
Enantioselective synthesis of α-(3-pyrrolyl)methanamines with an aza-tetrasubstituted center under metal-free conditions†
Atul Jankiram
Dolas
a,
Jyothi
Yadav
a,
Yadav Kacharu
Nagare
 a,
Krishnan
Rangan
b,
Eldhose
Iype
c and
Indresh
Kumar
*a
a,
Krishnan
Rangan
b,
Eldhose
Iype
c and
Indresh
Kumar
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani 333031, Rajasthan, India. E-mail: indresh.chemistry@gmail.com; indresh.kumar@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science, Hyderabad Campus, Hyderabad 500078, Telangana, India
cCollege of Engineering and Technology, American University of the Middle East, Egaila 54200, Kuwait
First published on 7th November 2024
Abstract
Construction of a chiral methanamine unit at the C3 position of pyrrole is highly desirable; nevertheless, it remains challenging due to its intrinsic electronic properties. Herein, we present an operationally straightforward and direct asymmetric approach for accessing α-(3-pyrrolyl)methanamines under benign organocatalytic conditions for the first time. The one-pot transformation proceeds smoothly through an amine-catalyzed direct Mannich reaction of succinaldehyde with various endo-cyclic imines, followed by a Paal–Knorr cyclization with a primary amine. Several N–H/alkyl/Ar α-(3-pyrrolyl)methanamines with an aza-tetrasubstituted center have been synthesized with good yields and excellent enantioselectivity.
Developing novel ways to insert suitably substituted pyrroles into a new structural framework is a continuous task that has received much interest in synthetic and medicinal chemistry.1 Furthermore, accessing enantiomerically rich pyrrole units can contribute to the growing demand for fresh drug candidates; thus, numerous approaches have been developed in this area.2 Specifically, enantioenriched (pyrrolyl)methanamines are significant constituents found in compounds of therapeutic importance.3 Depending on the pyrrole substitution position, these compounds can be classified as α-(2-pyrrolyl)methanamines and α-(3-pyrrolyl)methanamines (Scheme 1a). Following this, the asymmetric aza-Friedel–Crafts reaction of pyrrole with imines4 and nucleophilic addition to pyrrolyl-2-imine5 are simple ways to obtain these units; however, they primarily provide the corresponding chiral α-(2-pyrrolyl)methanamines. Meanwhile, using existing methods, obtaining similarly functionalized α-(3-pyrrolyl)methanamine units is difficult. Alternative strategies for developing chirality at the C3-position of pyrrole by blocking the more reactive site are effective,6 but none of these methods offers α-(3-pyrrolyl)methanamines asymmetrically. Johannsen reported the only instance of the metal-catalyzed asymmetric aza-Friedel–Crafts reaction of pre-functionalized 2-acylpyrrole with N-sulfonylimine with a particular example.7 Recently, Katukojvala and colleagues devised a non-conventional multicomponent reaction for the racemic synthesis of α-(3-pyrrolyl)benzylamines using diazoenals, arylamines, and aryl aldehydes under cooperative catalysis.8 Despite the significance of (3-pyrrolyl)methanamines as medicinally beneficial compounds (Scheme 1b),9 efforts to access this unit, especially in a direct asymmetric fashion, have not been made so far. Thus, due to the scarcity of conventional methods and other significant challenges associated with this unit, developing a new and direct method for substituting α-(3-pyrrolyl)methanamines asymmetrically will be an exciting task to study.
 | ||
| Scheme 1 (a) Background on (pyrrolyl)methanamines; (b) representative medicinally important compounds; (c) present work to access this unit. | ||
On the other hand, succinaldehyde is a simple carbonyl used in the universal Paal–Knorr reaction to access pyrrole,10 as well as in Robinson's protocol for tropinone synthesis,11 and has recently been utilized in many other transformations under amine catalysis.12 In this direction, we have been utilizing succinaldehyde to access pyrrole derivatives under metal-free conditions,13 while our recent development on the asymmetric access to C3-hydroxyalkylated pyrroles under chiral amine catalysis with a slight variation in the Paal–Knorr reaction has motivated us to explore more such transformations.14 Thus, during the investigation, we envisioned our idea to use reactive imines for the amine-catalyzed direct Mannich reaction in such a way that the resulting intermediate bicarbonylamine could be trapped further with a primary amine through the Paal–Knorr cyclization, eventually furnishing regiospecific access to α-(3-pyrrolyl)methanamine derivatives (Scheme 1c). The focal theme is based on harnessing the potential of the nucleophilic enamine intermediate, in situ generated from succinaldehyde under amine catalysis, with a suitable imine before the Paal–Knorr cyclization. Herein, we would like to report the realization of this idea for the asymmetric synthesis of α-(3-pyrrolyl)methanamines, which is the first direct access as per our knowledge.
Having experience in this direction, we quickly investigated the transformation utilizing succinaldehyde 2, 2-phenyl-3H-indol-3-one 3a and benzylamine 4a as model substrates under amine catalysis (Table 1). Here, we selected the more challenging ketamine indol-3-one,15 which has emerged as an appropriate electrophile for several enantioselective reactions,16 forming a pseudoindoxyl unit essential to many naturally occurring scaffolds.17 Pleasingly, the formation of product 5aa was observed (73%) with excellent enantioselectivity (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er) when step-I was carried out at 10 °C, followed by the Paal–Knorr cyclization with amine 4a at room temperature (entry 1, Table 1). The reaction outcome was inferior while using other conditions and catalysts 1b, 1c and 1d (entries 2–5, Table 1). Further lowering the reaction temperature did not improve the reaction outcome (entries 6, Table 1), and the best conditions were observed after several experiments with varying conditions (for details, see ESI Table S1†) applied to the remaining set of experiments.
:1 er) when step-I was carried out at 10 °C, followed by the Paal–Knorr cyclization with amine 4a at room temperature (entry 1, Table 1). The reaction outcome was inferior while using other conditions and catalysts 1b, 1c and 1d (entries 2–5, Table 1). Further lowering the reaction temperature did not improve the reaction outcome (entries 6, Table 1), and the best conditions were observed after several experiments with varying conditions (for details, see ESI Table S1†) applied to the remaining set of experiments.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions (step-I/step-II) | Yieldb (%) 5aa/6aa | erc for 5aa |
| a The reaction was carried out with 2 (3.0 M sol., 0.6 mmol), 3a (0.3 mmol), catalyst 1 (0.06 mmol), DMF (2.0 mL) at 10 °C, 7 h (step-I), then 4a (0.6 mmol), rt, 10 h (step-II). b Isolated yield of 5aa refers to 3a. c Determined using stationary chiral columns. d PhCO2H (20 mol%) was used as an additive. | |||
| 1 | None | 73/<10 | 99:1 |
| 2 | Catalyst 1a, rt, 6 h/— | 67/20 | 92:8 |
| 3 | Catalyst 1b, rt, 14 h/— | <10/50 | n.d. |
| 4 | Catalyst 1c, rt, 10 h/— | 42/30 | 88:12 |
| 5d | Catalyst 1d, rt, 10 h/— | 55/25 | 85:15 |
| 6 | 0 °C, 10 h/— | 62/<10 | 99:1 |
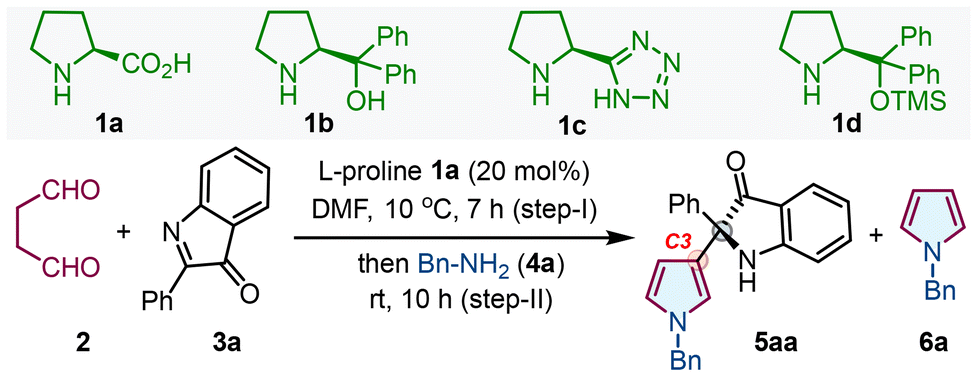
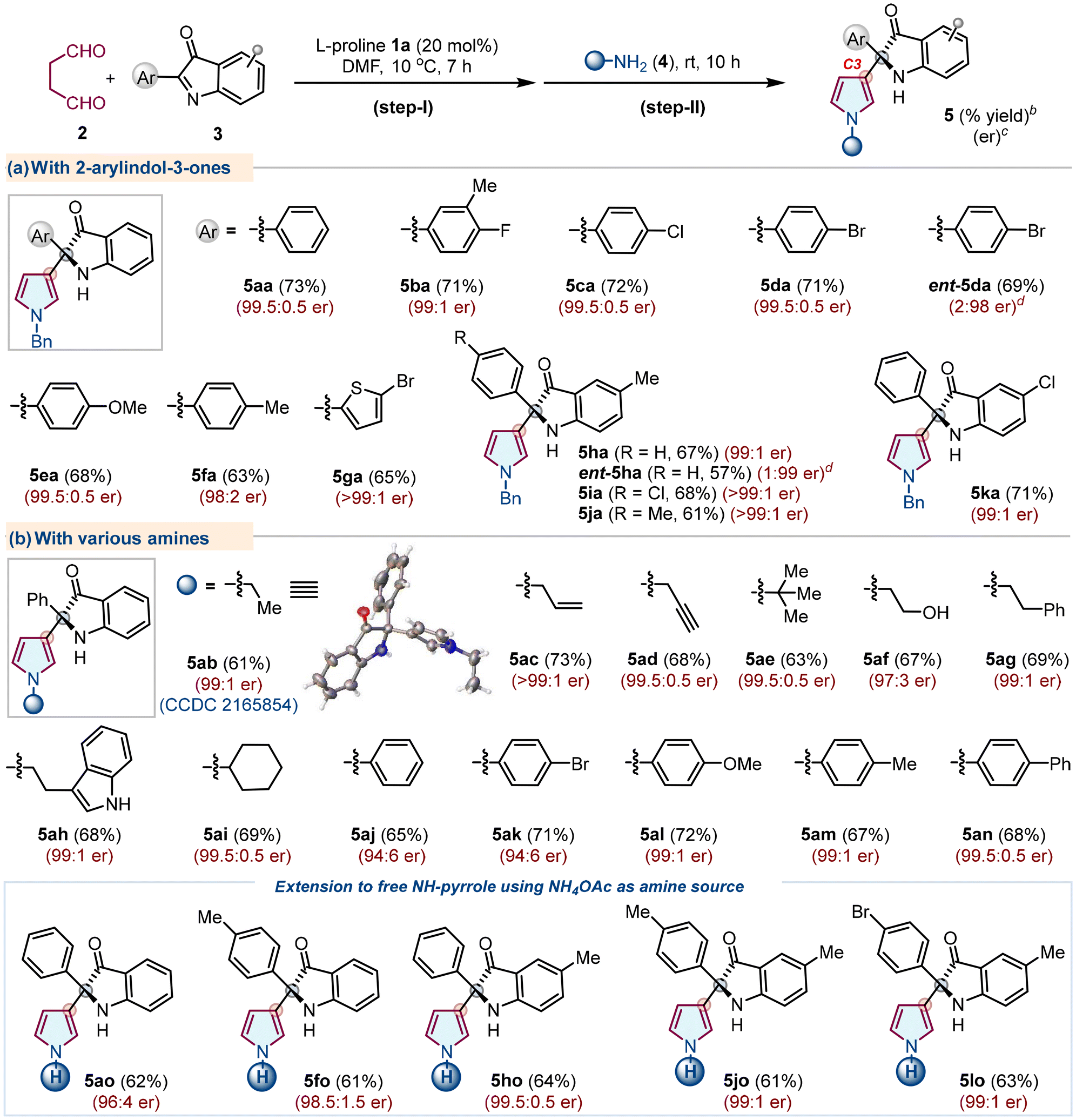
Under the optimized conditions, the reaction scope was checked with various 2-aryl-3H-indol-3-ones 3 with primary amine 4 (Table 2). Initially, a series of 2-aryl-3H-indol-3-ones 3a–3f having different substituents like –F, –Cl, –Br, –OCH3 and –CH3 on the aromatic ring at the C2-position and 2-heteroaryl-3H-indol-3-ones 3g were tested with succinaldehyde 2 and benzylamine 4a, furnishing products 5aa–5fa and 5ga with good yields and high enantioselectivity (up to >99:1 er). Besides, products 5ha–5ka were obtained with similar results when 3h–3k had different substituents on either of the aryl systems. Interestingly, opposite enantiomers ent-5da and ent-5ha were prepared with comparable outcomes using ent-1a under optimal conditions. Next, various aliphatic/aromatic primary amines 4b–4n also furnished the corresponding products 5ab–5an with good yields and excellent enantioselectivity. Excitingly, the more difficult free NH-pyrroles 5ao, 5fo, 5ho, 5jo, and 5lo were directly accessible utilizing NH4OAc as an amine source, demonstrating the method's uniqueness. The single crystal X-ray analysis of 5ab confirms the absolute configuration to be (R), and the stereochemistry of other products was assigned by analogy.18 The protocol failed to produce the corresponding products with hydrazines/hydroxylamines as amine sources (Fig. S2, see the ESI†).
| a The reaction was carried out using 2 (3.0 M sol., 0.6 mmol), 3 (0.3 mmol), L-proline 1a (0.06 mmol, 20 mol%), DMF (2.0 mL), 10 °C, 7 h (step-I); then 4 (0.6 mmol), rt, 10 h (step-II). b Isolated yield of 5 refers to 3 (≤10% of N–H/alkyl/aryl-pyrroles 6 were obtained in all the cases). c Determined using stationary chiral columns. d D-Proline (ent-1a) was used as a catalyst. |
|---|
|
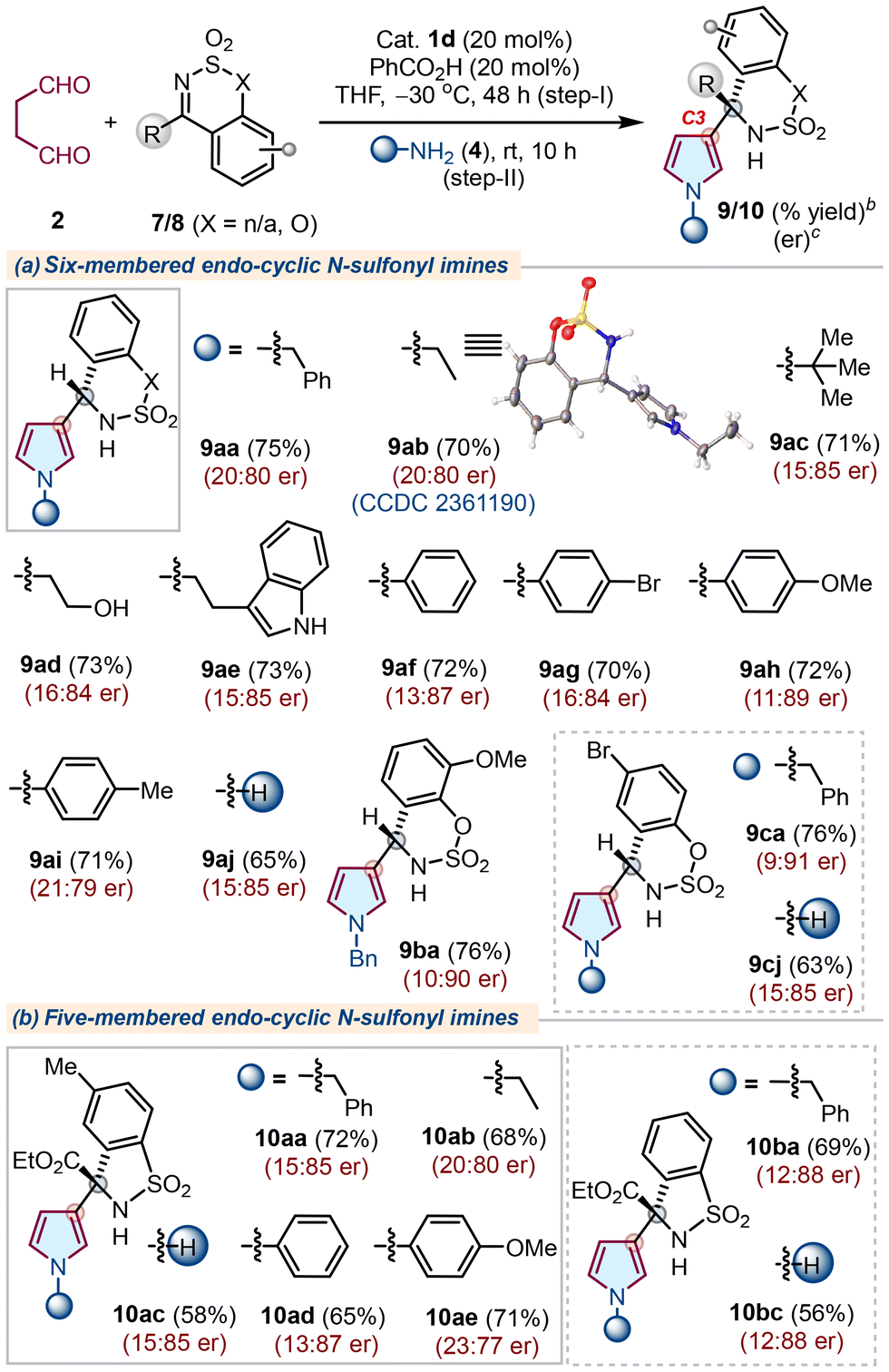
Next, we challenge ourselves to extend the scope of the developed protocol with more electrophilic cyclic imines, such as endo-cyclic N-sulfimines. Notably, cyclic N-sulfimines are suitable substrates for several asymmetric transformations, such as nucleophilic additions and annulation reactions.19 Moreover, the aza-Friedel–Crafts reaction of N-sulfimines with pyrrole to access chiral α-(2-pyrrolyl)methanamines has been developed;20 however, a similar strategy to access α-(3-pyrrolyl)methanamines remained elusive. In this direction, our initial efforts with cyclic N-sulfimine 7 did not provide the best results using earlier optimized conditions. Thus, the reaction conditions were again established to obtain improved results with catalyst 1d (Table S2, see the ESI†). Next, the reaction scope was tested with various N-sulfonyl imines 7/8 and primary amines 4 under optimized conditions (Table 3). Initially, cyclic N-sulfimine 7a furnished the corresponding products 9aa–9ai with overall good yields and enantioselectivity when treated with succinaldehdye 2 using catalyst 1d, followed by cyclization with several primary amines 4. Similarly, other substituted N-sulfimines 7b and 7c also yielded products 9ba and 9ca with comparable outcomes. Next, we evaluated five-membered N-sulfonyl α-ketiminoesters 8a and 8b for a similar set of reactions and the corresponding C3-substituted pyrroles 10aa–10bc were obtained directly with good yields and enantioselectivity (Table 3b), whereas a few N-sulfimines failed to produce the expected outcomes (Fig. S2, see the ESI†). Pleasingly, the corresponding –NH pyrroles 9aj, 9cj, 10ac, and 10bc were obtained with similar yields and enantioselectivities using NH4OAc as an amine source. The absolute configuration of 9ab was confirmed as (R) by X-ray analysis, and those of other products were assigned by analogy.18
| a The reaction was carried out using succinaldehyde 2 (3.0 M sol., 0.6 mmol), N-sulfonyl imine 7/8 (0.3 mmol), 1d (0.06 mmol), PhCO2H (0.06 mmol), THF (2.0 mL), −30 °C, 48 h (step-I); then 4 (0.6 mmol), rt, 10 h (step-II). b Isolated yield of 9/10 refers to 7/8 (≤10% of N–H/alkyl/aryl-pyrroles 6 were obtained in all the cases). c Determined using stationary chiral columns. |
|---|
|
Based on the reaction outcome and earlier reports, a reaction mechanism has been proposed for the developed protocol (Schemes S1 and S2, see the ESI†). Additionally, the synthetic utility of the method was shown in the gram-scale preparation of 5aa (Scheme 2a). The highly diastereoselective reduction of 5aa with LiBH4 was carried out under standard conditions to furnish 11aa (66%) as a single diastereomer, and stereochemistry (R,R) was confirmed by X-ray analysis (Scheme 2b).18 The rearranged product 12aa (76%) was obtained later when 11aa was exposed to an acid at 50 °C (Scheme 2c). Moreover, late-stage functionalization for the Vilsmeier–Haack reaction on 5ao was carried out under standard conditions to furnish the resulting regioselective C5-formylated pyrrole 13ao (72%) (Scheme 2d) without affecting the chirality.
 | ||
| Scheme 2 Gram-scale access to 5aa; synthetic transformations with 5aa and 5ao. | ||
In summary, we achieved a regiospecific synthesis of α-(3-pyrrolyl)methanamines asymmetrically with the aza-tetrasubstituted core in a one-pot manner under mild metal-free conditions. The suitability of endo-cyclic imines has been tested for the amine-catalyzed direct Mannich reaction with succinaldehyde before the Paal–Knorr cyclization with primary amines for the first time. A broad and readily available substrate scope, mild reaction conditions, regiospecific and highly enantioselective access to both enantiomers, gram-scale access, and late-stage synthetic applicability are the salient features of the developed protocol. Further investigation of more such asymmetric transformations is underway.
Date availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
AJD thanks BITS Pilani and DST-SERB, New Delhi, for the research fellowship. This work was financially supported by DST-SERB, New Delhi (CRG-2020/003424). The authors are also grateful for generous support from the DST-FIST (SR/FST/CSI-270/2015) for providing the Department of Chemistry at BITS-Pilani with the HRMS facility.References
- (a) A. Palmieri and M. Petrini, Org. Biomol. Chem., 2020, 18, 4533 RSC; (b) M. Leonardi, V. Estévez, M. Villacampa and J. C. Menéndez, Adv. Synth. Catal., 2019, 361, 2054 CrossRef CAS; (c) W. J. Olivier, J. A. Smith and A. C. M. Bissember, Org. Biomol. Chem., 2018, 16, 1216 RSC; (d) E. I. Martínez-Mora, M. A. Caracas, C. H. Escalante, C. Espinosa-Hicks, E. Quiroz-Florentino, F. Delgado and J. Tamariz, Synthesis, 2016, 1055 Search PubMed.
- (a) M. K. Hunjan, S. Panday, A. Gupta, J. Bhaumik, P. Das and J. K. Laha, Chem. Rec., 2021, 21, 715 CrossRef CAS PubMed; (b) S. C. Philkhana, F. O. Badmus, I. C. Dos Reis and R. Kartika, Synthesis, 2021, 1531 CrossRef CAS; (c) B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585 RSC; (d) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 2709 CrossRef CAS.
- (a) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801 RSC; (b) S. Thirumalairajan, B. M. Pearce and A. Thompson, Chem. Commun., 2010, 46, 1797 RSC.
- (a) G. Li, G. B. Rowland, E. B. Rowland and J. C. Antilla, Org. Lett., 2007, 9, 4065 CrossRef CAS PubMed; (b) K. Wu, M.-H. Zhuo, D. Sha, Y.-S. Fan, D. An, Y.-J. Jiang and S. Zhang, Chem. Commun., 2015, 51, 8054 RSC; (c) X. Shen, Y. Wang, T. Wu, Z. Mao and X. Lin, Chem. – Eur. J., 2015, 21, 9039 CrossRef CAS PubMed; (d) S. Nakamura, N. Matsuda and M. Ohara, Chem. – Eur. J., 2016, 22, 9478 CrossRef CAS; (e) M. Hatano, K. Toh and K. Ishihara, Org. Lett., 2020, 22, 9614 CrossRef CAS PubMed.
- G. Alvaro, R. Di Fabio, A. Gualandi and D. Savoia, Eur. J. Org. Chem., 2007, 5573 CrossRef CAS.
- (a) J. Majer, P. Kwiatkowski and J. Jurczak, Org. Lett., 2011, 13, 5944 CrossRef CAS PubMed; (b) Y. Lian and H. M. Davies, Org. Lett., 2012, 14, 1934 CrossRef CAS PubMed; (c) H. Oyama and M. Nakada, Tetrahedron: Asymmetry, 2015, 26, 195 CrossRef CAS; (d) Y. Zhang, N. Yang, X. Liu, J. Guo, X. Zhang, L. Lin, C. Hu and X. Feng, Chem. Commun., 2015, 51, 8432 RSC; (e) Q. Ma, L. Gong and E. Meggers, Org. Chem. Front., 2016, 3, 1319 RSC; (f) H. Q. Shen, C. Liu, J. Zhou and Y. G. Zhou, Org. Chem. Front., 2018, 5, 611 RSC.
- M. Johannsen, Chem. Commun., 1999, 21, 2233 RSC.
- S. G. Dawande, V. Kanchupalli, B. S. Lad, J. Rai and S. Katukojvala, Org. Lett., 2014, 16, 3700 CrossRef CAS PubMed.
- (a) F. Cerreto, A. Villa, A. Retico and M. Scalzo, Eur. J. Med. Chem., 1992, 27, 701 CrossRef CAS; (b) A. Thurkauf, J. Yuan, N. Chen, J. W. F. Wasley, R. Meade, K. H. Woodruff, K. Huston and P. C. Ross, J. Med. Chem., 1995, 38, 4950 CrossRef CAS PubMed; (c) M. Biava, G. C. Porretta, D. Deidda, R. Pompei, A. Tafi and F. Manetti, Bioorg. Med. Chem., 2004, 12, 1453 CrossRef CAS PubMed; (d) M. Biava, G. C. Porretta, G. Poce, S. Supino, D. Deidda, R. Pompei, P. Molicotti, F. Manetti and M. Botta, J. Med. Chem., 2006, 49, 4946 CrossRef CAS PubMed; (e) A. U. Wunnava, S. P. Kurati, K. E. Kumar and M. K. K. Muthyala, RSC Med. Chem., 2023, 14, 393 RSC.
- (a) C. Paal, Ber. Dtsch. Chem. Ges., 1984, 17, 2756 CrossRef; (b) L. Knorr, Ber. Dtsch. Chem. Ges., 1984, 17, 2863 CrossRef; (c) H. Cho, R. Madden, B. Nisanci and B. Török, Green Chem., 2015, 17, 1088 RSC.
- R. Robinson, J. Chem. Soc., 1917, 111, 762 RSC.
- I. Kumar, P. Ramaraju, N. A. Mir and A. Singh, Org. Biomol. Chem., 2015, 13, 1280 RSC.
- (a) A. P. Pawar, J. Yadav, N. A. Mir, E. Iype, K. Rangan, S. Anthal, R. Kant and I. Kumar, Chem. Commun., 2021, 57, 251 RSC; (b) S. Choudhary, J. Yadav, Mamta, A. P. Pawar, S. Vanaparthi, N. A. Mir, E. Iype, D. K. Sharma, R. Kant and I. Kumar, Org. Biomol. Chem., 2020, 18, 1155 RSC; (c) S. Choudhary, A. Singh, J. Yadav, N. A. Mir, S. Anthal, R. Kant and I. Kumar, New J. Chem., 2019, 43, 953 RSC.
- A. P. Pawar, J. Yadav, A. J. Dolas, Y. K. Nagare, E. Iype, K. Rangan and I. Kumar, Org. Lett., 2022, 24, 7549 CrossRef CAS PubMed.
- (a) A. Pareek, G. Singh and S. Yaragorla, Asian J. Org. Chem., 2022, 11, e202200395 CrossRef CAS; (b) R. Dalpozzo, Adv. Synth. Catal., 2017, 359, 1772 CrossRef CAS.
- (a) X.-X. Qiao, Y. He, T. Ma, C.-P. Zou, X.-X. Wu, G. Li and X.-J. Zhao, Chem. – Eur. J., 2023, e202203914 CrossRef CAS PubMed; (b) Q. Zhao, Y. Li, Q.-X. Zhang, J.-P. Cheng and X. Li, Angew. Chem., Int. Ed., 2021, 60, 17608 CrossRef CAS PubMed; (c) F. Xu and M. W. Smith, Chem. Sci., 2021, 12, 13756 RSC; (d) Q.-X. Zhang, Y. Li, J. Wang, C. Yang, C.-J. Liu, X. Li and J.-P. Cheng, Angew. Chem., Int. Ed., 2020, 59, 4550 CrossRef CAS; (e) J. Yadav, A. T. Dolas, E. Eldhose, R. Krishnan, J. Ohshita, D. Kumar and I. Kumar, J. Org. Chem., 2021, 86, 17213 CrossRef CAS PubMed; (f) X. Yuan, X. Wu, P. Zhang, F. Peng, C. Liu, H. Yang, C. Zhu and H. Fu, Org. Lett., 2019, 21, 2498 CrossRef CAS PubMed; (g) H. Zheng, X. Liu, C. Xu, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958 CrossRef CAS.
- For reviews, see. (a) P. S. Dhote, P. Patel, K. Vanka and C. Ramana, Org. Biomol. Chem., 2021, 19, 7970 RSC; (b) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC.
- The X-ray crystallographic structures for 5ab (CCDC 2165854), 9ab (2361190), and 11aa (2380259) have been deposited at the Cambridge Crystallographic Data Centre (CCDC) (for more details, see the ESI†).
- (a) Y.-H. Chen, D.-H. Li and Y.-K. Liu, ACS Omega, 2018, 3, 16615 CrossRef CAS; (b) A. Skrzyńska, S. Frankowski and Ł. Albrecht, Asian J. Org. Chem., 2020, 9, 1688 CrossRef; (c) W. Hao, L. Wang, J. Zhang, D. Teng and G. Cao, Beilstein J. Org. Chem., 2024, 20, 280 CrossRef CAS; (d) T. Nishimura, Y. Ebe and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 2092 CrossRef CAS PubMed; (e) K. N. Reddy, M. V. K. Rao, B. Sridhar and B. V. S. Reddy, Eur. J. Org. Chem., 2017, 4085 CrossRef; (f) Q. Li, X. Yuan, B. Li and B. Wang, Chem. Commun., 2020, 56, 1835 RSC; (g) D.-J. Cheng and Y.-D. Shao, ChemCatChem, 2019, 11, 2575 CrossRef CAS.
- S. Nakamura, Y. Sakurai, H. Nakashima, N. Shibata and T. Toru, Synlett, 2009, 1639 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2165854, 2361190 and 2380259. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01729c |
| This journal is © The Royal Society of Chemistry 2025 |