
Highly oxygenated steroids with immunosuppressive activity from Solanum undatum†
Shu-Shuai
Chen
ab,
Cheng-Yu
Zheng
a,
Guan-Zhou
Yang
a,
Jun-Su
Zhou
 a,
Shi-Jun
He
*c,
Yao-Yue
Fan
*ab and
Jian-Min
Yue
*ab
a,
Shi-Jun
He
*c,
Yao-Yue
Fan
*ab and
Jian-Min
Yue
*ab
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: jmyue@simm.ac.cn; yyfan@simm.ac.cn
bShandong Laboratory of Yantai Drug Discovery, Bohai Rim Advanced Research Institute for Drug Discovery, Yantai, Shandong 264117, China
cInnovation Research Institute of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, 1200 Cailun Road, Shanghai 201203, China. E-mail: heshijun@shutcm.edu.cn
First published on 5th November 2024
Abstract
Solanum undatum is a medicinal plant used for the treatment of oedema, rheumatoid arthritis and toothache, from which seven highly oxygenated steroids (1–7), including three new ones (1–3), have been characterized. Compound 1 is a new steroidal carboxylic acid featuring a cyclohexa-2,5-dien-1-one moiety and compounds 2 and 3 are new withanolide analogs with a 1,6-dimethyl-3,7-dioxabicyclo[4.1.0]heptan-2-ol terminus. Their structures and absolute configurations were determined by a combination of spectroscopic data, quantum chemical calculations, single-crystal X-ray diffraction, and the NMR-based phenylglycine methyl ester (PGME) method. An immunosuppressive activity assay revealed that compounds 2–7 exhibited substantial activities against the proliferation of T and B lymphocytes in vitro, with IC50 values ranging from 1.60 to 7.89 μM and 0.90 to 6.90 μM, respectively. Notably, compound 6 showed selective inhibitory effect toward B cells with the highest selective index (SI = 40.5). Preliminary structure–activity relationships of compounds 1–7 suggest that the terminal 1,6-dimethyl-3,7-dioxabicyclo[4.1.0]heptan-2-ol or 5,5-spiroacetal moiety is critical for immunosuppressive activity. Our study indicated that they could be promising lead compounds for immunosuppressive agents.
Introduction
Immunosuppressive therapies improve quality of life for people with a range of conditions, including autoimmune diseases, organ transplants, cancer and stem cell transplants. Despite the success and utility of immunosuppressants, there is still a need for new drug candidates with high efficacy and low side effects. Terrestrial plants are an attractive source of structurally novel immunosuppressive compounds.1–4 The genus Solanum (Solanaceae) is widespread across temperate to tropical regions,5 with many species being used in traditional medicine.6 Previous phytochemical and pharmacological studies on the genus Solanum have identified a number of natural products, including flavonoids, lignans, steroids, alkaloids and various glycosides, which exhibit diverse biological activities such as cytotoxic, antiviral, antimalarial, anti-inflammatory, antioxidant and antifungal activities.7–17Solanum undatum is an herb or a shrub often used in folk medicine to treat oedema, rheumatoid arthritis and toothache.18 Previous chemical investigations of S. undatum have led to the discovery of a variety of compounds such as steroids, flavonoids, lignans, amides, and various glycosides.18–20 In continuation of our search for steroids from medicinal plants with interesting structural and biological properties,21–23 the whole plant of S. undatum, collected from Hainan Province, China, was chemically investigated, from which three new steroids (1–3) and four known related congeners (4–7) were isolated (Fig. 1). Compound 1 is a steroid carboxylic acid with a cyclohexa-2,5-dien-1-one moiety. Compounds 2 and 3 are withanolide analogues featuring a unique 1,6-dimethyl-3,7-dioxabicyclo [4.1.0]heptan-2-ol terminus. The structures of the new compounds have been determined by analysis of spectroscopic data, ECD/NMR calculations, single crystal X-ray diffraction and the PGME method. Compounds 1–7 were tested for their immunosuppressive effects against the ConA-induced proliferation of T lymphocytes and the lipopolysaccharide (LPS)-induced proliferation of B lymphocytes. Compounds 2–7 exhibited moderate to high inhibitory activities against T cells with IC50 values ranging from 1.60 to 7.89 μM and against B cells with IC50 values ranging from 0.90 to 6.90 μM, respectively. It is noteworthy that compound 6 showed selective immunosuppressive activity against B cells with an IC50 value of 4.65 μM and the highest SI value of 40.5. The preliminary structure–activity relationship of this class of compounds was also investigated. Herein, we report the isolation, structural elucidation and immunosuppressive activities of these compounds.
 | ||
| Fig. 1 Structures of compounds 1–7. | ||
Results and discussion
Solaundaic acid A (1), obtained as a pale yellow oil, was assigned the molecular formula C27H36O5 with ten indices of hydrogen deficiency (IHDs) from the HRESIMS ion peak at m/z 439.2497 [M − H]− and its 13C NMR data. The IR absorptions at 2929, 1732, 1661, and 1603 cm−1 suggested the presence of carboxyl, carbonyl, and double bond groups. The 1H NMR data of 1 (Table 1) exhibited characteristic signals for three olefinic protons [δH 6.10 (br s), 6.27 (dd, J = 10.1, 1.9 Hz), and 7.04 (d, J = 10.1 Hz)] and four methyls [δH 0.85 (s), 1.04 (d, J = 5.9 Hz), 1.23 (d, J = 7.0 Hz), and 1.26 (s)]. The 27 carbon resonances in the 13C NMR (Table 1) spectrum were classified using DEPT and HSQC spectra into four methyls, seven methylenes, nine methines (three olefinic), and seven non-proton-bearing carbons (three carbonyls, one carboxyl, and one olefinic). The above functionalities accounted for six IHDs, and the remaining four IHDs suggested a tetracyclic structure for 1. The planar structure of 1 was deduced by 2D NMR analysis (Fig. 2A), starting with the identification of the four spin systems, highlighted by bold bonds, by 1H–1H COSY correlations. The connectivity of the four spin systems with the quaternary carbons was established by HMBC cross-peaks from H3-18 to C-12, C-13, C-14, and C-17; from H3-19 to C-1, C-5, C-9, and C-10; from H3-21 to C-17, C-20, and C-22 (δC 212.7); from H3-27 to C-24, C-25, and C-26 (δC 180.0); from H-1 to C-3 (δC 186.2) and C-5; from H-4 to C-2, C-6, and C-10; from H2-6 to C-5 and C-8; from H2-11 to C-8; from H2-15 to C-8 and C-16 (δC 217.3); from H-20 to C-16; and from H2-23 to C-22 to establish the 6/6/6/5-fused tetracyclic framework and the terminal 8-carbon acyclic segment (C-20–C-27). Compound 1 was therefore identified as a steroid carboxylic acid containing a cyclohexa-2,5-dien-1-one moiety (A-ring).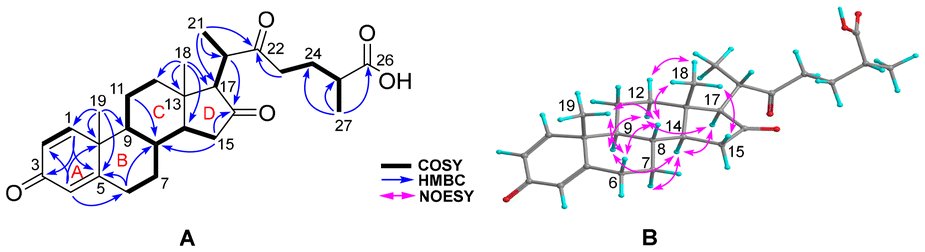 | ||
| Fig. 2 (A) Key COSY (bold bonds), HMBC, and (B) NOESY correlations of 1. | ||
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| No. | δ H (mult, J, Hz) | δ C | δ H (mult, J, Hz) | δ C | δ H (mult, J, Hz) | δ C |
| a Measured in CDCl3 at 600 MHz (1H) and 150 MHz (13C). b Measured in CDCl3 at 600 MHz (1H) and 125 MHz (13C). c Measured in CD3OD at 600 MHz (1H) and 125 MHz (13C). | ||||||
| 1 | 7.04, d (10.1) | 155.0 | 204.7 | 212.9 | ||
| 2 | 6.27, dd (10.1, 1.9) | 127.8 | 5.89, dd (10.0, 2.1) | 128.3 | 2.66, dd (20.0, 4.7) | 40.7 |
| 3.37, br d (20.0) | ||||||
| 3 | 186.2 | 6.80, ddd (10.0, 4.9, 2.5) | 145.6 | 5.63, ddd (9.8, 4.7, 2.7) | 122.8 | |
| 4 | 6.10, br s | 124.2 | 2.87, dd (21.1, 4.9) | 33.9 | 6.06, dd (9.8, 2.9) | 130.4 |
| 3.32, m | ||||||
| 5 | 168.0 | 136.0 | 142.6 | |||
| 6 | α 2.40, m; β 2.48, m | 32.4 | 5.64, br d (6.0) | 125.6 | 5.68, dd (5.8, 2.5) | 128.1 |
| 7 | α 1.16, m | 33.4 | α 1.95, overlap | 31.6 | α 1.66, overlap | 32.3 |
| β 1.89, overlap | β 2.30, overlap | β 2.18, m | ||||
| 8 | 1.79, overlap | 34.4 | 1.85, m | 32.4 | 1.60, overlap | 33.3 |
| 9 | 1.28, m | 51.6 | 1.77, m | 43.8 | 1.73, overlap | 42.3 |
| 10 | 43.3 | 51.0 | 53.4 | |||
| 11 | 1.80, overlap | 22.2 | α 2.33, overlap | 23.5 | 1.69, overlap | 37.8 |
| β 1.61, overlap | 2.02, m | |||||
| 12 | α 1.56, overlap | 38.2 | 1.65, overlap | 35.3 | 1.61, overlap | 33.4 |
| β 2.06, m | 1.78, overlap | |||||
| 13 | 41.9 | 47.8 | 49.5 | |||
| 14 | 1.61, overlap | 50.1 | 1.32, m | 65.9 | 1.75, overlap | 51.6 |
| 15 | α 2.22, dd (18.5, 7.7) | 37.1 | 4.59, d (8.8) | 78.7 | α 1.71, overlap | 24.6 |
| β 1.77, overlap | β 1.18, m | |||||
| 16 | 217.3 | 5.44, br s | 128.4 | 1.80, overlap | 23.4 | |
| 17 | 2.62, overlap | 66.1 | 158.3 | 86.7 | ||
| 18 | 0.85, s | 13.1 | 0.89, s | 19.6 | 0.81, s | 15.2 |
| 19 | 1.26, s | 18.7 | 1.29, s | 19.4 | 1.38, s | 20.9 |
| 20 | 2.61, overlap | 43.2 | 2.21, m | 36.6 | 1.95, m | 44.5 |
| 21 | 1.04, d (5.9) | 15.3 | 1.05, d (7.0) | 17.3 | 0.95, d (7.1) | 9.8 |
| 22 | 212.7 | 3.76, ddd (11.1, 6.8, 2.6) | 66.6 | 4.14, dt (11.4, 2.8) | 67.8 | |
| 23 | 2.68, m; 2.79, m | 39.8 | 1.66, overlap | 34.0 | 1.74, overlap | 34.1 |
| 1.92, overlap | 2.14, dd (14.7, 2.5) | |||||
| 24 | 1.90, overlap | 27.0 | 65.4 | 64.4 | ||
| 25 | 2.54, m | 38.3 | 64.2 | 64.0 | ||
| 26 | 180.0 | 5.01, s | 92.0 | 5.00, s | 93.0 | |
| 27 | 1.23, d (7.0) | 17.3 | 1.44, s | 16.9 | 1.36, s | 17.0 |
| 28 | 1.43, s | 19.3 | 1.36, s | 18.9 | ||
The partial relative configuration of 1 was determined on the basis of NOESY correlations (Fig. 2B). The NOESY interactions of H-8/H-6β, H3-18 and H3-19, and H3-18/H-12β and H-15β indicated a β-orientation. The NOESY cross-peaks of H-9/H-12α and H-14, H-14/H-7α and H-17, and H-17/H-12α revealed that they are cofacial and located in an α-orientation.
Before the identification of the relative configuration of C-20, we first determined the absolute configuration of C-25 in the side chain by means of the NMR-based PGME method.24 In detail, the carboxyl group of 1 was derivatized with (S)- and (R)-phenylglycine methyl esters to generate (S)- and (R)-PGME amides (1a and 1b), and the Δδ values (Δδ = δS − δR) obtained from the 1H NMR data of 1a and 1b suggested that the absolute configuration of C-25 was R (Fig. 3).24 The relative configuration of C-20 was then identified by GIAO25 NMR calculations and DP4+ analysis.26 Quantum chemical calculations on the 1D NMR data of two possible isomers (20R* and 20S*) were performed (Fig. S2†), and it was found that the 20S* isomer was the likely structure with a probability of 100% (Fig. S3†). To define the absolute configuration of 1, ECD calculations for 1 and ent-1 were performed and compared with the experimental ECD spectrum (Fig. 4), allowing the absolute configuration to be explicitly assigned as 8S, 9S, 10R, 13S, 14S, 17R, 20S, 25R.
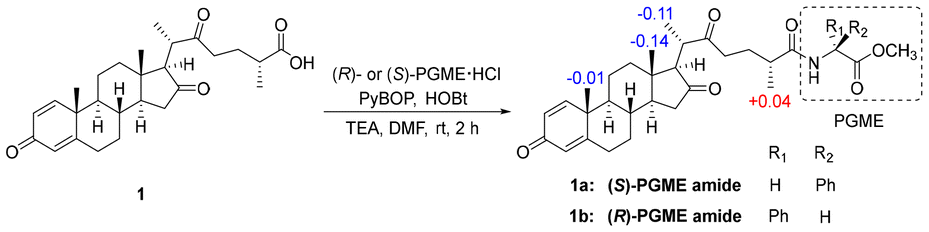 | ||
| Fig. 3 Δδ values [Δδ (in ppm) = δS − δR] obtained for the (S)- and (R)-PGME amides (1a and 1b) of Solaundaic acid A (1). | ||
 | ||
| Fig. 4 Experimental and calculated ECD spectra of 1. | ||
Solaundalide A (2) was deduced to have the molecular formula C28H38O5 based on the HRESIMS ion at m/z 472.3061 [M + NH4]+ and the 13C NMR data. An initial examination of the NMR data suggested that 2 was a structural analogue of 6,27 with distinct differences being the presence of an additional trisubstituted double bond and an extra oxygenated methine and the absence of a methoxy group in 2. The 1H–1H COSY correlations (Fig. 5A) of H-14/H-15 (δH 4.59)/H-16 (δH 5.44), combined with the HMBC correlations (Fig. 5A) from H-15 to C-8; from H-16 to C-13, C-17 (δC 158.3), and C-20, and from H3-18 and H3-21 to C-17, indicated a hydroxy group at C-15 and a Δ16 double bond in compound 2. The relatively upfield chemical shift of C-26 (92.0 ppm) compared with that of 6 (99.8 ppm)27 disclosed that the acetal of C-26 in 6 is demethylated to the hemiacetal in 2, which was further verified by the molecular formula. The above conclusions revealed compound 2 to be a withanolide-type steroid.
 | ||
| Fig. 5 (A) Key COSY (bold bonds), HMBC, and (B) NOESY correlations of 2. | ||
The NOESY correlations (Fig. 5B) of H-8/H-15, H3-18 and H3-19, H-15/H-7β and H3-18, H3-18/H-11β, and H3-19/H-11β revealed that they were cofacial and arbitrarily assigned a β-orientation. In contrast, the NOESY cross-peaks of H-9/H-7α, and H-14, and H-14/H-7α suggested an α-orientation. The relative configurations of the chiral carbons C-20, C-22, C-24, C-25 and C-26 were deduced to be the same as those of 6 on the basis of the comparable NMR data and biogenetic consideration. Meanwhile, the single-crystal X-ray diffraction analysis of the congener 6 [Flack parameter: 0.07 (4)] (Fig. S1†) further confirmed the above deduction. Finally, the absolute configurations of 2 was assigned as 8R, 9S, 10R, 13S, 14S, 15S, 20S, 22R, 24S, 25S, 26R by comparing the computational and experimental ECD spectra (Fig. 6). The structure of solaundalide A (2) was therefore unambiguously established.
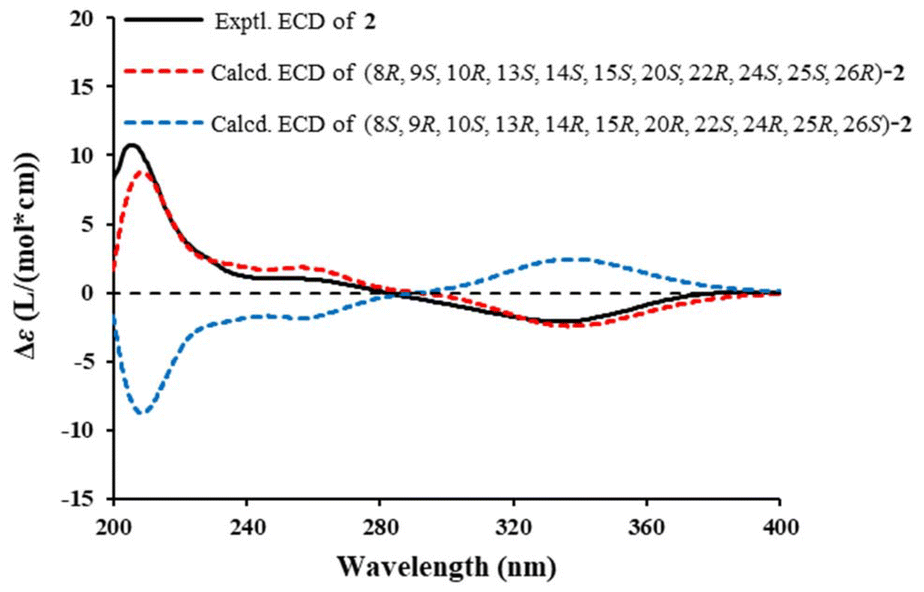 | ||
| Fig. 6 Experimental and calculated ECD spectra of 2. | ||
Solaundaolide B (3), a pale yellow powder, shared the molecular formula C28H40O5 with 5,27 according to the HRESIMS ion at m/z 479.2768 [M + Na]+ and the 13C NMR data. The 1H and 13C NMR data of 3 (Table 1) showed close similarities to those of 5, and the only difference was attributed to the position of the double bond in ring A. Conclusive HMBC correlations (Fig. 7A) from H-3 (δH 5.63) to C-1 (δC 212.9) and C-5 (δC 142.6) and from H-4 (δH 6.06) to C-2 (δC 40.7), C-6 (δC 128.1), and C-10 revealed a disubstituted double bond at C-3 and C-4 in 3. The NOESY correlations, as shown in Fig. 7B, in combination with the similar NMR data and coupling constants of 3 and 5, indicated that 3 had identical relative configuration to 5. By comparing the experimental and calculated ECD spectra of 3, its absolute configuration was determined to be 8S, 9S, 10R, 13S, 14S, 17S, 20R, 22R, 24S, 25S, 26R (Fig. 8).
 | ||
| Fig. 7 (A) Key COSY (bold bonds), HMBC, and (B) NOESY correlations of 3. | ||
 | ||
| Fig. 8 Experimental and calculated ECD spectra of 3. | ||
Four known compounds, 1-dehydronuatigenone (4),28 cilistol a (5),27 cilistol d (6),27 and cilistepoxide (7),29 were identified by comparison of the NMR data with the reported spectroscopic data.
The immunosuppressive activities of compounds 1–7 on murine lymphocyte proliferation stimulated by ConA or LPS were tested in vitro. Compounds 2–5 and 7 exhibited substantial inhibition against ConA-induced T-cell proliferation with IC50 values ranging from 1.60 to 7.89 μM and against LPS-induced B-cell proliferation with IC50 values ranging from 0.90 to 6.90 μM, respectively (Table 2 and Fig. S49–S51†). In particular, compound 6 was a selective inhibitor of LPS-induced B cell proliferation with an IC50 value of 4.65 and the highest selectivity index (SI = 40.5). A general structure–activity relationship for this class of compounds can be described as follows. (1) The terminal 1,6-dimethyl-3,7-dioxabicyclo[4.1.0]heptan-2-ol or 5,5-spiroacetal moiety is critical for immunosuppressive activity, as only compound 1 showed no activity. (2) Structure–activity analysis of 5 and 6 showed that methylation of the 26-OH group could reduce the cytotoxicity and increase the selectivity of the inhibition against B-cell proliferation. (3) The results from 5 and 7 indicated that the presence of a 5,6-epoxy segment in 7 could enhance the immunosuppressive effects.
| CC50 (μM) | ConA-induced T-cell proliferation | LPS-induced B-cell proliferation | |||
|---|---|---|---|---|---|
| Compd | IC50 (μM) | SIa | IC50 (μM) | SIa | |
| a The selectivity index (SI) is determined as the ratio of the concentration of the compound that reduced cell viability to 50% (CC50) to the concentration of the compound needed to inhibit the proliferation by 50% relative to the control value (IC50). b Cyclosporin A (CsA) was used as the positive control. | |||||
| 1 | 157.90 | 79.02 | 2.0 | 58.51 | 2.7 |
| 2 | 12.60 | 7.89 | 1.6 | 1.10 | 11.5 |
| 3 | 29.30 | 4.49 | 6.5 | 6.90 | 4.2 |
| 4 | 27.28 | 1.60 | 17.1 | 3.79 | 7.2 |
| 5 | 9.98 | 5.35 | 1.9 | 1.97 | 5.1 |
| 6 | 188.40 | 49.76 | 3.8 | 4.65 | 40.5 |
| 7 | 4.10 | 2.39 | 1.7 | 0.90 | 4.6 |
| CsA | 4.69 | 0.02 | 213.3 | 0.18 | 24.6 |
Conclusions
In summary, three previously uncharacterized compounds (1–3), including one steroidal carboxylic acid and two withanolides, and four related known compounds (4–7) were isolated from S. undatum. Compounds 2–7 exhibited potent immunosuppressive activities. Structure–activity relationships for this class of compounds showed that the cyclization of the terminal side chain is key to the immunosuppressive activity. These findings provide new chemical templates for developing new immunosuppressive drugs to treat autoimmune diseases.Experimental
General experimental procedures
Optical rotations (Na lamp, 589 nm) were measured on an Autopol VI polarimeter at room temperature; concentrations are reported in g per 100 mL. UV and ECD spectra were obtained on a JASCO J-810 spectrometer using a 0.1 cm path length sample cell. IR spectra were obtained on a Thermo Scientific Nicolet iS5 FT-IR Spectrometer with KBr panels. 1D and 2D NMR spectra were obtained on a Bruker AM-500 or 600 MHz NMR spectrometer with TMS as the internal standard. ESIMS and HRESIMS were measured on a Thermo Fisher Finnigan LTQ mass spectrometer and an Agilent G6520 Q-TOF mass spectrometer, respectively. X-ray crystallography was performed on a Bruker D8 Venture diffractometer equipped with graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Semi-preparative HPLC purification was performed on a Waters 2695 binary pump system equipped with a Waters 2489 detector using a YMC-Triart ODS-A column (250 × 10 mm, S-5 μm, 12 nm). Silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd), CHP20P MCI gel (75–150 μm, Mitsubishi Chemical Corporation), D101-macroporous absorption resin (Shanghai Hualing Resin Co., Ltd), and Sephadex LH-20 gel (Amersham Biosciences) were used for column chromatography (CC). Precoated silica gel GF254 plates (Qingdao Haiyang Chemical Co., Ltd) were used for TLC detection. All solvents used for CC were of analytical grade (Shanghai Chemical Reagents Co., Ltd), and solvents used for HPLC were of HPLC grade (J & K Scientific Ltd).Plant material
Whole plants of Solanum undatum were collected from Haikou, Hainan Province, People's Republic of China, in July 2014 and identified by Prof. Shi-Man Huang at Hainan University. A voucher specimen has been deposited at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (accession number: Chlosp-2017-1Y).Extraction and isolation
The dried whole plants of Solanum undatum (10.0 kg) were percolated with 95% EtOH (3 × 25 L, rt) to yield a crude extract (740 g). The crude extract was dissolved in water and then extracted with EtOAc to afford an EtOAc-soluble extract (230 g), which was then subjected to CC (D101-macroporous absorption resin) and eluted with 30%, 50%, 80% and 95% EtOH/H2O to yield four fractions A–D. Fraction C (105 g) was partitioned using an MCI gel column and eluted with a stepped gradient of MeOH/H2O (30%–100%) to give five fractions, C1–C5. Fraction C2 (3.2 g) was separated by silica gel CC with CH2Cl2/MeOH (300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1) as the eluent to give six subfractions (C2a–C2f). Fraction C2d (542.4 mg) was subjected to Sephadex LH-20 column elution with MeOH to obtain four subfractions (C2d1–C2d4). Fraction C2d2 (221.1 mg) was further fractionated via Sephadex LH-20 column elution with CH2Cl2/MeOH (1:1) to afford three subfractions (C2d2a–C2d2c). Fraction C2d2a (18.6 mg) was further separated by semipreparative HPLC using a YMC-Triart ODS-A column (45% MeCN in H2O, 3 mL min−1) to yield compound 1 (2.6 mg, tR = 18.0 min). Fraction C3 (14.1 g) was subjected to silica gel column chromatography and eluted with CH2Cl2/MeOH (300:1 to 10:1) to give six subfractions (C3a–C3f). Fraction C3c (1.7 g) was subjected to separation over a Sephadex LH-20 column (EtOH) to afford five subfractions (C3c1–C3c5). Fraction C3c2 (255.3 mg) was subjected to silica gel CC with petroleum CH2Cl2/MeOH (300:1 to 10:1) as the eluent to obtain three subfractions (C3c2a–C3c2c). Fraction C3c2b (64.8 mg) was further purified by semi-preparative HPLC (67% MeCN in H2O, 3 mL min−1) to provide compounds 5 (22.6 mg, tR = 14.0 min) and 6 (2.8 mg, tR = 30.0 min). Fraction C3c4 (133 mg) was separated using a Sephadex LH-20 column (CH2Cl2/MeOH, 1:1) and further purified by semi-preparative HPLC (45% MeCN in H2O, 3 mL min−1) to obtain compounds 2 (3.3 mg, tR = 20.0 min) and 7 (2.6 mg, tR = 36.0 min). Fraction C4 (12.5 g) was chromatographed on a silica gel column and eluted with CH2Cl2/MeOH (300:1 to 10:1) to provide five subfractions (C4a–C4e). Fraction C4b (360 mg) was subjected to passage over a Sephadex LH-20 column (MeOH) to give fractions C4b1–C4b4. Fraction C4b3 (65.4 mg) was further purified using semi-preparative HPLC (65% MeCN in H2O, 3 mL min−1) to obtain compounds 3 (11.5 mg, tR = 17.0 min) and 4 (7.3 mg, tR = 21.0 min).
:1 to 10:1) as the eluent to give six subfractions (C2a–C2f). Fraction C2d (542.4 mg) was subjected to Sephadex LH-20 column elution with MeOH to obtain four subfractions (C2d1–C2d4). Fraction C2d2 (221.1 mg) was further fractionated via Sephadex LH-20 column elution with CH2Cl2/MeOH (1:1) to afford three subfractions (C2d2a–C2d2c). Fraction C2d2a (18.6 mg) was further separated by semipreparative HPLC using a YMC-Triart ODS-A column (45% MeCN in H2O, 3 mL min−1) to yield compound 1 (2.6 mg, tR = 18.0 min). Fraction C3 (14.1 g) was subjected to silica gel column chromatography and eluted with CH2Cl2/MeOH (300:1 to 10:1) to give six subfractions (C3a–C3f). Fraction C3c (1.7 g) was subjected to separation over a Sephadex LH-20 column (EtOH) to afford five subfractions (C3c1–C3c5). Fraction C3c2 (255.3 mg) was subjected to silica gel CC with petroleum CH2Cl2/MeOH (300:1 to 10:1) as the eluent to obtain three subfractions (C3c2a–C3c2c). Fraction C3c2b (64.8 mg) was further purified by semi-preparative HPLC (67% MeCN in H2O, 3 mL min−1) to provide compounds 5 (22.6 mg, tR = 14.0 min) and 6 (2.8 mg, tR = 30.0 min). Fraction C3c4 (133 mg) was separated using a Sephadex LH-20 column (CH2Cl2/MeOH, 1:1) and further purified by semi-preparative HPLC (45% MeCN in H2O, 3 mL min−1) to obtain compounds 2 (3.3 mg, tR = 20.0 min) and 7 (2.6 mg, tR = 36.0 min). Fraction C4 (12.5 g) was chromatographed on a silica gel column and eluted with CH2Cl2/MeOH (300:1 to 10:1) to provide five subfractions (C4a–C4e). Fraction C4b (360 mg) was subjected to passage over a Sephadex LH-20 column (MeOH) to give fractions C4b1–C4b4. Fraction C4b3 (65.4 mg) was further purified using semi-preparative HPLC (65% MeCN in H2O, 3 mL min−1) to obtain compounds 3 (11.5 mg, tR = 17.0 min) and 4 (7.3 mg, tR = 21.0 min).
Solaundaic acid A (1)
Pale yellow oil; [α]20D = −70.8 (c 0.11, MeOH); UV (MeOH) λmax (logε) 195 (2.77), 243 (2.95) nm; ECD (MeOH) λ (Δε) 200 (−4.89), 236 (+1.81), 265 (−2.97), 279 (−2.82), 292 (−3.20), 330 (−0.28) nm; IR (KBr) νmax 2929, 2853, 1732, 1661, 1603, 1457, 887, 739 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 439.2497 [M − H]− (calcd for C27H35O5, 439.2490).
Solaundalide A (2)
Yellow solid; [α]20D = +20.8 (c 0.13, MeOH); UV (MeOH) λmax (logε) 199 (2.99), 228 (2.69) nm; ECD (MeOH) λ (Δε) 193 (−1.17), 204 (+10.93), 243 (+1.07), 252 (+1.08), 334 (−2.09), 370 (−0.35) nm; IR (KBr) νmax 3416, 2967, 2923, 2851, 1748, 1683, 1665, 1453, 1260, 1090, 1036 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 472.3061 [M + NH4]+ (calcd for C28H42NO5, 472.3057).
Solaundaolide B (3)
Pale yellow powder; [α]20D = −19.2 (c 0.13, MeOH); UV (MeOH) λmax (logε) 197 (2.68), 231 (2.70) nm; ECD (MeOH) λ (Δε) 206 (−4.07), 242 (+1.14), 305 (−1.09), 346 (−0.18) nm; IR (KBr) νmax 3459, 2929, 1715, 1458, 1266, 1090, 1034, 961, 737 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 479.2768 [M + Na]+ (calcd for C28H40NaO5, 479.2768).
X-ray crystallographic analysis
Colorless crystals of compound 6 were obtained by recrystallisation from MeOH at room temperature. X-ray data were collected on a Bruker D8 Venture diffractometer with Cu Kα radiation (λ = 1.54178 Å). Using Olex2, the structures were solved with the ShelXT structure solution program using intrinsic phasing methods and refined with the ShelXL refinement package using least-squares minimization.30–32 The crystallographic data for compound 6 (CCDC 2255800†) in standard CIF format have been deposited at the Cambridge Crystallographic Data Centre.Crystal data for compound 6
C29H42O5, M = 470.62 g mol−1, triclinic, size 0.15 × 0.08 × 0.05 mm3, a = 9.6163(3) Å, b = 7.9028(2) Å, c = 17.3878(5) Å, α = 90°, β = 103.8880(10)°, γ = 90°, V = 1282.77(6) Å3, T = 170.0 K, space group P21, Z = 2, μ(Cu Kα) = 0.648 mm−1, F (000) = 512.0. A total of 16663 reflections were measured in the range 5.236° ≤ 2θ ≤ 148.974°, containing 5052 independent reflections (Rint = 0.0400, Rsigma = 0.0360). The final R1 value was 0.0309 (I ≥ 2σ(I)) and ωR2 value was 0.0822 (all data). The goodness of fit on F2 was 1.053. Flack parameter = 0.07(4). The crystallographic data in the standard CIF format have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2255800†).
PGME derivatization of 1
To a solution of 1 (0.5 mg, 1.22 μmol) in DMF (10 μL) were added (R)-PGME·HCl (0.6 mg, 2.99 μmol), PyBOP (1.2 mg), HOBt (0.5 mg), and TEA (200 μL) at room temperature. The reaction mixture was stirred for 2 h and then concentrated in vacuo. The dried residue was directly purified by HPLC (YMC-Triart ODS-A column, 30% MeCN to 100% MeCN) to afford the (R)-PGME amide 1a (0.3 mg). The same procedure was applied to prepare the (S)-PGME amide 1b (0.3 mg from 0.5 mg of 1) using (S)-PGME·HCl.Conformational search, geometry optimization, and frequency calculations
Conformational searches were conducted using the Monte Carlo multiple minimum (MCMM) method33 under OPLS3 force field using the Macromodel 10.2 program (Schrödinger Release 2015-2: MacroModel, Schrödinger, LLC, New York, NY, USA),34 with the energy window set as 12.6 kJ mol−1. Conformations with favourable relative potential energies and observed frequencies were selected as candidate conformers. All the candidate conformers were subjected to geometry optimization at the B3LYP/6-31G (d) level of theory in corresponding solvents with the IEFPCM model, followed by frequency calculations to compute the Gibbs free energies and ensure that all geometries were at local minima. All quantum chemical calculations were executed using the Gaussian 16 program package.35NMR and DP4+ calculations
Geometry optimization and frequency calculation results for the two possible isomers of compound 1, isomer-1-1, and isomer-1-2, are shown in Tables S3 and S4,† while the corresponding Cartesian coordinates of the optimized conformers can be found in Tables S5 and S6.† NMR shielding tensors were computed at the PCM/mPW1PW91/6-31 + G(d,p) level in CHCl3, with the GIAO25 (gauge-independent atomic orbital) method. The shielding tensors were subjected to Boltzmann averaging over conformers to calculate the NMR shifts. After Boltzmann weighting conversion of the NMR shielding tensors, the unscaled chemical shifts were computed using TMS as the reference standard. The unscaled chemical shifts were subjected to DP4+ calculations (Fig. S2 and S3†).26ECD calculations
The conformational search and geometry optimization results of compounds 1–3 are shown in Tables S7–S9,† while the corresponding Cartesian coordinates of optimized conformers are included in Tables S10–S12.† The theoretically calculated ECD spectra of compounds 1–3 were established using the time-dependent density functional theory (TDDFT) method at the ωB97XD/6-311G(d,p) level of theory in MeOH. The Boltzmann-averaged ECD spectra were simulated by the overlapping Gaussian function with the aid of SpecDis 1.71.36–38 The σ and UV correction values applied for the final calculated ECD spectra are (0.30 eV and 16 nm), (0.30 eV and 15 nm), and (0.25 eV and 2 nm) for compounds 1–3, which are within a reasonable range.Immunosuppressive assay
Compounds 1–7 were evaluated for their immunosuppressive activities according to previously described methods.3,38Author contributions
S.-S. Chen, S.-J. He, Y.-Y. Fan, and J.-M. Yue conceived the project and designed the experiments. S.-S. Chen, C.-Y. Zheng, G.-Z. Yang, and J.-S. Zhou performed compound isolation and structure identification. S.-J. He contributed to the bioactive evaluation. S.-S. Chen, S.-J. He, Y.-Y. Fan, and J.-M. Yue supervised the project and provided comments on the manuscript. S.-S. Chen and Y.-Y. Fan wrote the manuscript. All authors discussed the results and commented on the manuscript.Data availability
The authors declare that the data supporting the findings of this study are available within the main text and its ESI.†The crystallographic data of compound 6 have been deposited at the Cambridge Crystallographic Data Centre under accession codes CCDC 2255800.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2023YFE0206100) and the National Natural Science Foundation (22237007 and 82122062) of the People's Republic of China. We also thank Prof. S.-M. Huang of Hainan University for the identification of the plant material.References
- Y. Y. Fan, H. Zhang, Y. Zhou, H. B. Liu, W. Tang, B. Zhou, J. P. Zuo and J. M. Yue, J. Am. Chem. Soc., 2015, 137, 138–141 CrossRef CAS.
- X. R. Li, B. C. Yan, K. Hu, S. J. He, H. D. Sun, J. P. Zuo and P. T. Puno, Org. Lett., 2021, 23, 5647–5651 CrossRef CAS.
- Y. Ren, X. Tong, Y. Zhao, S. J. He, Y. Y. Fan and J. M. Yue, J. Nat. Prod., 2022, 85, 1581–1590 CrossRef CAS PubMed.
- X. C. Shao, Z. H. Chen, S. S. Liu, F. Wu, H. Y. Mu, W. H. Wei, Y. Feng, J. P. Zuo, J. Q. Zhang, S. J. He and W. M. Zhao, Bioorg. Chem., 2021, 108, 104641 CrossRef CAS.
- Z. Wu and P. H. Raven, Flora Reipublicae Popularis Sinicae, Science Press, Beijing, 1994, 44, 131 Search PubMed.
- Jiangsu New Medical College, Dictionary of Chinese Material Medica, Shanghai Scientific and Technological Publishers, Shanghai, 1998, pp. 327 Search PubMed.
- D. Arthan, J. Svasti, P. Kittakoop, D. Pittayakhachonwut, M. Tanticharoen and Y. Thebtaranonth, Phytochemistry, 2002, 59, 459–463 CrossRef CAS PubMed.
- A. K. Chakravarty, S. Mukhopadhyay, S. Saha and S. C. Pakrashi, Phytochemistry, 1996, 41, 935–939 CrossRef CAS.
- X. Y. Gu, X. F. Shen, L. Wang, Z. W. Wu, F. Li, B. Chen, G. L. Zhang and M. K. Wang, Phytochemistry, 2018, 147, 125–131 CrossRef CAS PubMed.
- Y. Y. Lu, J. G. Luo, X. F. Huang and L. Y. Kong, Steroids, 2009, 74, 95–101 CrossRef CAS.
- Y. Y. Lu, J. G. Luo and L. Y. Kong, Phytochemistry, 2011, 72, 668–673 CrossRef CAS.
- J. Saez, W. Cardona, D. Espinal, S. Blair, J. Mesa, M. Bocar and A. Jossang, Tetrahedron, 1998, 54, 10771–10778 CrossRef CAS.
- O. M. Singh, K. Subharani, N. I. Singh, N. B. Devi and L. Nevidita, Nat. Prod. Res., 2007, 21, 585–590 CrossRef CAS PubMed.
- S. Sudheesh, C. Sandhya, A. S. Koshy and N. R. Vijayalakshmi, Phytother. Res., 1999, 13, 393–396 CrossRef CAS.
- W. J. Syu, M. J. Don, G. H. Lee and C. M. Sun, J. Nat. Prod., 2001, 64, 1232–1233 CrossRef CAS PubMed.
- A. Zamilpa, J. Tortoriello, V. Navarro, G. Delgado and L. Alvarez, J. Nat. Prod., 2002, 65, 1815–1819 CrossRef CAS.
- X. L. Zhou, X. J. He, G. H. Wang, H. Gao, G. X. Zhou, W. C. Ye and X. S. Yao, J. Nat. Prod., 2006, 69, 1158–1163 CrossRef CAS.
- X. J. Qin, P. K. Lunga, Y. L. Zhao, Y. P. Liu and X. D. Luo, Chin. J. Nat. Med., 2016, 14, 308–312 CAS.
- Q. D. Li, H. F. Jiang, L. Chen, F. L. Zhang and B. J. Shi, J. Chin. Med. Mater., 2015, 38, 1206–1208 CAS.
- W. N. Zhang, China J. Chin. Mater. Med., 2015, 40, 264–268 Search PubMed.
- P. P. An, Y. S. Cui, Q. Y. Shi, Y. H. Ren, P. Q. Wu, Q. F. Liu, H. C. Liu, B. Zhou and J. M. Yue, Tetrahedron Lett., 2022, 93, 153691 CrossRef CAS.
- Y. Z. Ge, B. Zhou, R. X. Xiao, X. J. Yuan, H. Zhou, Y. C. Xu, M. A. Wainberg, Y. S. Han and J. M. Yue, Sci. China: Chem., 2018, 61, 1430–1439 CrossRef CAS.
- Y. H. Ren, Q. F. Liu, L. Chen, S. J. He, J. P. Zuo, Y. Y. Fan and J. M. Yue, Org. Lett., 2019, 21, 1904–1907 CrossRef CAS PubMed.
- T. Yabuuchi and T. Kusumi, J. Org. Chem., 2000, 65, 397–404 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS PubMed.
- X. H. Zhu, M. Takagi, S. Ikeda, K. Midzuki and T. Nohara, Phytochemistry, 2001, 56, 741–745 CrossRef CAS PubMed.
- F. Q. Wang, C. G. Zhang, B. Li, D. Z. Wei and W. Y. Tong, Environ. Sci. Technol., 2009, 43, 5967–5974 CrossRef CAS PubMed.
- R. Niero, I. T. Da Silva, G. C. Tonial, B. D. S. Camacho, E. Gacs-Baitz, G. Delle Monache and F. Delle Monache, Nat. Prod. Res., 2006, 20, 1164–1168 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef.
- G. Chang, W. C. Guida and W. C. Still, J. Am. Chem. Soc., 1989, 111, 4379–4386 CrossRef CAS.
- E. Harder, W. Damm, J. Maple, C. J. Wu, M. Reboul, J. Y. Xiang, L. L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Jr. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Pescitelli, SpecDis 1.71, Berlin, Germany, 2017 Search PubMed.
- S. S. Chen, X. Tong, X. Y. Liu, C. C. Zheng, J. S. Zhou, Y. Y. Fan, S. S. He, B. Zhou and J. M. Yue, J. Org. Chem., 2023, 88, 455–461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2255800. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01642d |
| This journal is © The Royal Society of Chemistry 2025 |