
Paired electrocatalysis enabled oxidative coupling of styrenes with alkyl radicals†
Dong
Li‡
ad,
Ling
Zhang‡
a,
Daixi
Li‡
bc,
Peng
Yu
*d and
Tao
Shen
*a
aFrontiers Science Center for Transformative Molecules (FSCTM), Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, ZhangJiang Institute for Advanced Study, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: taoshen@sjtu.edu.cn
bDepartment of Respiratory and Critical Care Medicine, Zhongshan Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China
cThe School of Clinical Medicine, Fujian Medical University, Fuzhou, China
dEastern Institute for Advanced Study, Eastern Institute of Technology, Ningbo, 315200, P. R. China. E-mail: pyu@eitech.edu.cn
First published on 30th October 2024
Abstract
A paired electrocatalysis strategy for intermolecular oxidative cross-dehydrocoupling between styrenes and ethers or p-methylphenol derivatives using ketone as a mild oxidant is described. This approach enables the generation of Csp3 carbon-centered radicals through anodic oxidation, followed by reductive coupling of ketones at the cathode, ultimately yielding valuable oxidative alkylation products.
Direct functionalization of alkenes, an inexpensive and readily available feedstock, represents one of the most appealing transformations in organic chemistry.1 Among these, the direct oxidative coupling of alkenes with alkyl radicals, following β-H elimination, stands out as the most ideal yet challenging strategy for obtaining valuable highly substituted olefins and analogues.2 In this context, cross-dehydrocoupling of alkenes and alkyl carbon-centered radicals generated via oxidation of Csp3–H bonds3 from various precursors has been developed.4 In particular, α-functionalized ether derivatives, which are prevalent in a wide range of natural products and biologically active molecules,5 have been synthesized via cross-dehydrocoupling of alkenes and ethers.4c,i,j Despite these advances, these elegant protocols usually require strong oxidants, transition-metal catalysts, high temperature and harsh conditions. Therefore, the development of a green, mild and metal-free method for direct oxidative coupling of olefins and alkyl radicals via oxidation of Csp3–H bonds remains challenging but highly desirable.
Utilizing the inherently safe and sustainable electrons as redox reagents, electrochemistry offers an eco-friendly approach for organic synthesis.6 Typically, a single half-electrode reaction is necessary, with either cathodic H2 evolution or the use of a sacrificial anode required to maintain electroneutrality. Paired electrolysis, which simultaneously activates reactants on both electrodes, has remained underexploited.7 Significant challenges arise with two-electrode reactions, particularly in matching the reaction scale and rate required for convergent synthesis. Although electrocatalyzed8 and photocatalyzed9 allylic C–H alkylation reactions with carbon nucleophiles have been reported, the substrates are limited to active, protic malonates and their derivatives. Thus, we hypothesized that paired electrolysis, wherein the anodic oxidation of active alkyl substrates such as ether or toluene derivatives for C(sp3)–H bond activation generates Csp3 radical intermediates, and the cathodic reduction of suitable oxidants, leads to the highly efficient oxidative alkylation of alkenes. Herein, we present a direct paired electrocatalysis strategy for intermolecular cross-dehydrocoupling between styrenes and alkyl radicals generated from ethers or p-methylphenol derivatives using ketone as a mild oxidant (Scheme 1). Notable features of this protocol include: (a) paired electrocatalysis, two types of valuable products were obtained simultaneously; (b) transition metal-free; (c) strong oxidant-free, ketone was used as a mild oxidant and was reduced at the cathode to deliver valuable pinacol rearrangement products; and (d) both ethers and p-methylphenol derivatives serve as Csp3 radical precursors.
 | ||
| Scheme 1 Oxidative coupling of styrenes with alkyl radicals. | ||
Initially, styrene (1a) was employed as the model substrate to examine various reaction conditions under a constant current for oxidative alkene alkylation with THF. We found that the alkylation product 4a was obtained in 53% isolated yield under a constant current (10 mA) in THF with 0.8 equivalents of ketone 3a under air conditions, LiClO4 as an electrolyte, a Pt plate as the anode and a Ni plate as the cathode (Table 1, entry 1). The control experiment showed that Pt was essential as the anode as other materials such as GF could not lead to the desired product (entry 2). Interestingly, the presence of I− anion species significantly improved the yield to 70% (entry 3), consistent with previous reports.4c Surprisingly, increasing the amount of ketone 3a to 2.0 equivalents did not significantly enhance the yield (entry 4), indicating that other species are being reduced at the cathode. Other ketones, such as benzophenone and cyclohexanone, produced the desired product in much lower yields (entries 5 and 6), suggesting that the redox potential of the ketone may be a key factor in this transformation. Control reactions conducted without ketone 3a (entry 7) resulted in very low product formation, while without current (entry 8), there was no product formation, demonstrating the essential roles of both current and ketone in the reaction.
|
|
||
|---|---|---|
| Entry | Deviations | Isolated yield of 4a |
| 1 | None | 53% |
| 2 | C(+)|Ni(−) | 0% |
| 3 | n Bu 4 NI (0.2 equiv.) | 70% |
| 4 | 2.0 equiv. 3a | 55% |
| 5 | 2.0 equiv. 3b | 31% |
| 6 | 2.0 equiv. 3c | 20% |
| 7 | No 3a | 14% |
| 8 | No current | 0% |

|
||

With the optimized reaction conditions in hand (entry 3, Table 1), we next explored the scope of this transformation (Scheme 2). Initially, various representative styrenes afforded the corresponding alkylation products in good yields (4a and 4b). To enhance the synthetic value of this green method, a gram-scale synthesis of 4a was conducted. 1,1-Diphenylethene derivatives with either electron-rich or electron-poor substituents on the benzene ring reacted effectively, producing the corresponding alkenylation products in good yields (4c to 4f). 1,3-Bis(1-phenylvinyl)benzene underwent efficient conversion to adduct 4g, with only one of the alkene groups being alkenylated. Cycloalkenes such as 1H-indene and 1,2-dihydronaphthalene, which serve as important cores of drugs, smoothly converted into the corresponding products 4h and 4i, respectively. 1-Aryl–1-alkyl olefins typically suffer from poor regioselectivity in previous methods4c due to two different elimination patterns, resulting in nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 isomer ratios. To our delight, much better regioselectivity was achieved for most 1-aryl–1-alkyl olefins (about 5:1, 4j to 4z). Alkyls, halogens, Bpin-, TBSO-, and Naph- remained intact under the standard conditions, demonstrating good functional group compatibility (4k to 4s). Besides α-methyl styrene, other alkyl groups such as Bn, Bu, and cyclopentyl also participated well in the transformation, yielding only small amounts of elimination isomers (4t to 4w). Trisubstituted 1-aryl–1-alkyl olefins smoothly converted into the desired products (4x and 4y), including a derivative of sertraline (4z). However, the presence of electron-withdrawing groups (EWGs) completely suppressed the reaction, resulting in no desired products. Notably, 1,2,2,2-tetrakis(4-methoxyphenyl)ethan-1-one (5a), formed by pinacol rearrangement following the reductive coupling of ketone 3a, was obtained in a nearly quantitative yield in the above reactions, demonstrating the high efficiency of this electroreductive process.
:1 isomer ratios. To our delight, much better regioselectivity was achieved for most 1-aryl–1-alkyl olefins (about 5:1, 4j to 4z). Alkyls, halogens, Bpin-, TBSO-, and Naph- remained intact under the standard conditions, demonstrating good functional group compatibility (4k to 4s). Besides α-methyl styrene, other alkyl groups such as Bn, Bu, and cyclopentyl also participated well in the transformation, yielding only small amounts of elimination isomers (4t to 4w). Trisubstituted 1-aryl–1-alkyl olefins smoothly converted into the desired products (4x and 4y), including a derivative of sertraline (4z). However, the presence of electron-withdrawing groups (EWGs) completely suppressed the reaction, resulting in no desired products. Notably, 1,2,2,2-tetrakis(4-methoxyphenyl)ethan-1-one (5a), formed by pinacol rearrangement following the reductive coupling of ketone 3a, was obtained in a nearly quantitative yield in the above reactions, demonstrating the high efficiency of this electroreductive process.
 | ||
| Scheme 2 Substrate scope for the oxidative coupling of styrenes with THF. Isolated yield. aNMR yield of another elimination product. | ||
Inspired by these results, we further extended this transformation to p-methylphenol derivatives to achieve new kinds of adducts of alkenes and benzylic radicals (Scheme 3). A range of substrates proved amenable to this transformation. Several styrenes were tested and they participated well in the transformation with BHT (7a to 7c). However, for 1-aryl–1-alkyl olefins such as α-methyl styrene, the regioselectivity was not good (7d and 7d′); in contrast, α-cyclopentyl styrene gave only one elimination isomer due to a kinetically controlled elimination process (7e). Internal alkenes like 1H-indene could also work well in this reaction (7f). Notably, this oxidative alkylation process was successfully applied to synthesize the estrone derivative 7g, which shows anti-tumor activity against A549 and Huh7 (see the ESI†). Furthermore, other p-methylphenol derivatives such as 2-bromo-4,6-dimethylphenol and 3,5-dimethyl-[1,1′-biphenyl]-2-ol were treated under the standard conditions and they produced the desired oxidative alkylation products in moderate yields (7h and 7i).
 | ||
| Scheme 3 Substrate scope for the oxidative coupling of styrenes with p-methylphenols. | ||
To gain insight into the mechanism of this transformation, several mechanistic experiments were conducted. To clarify the possible radical mechanism, radical-trapping experiments were performed using TEMPO and BHT (Fig. S3†). The formation of the desired product was completely suppressed; meanwhile the radical trapping product 6a was isolated in 54% yield (also see Scheme 3), indicating that a radical process is most likely involved in C–H cleavage of ethers and p-methylphenols. When substrate 3a was directly treated under the standard reaction conditions without 1a, the pinacol rearrangement product 5a was obtained in 62% yield (Fig. S3†), demonstrating that reduction of 3a on the cathode was involved. However, using only 0.8 equivalents of ketone 3a was insufficient to complete the entire electron transfer in this transformation. Therefore, there must be other species being reduced at the cathode. Further GC analysis detected the generation of H2 under the standard conditions (Fig. S6†), demonstrating that proton reduction was involved at the cathode to balance the electron transfer of the entire reaction. Lastly, the CV test showed that the redox potential of TBAI was 0.3 V vs. SCE (Fig. S5†), indicating that TBAI undergoes direct oxidation and reduction at the anode and cathode; both I2 and I− anion may promote the elimination of benzylic radical or cation intermediates.4c
Based on the above studies and previous reports,4 a plausible paired electrocatalysis mechanism was proposed (Scheme 4). Initially, THF (A) or p-methylphenol derivatives (A′) undergo anodic oxidation, resulting in the formation of a Csp3 radical intermediate B or B′,7c,10 which is subsequently intercepted by olefins to yield the benzylic radical intermediate C. Radical C is further subjected to anodic oxidation, leading to the formation of its carbocation intermediate D. Following elimination with I− or w/o I− anion, the carbocation intermediate D generates the final product E or E′. In another pathway, the benzylic radical intermediate C might generate benzylic iodide species with the assistance of I2 oxidized on the anode, followed by elimination of HI to deliver the final product E or E′. At the cathode, the reduction of three species was involved based on experiments: (a) reductive coupling of 5a to deliver the pinacol intermediate F, followed by pinacol rearrangement to deliver ketone G; this process was proved to be paired with oxidation of THF or p-methylphenol derivatives at the anode; (b) the reduction of I2 to I− facilitated the catalytic cycle of TBAI, which was a key factor in improving the efficiency of the entire transformation and (c) lastly, proton reduction to generate H2.
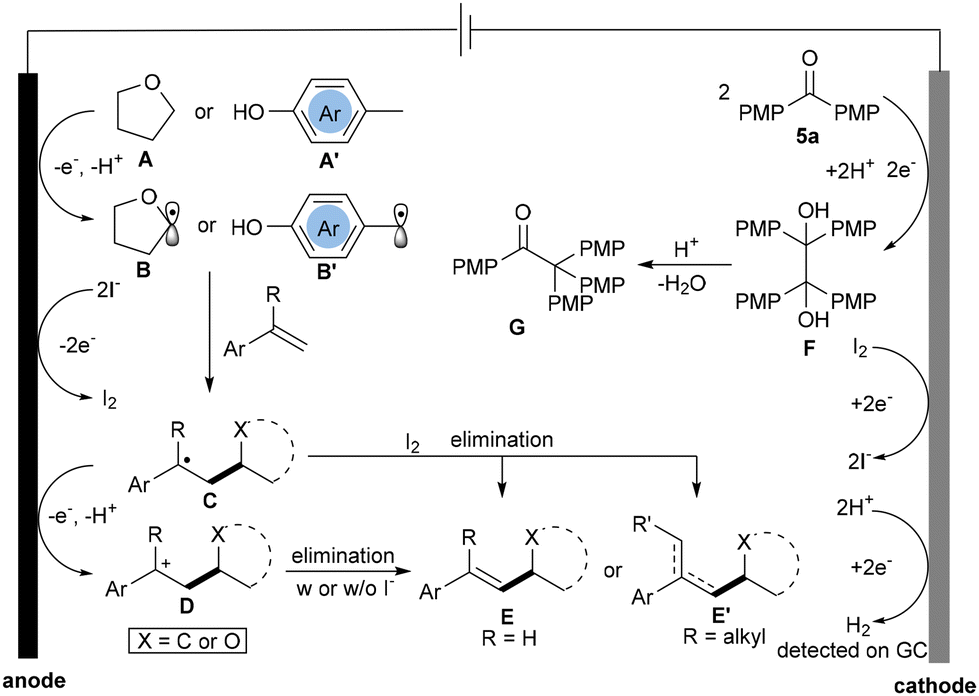 | ||
| Scheme 4 Plausible reaction mechanisms. | ||
Conclusions
In conclusion, a paired electrocatalysis strategy for oxidative coupling of styrenes with alkyl radicals generated from oxidation of ethers or p-methylphenol derivatives using ketone as a mild oxidant was developed. The aforementioned protocol functions as an efficient synthesis tool capable of generating valuable desaturated alkanes with good regio- and chemo-selectivity, ultimately opening a new pathway for the development of paired electrochemical reactions.Author contributions
Dong Li: methodology, data curation, investigation, formal analysis, and writing – original draft. Ling Zhang: investigation and formal analysis. Daixi Li: investigation, writing – review & editing, and funding acquisition. Peng Yu: writing – review & editing and supervision. Tao Shen: writing – review & editing, writing – original draft, validation, supervision, resources, project administration, funding acquisition, data curation, and conceptualization.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from National Science Foundation of China (Grant No. 22301179, No. 22471156), Fundamental Research Funds for the Central Universities (24X010301678, 23X010301599), Excellent Young Scientists Fund Program (overseas), and Shanghai Jiao Tong University 2030 Initiative, Fujian Province Natural Science Foundation (2021J05280) and Fujian Province Talent Introduction Program Foundation (01102801). TS is a Xiaomi Young Scholar and are grateful for financial support from Xiaomi Corporation.References
- (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (b) S. Tang, K. Liu, C. Liu and A. Lei, Chem. Soc. Rev., 2015, 44, 1070–1082 RSC.
- For reviews, see: (a) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed; (b) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (c) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697–4706 CrossRef CAS.
- For reviews on oxidation of C–H bonds, see: (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (c) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC; (d) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (e) C.-J. Li, Acc. Chem. Res., 2008, 42, 335–344 CrossRef PubMed; (f) W.-J. Yoo and C.-J. Li, in C-H Activation, ed. J.-Q. Yu and Z. Shi, Springer, Berlin, Heidelberg, 2010, vol. 292, pp. 281–302 Search PubMed.
- (a) C. Liu, S. Tang, D. Liu, J. Yuan, L. Zheng, L. Meng and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 3638–3641 CrossRef CAS PubMed; (b) B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727–12731 CrossRef CAS; (c) D. Liu, C. Liu, H. Li and A. Lei, Chem. Commun., 2014, 50, 3623–3626 RSC; (d) X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738–743 CrossRef CAS; (e) C. Wang, R.-H. Liu, M.-Q. Tian, X.-H. Hu and T.-P. Loh, Org. Lett., 2018, 20, 4032–4035 CrossRef CAS PubMed; (f) X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410–16143 CrossRef CAS PubMed; (g) J. M. Howell, W. Liu, A. J. Young and M. C. White, J. Am. Chem. Soc., 2014, 136, 5750–5754 CrossRef CAS PubMed; (h) X. Wu, J. Riedel and V. M. Dong, Angew. Chem., Int. Ed., 2017, 56, 11589–11593 CrossRef CAS; (i) L. Chen, K. Jiang, G. Zeng and B. Yin, Org. Lett., 2022, 24, 9071–9075 CrossRef CAS PubMed; (j) G. Kibriya, D. Ghosh and A. Hajra, Sci. China: Chem., 2020, 63, 42–46 CrossRef CAS , and references therein.
- (a) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC; (b) V. Mammoli, A. Bonifazi, F. Del Bello, E. Diamanti, M. Giannella, A. L. Hudson, L. Mattioli, M. Perfumi, A. Piergentili, W. Quaglia, F. Titomanlio and M. Pigini, Bioorg. Med. Chem., 2012, 20, 2259–2265 CrossRef CAS.
- For recent reviews, see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 CrossRef CAS PubMed; (c) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed; (d) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (e) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS; (f) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (g) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (h) Y. Liu, P. Li, Y. Wang and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202306679 CrossRef CAS PubMed; (i) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS.
- (a) J. Lu, Y. Yao, L. Li and N. Fu, J. Am. Chem. Soc., 2023, 145, 26774–26782 CrossRef CAS; (b) D. Liu, Z.-R. Liu, Z.-H. Wang, C. Ma, S. Herbert, H. Schirok and T.-S. Mei, Nat. Commun., 2022, 13, 7318–7326 CrossRef CAS PubMed; (c) Y. Tao, W. Ma, R. Sun, C. Huang and Q. Lu, Angew. Chem., Int. Ed., 2024, 63, e202409222 CrossRef CAS; (d) Y.-J. Chen, W.-H. Deng, J.-D. Guo, R.-N. Ci, C. Zhou, B. Chen, X.-B. Li, X.-N. Guo, R.-Z. Liao, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2022, 144, 17261–17268 CrossRef CAS.
- M. Chen, Z.-J. Wu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2022, 61, e202115954 CrossRef CAS PubMed.
- M.-Y. Dong, C.-Y. Han, D.-S. Li, Y. Hong, F. Liu and H.-P. Deng, ACS Catal., 2022, 12, 9533–9539 CrossRef CAS.
- K. Hayashi, J. Griffin, K. C. Harper, Y. Kawamata and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 5762–5768 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01605j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |