying ESI.‡O-Directed hydrostannation of 15: synthesis of vinyl triphenyltin 16
Inside a sealed glove bag filled with an N2 atmosphere, Ph3SnH (5.11 g, 14.56 mmol, 1.7 equiv.) was quickly weighed into a pear-shaped flask fitted with a rubber septum. When the weighing process was complete, the septum-sealed flask was removed from the glove bag and fitted with an N2-filled balloon connected to a Luer-locked needle. Dry PhMe (8.5 mL) was added to the neat Ph3SnH and that solution was cannulated into alkynol 15 (2.98 g, 8.57 mmol) under N2 at rt. A solution of Et3B (1.0 M in hex, 1.30 mL, 1.30 mmol, 0.15 equiv.) was then added dropwise to the two reactants. Air (20 mL) was thereafter injected into the reaction mixture twice at 5 min and then at 1 h. The reaction mixture was stirred at rt for 19 h with the N2 atmosphere being maintained throughout. The reaction mixture was then diluted with EtOAc (20 mL) and quenched with H2O (20 mL). The aqueous layer was extracted with EtOAc (20 mL × 5). The combined organic extracts were then dried using MgSO4, filtered and concentrated under reduced pressure. The crude residue, which consisted essentially of a 25
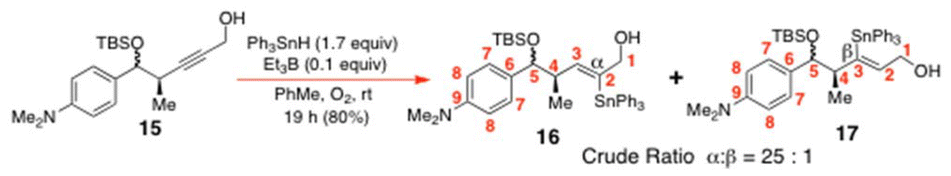 :1 α:β mixture of 16:17, was purified by gradient elution SiO2 flash chromatography initially using petrol:CH2Cl2 (3:1 → 2:1 → 1:1) to remove the tin residues and other impurities. Gradient elution with petrol:EtOAc (18:1 → 15:1) thereafter gave the entire product 16 (4.76 g, 80%) as a mixture of C5-epimers and as a colourless oil. A small amount of the β-vinyltin adduct 17 was subsequently eluted with petrol:EtOAc (12:1 → 8:1) as the eluent. It was characterised and identified by 1H NMR spectroscopy (see the ESI‡ for a copy of the 600 MHz 1H NMR spectrum of 17). Data for 16: IR of 16 (neat film): 3423 (m), 3063 (m), 3050 (m), 2959 (s), 2926 (s), 2856 (s), 1730 (w), 1615 (s), 1524 (s), 1431 (s), 1385 (m), 1254 (m), 1073 (s), 864 (m), 837 (m), 779 (m), 731 (s), 698 (s) cm−1.
:1 α:β mixture of 16:17, was purified by gradient elution SiO2 flash chromatography initially using petrol:CH2Cl2 (3:1 → 2:1 → 1:1) to remove the tin residues and other impurities. Gradient elution with petrol:EtOAc (18:1 → 15:1) thereafter gave the entire product 16 (4.76 g, 80%) as a mixture of C5-epimers and as a colourless oil. A small amount of the β-vinyltin adduct 17 was subsequently eluted with petrol:EtOAc (12:1 → 8:1) as the eluent. It was characterised and identified by 1H NMR spectroscopy (see the ESI‡ for a copy of the 600 MHz 1H NMR spectrum of 17). Data for 16: IR of 16 (neat film): 3423 (m), 3063 (m), 3050 (m), 2959 (s), 2926 (s), 2856 (s), 1730 (w), 1615 (s), 1524 (s), 1431 (s), 1385 (m), 1254 (m), 1073 (s), 864 (m), 837 (m), 779 (m), 731 (s), 698 (s) cm−1.
1H NMR of pure 16 (pure α-diastereoisomer 1) (399.9 MHz, CDCl3): δ 7.60 (m, 6H, 3J119/117Sn–1H = ca. 48.0 Hz, o-C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , –SnPh3), 7.35 (m, 9H, p- and m-C, –SnPh3), 6.78 (d, J = 8.4 Hz, 2H, H7), 6.35 (d, J = 8.7 Hz, 2H, H8), 6.43 (d, J = 10.0 Hz, 3J119Sn–1H = 173.6 Hz, 3J117Sn–1H = 153.6 Hz, 1H, H3), 4.32 (s, 2H, CH2, 3J119/117Sn–1H = 54.0 Hz, H1a,b), 4.22 (d, 1H, J = 6.8 Hz, H5), 2.92 (s, 6H, N(C3)2), 2.47 (m, 1H, J = 6.8 Hz, H4), 1.32 (br, 1H, O), 0.83 (s, 9H, t-Bu of OTBS), 0.56 (d, J = 6.8 Hz, C4-
, –SnPh3), 7.35 (m, 9H, p- and m-C, –SnPh3), 6.78 (d, J = 8.4 Hz, 2H, H7), 6.35 (d, J = 8.7 Hz, 2H, H8), 6.43 (d, J = 10.0 Hz, 3J119Sn–1H = 173.6 Hz, 3J117Sn–1H = 153.6 Hz, 1H, H3), 4.32 (s, 2H, CH2, 3J119/117Sn–1H = 54.0 Hz, H1a,b), 4.22 (d, 1H, J = 6.8 Hz, H5), 2.92 (s, 6H, N(C3)2), 2.47 (m, 1H, J = 6.8 Hz, H4), 1.32 (br, 1H, O), 0.83 (s, 9H, t-Bu of OTBS), 0.56 (d, J = 6.8 Hz, C4-![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) ), −0.09 (s, 3H, C3Si), −0.27 (s, 3H, C3Si) ppm. 13C NMR of 16 (pure α-diastereoisomer 1) (100.57 MHz, CDCl3): δ 149.6 (C9), 149.2 (C3, 2J119/117Sn–13C = 32.2 Hz), 139.7 (C2), 139.4 (quaternary C, –SnPh3), 137.2 (o-
), −0.09 (s, 3H, C3Si), −0.27 (s, 3H, C3Si) ppm. 13C NMR of 16 (pure α-diastereoisomer 1) (100.57 MHz, CDCl3): δ 149.6 (C9), 149.2 (C3, 2J119/117Sn–13C = 32.2 Hz), 139.7 (C2), 139.4 (quaternary C, –SnPh3), 137.2 (o-![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) , –SnPh3, 3J119/117Sn–13C = 38.2 Hz), 131.4 (C6), 128.7 (p-, –SnPh3, 4J119/117Sn–13C = 12.1 Hz), 128.5 (m-, –SnPh3, 3J119Sn–13C = 52.3 Hz), 127.9 (C7), 111.8 (C8), 79.3 (C5), 70.4 (C1, 2J119/117Sn–13C = 46.3 Hz), 47.4 (C4, 3J119/117Sn–13C = 38.2 Hz), 40.6 (N(H3)2), 25.9 ((H3)3CSi), 18.3 ((CH3)3Si), 17.6 (C4-Me), −4.4 (H3Si), −5.1 (H3Si) ppm.
, –SnPh3, 3J119/117Sn–13C = 38.2 Hz), 131.4 (C6), 128.7 (p-, –SnPh3, 4J119/117Sn–13C = 12.1 Hz), 128.5 (m-, –SnPh3, 3J119Sn–13C = 52.3 Hz), 127.9 (C7), 111.8 (C8), 79.3 (C5), 70.4 (C1, 2J119/117Sn–13C = 46.3 Hz), 47.4 (C4, 3J119/117Sn–13C = 38.2 Hz), 40.6 (N(H3)2), 25.9 ((H3)3CSi), 18.3 ((CH3)3Si), 17.6 (C4-Me), −4.4 (H3Si), −5.1 (H3Si) ppm.
1H NMR of the other diastereoisomer of 16 (α-diastereoisomer 2) (399.9 MHz, CDCl3). Resonances and multiplicities have been reported, where determinable, from the purified but diastereomerically enriched 3:1 mixture of C5 epimers: δ 7.61 (m, 6H, o-C, –SnPh3, 3J119/117Sn–1H = 48.4 Hz), 7.38 (m, 9H, –SnPh3), 6.44 (d, J = 8.8 Hz, 2H, H7) superimposed upon 6.42 (m, 1H, H3), 6.40 (d, J = 8.8 Hz, 2H, H8), 4.43 (d, J = 3.6 Hz, 1H, H5), 4.32 (s, 2H, H1), 2.88 (s, 6H, N(C3)2), 2.27 (m, 1H, H4), 1.32 (br, 1H, O), 0.91 (s, 9H, t-Bu of OTBS), 0.74 (d, J = 6.8 Hz, 3H, C4-), −0.00 (s, 3H, C3Si), −0.23 (s, 3H, C3Si) ppm. 13C NMR (100.57 MHz, CDCl3) of the other diastereoisomer of 16 (α-diastereoisomer 2). Resonances and J values have been reported, where determinable, from the purified but diastereomerically enriched 3:1 mixture: δ 150.1 (C9), 149.2 (C3), 139.2 (Sn––CH, –SnPh3), 138.8 (C2), 137.1 (o-, –SnPh3, 3J119/117Sn–13C = 38.2 Hz), 131.6 (C6), 128.9 (p-, –SnPh3, 4J119/117Sn–13C = 12.1 Hz), 128.6 (m-, –SnPh3, 4J119/117Sn–13C = 52.3 Hz), 126.8 (C7), 111.7 (C8), 77.1 (C5), 70.3 (C1, 2J119/117Sn–13C = 46.3 Hz), 47.12 (CH-Me), 40.7 (N(H3)2), 25.9 ((H3)3CSi), 18.3 ((CH3)3Si), 13.8 (CH–H3), −4.5 (H3Si), −5.0 (H3Si) ppm.
TOF ES+ HRMS of 16: calcd for C38H50NO2SiSn [M + H]+: 700.2640. Found: 700.2611.
I–Sn exchange of 16: preparation of vinyl iodide 18
Before commencing this experiment, the reaction flask was covered with Al-foil to protect it from the adverse effects of light. Thereafter, to a stirred −78 °C solution of the vinyl triphenyltin 16 (4.76 g, 6.81 mmol) in dry CH2Cl2 (68 mL) under N2 was added I2 (2.42 g, 9.52 mmol, 1.4 equiv.) in one portion. Stirring was continued at −78 °C for 10 min, after which the cooling bath was removed, and the reactants were allowed to stir at rt for a further 1 h. The reaction mixture was then diluted with CH2Cl2 (50 mL) and quenched with H2O (50 mL). The aqueous layer was extracted with EtOAc (50 mL × 3) and the combined organic extracts were successively washed with saturated aq. Na2S2O3 (100 mL) and H2O (50 mL × 2). The organic extract was then dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by gradient elution SiO2 flash chromatography using petrol
 :CH2Cl2 (3:1 → 2:1 → 1:1) as the eluent to remove tin residues, and thereafter petrol:EtOAc (15:1 → 10:1) to elute iodide 18 (3.0 g, 93%) as a C5-mixture of epimers; it was obtained as an amber oil. IR for the 18 mixture (neat film): 3383 (m), 3101 (w), 3075 (w), 2959 (s), 2927 (s), 2856 (s), 2800 (m), 1738 (w), 1617 (s), 1521 (s), 1471 (m), 1357 (m), 1254 (m), 1074 (s), 941 (m), 876 (s), 835 (s), 775 (s) cm−1.
:CH2Cl2 (3:1 → 2:1 → 1:1) as the eluent to remove tin residues, and thereafter petrol:EtOAc (15:1 → 10:1) to elute iodide 18 (3.0 g, 93%) as a C5-mixture of epimers; it was obtained as an amber oil. IR for the 18 mixture (neat film): 3383 (m), 3101 (w), 3075 (w), 2959 (s), 2927 (s), 2856 (s), 2800 (m), 1738 (w), 1617 (s), 1521 (s), 1471 (m), 1357 (m), 1254 (m), 1074 (s), 941 (m), 876 (s), 835 (s), 775 (s) cm−1.
Data for pure 18: 1H NMR of 18 (pure isomer 1) (399.9 MHz, CDCl3): δ 7.16 (d, J = 8.6 Hz, 2H, H7), 6.68 (d, J = 8.6 Hz, 2H, H8), 5.81 (d, J = 8.9 Hz, 1H, H3), 4.64 (d, 1H, J = 4.2 Hz, H5), 4.19 (s, 2H, H1a,b), 2.94 (s, 6H, N(C3)2), 2.65 (m, 1H, H4), 1.79 (br, 1H, O), 0.94 (d, J = 7.0 Hz, 3H, C4-Me), 0.90 (s, 9H, t-![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif)
![[u with combining low line]](https://www.rsc.org/images/entities/char_0075_0332.gif) of OTBS), 0.02 (s, 3H, C3Si), −0.20 (s, 3H, C3Si) ppm. 13C NMR of 18 (pure isomer 1) (100.57 MHz, CDCl3): δ 149.8 (C9), 139.4 (C3), 130.9 (C6), 127.6 (C7), 111.9 (C8), 107.8 (C2), 76.8 (H-OTBS), 71.9 (C1), 49.3 (C4), 40.7 (N(H3)2), 25.8 ((H3)3CSi), 18.2 ((CH3)3Si), 15.3 (C4-Me), −4.5 (H3Si), −5.0 (H3Si) ppm.
of OTBS), 0.02 (s, 3H, C3Si), −0.20 (s, 3H, C3Si) ppm. 13C NMR of 18 (pure isomer 1) (100.57 MHz, CDCl3): δ 149.8 (C9), 139.4 (C3), 130.9 (C6), 127.6 (C7), 111.9 (C8), 107.8 (C2), 76.8 (H-OTBS), 71.9 (C1), 49.3 (C4), 40.7 (N(H3)2), 25.8 ((H3)3CSi), 18.2 ((CH3)3Si), 15.3 (C4-Me), −4.5 (H3Si), −5.0 (H3Si) ppm.
1H NMR of 18 (isomer 2) (399.9 MHz, CDCl3). Resonances and J values have been reported, where determinable, from the purified but diastereomerically enriched 3:1 mixture: δ 7.10 (d, J = 8.8 Hz, 2H, H7), 6.66 (d, J = 8.0 Hz, 2H, H8), 5.72 (d, J = 8.8 Hz, 1H, H3), 4.49 (d, 1H, J = 6.0 Hz, H5), 4.19 (s, 2H, H1), 2.94 (s, 6H, N(C3)2), 2.74 (m, 1H, H4), 1.58 (br, 1H, O), 0.89 (d, J = 6.8 Hz, 3H, C4-), 0.87 (s, 9H, t- of OTBS), −0.02 (s, 3H, C3Si), −0.20 (s, 3H, C3Si) ppm. 13C NMR of 18 (isomer 2) (100.57 MHz, CDCl3): δ 149.8 (C9), 139.4 (C3), 130.9 (C6), 127.6 (C7), 111.9 (C8), 107.7 (C2), 77.5 (C5), 71.9 (C1), 49.3 (C4), 40.7 (N(H3)2), 25.9 ((H3)3CSi), 18.2 ((CH3)3Si), 15.3 (C4-), −4.5 (H3Si), −5.0 (H3Si) ppm.
TOF ES+ HRMS for 18: calcd for C20H35NO2ISi [M + H]+: 476.1482. Found: 476.1500.
Preparation of allylic alcohol 19
To the iodovinylic alcohol 18 (0.3065 g, 0.645 mmol) inside a small pear-shaped flask capped with a rubber septum and an N2-filled balloon was added dry DMF (1.28 mL) via a syringe. The outside of the flask was wrapped in Al-foil to protect it from daylight. While maintaining the N2 atmosphere inside the flask, Me4Sn (1.34 mL, 9.67 mmol, 15 equiv.) was added in one portion via a syringe. CsF (0.2448 g, 1.6 mmol, 2.5 equiv.), CuI (30.7 mg, 0.16 mmol, 0.25 equiv.), and (Ph3P)4Pd (37.2 mg, 0.032 mmol, 0.05 equiv.) were then sequentially and successively added to the reaction flask in that order, maintaining the N2 atmosphere throughout the various additions. The reaction mixture was then left to stir at rt for 22 h, whereafter TLC analysis revealed that two C5-diastereomeric products 19 had formed that had exactly the same TLC mobility (in 5
 :1 hexane:EtOAc) as the starting iodovinylic alcohol 18. Staining and heating of this TLC plate with the anisaldehyde/H2SO4 stain did, however, show that the two newly formed products of structure 19 both stained with a purple/blue colour, which allowed them to be readily distinguished from their precursor 18; this revealed that the reaction was complete, and that no starting 18 remained. The reaction mixture was thereupon diluted with EtOAc (10 mL), transferred to a separatory funnel and H2O (15 mL) was added. After extraction and separation, the aqueous later was extracted with additional EtOAc (6 × 20 mL), and the combined organic extracts were washed with H2O (2 × 50 mL). The organic layer was then dried over MgSO4, filtered, and concentrated in vacuo. The resulting oil was then purified by gradient elution SiO2 flash chromatography with petrol:EtOAc 20:1 → 15:1 initially, to remove reagent-related impurities, followed by petrol:EtOAc 12:1 → 10:1 to secure the entire allylic alcohol 19 (0.189 g, 80%) product as an oil, and as a C5-mixture of epimers.
:1 hexane:EtOAc) as the starting iodovinylic alcohol 18. Staining and heating of this TLC plate with the anisaldehyde/H2SO4 stain did, however, show that the two newly formed products of structure 19 both stained with a purple/blue colour, which allowed them to be readily distinguished from their precursor 18; this revealed that the reaction was complete, and that no starting 18 remained. The reaction mixture was thereupon diluted with EtOAc (10 mL), transferred to a separatory funnel and H2O (15 mL) was added. After extraction and separation, the aqueous later was extracted with additional EtOAc (6 × 20 mL), and the combined organic extracts were washed with H2O (2 × 50 mL). The organic layer was then dried over MgSO4, filtered, and concentrated in vacuo. The resulting oil was then purified by gradient elution SiO2 flash chromatography with petrol:EtOAc 20:1 → 15:1 initially, to remove reagent-related impurities, followed by petrol:EtOAc 12:1 → 10:1 to secure the entire allylic alcohol 19 (0.189 g, 80%) product as an oil, and as a C5-mixture of epimers.
Data for pure 19: 1H NMR of 19 (pure isomer 1) (600.13 MHz, CDCl3): δ 7.09 (d, J = 8.4 Hz, 2H, H7), 6.66 (d, J = 7.8 Hz, 2H, H8), 5.26 (ddd, J = 9.6 Hz, 1H, H3), 4.35 (d, J = 6.0 Hz, 1H, H5), 3.98 (s, 2H, H1a,b), 2.93 (s, 6H, N(C3)2), 2.616 (m, 1H, H4), 1.557 (d, J = 1.2 Hz, C2-), 1.25 (br s, 1H, OH), 0.85 (s, 9H, t- of OTBS), 0.82 (d, J = 6.6 Hz, 3H C4-Me), −0.02 (s, 3H, C3Si), −0.22 (s, 3H, C3Si) ppm. 13C NMR of 19 (pure isomer 1) (150.92 MHz, CDCl3): δ C9 not detected, 134.5 (C3), 129.9 (C6), 127.6 (C7 and C2), 111.9 (C8), 78.9 (C5), 69.4 (C1), 41.1 (N(H3)2), 40.7 (C4), 25.8 ((H3)3CSi), 18.2 ((CH3)3Si), 17.0 (C2-), 13.9 (C4-), −4.5 (H3Si), −5.0 (H3Si) ppm.
1H NMR of 19 (pure isomer 2) (600.13 MHz, CDCl3): δ 7.09 (m, 2H, H7), 6.64 (m, 2H, H8), 5.23 (ddd, J = 10.2, 3.0, 1.2 Hz, 1H, H3), 4.35 (d, J = 6.0 Hz, 1H, H5), 3.89 (s, 2H, H1a,b), 2.92 (s, 6H, N(C3)2), 2.59 (m, J = 9.6, 6.0 Hz, 1H, H4), 1.464 (d, J = 1.2 Hz, C2-), 1.25 (br s, 1H, OH), 0.96 (d, J = 7.2 Hz, C4-), 0.85 (s, 9H, t- of OTBS), −0.013 (s, 3H, C3Si), −0.21 (s, 3H, C3Si) ppm. 13C NMR of 19 (pure isomer 2) (150.92 MHz, CDCl3): δ 149.4 (C9), 134.2 (C3), 130.0 (C6), 127.5 (C7 and C2), 111.7 (C8), 78.7 (C5), 69.3 (C1), 41.2 (N(H3)2), 40.7 (C4), 25.9 ((H3)3CSi), 18.3 ((CH3)3Si), 16.0 (C2-), 13.8 (C4-), −4.5 (H3Si), −5.1 (H3Si) ppm.
TPAP/NMO oxidation19 of iodo-allylic alcohol 18 to iodoenal 20
Before commencing this experiment, the reaction flask was covered with Al-foil to protect it from the adverse effects of visible light. Thereafter, to a stirred rt solution of the iodovinylic alcohol 18 (3.55 g, 7.47 mmol) in dry CH2Cl2 (74.7 mL) under N2 was successively added powdered 4 Å molecular sieves (flame dried, 3.60 g), N-methylmorpholine-N-oxide (NMO, 1.75 g, 14.93 mmol, 2 equiv.) and tetrapropylammonium perruthenate19 (TPAP, n-Pr4NRuO4, 0.13 g, 0.37 mmol, 0.05 equiv.), each in one portion. Stirring was continued at rt for 15 min. The reaction mixture was then filtered to remove the sieves, the filtrate was concentrated in vacuo, and the resulting crude residue was purified by SiO2 flash chromatography with 40
 :1 petrol:EtOAc as the eluent. Following purification, aldehyde 20 (2.10 g, 59%) was obtained as a C5-epimeric mixture (ca. 3:1) as an amber oil.
:1 petrol:EtOAc as the eluent. Following purification, aldehyde 20 (2.10 g, 59%) was obtained as a C5-epimeric mixture (ca. 3:1) as an amber oil.
Data for 20 (ca. 3:1 mixture of isomers of tentatively assigned stereochemistry at C5): IR of the 20 mixture (neat film): 3393 (w), 2959 (s), 2929 (s), 2856 (s), 2806 (w), 1701 (s), 1615 (s), 1524 (s), 1461 (m), 1388 (s), 1355 (m), 1251 (m), 1186 (w), 1168 (m), 1080 (s), 1027 (m), 1007 (m), 944 (w), 858 (m), 838 (m), 780 (m) cm−1.
1H NMR of 20 (minor isomer 1 – anti) (399.9 MHz, CDCl3): δ 8.64 (s, 1H, H1), 7.10 (d, J = 8.4 Hz, 2H, H7), 7.09 (d, J = 9.2 Hz, 1H, H3), 6.66 (d, J = 8.4 Hz, 2H, H8), 4.61 (d, 1H, J = 6.0 Hz, H5), 3.18 (m, 1H, H4), 2.94 (s, 6H, N(C3)2), 1.02 (d, J = 6.8 Hz, 3H, C4-), 0.86 (s, 9H, t- of OTBS), 0.028 (s, 3H, C3Si), −0.20 (s, 3H, C3Si) ppm. 13C NMR of 20 (minor isomer 1 – anti) (100.57 MHz, CDCl3): δ 188.1 (C1), 165.3 (C3), 150.0 (C9), 130.1 (C6), 127.33 (C7), 112.0 (C8), 111.3 (C2), 77.6 (C5), 50.2 (C4), 40.5 (N(H3)2), 25.79 ((H3)3CSi), 18.1 ((CH3)3Si), 15.2 (C4-), −4.5 (H3Si), −5.1 (H3Si) ppm.
1H NMR of 20 (major isomer 2 – syn) (399.9 MHz, CDCl3): δ 8.59 (s, 1H, H1), 7.16 (d, J = 8.4 Hz, 2H, H7), 7.05 (d, J = 9.6 Hz, 1H, H1), 6.68 (d, J = 8.4 Hz, 2H, H8), 4.73 (d, 1H, J = 4.4 Hz, H5), 3.12 (m, 1H, H4), 2.95 (s, 6H, N(C3)2), 1.07 (d, J = 6.8 Hz, 3H, C4-), 0.90 (s, 9H, t- of OTBS), −0.012 (s, 3H, C3Si), −0.17 (s, 3H, C3Si) ppm. 13C NMR of 20 (major isomer 2) (100.57 MHz, CDCl3): δ 188.09 (C1), 165.3 (C3), 149.9 (C9), 129.8 (C6), 127.29 (C7), 111.9 (C8), 110.4 (C2), 76.4 (C5), 49.7 (C4), 40.5 (N(H3)2), 25.84 ((H3)3CSi), 18.2 ((CH3)3Si), 13.3 (C4-), −4.5 (H3Si), −5.1 (H3Si) ppm.
TOF ES+ HRMS of 20: calcd for C20H33NO2ISi [M + H]+: 474.1325. Found: 474.1317.
Baldwin–Lee Stille cross-coupling of iodo enal 20 to obtain trisubstituted enal 4
A solution of the iodoenal 20 (0.98 g, 2.06 mmol) in dry DMF (4.12 mL) was transferred via a cannula into a Teflon-screw-capped sealed tube temporarily fitted with a rubber septum under N2. Me4Sn (4.29 mL, 30.90 mmol, 15 equiv.), CsF (0.78 g, 5.15 mmol, 2.5 equiv.), CuI (0.10 g, 0.52 mmol, 0.25 equiv.), and (Ph3P)4Pd (0.12 g, 0.10 mmol, 0.05 equiv.) were then successively added in that order, and the septum was replaced with the Teflon-screw cap, maintaining the N2 atmosphere throughout the addition and sealing process. The sealed tube was then covered with Al-foil, placed inside an oil bath, and heated at 60 °C overnight (18 h) with vigorous magnetic stirring. The reaction mixture was then cooled to rt, carefully opened, and diluted with EtOAc (5 mL) before being fully quenched with H2O (10 mL). The sealed tube was rinsed multiple times with EtOAc (20 mL × 5) and H2O (20 mL × 3) and the combined reaction washings were filtered through Celite, before being fractionated in a separatory funnel. The aqueous layer was further extracted with EtOAc (50 mL × 3), and the combined organic extracts were washed with H2O (100 mL × 2), dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by SiO2 flash chromatography using 45
 :1 petrol:EtOAc as an eluent to afford 4 (0.59 g, 79%) as a mixture of C5-epimers and as a colourless oil. IR of the 4 mixture (neat film): 3353 (w), 2959 (s), 2932 (s), 2858 (s), 2808 (m), 2707 (w), 1690 (s), 1617 (s), 1524 (s), 1466 (m), 1385 (m), 1355 (m), 1257 (m), 1186 (w), 1072 (m), 1022 (m), 949 (w), 863 (m), 835 (m), 777 (m), 671 (w), 565 (w) cm−1.
:1 petrol:EtOAc as an eluent to afford 4 (0.59 g, 79%) as a mixture of C5-epimers and as a colourless oil. IR of the 4 mixture (neat film): 3353 (w), 2959 (s), 2932 (s), 2858 (s), 2808 (m), 2707 (w), 1690 (s), 1617 (s), 1524 (s), 1466 (m), 1385 (m), 1355 (m), 1257 (m), 1186 (w), 1072 (m), 1022 (m), 949 (w), 863 (m), 835 (m), 777 (m), 671 (w), 565 (w) cm−1.
Data for 4: 1H NMR of 4 (ca. 3:1 mixture of isomers at C5) (isomer 1 – major) (399.9 MHz, CDCl3): δ 9.40 (s, 1H, H1), 7.09 (d, J = 8.8 Hz, 2H, H7), 6.65 (d, J = 8.8 Hz, 2H, H8), 6.43 (dd, J = 10.0 Hz, 1.2 Hz, 1H, H3), 4.47 (d, 1H, J = 6.0 Hz, H5), 2.93 (br m, 1H, H4) superimposed upon 2.93 (s, 6H, N(C3)2), 1.62 (d, J = 1.2 Hz, 3H, C2-), 0.95 (d, J = 6.8 Hz, 3H, C4-), 0.84 (s, 9H, t- of OTBS), −0.011 (s, 3H, C3Si), −0.23 (s, 3H, C3Si) ppm. 13C NMR of 4 (isomer 1 – major) (100.57 MHz, CDCl3): δ 195.6 (C1), 158.0 (C3), 149.9 (C9), 139.1 (C2), 130.9 (C6), 127.3 (C7), 112.0 (C8), 78.6 (C5), 42.9 (C4), 40.55 (N(H3)2), 25.75 ((H3)3CSi), 18.1 ((CH3)3Si), 16.5 (C4-), 9.31 (C2-), −4.5 (H3Si), −5.1 (H3Si) ppm.
1H NMR of 4 (isomer 2 – minor) (399.9 MHz, CDCl3): δ 9.31 (s, 1H, H1), 7.08 (d, J = 8.8 Hz, 2H, H7), 6.63 (d, J = 8.7 Hz, 2H, H8), 6.30 (dd, J = 10.0 Hz, 1.2 Hz, 1H, H3), 4.50 (d, 1H, J = 5.6 Hz, H5), 2.93 (br m, 1H, H4) superimposed upon 2.93 (s, 6H, N(C3)2), 1.61 (d, J = 1.6 Hz, 3H, C2-Me), 1.07 (d, J = 6.4 Hz, 3H, C4-Me), 0.89 (s, 9H, t- of OTBS), −0.04 (s, 3H, C3Si), −0.20 (s, 3H, C3Si) ppm. 13C NMR of 4 (isomer 2 – minor) (100.57 MHz, CDCl3): δ 195.6 (C1), 157.4 (C3), 149.8 (C9), 138.5 (C2), 130.6 (C6), 127.3 (C7), 111.8 (C8), 77.8 (C5), 42.6 (C4), 40.53 (N(H3)2), 25.83 ((H3)3CSi), 18.2 ((CH3)3Si), 15.3 (C4-), 9.3 (C2-), −4.5 (H3Si), −5.1 (H3Si) ppm.
TOF ES+ HRMS of 4: calcd for C21H36NO2Si [M + H]+: 362.2515. Found: 362.2517.
Wittig olefination of enal 4 to obtain dienoate 3
To a stirred rt solution of the enal 4 (0.5777 g, 1.60 mmol) in dry CH2Cl2 (1.6 mL) and PhMe (2 mL) under N2 was added carbethoxymethylenetriphenylphosphorane (0.835 g, 2.40 mmol, 1.5 equiv.) and the reactants were stirred at 38 °C for 10 d. The reaction mixture was thereafter evaporated to dryness before petrol (5 mL) was added and the resulting mixture was stirred for 2 h. The reaction mixture was filtered to remove Ph3P
 O and concentrated under reduced pressure. The crude residue was purified by SiO2 flash chromatography using petrol:EtOAc (50:1) as the eluent to give 3 (0.62 g, 90%) as a colorless oil and mixture of C7-epimers (N.B.: (+)-(R)-trichostatin A numbering is now being used here onwards). IR of the 3 mixture (neat film): 2959 (m), 2934 (m), 2856 (m), 1721 (m), 1620 (s), 1521 (m), 1458 (m), 1388 (s), 1310 (w), 1259 (w), 1168 (m), 1097 (w), 1070 (w), 1029 (w), 871 (w), 780 (w) cm−1.
O and concentrated under reduced pressure. The crude residue was purified by SiO2 flash chromatography using petrol:EtOAc (50:1) as the eluent to give 3 (0.62 g, 90%) as a colorless oil and mixture of C7-epimers (N.B.: (+)-(R)-trichostatin A numbering is now being used here onwards). IR of the 3 mixture (neat film): 2959 (m), 2934 (m), 2856 (m), 1721 (m), 1620 (s), 1521 (m), 1458 (m), 1388 (s), 1310 (w), 1259 (w), 1168 (m), 1097 (w), 1070 (w), 1029 (w), 871 (w), 780 (w) cm−1.
1H NMR of 3 (isomer 1 – minor) (399.9 MHz, CDCl3): δ 7.32 (dd, J = 15.6, 0.4 Hz, 1H, H2), 7.07 (d, J = 8.8 Hz, 2H, H9), 6.64 (d, J = 8.8 Hz, 2H, H10), 5.79 (d, 1H, J = 10.0 Hz, H5), 5.75 (d, J = 15.2 Hz, 1H, H3), 4.39 (d, J = 6.0 Hz, 1H, H7), 4.20 (m, J = 7.2 Hz, 2H, C2 of OEt), 2.93 (s, 6H, N(C3)2), 2.76 (m, 1H, H6), 1.64 (d, J = 1.2 Hz, 3H, C4-Me), 1.30 (t, J = 7.2 Hz, 3H, of OEt), 0.87 (d, C6-Me) 0.83 (s, 9H, t- of OTBS), −0.03 (s, 3H, C3Si), −0.23 (s, 3H, C3Si) ppm. 13C NMR of 3 (isomer 1 – minor) (100.57 MHz, CDCl3): δ 167.8 (C1), 150.0 (C2), 149.8 (C11), 145.4 (C5), 132.6 (C4), 131.6 (C8), 127.5 (C9), 115.4 (C3), 111.94 (C10), 78.8 (C7), 60.1 (H2 of OEt), 42.4 (C6), 40.6 (N(H3)2), 25.8 ((H3)3CSi), 18.1 ((CH3)3Si), 16.9 (C6-), 14.3 ( of OEt), 12.4 (C4-), −4.6 (H3Si), −5.1 (H3Si) ppm.
1H NMR of 3 (isomer 2 – major) (399.9 MHz, CDCl3): δ 7.23 (d, J = 15.6, 0.4 Hz, 1H, H2), 7.06 (d, J = 8.8 Hz, 2H, H9), 6.63 (d, J = 8.4 Hz, 2H, H10), 5.71 (d, J = 15.6 Hz, 1H, H3), 5.70 (d, 1H, J = 10.0 Hz, H5), 4.41 (d, J = 6.0 Hz, 1H, H7), 4.20 (m, J = 7.2 Hz, 2H, C2 of OEt), 2.92 (s, 6H, N(C3)2), 2.76 (m, 1H, H6), 1.60 (d, J = 1.2 Hz, 3H, C4-Me), 1.29 (t, J = 7.2 Hz, 3H, of OEt), 1.00 (d, J = 6.8 Hz, 3H, C6-), 0.88 (s, 9H, t- of OTBS), −0.015 (s, 3H, C3Si), −0.22 (s, 3H, C3Si) ppm. 13C NMR of 3 (isomer 2 – major) (100.57 MHz, CDCl3): δ 167.7 (C1), 149.9 (C2), 149.7 (C11), 145.0 (C5), 132.1 (C4), 131.4 (C8), 127.4 (C9), 115.5 (C3), 111.9 (C10), 78.3 (C7), 60.1 (H2 of OEt), 42.4 (C6), 40.6 (N(H3)2), 25.9 ((H3)3CSi), 18.2 ((CH3)3Si), 15.9 (C6-), 14.3 ( of OEt), 12.3 (C4-), −4.5 (H3Si), −5.1 (H3Si) ppm.
TOF ES+ HRMS of 3: calcd for C25H42NO3Si [M + H]+: 432.2934. Found: 432.2942.
Preparation of 7-OTBS trichostatic acid 21
To a stirred rt solution of ester 3 (1.06 g, 2.46 mmol) in THF/H2O/MeOH (2 mL
 :2 mL:2 mL) was added LiOH monohydrate (0.41 g, 9.82 mmol, 4 equiv.) and the mixture was thereafter allowed to stir vigorously for 48 h. The reaction mixture was then diluted with EtOAc (10 mL) and acidified with 10% aq. HCl until pH 5 was attained. The aqueous layer was extracted with EtOAc (20 mL × 3). The combined organic layer was washed with H2O (50 mL × 2), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by gradient elution SiO2 flash chromatography with 6:1 → 4:1 petrol:EtOAc as an eluent to obtain 21 (0.97 g, 98%) as a colourless oil.
:2 mL:2 mL) was added LiOH monohydrate (0.41 g, 9.82 mmol, 4 equiv.) and the mixture was thereafter allowed to stir vigorously for 48 h. The reaction mixture was then diluted with EtOAc (10 mL) and acidified with 10% aq. HCl until pH 5 was attained. The aqueous layer was extracted with EtOAc (20 mL × 3). The combined organic layer was washed with H2O (50 mL × 2), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by gradient elution SiO2 flash chromatography with 6:1 → 4:1 petrol:EtOAc as an eluent to obtain 21 (0.97 g, 98%) as a colourless oil.
Data for 21 (for a 3:1 mixture of C7-epimers): IR of the 21 mixture (neat film): 3376 (extremely br), 2959 (s), 2932 (s), 2858 (m), 2806 (w), 1688 (s), 1617 (s), 1524 (s), 1468 (m), 1383 (m), 1282 (m), 1254 (m), 1209 (w), 1072 (s), 1029 (m), 987 (w), 871 (s), 777 (m) cm−1.
1H NMR of 21 (isomer 1 – minor) (399.9 MHz, CDCl3): δ 10.58 (very br, 1H, –CO2), 7.41 (d, J = 15.6 Hz, 1H, H2), 7.08 (d, 2H, H9), 6.65 (d, 2H, H10), 5.86 (d, 1H, J = 9.6 Hz, H5), 5.72 (d, J = 15.6 Hz, 1H, H3), 4.40 (d, J = 6.0 Hz, 1H, H7), 2.93 (s, 6H, N(C3)2), 2.78 (m, 1H, H6), 1.66 (s, 3H, C4-), 0.89 (d, 3H, C6- obscured by a large singlet for t-Bu for the major isomer of 21), 0.84 (s, 9H, t- of OTBS), −0.02 (s, 3H, C3Si), −0.23 (s, 3H, C3Si) ppm. 13C NMR of 21 (isomer 1 – minor) (100.57 MHz, CDCl3): δ 172.9 (C1), 152.3 (C2), 149.8 (C11), 146.8 (C5), 132.7 (C4), 131.6 (C8), 127.5 (C9), 114.5 (C3), 112.0 (C10), 78.8 (C7), 42.6 (C6), 40.7 (N(H3)2), 25.8 ((H3)3CSi), 18.1 ((CH3)3Si), 16.9 (C6-), 12.4 (C4-), −4.6 (H3Si), −5.1 (H3Si) ppm.
1H NMR of 21 (isomer 2 – major) (399.9 MHz, CDCl3): δ 10.58 (very br, 1H, –CO2), 7.32 (d, J = 15.6 Hz, 1H, H2), 7.07 (d, J = 8.4 Hz, 2H, H9), 6.65 (d, J = 8.4 Hz, 2H, H10), 5.76 (d, 1H, J = 10.4 Hz, H5), 5.71 (d, J = 15.6 Hz, 1H, H3), 4.43 (d, J = 6.0 Hz, 1H, H7), 2.93 (s, 6H, N(C3)2), 2.78 (m, 1H, H6), 1.62 (s, 3H, C4-Me), 1.01 (d, J = 6.8 Hz, 3H, C6-), 0.89 (s, 9H, SiC(CH3)3), −0.01 (s, 3H, C3 Si), −0.21 (s, 3H, C3Si) ppm. 13C NMR of 21 (isomer 2 – major) (100.57 MHz, CDCl3): δ 172.8 (O), 152.2 (C2), 149.7 (C11), 146.4 (C5), 132.2 (C4), 131.4 (C8), 127.4 (C9), 114.7 (C3), 112.0 (C10), 78.2 (C7), 42.5 (C6), 40.7 (N(H3)2), 25.9 ((H3)3CSi), 18.2 ((CH3)3Si), 15.8 (C6-), 12.3 (C4-), −4.5 (H3Si), −5.1 (H3Si) ppm.
TOF ES+ HRMS of 21: calcd for C23H38NO3Si [M + H]+: 404.2621. Found: 404.2613.
Conversion of 7-OTBS trichostatic acid 21 into (+)-(R)-trichostatic acid (22)
To a stirred 0 °C solution of 7-OTBS trichostatic acid 21 (0.61 g, 1.51 mmol) in CH2Cl2/H2O (20
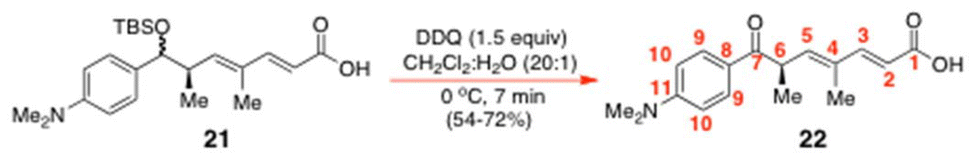 :1) (9.88 mL; made up from 9.41 mL CH2Cl2: 0.47 mL H2O) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 0.51 g, 2.26 mmol, 1.5 equiv.) in one portion and the cold reaction mixture was vigorously stirred for just 7 min (important note: stirring for longer than this time caused significant decomposition). The reaction mixture was then diluted with CH2Cl2 (10 mL) and quenched with H2O (10 mL). The aqueous layer was extracted with CH2Cl2 (50 mL × 3). The combined organic extracts were washed with H2O multiple times (50 mL × 8), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by gradient elution SiO2 flash chromatography with petrol:EtOAc (1:1 → 1:2) as an eluent with 21 (0.31 g, 71%) being obtained as an amber oil.
:1) (9.88 mL; made up from 9.41 mL CH2Cl2: 0.47 mL H2O) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 0.51 g, 2.26 mmol, 1.5 equiv.) in one portion and the cold reaction mixture was vigorously stirred for just 7 min (important note: stirring for longer than this time caused significant decomposition). The reaction mixture was then diluted with CH2Cl2 (10 mL) and quenched with H2O (10 mL). The aqueous layer was extracted with CH2Cl2 (50 mL × 3). The combined organic extracts were washed with H2O multiple times (50 mL × 8), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by gradient elution SiO2 flash chromatography with petrol:EtOAc (1:1 → 1:2) as an eluent with 21 (0.31 g, 71%) being obtained as an amber oil.
Data for 22: [α]D = +150.7° (c 0.416, CH2Cl2), mp 89–90 °C [literature mp17 88–89 °C].
IR of 22 (neat film): 3600–2300 (extremely br), 2980 (m), 2932 (m), 2874 (m), 2669 (w), 2596 (w), 1685 (s), 1597 (s), 1552 (m), 1438 (m), 1380 (s), 1272 (m), 1244 (m), 1191 (m), 1171 (m), 1004 (w), 979 (w), 858 (w), 828 (w), 737 (m) cm−1.
1H NMR of pure 22 (400.11 MHz, CDCl3): δ 7.84 (d, J = 9.2 Hz, 2H, H9), 7.36 (d, J = 15.6 Hz, 1H, H2), 6.64 (d, J = 9.2 Hz, 2H, H10), 6.06 (d, J = 9.6 Hz, 1H, H5), 5.85 (d, 1H, J = 15.6 Hz, H3), 4.38 (m, 1H, H6), 3.05 (s, 6H, N(C3)2), 1.92 (d, J = 1.2 Hz, 3H, C4-Me), 1.31 (d, J = 6.8 Hz, 3H, C6-) ppm. 13C NMR of 22 (100.57 MHz, CDCl3): δ 198.3 (C7), 171.7 (C1), 153.5 (C11), 151.4 (C2), 143.0 (C5), 132.6 (C4), 130.6 (C9), 123.9 (C8), 115.7 (C3), 110.8 (C10), 40.9 (C6), 40.0 (N(H3)2), 17.7 (C6-), 12.5 (C4-) ppm.
TOF ES+ HRMS of 22: calcd for C17H22NO3 [M + H]+: 288.1600. Found: 288.1584.
Conversion of (+)-(6R)-trichostatic acid (22) into (+)-(6R)-trichostatin A (1a)
Following Helquist's procedure,17e a stirred 0 °C solution of (+)-(6R)-trichostatic acid 22 (56.2 mg, 0.195 mmol) in dry THF (1 mL) under N2 was treated with freshly distilled dry Et3N (0.03 mL, 0.23 mmol, 1.2 equiv.) followed by dropwise addition of ethyl chloroformate (0.04 mL, 0.40 mmol, 2 equiv.). The reactants were allowed to stir at 0 °C for 2 h, whereafter a solution of TBSONH2 (57.6 mg, 0.39 mmol, 1.95 equiv.) in dry THF (0.5 mL) was added via a microcannula. The reaction mixture was stirred at 0 °C for 0.5 h and then warmed to rt for 1.5 h. It was then diluted with EtOAc (1 mL) and quenched with H2O (1 mL). The aqueous layer was extracted with EtOAc (10 mL × 3). The organic layer was washed with H2O (10 mL × 2), dried over MgSO4, filtered and concentrated under reduced pressure and the crude residue of 24 was used directly without further purification.

To a stirred rt solution of the aforementioned TBS-protected crude trichostatin A 24 in dry MeOH (2 mL) under N2 was added solid CsF (35.6 mg, 0.23 mmol, 1.2 equiv.) (pre-dried at 120 °C under high vacuum for 2 h), and the reactants were allowed to stir for 1 h. The reaction mixture was then diluted with CH2Cl2 (5 mL) and washed with H2O (2 mL). The aqueous layer was then further extracted with CH2Cl2 (10 mL × 5). The combined organic extracts were washed with H2O (20 mL × 1), dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by gradient elution SiO2 flash chromatography with CH2Cl2/MeOH (25:1 → 20:1) to obtain (+)-(R)-trichostatin A (1a) (35 mg, 59% over 2 steps) as a white solid.
Data for (R)-(+)-trichostatin A (1a): [α]D = +88.8° (c 0.26, MeOH), mp 140–143 °C [lit.17 140–143 °C]. IR of (+)-TSA (1a) (neat film): 3234 (br, s), 2927 (s), 2853 (s), 1650 (m), 1595 (s), 1547 (m), 1383 (s), 1249 (m), 1189 (s), 1059 (m), 976 (m) cm−1.
1H NMR of (+)-TSA (1a) (600.13 MHz, CD3OD): δ 7.87 (d, J = 9.0 Hz, 2H, H9), 7.18 (d, J = 15.0 Hz, 1H, H2), 6.72 (d, J = 9.6 Hz, 2H, H10), 5.91 (d, J = 9.6 Hz, 1H, H5), 5.87 (d, 1H, J = 15.6 Hz, H3), 4.53 (dq, J = 9.0, 6.6 Hz, 1H, H6), 3.06 (s, 6H, N(C3)2), 1.92 (s, 3H, C4-), 1.27 (d, J = 6.6 Hz, 3H, C6-) ppm.
13C NMR of (+)-TSA (1a) (150.92 MHz, CD3OD): δ 201.4 (C7), 166.8 (C1), 155.5 (C11), 145.9 (C2), 141.3 (C5), 134.3 (C8), 131.9 (C9), 124.7 (C4), 117.1 (C3), 111.9 (C10), 41.7 (C6), 40.1 (N(H3)2), 18.3 (C6-), 12.7 (C4-Me) ppm.
TOF ES+ HRMS of (+)-TSA (1a): [M + H]+ calcd for C17H23N2O3: 303.171. Found: 303.177.
Data availability
All data supporting this article are included in the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the ACS and QUB for financial support of this work.References
- Review: K. J. Hale, S. Manaviazar and H. A. Watson, Chem. Rec., 2019, 19, 238 CAS.
- (a) P. Dimopoulos, A. Athlan, S. Manaviazar, J. George, M. Walters, L. Lazarides, A. Aliev and K. J. Hale, Org. Lett., 2005, 7, 5369 CrossRef CAS PubMed; (b) R. Willem, A. Delmotte, I. De Borger, I. Biesemans, M. Gielen and F. Kayser, J. Organomet. Chem., 1994, 480, 255 CrossRef CAS; (c) P. Dussault, C. T. Eary, R. J. Lee and U. R. Zope, J. Chem. Soc., Perkin Trans. 1, 1999, 2189 RSC.
- Mechanistic studies: (a) P. Dimopoulos, J. George, D. A. Tocher, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5377 CrossRef CAS PubMed; (b) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Chem. Commun., 2019, 55, 14454 RSC; (c) H. A. Watson, S. Manaviazar, H. G. Steeds and K. J. Hale, Tetrahedron, 2020, 76, 131061 CrossRef CAS; (d) H. A. Watson, A. J. Fielding and K. J. Hale, Chem. Commun., 2021, 57, 7449 CAS.
- (a) E. J. Corey and R. H. Wollenberg, J. Org. Chem., 1975, 40, 2265 CrossRef CAS; (b) H. E. Ensley, R. R. Buescher and K. Lee, J. Org. Chem., 1982, 47, 404 CrossRef CAS; (c) C. Nativi and M. Taddei, J. Org. Chem., 1988, 58, 820 CrossRef; (d) M. Lautens and A. H. Hudson, Tetrahedron Lett., 1990, 31, 3105 CrossRef CAS.
- (a) K. Pati, G. dos Passos Gomes, T. Harris, A. Hughes, H. Phan, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 1165 CrossRef CAS PubMed; (b) N. P. Tsvetkov, E. Gonzales-Rodriguez, A. Hughes, G. dos Passos Gomes, F. D. White, F. Kuriakose and I. V. Alabugin, Angew. Chem., Int. Ed., 2018, 57, 3651 CrossRef CAS PubMed; (c) E. Gonzalez-Rodriguez, M. A. Abdo, G. dos Passos Gomes, S. Ayad, F. D. White, N. P. Tsvetkov, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2020, 142, 8352 CrossRef CAS PubMed; (d) C. Hu, L. Kuhn, F. D. Makurvet, E. S. Knorr, X. Lin, R. K. Kawade, F. Mentink-Vigier, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2024, 146, 4187 CrossRef CAS PubMed; (e) I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed; (f) I. V. Alabugin, K. M. Gilmore and P. W. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109 CAS; (g) I. V. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons Ltd, Chichester, UK, 2016, ch. 3, p. 42 CrossRef; (h) C. Hu, J. Mena and I. V. Alabugin, Nat. Rev. Chem., 2023, 7, 405 CrossRef PubMed.
- S. Manaviazar, K. J. Hale and A. LeFranc, Tetrahedron Lett., 2011, 52, 2080 CrossRef CAS.
- K. J. Hale, M. Grabski, S. Manaviazar and M. Maczka, Org. Lett., 2014, 16, 1164 CrossRef CAS PubMed.
- K. J. Hale, M. Maczka, A. Kaur, S. Manaviazar, M. Ostovar and M. Grabski, Org. Lett., 2014, 16, 1168 CrossRef CAS PubMed.
- K. Micoine, P. Persich, J. Llaveria, M.-H. Lam, A. Merderna, F. Loganzo and A. Furstner, Chem. – Eur. J., 2013, 19, 7370 CrossRef CAS PubMed.
- P. Dimopoulos, A. Athlan, S. Manaviazar and K. J. Hale, Org. Lett., 2005, 7, 5373 CrossRef CAS PubMed.
- (a) S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132 CrossRef CAS PubMed; (b) V. Lee, Org. Biomol. Chem., 2019, 17, 9095 CAS.
- V. Farina, S. Kapadia, B. Krishnan, C. Wang and L. S. Liebeskind, J. Org. Chem., 1994, 59, 5909 Search PubMed.
- J. A. Marshall and S. Xie, J. Org. Chem., 1995, 60, 723 CrossRef.
- (a) N. Tsuji, M. Kobayashi, K. Nagashima, Y. Wakisawa and K. Koizumi, J. Antibiot., 1976, 29, 1 CAS; (b) D. M. Vigushin, S. Ali, N. Mirsaidi, K. Ito, I. Adcock and R. C. Coombes, Clin. Cancer Res., 2001, 7, 971 CAS; (c) L. Sanderson, G. W. Taylor, E. O. Aboagye, J. P. Alao, J. R. Latigo, R. C. Coombes and D. M. Vigushin, Drug Metab. Dispos., 2004, 32, 1132 CAS; (d) A. M. Grabiec, S. Krausz, W. de Jager, T. Burakowski, D. Groot, M. E. Sanders, B. J. Prakken, W. Maslinski, E. Eldering, P. P. Tak and K. A. Reedquist, J. Immunol., 2010, 184, 2718 CAS.
- C. Mozzetta, S. Consalvi, V. Saccone, M. Tierney, A. Diamantini, K. J. Mitchell, G. Marazzi, G. Borsellino, L. Battistini, D. Sassoon, A. Sacco and P. L. Puri, EMBO Mol. Med., 2013, 5, 626 CAS.
- C. Wang, L. Lei, Y. Xu, Y. Li, J. Zhang, Y. Xu and S. Si, Pharmaceuticals, 2024, 17, 425 CAS.
- For previous trichostatin A total syntheses, see: (a) I. Fleming, J. Iqbal and E.-P. Krebs, Tetrahedron, 1983, 39, 841 CrossRef CAS; (b) K. Mori and K. Koseki, Tetrahedron, 1988, 44, 6013 CrossRef CAS; (c) S. Zhang, W. Duan and W. Wang, Adv. Synth. Catal., 2006, 348, 1228 CrossRef CAS; (d) C. C. Cosner and P. Helquist, Org. Lett., 2011, 13, 3564 CrossRef CAS PubMed; (e) C. C. Cosner, V. B. R. Iska, A. Chatterjee, J. T. Markiewicz, S. J. Corden, J. Lofstedt, T. Anker, J. Richer, T. Hulett, D. J. Schauer, O. Wiest and P. Helquist, Eur. J. Org. Chem., 2013, 162 CrossRef CAS; (f) Trichostatin D total synthesis: S. Hosokawa, T. Ogura, H. Togashi and K. Tatsuta, Tetrahedron Lett., 2005, 46, 333 CrossRef CAS; (g) Synthesis of trichostatic acid (22): J. T. Markiewicz, D. J. Schauer, J. Lofstedt, S. J. Corden, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 2061 CrossRef CAS PubMed.
- For a related allenyl-Pd(II) intermediate also being prevented from undergoing transmetallation by an α-allylic OPv, see: J. A. Marshall and J. J. Mulhearn, Org. Lett., 2005, 7, 1593 CrossRef CAS PubMed.
- (a) W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 21, 1625 RSC; (b) S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639 CrossRef; (c) For a previous application of the Ley and Griffith cat. TPAP/NMO oxidation system in the total synthesis of the macrolide immunosuppressants (+)-prunustatin A and (+)-SW-163A, see: S. Manaviazar, P. Nockemann and K. J. Hale, Org. Lett., 2016, 18, 2902 CrossRef CAS PubMed.
- (a) N. Tsuji and M. Kobayashi, J. Antibiot., 1978, 31, 939 CrossRef CAS PubMed; (b) R. R. Hughes, K. A. Shaaban, J. Zhang, H. Cao, G. N. Phillips and J. S. Thorsen, ChemBioChem, 2017, 18, 363 CrossRef CAS PubMed; (c) The details of our synthesis of (+)-trichostatin C from (+)-trichostatic acid (22) will be reported in due course.
- A reviewer of our paper has requested that we briefly compare our new synthetic route with the other total syntheses of trichostatin A that are listed in ref. 17. We will duly do this here. (a) The first total synthesis of (±)-trichostatin A to be achieved was that of Fleming and coworkers in 1983. It provided the natural product in racemic form in 5 steps (ref. 17a), but according to Mori and Koseki (ref. 17b), its final KOH/NH2OH-mediated ester N-hydroxy-ammonolysis step cannot be used to secure biologically-active (+)-trichostatin A;; (b) Although Mori and Koseki's later synthesis of (+)-trichostatin A (1a) (ref. 17b) did deliver the natural product in ≥98% ee from (R)-(−)-3-hydroxy-2-methyl-propionate, it did require 18 steps to be implemented overall, and it had a longest linear sequence of 16 steps. While Mori's synthesis was perfectly stereocontrolled, with respect to installation of the (6R)-stereocentre and the C(2)–C(5)-dienoate array, it did encounter low yields during its final stages; its penultimate steps 14 and 15 proceeded with a combined yield of 11%;; (c) Helquist later published three synthetic routes to (±)-trichostatin A (ref. 17d, e and g), one of which (ref. 17e) was subsequently rendered enantioselective. The latter route required 17 steps to be implemented overall, when the more reliable and higher yielding 7-step (S)-ethyl lactate pathway was used to access its key (S)-3-butyn-2-yl O-mesylate starting material. Importantly, Helquist's asymmetric route to 1a had a longest linear sequence of 10 steps, and its yields were largely good throughout. It was also fully stereocontrolled with regard to installation of the C(6)–Me group and the C(2)–C(5)-dienyl array. It did, however, produce (+)-(R)-trichostatin A (1a) in only 81% ee;; (d) Contrastingly, our new O-directed hydrostannylative route to (+)-trichostatin A proceeds in 18 steps overall (which is one step more than Helquist's), and it has a longest linear sequence of 12 steps. It also proceeds with a maximal overall yield of 8%. However, most critically, it does provide (+)-(R)-trichostatin A in ≥98% ee, as evidenced by our conversion of 1a to (+)-trichostatin C, without the accompanying formation of the C(6)-(S)-β-glycoside diastereoisomer. Importantly, our prior derivatisation of 15 to obtain 25 (see ESI) also confirmed that 15 was of ≥98% ee;; (e) Although Wang's 2006 report (ref. 17c) did describe a 10 step L-proline-catalysed aldol route to (+)-(R)-trichostatin A, which delivered a material of ≥99% ee in an apparently good overall yield (17.4%), its final benzylic alcohol oxidation step had to be conducted with just 0.59 equiv. of DDQ in dioxane. Presumably this was done to minimise or prevent competing N-oxidation of the hydroxamic acid unit to give a highly reactive N-acyl-nitroso intermediate, which would almost certainly self-condense, if generated. We note here that the ref. 17c team were unable to recover any of their unreacted starting hydroxamic acid amide precursor from this DDQ oxidation, which proceeded with 49% yield for these last two steps. Contrastingly, each of the other enantioselective routes to (+)-trichostatin A have utilised an O-alkoxy/siloxyamine coupling strategy with a mixed anhydride derived from (+)-trichostatic acid to install the hydroxamic acid motif of the natural product.
- (a) For a recent outstanding review on alkyne hydrometallation with Group IV metal hydrides, see the following book chapter by: T. Wiesner and M. Haas, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2024, DOI:10.1016/B978-0-323-96025-0.00125-3 (b) For McLaughlin and Roberts’ 2023 report on the highly regiocontrolled PtCl2/XPhos-catalysed hydrostannation of terminal aryl acetylenes and propargylic alcohols, see: D. D. Roberts and M. G. McLaughlin, Adv. Synth. Catal., 2023, 365, 1602 CrossRef CAS. This paper lists much valuable new metal-catalysed hydrostannation literature that has recently appeared; (c) For McLaughlin's landmark application of the PtCl2/XPhos/Et3SiH-catalyst system to mediate an analogous highly regiocontrolled hydroboration of terminal alkyl, aryl and heteroaryl acetylenes with HBPin, see: K. L. E. Hale, D. D. Roberts and M. G. McLaughlin, Eur. J. Org. Chem., 2025, e202401355 CrossRef CAS.
Footnotes |
| † Dedicated with admiration and respect to the memory of the great Professor Amos B. Smith III (2014 William H. Nichols Gold Medallist, 2015 RSC Perkin Prize Winner, 2009 RSC Simonsen Medallist, and 2002 RSC Centenary Prize Medallist) in recognition of his numerous magnificent achievements in complex natural product total synthesis, new synthetic methodology development, materials science, and his rational design of a totally new class of HIV-1-neutralising drugs. Sadly, Professor Smith passed away on the morning of Monday February 3rd, 2025, aged 80 years. His landmark contributions to the fields of organic synthesis and medicinal chemistry will continue to serve as a source of much future inspiration to us all. He will forever be missed by his many friends, former students, postdoctoral fellows, Associate Editors, and admirers within the world of organic chemistry. Professor Smith was a recipient of the Order of the Rising Sun of Japan. |
| ‡ Electronic supplementary information (ESI) available: experimental procedures for the synthesis of 15, copies of the NMR, IR, and HR mass spectra of all compounds, TLC photos, and additional experimental discussions. See DOI: https://doi.org/10.1039/d4ob01848f |
| § Current address: Halazar Pharma Ltd, Edgware, Middlesex, HA8 7RB, UK. |
| This journal is © The Royal Society of Chemistry 2025 |