
Recent advances in site-selective transformations of β-enaminones via transition-metal-catalyzed C–H functionalization/annulation
Prasanta
Roy†
 ,
Karuna
Mahato†
,
Divya
Shrestha†
,
Sonaimuthu
Mohandoss†
,
Seung Woo
Lee
* and
Yong Rok
Lee
*
,
Karuna
Mahato†
,
Divya
Shrestha†
,
Sonaimuthu
Mohandoss†
,
Seung Woo
Lee
* and
Yong Rok
Lee
*
School of Chemical Engineering, Yeungnam University, Gyeongsan 38541, Republic of Korea. E-mail: leesw1212@ynu.ac.kr; yrlee@yu.ac.kr; Fax: +82-53-810-4631
First published on 4th November 2024
Abstract
β-Enaminone transformation strategies are widely employed in the synthesis of numerous biologically active drugs and natural products, highlighting their significance in medicinal chemistry. In recent years, various strategies have been developed for synthesizing several five- and six-membered heterocycles, as well as substituted polyaromatic scaffolds, which serve as crucial synthons in drug development, from β-enaminones. Among these approaches, site-selective transformations of β-enaminones via C–H activation and annulation have been particularly well explored. This review summarizes the most recent literature (over the past eight years) on β-enaminone transformations for developing bioactive scaffolds through site-selective C–H bond functionalization and annulation.
 Prasanta Roy | Dr Prasanta Roy is from West Bengal, India. He pursued his Ph.D. under the supervision of Prof. Abu T. Khan and Prof. B. K. Patel from the Department of Chemistry, Indian Institute of Technology, Guwahati, India, in 2016. He worked as a postdoctoral fellow at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, China (2016–2018), Yunnan University, China (2019–2021), Indian Institute of Technology, Kanpur, India (2021–2022), and Yeungnam University, South Korea (2022–2023). Presently, he is International Research Professor (Assistant Professor) at the School of Chemical Engineering, Yeungnam University, South Korea. His research interests include C–H activation, Asymmetric Organic Synthesis, Click Chemistry, Multi-component Reaction and Medicinal Chemistry. |
 Karuna Mahato | Dr Karuna Mahato received her Ph.D. in organic synthesis under the supervision of Prof. Abu T. Khan and Prof. Bhubaneswar Mandal from the Department of Chemistry, IIT Guwahati, in 2018. She continued her scientific training as a Postdoctoral Fellow under Prof. Aiwen Lei at Wuhan University, China, in the field of organic electrosynthesis. In 2021, she joined as IPDF at the Department of Chemistry at IIT Kanpur. Currently she is working as Assistant Professor of Chemistry (international faculty member) at the School of Chemical Engineering, Yeungnam University, Korea. Her current research interests are focused on the transition metal-catalyzed annulation reactions. |
 Divya Shrestha | Ms Divya Shrestha is from Agra, Uttar Pradesh, India. She obtained her B.Sc. and M.Sc. from Dayalbagh Educational Institute, Agra, Uttar Pradesh, India, in 2017 and 2019, respectively. She is pursuing a Ph.D. under Prof. Yong Rok Lee from the School of Chemical Engineering, Yeungnam University, South Korea. Her research interests include Organic Synthesis, C–H activation and annulation reactions. |
 Sonaimuthu Mohandoss | Dr S. Mohandoss received a doctoral degree from Alagappa University and has 10 years of research experience in Materials Chemistry, Supramolecular Chemistry, Organic Synthesis for Chemical Sensors, Computational Chemistry & Bioimaging Applications. He is the recipient of several awards including Basic Scientific Research-Junior Research Fellow (BSR-JRF) and extended Senior Research Fellow (BSR-SRF), University Grant Commission (UGC), India (2011–2015). He received the award (Ministry of Science and Technology) MOST Fellowship for Post-Doctoral Fellow at the Department of Chemistry, National Sun Yat-Sen University, Taiwan (2017–2018). He worked as a Post-Doctoral Researcher in the School of Chemical Engineering, Yeungnam University, Republic of Korea (2018–2020). Currently, he is working as a Research Professor at the School of Chemical Engineering, Yeungnam University, Republic of Korea. His research interests are focused on organic materials and small molecules-derived carbon nanomaterials for Chemosensor, environmental, and live cell imaging. |
 Seung Woo Lee | Prof. Seung Woo Lee received his M.S. (1999) and Ph.D. (2003) from Pohang University of Science and Technology, South Korea, under the supervision of Prof. Moonhor Ree in polymer chemistry. He worked as a postdoctoral fellow with Prof. Chad Mirkin (2004–2005) at Northwestern University. He has received awards from the Polymer Society of Korea (2013, 2018) for his outstanding research in polyimide synthesis and characterization. Currently, his research is focused on organic and bio-polymer chemistry, high-temperature polymers, and polymers for fuel cells and Li ion battery systems. |
 Yong Rok Lee | Prof. Yong Rok Lee received his M.S. (1984) and Ph.D. (1992) from Seoul National University (South Korea) under the supervision of Prof. Eun Lee in organic chemistry. He worked as a postdoctoral fellow with Prof. M. C. Pirrung (1993–1994) at Duke University and Prof. L. Paquette (1995) at Ohio State University. He was also a visiting professor (2000) in Prof. W. Wulff's laboratory at Michigan State University. He has received awards from the Minister of Education in South Korea (2015) and the organic division of the Korean Chemical Society (2017) for his outstanding research in organic synthesis. Currently, his research is focused on organic and natural product syntheses, applications, drug developments, and nanomaterials. |
1. Introduction
Enaminones and their analogs are highly attractive building blocks in organic synthesis1 owing to their strongly polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC bonds, making them ideal synthons for C–H activation. The carbonyl and amine groups in enaminones serve as directing groups, facilitating the functionalization of C–H bonds at the aryl, α-, and β-positions. These functionalization processes often lead to site-selective transformations of enaminones, yielding annulated products depending on the reaction conditions and the stability of the cyclized products. The versatility of enaminones originates from their susceptibility to both electrophilic and nucleophilic attacks, which broadens their applications in organic synthesis.2 To date, synthetic chemists have developed several methodologies for transforming β-enaminones into a wide array of organic compounds, including pyrroles, oxazoles, imidazoles, thiazoles, triazoles, indoles, pyridinones, quinolones, pyrimidines, triazines, and other nitrogen-containing compounds.3 The past decade has witnessed notable advancements in the C–H functionalization of β-enaminones at the aryl, α-, and β-positions and their subsequent annulation via transition-metal-catalyzed,4 electrochemical,5 photocatalytic,6 or metal-free approaches.7 Among this recent progress, the C–H functionalization and annulation of β-enaminones with various coupling partners using transition-metal catalysts have proved valuable for drug development. Considering the importance of these transformations in addressing issues of regioselectivity in synthetic chemistry, numerous research groups have reviewed various related synthetic strategies. However, site-selective transformations of enaminones via C–H functionalization and annulation have received little attention.8
O and CC bonds, making them ideal synthons for C–H activation. The carbonyl and amine groups in enaminones serve as directing groups, facilitating the functionalization of C–H bonds at the aryl, α-, and β-positions. These functionalization processes often lead to site-selective transformations of enaminones, yielding annulated products depending on the reaction conditions and the stability of the cyclized products. The versatility of enaminones originates from their susceptibility to both electrophilic and nucleophilic attacks, which broadens their applications in organic synthesis.2 To date, synthetic chemists have developed several methodologies for transforming β-enaminones into a wide array of organic compounds, including pyrroles, oxazoles, imidazoles, thiazoles, triazoles, indoles, pyridinones, quinolones, pyrimidines, triazines, and other nitrogen-containing compounds.3 The past decade has witnessed notable advancements in the C–H functionalization of β-enaminones at the aryl, α-, and β-positions and their subsequent annulation via transition-metal-catalyzed,4 electrochemical,5 photocatalytic,6 or metal-free approaches.7 Among this recent progress, the C–H functionalization and annulation of β-enaminones with various coupling partners using transition-metal catalysts have proved valuable for drug development. Considering the importance of these transformations in addressing issues of regioselectivity in synthetic chemistry, numerous research groups have reviewed various related synthetic strategies. However, site-selective transformations of enaminones via C–H functionalization and annulation have received little attention.8
This review aims to bridge the aforementioned gap by focusing exclusively on the site-selective transformations of β-enaminones. Specifically, the review presents a systematic summary of recent advancements in the site-selective C–H functionalization and annulation of β-enaminones using transition-metal catalysts, with a special emphasis on elucidating the scope and mechanistic aspects of these transformations. Some of the transformation skeletons of β-enaminones based on site-selective functionalization of C–H bond/annulation via transition metal-catalyzed reactions are shown in Scheme 1. This review is organized into two sections based on the site selectivity of C–H functionalization and annulation: (1) transition-metal-catalysed selective transformations of β-enaminones via aryl-selective C–H functionalization and annulation and (2) transition-metal-catalysed transformations of β-enaminone via α/β-selective C–H functionalization and annulation. Representative examples from recent years are systematically summarized, along with detailed explanations of the reaction conditions, substrate scope, synthetic transformations, limitations, and mechanistic aspects of the transformations. Furthermore, existing challenges and personal perspectives for future developments in the field are detailed at the end.
 | ||
| Scheme 1 Some of the transformation skeletons of β-enaminones based on site-selective functionalization of C–H bond/annulation. | ||
2. Transition-metal-catalyzed transformation of β-enaminone via aryl selective-C–H functionalization/annulation
The transition metal-catalyzed C–H functionalization and annulation reaction is a dominant strategy that improves the proficiency of the transformations for bioactive complex molecules and provides a platform for chemists to approach versatile synthetic challenges.9 Regioselectivity can be stimulated by vicinity of C–H bond and reactive metal center, and substrate–catalyst interactions promote the reaction significantly through coordinating heteroatoms and transition metal catalysts.10 Therefore, the transition metal coordinating with the directing group is typically reflected as a vital step in these types of catalytic C–H bond functionalization methodologies. The functionalization of the aryl C–H bond of β-enaminones with transition metal and numerous coupling partners is well established. In this part, some illustrative examples of directing group-aided selective aryl functionalization of C–H bonds/annulation of β-enaminones using diverse coupling partners based on different types of bond cleavage of enaminones such as C–C, C–N, and CC double bond cleavage are discussed.
2.1. Functionalization of aryl C–H bond of β-enaminones via transition metal catalyst-based C–N bond cleavage
In 2016, Zhu's group described a useful method for creating 1-hydroxy-naphthaldehydes through Rh-catalyzed directing group-sponsored aryl C–H functionalization/annulation of β-enaminones with alkynes or diazo compounds by virtue of C–N bond cleavage of β-enaminones.11 The ortho-C–H bond of the phenyl ring was activated via the significant chelation of enaminones 1 carbonyl group with Rh. The synthesized naphthalene derivatives 3a had formyl and OH functional groups which were used for further transformations. The aldehyde was reacted with an aniline to afford imine product 7, whereas, the hydroxy group was converted to the corresponding ethers through reactions with bromo-containing molecules to form 6 and 8.12a Significantly, participation of both hydroxy groups and aldehyde with dimethyl malonate in the company of piperidine gave an extra ring structure on the parent naphthalene framework12b,c9 (Scheme 2). | ||
| Scheme 2 Rh(III)-catalyzed reaction for synthesizing naphthalenes via directed ortho-selective C–H functionalization/annulation of enaminones 1. | ||
A catalytic cycle for the product 3a is depicted in Scheme 3: the active [Cp*RhOAc]+I species was formed by reacting [RhCp*Cl2]2 with AgSbF6 and AgOAc which coordinated with 1a giving an intermediate shown as II. Next, intermediate II reacted with alkyne 2a forming III that underwent migratory insertion leading to a 7-member-containing rhodacycle IV. Successive migratory insertion of intermediate IV formed V which was released to give intermediate 3a-N along with [Cp*RhH]+ by β-hydride elimination. The oxidation of [Cp*RhH]+ with Ag(I) oxidant in air gave the active [Cp*RhOAc]+I species. Finally, the hydrolysis of 3a-N with the water led to the desired formyl product 3a along with release of Me2NH by C–N bond cleavage. For compound 5, a reasonable reaction mechanism was projected according to literature patterns.13
 | ||
| Scheme 3 Proposed mechanism for the formation of naphthalene derivative 3a. | ||
In 2017, Li and Zhu et al. separately described an efficient methodology for the construction of quinolones followed by ortho-selective amidations via Co(III) and Rh(III)-catalyst-directed functionalization of C–H bond of enaminone 1 using various dioxazolones 10.14 The enaminone alkene skeleton acted as an electrophilic directing group for the hydrolysis cyclization reactions of amidation to furnish diverse quinolone skeletons followed by deacylation of amide group and C–N bond cleavage (Scheme 4).
 | ||
| Scheme 4 Co(III) and Rh(III) catalysts directed o-selective C–H amidation of enaminone for the access to quinolones followed by cleavage of C–N bond. | ||
A catalytic cycle leading to the construction of product 11a is proposed in Scheme 5:11 the active cationic [Cp*Co]2+I species was spawned in situ from the reaction of [CoCp*COl2] with AgBF4/AgSbF6 and AgOAc/KOAc which coordinated to the carbonyl group of 1a and activated the C–H bond at the ortho position giving a 5-member metallocyclic intermediate II. Subsequently, 10a reacted with II to form III which converted to IV with the release of CO2 followed by migratory insertion. Then intermediate IV underwent proto-demetalation to give desired product 11a and released I for a new catalytic cycle. Finally, the hydrolysis–cyclization reaction of amidation product 11a with aq. HCl delivered the target N-heterocyclic product 12a and Me2NH via C–N bond cleavage.
 | ||
| Scheme 5 Proposed catalytic sequence for the formation of product 11a. | ||
In 2018, Loh and co-workers developed [5 + 1] annulation reactions of enaminones 1 and vinyl esters 13 using Rh(III) as catalyst, leading to valuable polyaromatic compounds with amino and formyl groups via ortho-selective C–H bond functionalization.15 The [5 + 1] annulation reactions happened owing to coordinating and di-functional β-enaminone directing-group via C–N bond fragmentation and C–N bond generation, respectively. The ionic pairing of the electron-deficient formyl group and electron-donating amino group in the reactive site of the obtained 1,4-substituted polyaromatic derivatives led to a push–pull electronic structure that could find application as an efficient fluorescent probe in materials and biological science.
Additionally, the reaction was performed at a large scale. The reaction on a 5.0 mmol scale of 1a gave a marginally lower yield (45%). The presence of the formyl and amino functional groups of the synthesized product 14a was used for further synthetic transformation. The formyl group reacted with dimethyl malonate,16a MePPh3Br16b and Grignard reagent16c giving products 15, 16 and 17, respectively. Moreover, the amino group was converted to NO/NO2 using reaction with TBN/TEMPO16d (Scheme 6).
 | ||
| Scheme 6 Rhodium-catalyzed [5 + 1] annulation of enaminones 1via ortho-selective C–H functionalization. | ||
A possible reaction pathway for the formation of product 14a is shown in Scheme 7: initially, in situ generation of the active [RhCp*X2] I species took place from the reaction of [RhCp*Cl2]2 with KOAc that activated the ortho-site of the aryl ring via feeble coordination through the keto position of enaminone 1a, leading to intermediate II. Then intermediate II reacted with vinyl pivalate 13a giving intermediate III, which transformed to intermediate IV by migratory insertion. Later, β-H elimination of IV formed intermediate V and [RhCp*] that was later oxidized by Cu(OAc)2 oxidant in air to regenerate the active [RhCp*X2] I species. After rotation of the C–C bond, intermediate V reacted with [RhCp*X2] to form complex VIvia pivalate-mediated C–H activation. Then, reductive elimination of complex VI gave six-membered cyclic intermediate VII. The Rh complex was released with the amino group from intermediate VII, and the resulting amino group condensed with the keto group giving intermediate VIII. Finally, intermediate VIII gave the desired product 14a by hydrolysis and aromatization.
 | ||
| Scheme 7 Proposed mechanism for the product 14a. | ||
In 2019, the Wang group reported Rh-catalyzed ortho-selective C–H functionalization/annulation of enaminones 1 with sulfoxonium ylides 19 leading to the synthesis of polysubstituted naphthalenes via C–N bond cleavage.17 Here enaminone acted as a directing as well as cyclization group for the application of sulfoxonium ylide in C–H functionalization. This method provided a wide range of multi-substituted naphthalenes having diverse functional group tolerance in good yields (Scheme 8).
 | ||
| Scheme 8 Rhodium-catalyzed annulation of enaminones 1 using sulfoxonium ylides 19via functionalization of ortho C–H bond. | ||
A possible reaction mechanism for the product 20a is shown in Scheme 9:18 initially, in situ-generated [RhCp*X2] I species from the reaction of [RhCp*Cl2]2 with AgSbF6 and HOAc activated the o-position of the phenyl ring of enaminone 1a through weak keto coordination to produce a rhodacyclic intermediate II. Intermediate II coordinated with 19a to form Rh(III) intermediate III followed by the α-elimination of DMSO giving intermediate IV. After this the migratory insertion of IV gave intermediate V.
 | ||
| Scheme 9 Proposed mechanism for substituted naphthalene 20a. | ||
Subsequently, protonolysis of intermediate V gave intermediate VI and released [RhCp*X2] I species, completing the cycle. In HOAc, intermediate VI was transformed to hexatomic ring VIIvia C–C coupling through Michael-type addition. Then, intermediate VIII was formed through the dehydration process of intermediate VII. The hydrolysis of intermediate VIII in the presence of HOAc gave the product 20a (Scheme 9).
In 2021, Zhou and co-workers developed a new approach for constructing diversely functionalized carbazole frameworks 23via Rh(III)-catalyzed selective C–H functionalization/annulation of indole enaminones 21 with diazo compounds 22.19 The reactions profited from good to excellent yields following attractive C–N bond cleavage and the release of N,N-dimethylacetamide via an unexpected [5 + 1] annulation pathway. In addition, gram-scale reactions were also carried out for further transformations and industrial applications. As an example of synthetic utility, the resulting carbazole derivatives were converted to the hydrolyzed product 24 by treating with KOH. In the meantime, 23a reacted with benzaldehyde, ethyl 2-cyanoacetate, triethyl amine and pyrrolidine resulting in the formation of pyrano[3,2-c]carbazole 25 having in vitro antiproliferative and antioxidant activity (Scheme 10).20
 | ||
| Scheme 10 Rh(III)-catalyzed [5 + 1] annulation of enaminones 21 with diazo compounds 22via selective Ar–C–H functionalization. | ||
A possible reaction pathway for the construction of product 23a is shown in Scheme 11:21 as usual, active [RhCp*X2] I species was produced in situ from the reaction of [RhCp*Cl2]2 with CF3COOAg and succinic acid which coordinated with indole–enaminone 21a through activation of C–H bond, yielding a rhodacyclic intermediate II.
 | ||
| Scheme 11 Proposed mechanism for the formation of product 23a. | ||
Next, intermediate II reacted with α-diazo-β-ketoester 22a giving rhodium carbene intermediate III. Then, rhodium carbene intermediate III transformed to give intermediate IVvia an intramolecular migratory insertion followed by intramolecular coordination to the olefinic bond. Subsequently, intermediate IV underwent another migratory insertion followed by protonolysis, leading to the 6-membered cyclic intermediate V. Later, reductive elimination of V gave six-membered cyclic intermediate VI with the discharge of RhCp*XNMe2 complex. Next, N,N-dimethylamine was liberated from the rhodium complex which reacted with VI to give VII by nucleophilic addition to the acetyl group. The desired product 23a was obtained by aromatization of intermediate VII with liberation of N,N-dimethylacetamide.
In 2022, Yan's group described a Rh-catalysed ortho-selective C–H bond activation and the vinylene bond transfer of the enaminones 1 with vinylene carbonate 26 leading to the synthesis of substituted 1-hydroxy-2-naphthaldehydes 27via C–N bond cleavage.22 This protocol provided a large-scale synthesis of hydroxy-naphthaldehyde 27a which was further used in hydroxyl-directed derivatization reactions generating bioactive heterocyclic molecules.
For example, the reaction of 27a, [RhCl2Cp*]2 and pyridine in Et3N at room temperature led to a rhodacycle compound 27a-Rh. Moreover, the hydroxyl group-directed rhodium-catalysed C–H activation of 27a was carried out under different reaction conditions to synthesize the various oxygen-containing bioactive compounds 28–30 (Scheme 12).
 | ||
| Scheme 12 Rh-catalyzed-annulation of enaminones 1 with vinylene carbonate 26via ortho-selective C–H bond functionalization. | ||
A conceivable reaction pathway for the formation of product 27a is shown in Scheme 13:11,17 initially, [Cp*Rh-(III)]2+I was produced in situ from the reaction of [RhCp*Cl2]2 with AgBF4, which coordinated with enaminone 1avia a carbonyl-directed C–H activation process to give intermediate II. Then, intermediate II reacted with vinylene carbonate 26via migratory insertion leading to a 7-membered rhodacycle III. Next, intermediate III was converted to 8-member rhodacycle intermediate IV and 6-member rhodacycle intermediate Vvia path a and path b, respectively. Next, intermediate IV or intermediate V further underwent protonolysis leading to open chain-containing intermediate VI and released Rh(III) active catalyst I. In the presence of HOAc, intermediate VI was transformed into oxonium salt VII, which was further converted to intermediate VIIIvia a Michael addition-type C–C coupling. Next, the dehydration of intermediate VIII gave intermediate IX. Finally, hydrolysis of intermediate IX gave the desired product 27a and released Me2NH via C–N bond cleavage.
 | ||
| Scheme 13 Proposed mechanism for the formation of product 27a. | ||
In 2022, Reddy and co-workers established an oxidative rhodium-catalyzed three-point two-fold annulation reaction of β-enaminones 1 with Propargyl alcohols 31via ortho-selective C–H bond functionalization and the C–N bond cleavage leading to the synthesis of arylnaphthalene-based derivatives of lignan.23 The propargylic alcohol was inserted regioselectively into the rhodacycle as a result of Rh and OH group coordination. Additionally, a large-scale reaction with 5 mmol of 1a and 31a was performed for the synthetic application and the anticipated product 32a was obtained in 53% yield. The compound 32a reacted with DIBAL-H unexpectedly to give the fused dihydrofuran 33.24 Further treatment of compound 32a with bromoethyl acetate in the company of potassium carbonate gave 34 (Scheme 14).
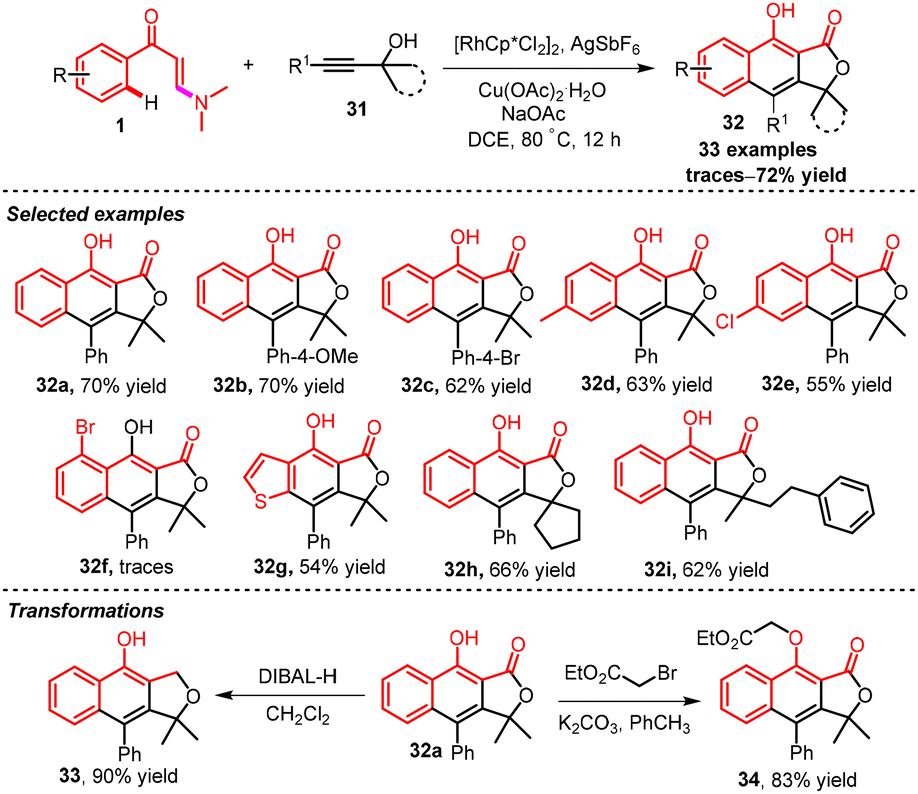 | ||
| Scheme 14 Rh-catalyzed double annulation of propargylic alcohols 31 with enaminones 1via ortho-selective C–H functionalization. | ||
A possible reaction pathway for product 32a is shown in Scheme 15:11 initially, the active catalyst [Cp*Rh(III)] I was formed in situ from the reaction of [RhCp*Cl2]2 with AgSbF6, Cu(OAc)2·H2O and NaOAc. Active catalyst [Cp*Rh(III)] I reacted with enaminone 1a to give a five-membered rhodacycle intermediate IIvia ortho C–H bond activation at the phenyl ring. Then, intermediate II coordinated with 31a to form intermediate III. Regioselective migratory insertion of the propargyl alcohol at the Rh–C bond gave a 7-membered rhodacycle intermediate IV that underwent intramolecular carbo-rhodation of the olefin of enamine to give intermediate V. Consequent reductive elimination of intermediate V gave Rh(I) with the hemiaminal ether VI. Intermediate VI was oxidized to iminium ion intermediate VII which went through hydrolysis to give desired product 32a. Subsequently, Rh(I) was oxidized to the active catalyst Rh(III) I to continue the cycle.
 | ||
| Scheme 15 Proposed mechanism for the formation of product 32a. | ||
In 2023, Wan's group developed25 a multi-component synthesis of various N-naphthyl pyrazoles by Rh(III)-catalyzed coupling of enaminones, aryl hydrazine hydrochlorides and internal alkynes via aryl selective C–H activation and annulation reaction. This synthetic methodology used simple substrates for synthesizing cyclic moieties containing a pyrazole ring and phenyl ring as shown in Scheme 16.
 | ||
| Scheme 16 Rh(III)-catalyzed one-step multi-component reaction of enaminones 1via aryl-selective C–H functionalization and annulation. | ||
The proposed reaction mechanism is shown in Scheme 17. Initially, the condensation of the enaminone 1 and hydrazine chloride 35 took place to form the pyrazole. This pyrazole moiety underwent C–H activation in the presence of active rhodium-III species formed by ligand exchange between Cu(OAc)2 and Cp*RhCl2 to form the intermediate I. The metalation of the intermediate I with the alkyne 2 gave the Rh(III) complex II, which upon further activation gave the metallocycle III. With NaOAc as base, in the presence of the alkyne, the cleavage of the carbon–metal bond took place to generate the intermediate IV or IV′. Finally, the reductive elimination gave the final product 36 and liberated Rh-I species, which upon oxidation by Cu(II) liberated the active Rh-III species for further catalytic cycling.
 | ||
| Scheme 17 Proposed mechanistic pathway for N-naphthyl pyrazoles 36. | ||
The same group reported26 a Rh-catalysed cascade dual-ring synthesis of pyrazolo[5,1-a]isoquinolines 38 with enaminones 1, internal alkynes 2 and hydrazine hydrochloride 37via aryl selective C–H activation and annulation reaction as shown in Scheme 18.
 | ||
| Scheme 18 Rh-catalysed cascade dual ring synthesis of the pyrazolo[5,1-a]isoquinolines via aryl selective C–H bond activation and annulation reaction. | ||
Scheme 19 depicts the plausible mechanistic pathway for the formation of product 38a. In the presence of acid, the first step in the cycle was the condensation between the enaminone 1 and hydrazine 37 to form pyrazole. The pyrazole underwent aryl C–H bond activation with active Rh-III species to form intermediate I. Intermediate I, upon coordination with the internal alkyne, formed the intermediate II going through the formation of TS-I.
 | ||
| Scheme 19 Proposed mechanism for product 38a. | ||
Lastly, intermediate II upon reductive elimination gave final pyrazolo[5,1-a]isoquinoline 38a through the formation of C–N bond, and Rh-I was oxidized to regenerate the active Rh-III species for participation in the next cycle.
In 2023, the Reddy group also described the synthesis of polycyclic naphthopyran scaffolds via Rh-catalysed double ortho-selective C–H bond functionalization and triple annulation of β-enaminones 1.27 This methodology had a good substrate scope with high regioselectivity, scalability and high functionality tolerance. Additionally, a large-scale experiment was carried out for applications in both academic and industrial laboratories. However, reactions performed using 5.0 mmol scale of 1a and 39a gave the desired product 40a in slightly lower yield (48%). Furthermore, Suzuki and Sonagashira coupling reactions using bromo handles were performed for synthetic application of this protocol, providing compounds 41 and 42, respectively (Scheme 20).
 | ||
| Scheme 20 Rh-catalyzed dual ortho-selective C–H functionalization for poly annulation of enaminones 1. | ||
The proposed plausible reaction pathway for the formation of product 40a is shown in Scheme 21:11,23 initially, the active catalyst-[Cp*Rh(III)] I was made in situ from the reaction of [RhCl2Cp*]2 with AgSbF6 and NaOAc. Active catalyst [Cp*Rh(III)] I coordinated with enaminone 1a giving a five-membered rhodacyclic intermediate IIvia ortho-selective C–H activation at the phenyl ring. Then a regioselective insertion of the hydroxy alkynoate 39a into the rhodacycle intermediate II produced a rhodacycle IV which after intramolecular carbo-rhodation of the enamine olefin formed intermediate V. Successive rhodacycle formation, reductive elimination and oxidation gave intermediate VIII, which led to rhodacycle intermediate IX after hydrolysis and peri-selective C–H activation. Insertion of second hydroxy alkynoate 39a into the rhodacycle followed by rhodium coordination and lactonization provided the final product 40a. The Rh(I) was oxidized to active catalyst [Cp*Rh(III)] I with silver salt or with air to continue the catalytic cycle.
 | ||
| Scheme 21 Proposed mechanism for the formation of product 40a. | ||
In 2023, Fan and Zhang's group reported28 a Rh(III)-catalyzed novel cascade reaction of enaminones 1 with diazo homophthalimides 43 leading to the synthesis of homophthalimide spironaphthalenones 44via aryl selective C–H activation and [5 + 1] spiro-annulation. The reaction profits from readily accessible substrates, mild reaction conditions, concise synthetic procedure, valuable products, and good functional group tolerance. Moreover, the usefulness of this method was further showcased by its suitability for scale-up synthetic scenarios and diverse transformation of products as shown in Scheme 22.
 | ||
| Scheme 22 Rh(III)-catalysed novel cascade reaction of enaminones 1 with diazo homophthalimides 43via aryl-selective C–H functionalization and annulation. | ||
A plausible reaction pathway accounting for the formation of 44a is proposed in Scheme 23. Initially, [RhCp*Cl2]2 reacted with AgNTf2 to give an active Rh(III)-catalyst that coordinated with 1a to form a five-membered-rhodacycle intermediate I. Then, intermediate I reacted with 43a to form carbene intermediate II by releasing N2. After that, intermediate II was converted to intermediate III which gave intermediate IV through migratory insertion. Finally, intermediate IV underwent β-elimination to give the desired product 44a with release of NHMe2.
 | ||
| Scheme 23 Proposed mechanism for 44a. | ||
Recently, Fan and co-workers reported a novel synthetic strategy for indanone-fused pyran derivatives 48 from the reaction of aryl enaminones 1 with cyclopropenones 47.29 The product was formed in a one-pot cascade fashion involving ortho-selective functionalization of aryl C–H bond and the cleavage of C–C bond of cyclopropane. The allylic alcohol, formed in situ, underwent 1,3 rearrangement followed by O-nucleophilic addition intramolecularly and C–N bond cleavage.
The advantages of this protocol included a one-pot cascade method with a comprehensive range of substrates, compatibility of different functional groups, and an attractive reaction mechanism. In addition, synthetic utility had also evolved to find wider applications in related areas. To demonstrate the synthetic application of compound 48a, it was treated with benzylamine to give the indeno[2,1-c]pyridine derivative 49. Furthermore, compound 48a reacted with hydrazine hydrate and NaBH4 to give 50 and 51, respectively. Subsequently, compound 48a was stirred for 12 h in DCE at 80 °C to afford product 52 (Scheme 24).
 | ||
| Scheme 24 Rh-catalyzed ortho-selective C–H functionalization/annulation of enaminones 1 through cleavage of C–N bond. | ||
A conceivable reaction mechanism for 48a is shown in Scheme 25:11,30 first, the active catalyst [Rh(OAc)2Cp*] I was formed in situ by the reaction of [Cp*RhCl2]2 with Zn(OAc)2. The active catalyst then reacted with 1avia C–H bond metalation to furnish intermediate II. Next, intermediate II coordinated with 47a to generate intermediates III and IV. Then, intermediate IV was converted to intermediate Vvia a migratory insertion. Subsequently, another migration of Rh(III) from the enone to the enamine moiety to give intermediate VI led to intramolecular nucleophilic attack to furnish intermediate VII. Intermediate VII was converted to the allylic alcohol VIII with regeneration of the active catalyst via proto-demetalation. Next, under the reaction conditions, the allylic alcohol moiety in the intermediate VIII underwent 1,3-rearrangement to give intermediate IXvia a cascade process that afforded an intramolecular O-nucleophilic addition to intermediate X. Finally, the elimination of the dimethyl–amine via C–N bond breakage afforded the final product 48a.
 | ||
| Scheme 25 Proposed reaction pathway for the formation of product 48a. | ||
2.2. Transition metal-catalyzed selective aryl–C–H functionalization of β-enaminones based on C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bond cleavage
C bond cleavage
In 2022, Zhu et al. reported a Rh(III)-catalysed ortho-selective C–H bond activation of enaminones with diazodicarbonyls for divergent synthesis of isocoumarins skeletons 54via C–C cleavage.31 The protocol for synthesizing naphthalene skeleton 56 depended upon the size of the diazodicarbonyl ring and pH of the reaction. The reactions of five-membered and six-membered ring diazodicarbonyls 53 gave isocoumarin scaffolds 54, depending on the nature and amount of additives used (HOAc, NaOAc, CsOAc). The reactions of seven-membered ring diazodicarbonyls 55 formed naphthalene scaffolds 56 under mildly basic (0.2 equiv. CsOAc) or acidic (HOAc) conditions, whereas isocoumarin scaffolds 54 were attained under stronger basic conditions (≥0.4 equiv. CsOAc). In addition, structural diversification of the isocoumarin skeleton was also carried out to produce pharmaceutically important scaffolds. For example, simple iodination reaction of synthesized product 54a with N-iodosuccinimide gave compound 57. Furthermore, the construction of the amide ring structure 58 was obtained from the reaction of ester 54a and benzylamine (Scheme 26).
 | ||
| Scheme 26 Rh-catalyzed selective ortho C–H functionalization/annulation of enaminones 1via C–C cleavage. | ||
The proposed reaction mechanism for the product 54a is portrayed in Scheme 27:32 active catalytic species I coordinated with 1avia ortho C–H bond activation gave intermediate II. Then, intermediate II on reaction with 53a formed Rh(III) carbene intermediate III, which changed to intermediate IV by 1,1-migratory insertion. Furthermore, intermediate IV converted to intermediate V by coordination with 1a which gave intermediate VIvia proto-demetalation along with the release of intermediate II in the cycle. Finally, with HOAc, intermediate VI was converted to enolate VII which was cyclized via C–C cleavage to form 54a.
 | ||
| Scheme 27 Plausible mechanism for the formation of product 54a. | ||
In 2022, the Li group reported Rh(III)-catalyzed C–H/C–C functionalization/annulation of 1 using iodonium ylides 59 for the synthesis of isocoumarin scaffolds 60.33 The transformation provided decent regioselectivity and high functional group acceptance through the novel [3 + 3] annulation with cleavage of the alkene C–C bond of the enaminone (Scheme 28).
 | ||
| Scheme 28 Rh(III)-catalyzed C–H/C–C annulation of 1via C–C bond cleavage. | ||
The proposed plausible reaction mechanism for the formation of product 60a was:34 dimeric [Cp*RhCl2]2 reacted with AgSbF6 and NaOAc forming active catalyst Cp*RhOAc I which coordinated with the oxygen atom of the enaminone 1avia ortho C–H activation forming rhodacyclic intermediate II. Next, the iodonium ylide 59a reacted with rhodacyclic intermediate II to give Rh-carbenoid intermediate III, which endured rapid migratory insertion to form rhodacyclic intermediate IV. Later combination of 1a with rhodacyclic intermediate IV formed intermediate V, which endured proto-demetalation forming intermediate VI with the discharge of intermediate II for the next cycle. Finally, intermediate VI underwent intramolecular attack of the nucleophilic enol oxygen of the dicarbonyl to the carbonyl of the enaminone to furnish the desired product 60a with the elimination of acetaldehyde and dimethylamine as byproducts (Scheme 29). Later, other research groups35 independently followed the same mechanistic route and reported different synthetic strategies using enaminones 1 with coupling partner iodonium ylides/in situ generation of iodonium ylides 59 to construct isocoumarin scaffolds via ortho-selective Rh-catalyzed C–H bond functionalization and annulation through C–C cleavage.
 | ||
| Scheme 29 Plausible mechanism for the product 60a. | ||
In 2023, Zhu's group developed cobalt homeostatic catalysis for ortho-selective C–H functionalization/annulation by the coupling partner oxadiazolones 61 to construct the bioactive quinazolinone skeleton 62via C–C bond cleavage.36 Homeostatic catalysis could persist in catalytic cycles after chemical transformations, enabling further expansion into pharmaceutically active agents. With this efficient catalysis, a wide structural scope for quinazolinones was achieved in good yields. Moreover, the synthesized quinazolinones could be used as starting constituents for further structural expansion into various pharmaceuticals. For example, treatment of the compound 62h with glycidol in Zn(ClO4)2·6H2O at 50 °C produced diproqualone 63, which acts against inflammation and as an analgesic drug.37 In addition, the reaction of synthesized compound 62h with isatin in the presence of acetic acid gave schizocommunin, a fungal alkaloid cytotoxic against murine lymphoma cells via activation of the aryl hydrocarbon receptor gene battery38 (Scheme 30).
 | ||
| Scheme 30 Cobalt homeostatic-catalyzed C–H bond functionalization/annulation of enaminones 1 by the coupling partner oxadiazolones 61via C–C cleavage. | ||
The proposed reaction mechanism for product 62a is shown in Scheme 31: iodide was abstracted from [CoCp*(CO)I2] by AgSbF6 forming [CoCp*]2+I which coordinated with enaminone 1a to give intermediate II. Alternatively, hydrolysis of the enaminone 1a produced 1a-OH which coordinated with [CoCp*]2+I to form intermediate II′. Intermediate II and intermediate II′ were equilibrated by dynamic covalent bonds.
 | ||
| Scheme 31 Plausible mechanism for formation of product 62a. | ||
Next, intermediate II reacted with 61a to give intermediate III which altered to intermediate IVvia 1,1-migratory insertion. Then protodemetalation of intermediate IV gave intermediate V and release of [CoCp*]2+I. Finally, intermediate V converted to product 62avia intermolecular C–N cyclization and C–C cleavage.
In 2023, Baidya and co-workers developed an enaminone-directed ruthenium(II)-catalysed ortho-selective C–H bond activation/annulation reaction of arenes using diazonaphthoquinones 65 or 67via C–C bond cleavage.39 In this protocol enaminone played a dual role in enaminone functionality and quinoid carbene species through the ruthenium(II)/(IV) pathway in the catalytic cycle. The protocol unveiled extensive substrate scope with functional group tolerance, and is useful in the modification of various pharmacophore-coupled compounds. In addition, synthetic transformations were further demonstrated through post-synthetic manipulation. For example, treatment of the polycyclic benzocoumarin 66b with Lawesson's reagent afforded the thiocoumarin analog 69 in excellent yield. Furthermore, NaBH4 reduction of product 66b gave polycyclic benzochromene 70 (Scheme 32).
 | ||
| Scheme 32 Ruthenium(II)-catalyzed enaminone directed C–H functionalization and annulation reaction of arenes with diazonaphthoquinones via C–C bond cleavage. | ||
A plausible mechanism for product 66a is shown in Scheme 33: initially, active cationic ruthenium complex I was formed by reacting [Ru(p-cymene)Cl2]2 with AgSbF6via a ligand-exchange event that initiated a C–H bond activation reaction with 1a forming five-member containing ruthenacycle II. Then, intermediate II coordinated with 65a to give carbenoid intermediate III, and the successive migratory insertion delivered six-membered intermediate IV. Protodemetalation and aromatization produced intermediate V which cyclized in the presence of K2CO3 leading to the desired product 66a and the byproducts acetaldehyde and dimethyl amine.
 | ||
| Scheme 33 Proposed reaction mechanism for the product 66a. | ||
Recently, our group developed a methodology for Rh-catalysed ortho C–H functionalization/annulation of enaminones 1 using coupling partners exomaleimides 71 and maleimides 73 to construct biologically interesting azaspirotetralones 72 and benzo[e]isoindoles 74 through CC bond cleavage.40 This protocol provided high yields of functionalized benzoisoindoles and aza-spirotetralones through one-pot [4 + 2] annulation reaction, with good scope of substrates, high tolerance of functional groups and regioselectivity. Moreover, some selected compounds were investigated for synthesizing pharmaceutically important scaffolds. For instance, 72a with hydroxylamine hydrochloride in the presence of K2CO3 in methanol gave oxime 75 and reaction of compound 72d with NaN3 in TFA formed aryl-migrated lactam 76 (Schmidt reaction). An additional treatment of compound 74a with p-TsCl in TEA and DMAP in CH2Cl2 gave tosylate product 77, while reaction of 74e with 3-methyl-2-butenal resulted in the pyran moiety-containing product 78 (Scheme 34).
 | ||
| Scheme 34 Rh-catalyzed C–H functionalization/annulation strategies of β-enaminones 1 through CC bond cleavage. | ||
The proposed reaction mechanism for the product 72a is shown in Scheme 35:11 initially, formation of Rh(III) complex I took place via the reaction of [Cp*RhCl2]2, CsOAc, and Cu(OAc)2·H2O, which coordinated with 1a forming II. Then, intermediate II upon coordination with 71a gave intermediate III, which converted to 7-member-containing rhodacycle IV by migratory insertion. Next, intermediate IV was transformed into intermediate V by another migratory insertion. Subsequently, intermediate V underwent E2 elimination to form intermediate VI and Cp*Rh(I) was liberated. The liberated Rh(I)Cp* was oxidized by Cu-II in air, releasing active rhodium complex I for the next catalytic cycle. Simultaneously, hydrolysis of intermediate VI with water gave intermediate VII, which got converted to desired compound 72a by the removal of DMF.
 | ||
| Scheme 35 Proposed reaction pathway for the formation of product 72a. | ||
Furthermore, a plausible pathway for the product 74a is shown in Scheme 36: Rh(III) species I coordinated with 1a and produced 5-member-containing rhodacycle II. Then, the intermediate II coordinated with 73a to give III′, which was converted to 7-member-containing rhodacycle ring IV′via migratory insertion. Next, intermediate IV′ transformed to V′via another migratory insertion, which gave intermediate VI′ and Cp*Rh-I was released via E2 elimination. Intermediate VI′ then reacted with water via a Michael-type addition reaction to form intermediate VII′, which released DMF and VIII′. Successive oxidation of VIII′ in air provided 74a, while Rh(I) was oxidized to release the active Rh-III species I by the Cu-II in air, for the next cycle.
 | ||
| Scheme 36 Proposed mechanism for product 74a. | ||
3. Transition-metal-catalyzed selective transformation of β-enaminone via α, β-C–H functionalization/annulation
In previous years, the selective transformation of β-enaminones via α,β-C–H bond functionalization/annulation has been applied in the synthesis of many organic products with varying central backbones, like aromatics, acyclic molecules, heterocycles, and natural products.41 According to the literature on synthesis based on enaminone moieties, enaminones consist of electrophilic carbonyl, nucleophilic α,β-carbons and amino groups that serve as the main reactive sites.42 However, research interest in organic chemistry using β-enaminones has driven the rapid advancement of synthetic methods toward bioactive molecules, and a wide variety of synthetic techniques, including organo/Lewis acid catalysts,43 visible-light,44 electrochemical45 and transition metal-catalysts46 have been successfully developed. In this section, representative examples of transition-metal-catalysed α/β selective C–H functionalization/annulation of enaminones with several coupling partners are discussed.In 2016, Wan and co-workers developed a copper-catalysed intramolecular oxidative amination of enaminone for synthesizing imidazo[1,2-a]pyridines via α-selective C–H functionalization/annulation.47 This protocol involved C(sp2)–H amination of N-pyridinyl enaminones intramolecularly, in aerobic conditions with good yields and relatively mild reaction conditions (Scheme 37).
 | ||
| Scheme 37 Cu-catalyzed α-selective C–H functionalization/annulation of enaminones 79. | ||
The proposed plausible reaction mechanism for product 80 is shown in Scheme 38:48 initially the enaminone 79 reacted with the copper(I) ion forming the cuprous intermediate I taking the help of N-chelating site of ring of pyridine.
 | ||
| Scheme 38 Proposed reaction mechanism for product 80. | ||
Then, intermediate I was converted to intermediate Cu(III) II by oxidative addition under O2 (air). The desired imidazo[1,2-a]pyridine product 80 was formed by reductive elimination of intermediate II, releasing the copper(I) ion for further catalytic processes.
Patil's group in 2016 established a gold-catalysed alkynylation/cyclization strategy of ortho-hydroxyaryl enaminones 81 to access 3-alkynylchromones 83via α,β-selective C–H functionalization/annulation.49 The reaction involved alkynylation of the enaminone moiety with gold as a catalyst and TIPS-EBX and then intramolecular cyclization. The utility of this method was established through the transformation of the de-silylated product to functionalized chromones. Initially, attempts to remove the TIPS group from product 83a in TBAF and camphor sulfonic acid led to 3-ethynylchromone 84. Next, the synthesized product 3-ethynylchromone 84 was converted to the corresponding triazole 85 by a copper-catalysed azide–alkyne cycloaddition reaction and the internal alkyne 86 by a Sonogashira reaction, respectively. Moreover, Sonogashira and intramolecular hydro-alkoxylation reaction of the synthesized product 3-ethynylchromone 84 gave 3-(benzofuran-2-yl)-chromone 87. Furthermore, treatment of 3-ethynylchromone 84 with the nitrogenating agent tBuONO and the oxidant 2-picoline-N-oxide gave 3-cyanochromone 88 (Scheme 39).50
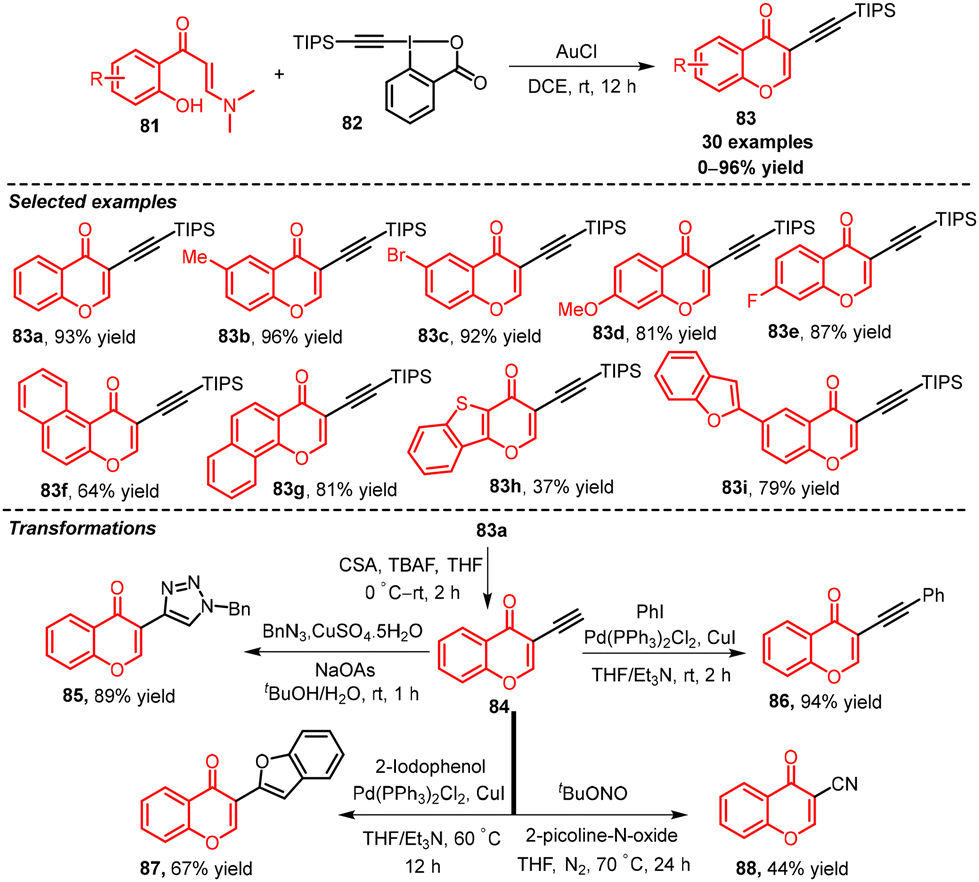 | ||
| Scheme 39 Au-catalyzed α,β-selective C–H bond functionalization/annulation of enaminones 81. | ||
The proposed plausible mechanisms for 83avia two pathways, path a and path b, are shown in Scheme 40: Michael addition followed by cyclization of ortho-hydroxy phenyl enaminone 81a formed intermediate I which reacted with TIPS-EBX forming intermediate IIIvia path a. Simultaneously, 81a reacted with TIPS-EBS to form alkynylation intermediate II that afforded cyclization intramolecularly, via pathway b, giving intermediate III. Lastly, liberation of N,N-dimethylamine from intermediate III afforded the desired product 3-alkynylchromone 83a.
 | ||
| Scheme 40 Proposed reaction mechanism for product 83a. | ||
In 2020, Liu's group established51 a copper-catalysed β-C(sp2)–H selective N-hetero-arylation of enaminones 81 with numerous nitrogen nucleophiles such as azoles and cyclic amines 89, for the construction of β-nitrogenated-chromones 90 as shown in Scheme 41.
 | ||
| Scheme 41 Cu-catalyzed β-selective C–H bond functionalization/annulation of enaminones 81 with numerous nitrogen nucleophiles. | ||
The authors reported a plausible mechanism for 90 as depicted in Scheme 42. Initially, ortho-hydroxy phenyl enaminone 81 was converted to 3-iodo-chromone intermediate I by the reaction with I2 and Ph(OAc)2. Then, intermediate I coordinated with Cu(I) to generate intermediate II which reacted with azole/amine 89 to form intermediate III. Finally, the desired β-nitrogenated chromones 90 were obtained by the selective elimination of HI from intermediate III in the presence of the base.
 | ||
| Scheme 42 Proposed reaction mechanism for product 90. | ||
Later in 2020, the same group reported52 a Pd-catalysed domino chromone annulation and a transient halogenation-mediated synthesis of 3-vinyl chromones 92via α-selective C–H alkenylation of o-hydroxyphenyl enaminones 81 with several alkenes 91 as shown in Scheme 43.
 | ||
| Scheme 43 α-Selective C–H alkenylation of o-hydroxyphenyl enaminones 81 with several alkenes 91. | ||
The authors reported a plausible mechanism for 92 as depicted in Scheme 44. Initially, iodine anion was oxidized in the presence of TBHP to give iodine radical by single-electron-transfer (SET). Then, iodine radical reacted with enaminone 81a to form free-radical intermediate I which combined with hydroxyl-free-radical to give intermediate II. Subsequently, intermediate II underwent a transetherification to deliver chromanone-intermediate III. Next, intermediate III was converted to intermediate IV by elimination of NHMe2. Finally, a Pd-catalysed Heck-type alkenylation of intermediate IV with alkene 91 gave the desired product 92. Recently, the same group also reported53 Pd-catalysed synthesis of 3-phosphonate-chromones via Arbuzov-type α-selective C–P coupling reaction of o-hydroxyphenyl enaminones 81 with phosphites following a similar mechanistic route.
 | ||
| Scheme 44 Proposed reaction mechanism for product 92. | ||
In 2020, Jin's group developed an Ag-catalysed regio- and chemoselective [4 + 1C]insert cascade reaction for the synthesis of pyrroles 95 from enaminones 93 and carbenes 94 through α-selective C–H bond functionalization/annulation.54 This protocol provided a convenient atom economical process for multi-substituted pyrroles and further pyrrole diversity for natural product lamellarin L 96 (Scheme 45).
 | ||
| Scheme 45 Ag-catalyzed α-selective C–H functionalization/annulation of enaminones 93. | ||
The proposed reaction mechanism for the product 95 is presented in Scheme 46:55 AgOTf reacted with the enaminone 93 to form the thermodynamic equilibrium cis-enaminone–Ag complex I, that further reacted with the diazoester 94 to give the electrophilic silver carbenoid II with the evolution of N2via C–C bond insertion cycle. Then, the silver carbenoid II was converted to the cyclopropanation intermediate III. The C–C bond at the α,β-site of the carbonyl group of intermediate III was regioselectively cleaved to give the silver enolate IV due to the presence of nitrogen lone pair and ring strain in the intermediate III. Then, in the presence of HOTf, protonation of the silver enolate IV produced the silver keto intermediate V which cyclized to give intermediate VI. Subsequently, intermediate VI was transformed into intermediate VII which further gave intermediate VIIIvia chemo- and regioselective thermal [1,5]-shift. Finally, the desired product 95 was obtained by dehydroaromatization of intermediate VIII.
 | ||
| Scheme 46 Proposed reaction pathway for the product 95. | ||
In 2020, Loh and co-workers developed a formal [3 + 3] annulation reaction of enaminones 97 with acrylates 98 for 2-pyridones 99 having N-substitution through Rh(III)-catalysed α-selective C–H bond functionalization.56 The methodology involving cross-coupling of C–C bond by C–H bond functionalization and aminolysis cyclization was suggested by control and deuterated experiments. This technique delivered a brief synthesis of the structural motifs of N-substituted 2-pyridones (Scheme 47).
 | ||
| Scheme 47 Rh-catalyzed α-selective C–H functionalization/annulation of enaminones 97. | ||
The proposed reaction mechanism for the product 99 is shown in Scheme 48:57 in the beginning, active catalyst rhodium acetate species I was formed by reacting [RhCp*Cl2]2 with AgOAc. Then, the active catalytic rhodium acetate species reacted with the (Z)-N-substituted enaminone 97via N–H deprotonation and C–H bond activation to give E configuration intermediate II, and subsequent conversion to the 4-membered rhodacycle intermediate III occurred. Next, III coordinated with methyl acrylate 98 to generate IV which transformed to the 6-member-containing rhodacycle intermediate V through migratory insertion. Subsequently, intermediate V underwent β-hydride elimination to give a Rh(III) hydride intermediate VI. Finally, intramolecular aminolysis followed by reductive elimination of intermediate VI gave 99 as the desired product with Rh(I)Cp*. Alternatively, aminolytic cyclization of intermediate V formed intermediate VII, which was converted to 99via β-hydride elimination by the release of Cp*Rh(I). Subsequently, Cu(OAc)2 oxidized the Cp*Rh(I) to regenerate Rh-III acetate species I for the next cycle.
 | ||
| Scheme 48 Proposed reaction mechanism for product 99. | ||
Wan's group, in 2022, described a Rh-catalysed [4 + 2] annulation reaction of N-pyridinyl enaminones 100 with internal alkynes 2 for the construction of iminopyranes 101via β-selective C–H functionalization.58 The protocol involved C(sp2)–H bond functionalization and N–H bond scission in β-enaminones. Moreover, DFT calculations gave a detailed understanding of the reaction mechanism. In addition, the pyridine ring of enaminones served as an auxiliary monodentate site for the annulation and could be easily removed by a simple hydrolysis to give pyrenones 102 (Scheme 49).
 | ||
| Scheme 49 Rh-catalyzed β-selective C–H functionalization/annulation of enaminones 100. | ||
The proposed reaction pathway for 101a is shown in Scheme 50:59 in the beginning, Rh-dimer was converted to active catalyst RhCp*Cl2, which reacted with enaminone 100a to form intermediate I. Then, intermediate I coordinated with PivOH in the presence of AgSbF6 to generate intermediate II. Next, intermediate II was converted to intermediate III, which coordinated with alkyne 2a to give intermediate IV. Intermediate IV underwent migratory insertion in a subsequent step to form intermediate V. Then, V was converted to intermediate VI, which cyclized to intermediate VII. Finally, the desired product 101a was furnished from intermediate VII by the release of Rh(I)Cp*.
 | ||
| Scheme 50 Proposed reaction pathway for product 101a. | ||
Reddy's group in 2022 demonstrated a Cu-catalysed tandem cyclization and α,β-C–H functionalization of β-enaminones using enynones 103 for the construction of multisubstituted furans 104 and furano-pyrroles 105.60 The tandem cyclization/annulation reactions were proposed to proceed through carbene insertion and aryl migration in the rearrangement step. This protocol was studied for its general scope and high scalability. Furthermore, the synthesized bis-heterocycles were applied for synthetic transformations to produce bioactive bis-heterocyclic derivatives. For example, reduction of synthesized product 105c with NaBH4 gave the analogous bis-heterocyclic alcohol 106. Furthermore, reaction of the synthesized product 105c in the presence of methoxyamine gave the corresponding oxime 107 (Scheme 51).
 | ||
| Scheme 51 Cu-catalyzed α,β-selective C–H bond functionalization and annulation of enaminones 1 & 93. | ||
The proposed plausible reaction route for products 104 and 105 is shown in Scheme 52: initially, Cu(OTf)2-activated enynones 103 afforded important furanyl copper carbenoids intermediate Ivia electrophilic cyclization. Then, intermediate I reacted with the enaminone to form cyclopropane intermediate II. The unstable cyclopropane ring intermediate II was opened up to give intermediate III, then hydrolysis of intermediate III led to the desired product 104 with the release of amine. Next, the dicarbonyl product 104 reacted with amine to give a cyclic iminium ion intermediate VIvia intermediate IV and V. Finally, aromatization of intermediate VI led to the desired furanyl pyrrole 105via rearrangement of the Ph-group.
 | ||
| Scheme 52 Proposed reaction mechanism for the products 104 and 105. | ||
Jiang and co-workers in 2023 reported an Au(III)/Ag(I) co-catalytic protocol for the design of cyclopentadienes 109via amine-release β-selective functionalization of C–H bond and annulation of β-enaminones 1 with alkynes 108.61 This bimetallic catalytic platform had compatibility with wide variety of substrates, and that too under mild reaction conditions. Furthermore, the synthesized products were allowed to undergo various synthetic processes and further stage modifications for the construction of complex molecules. For example, the ester group of synthesized spiro-cyclo-pentadiene 109g could easily undergo hydrolysis under the acidic conditions forming 1,4-diketone 110, and the carbonyl group of 109g could be reduced by NaBH4 forming the hydroxyl group as 111 (Scheme 53).
 | ||
| Scheme 53 Au(III)/Ag(I) co-catalytic protocol for the synthesis of cyclopentadienes 109. | ||
The proposed reaction route for product 109 is shown in Scheme 54: propargyl ester 108 reacted with Au-catalyst to form the vinyl carbene complex Ivia migration.62 The product formation might have taken two possible pathways. Vinyl aminocyclopropane intermediate II might have been formed through direct cyclopropanation in path-I. Due to the effect of π-conjugation, the ring opening of the unstable cyclopropane ring via selective C–C bond cleavage afforded zwitterion intermediate III. It is to be noted that the intermediates (II and III) could undergo hydrolysis leading to keto aldehyde with no substitution of acetate group of the vinyl moiety according to previous literature reports.63 However, it was observed that the intermediate III had a preference for cyclizing to amino cyclopentene V in an intramolecular fashion, which underwent a deaminative process to produce the desired product 109. Contrary to that, possibly, enaminone 1 also attacked vinyl carbene complex I nucleophilically to produce intermediate IVvia pathway II. The intermediate IV could also synthesize amino cyclopentene V through intramolecular cyclization. Lastly, amino cyclopentene V underwent a deaminative process generating desired product 109.
 | ||
| Scheme 54 Reaction pathway for product 109. | ||
In 2023, Baell et al. reported a copper-catalysed α-selective C–H functionalization and annulation protocol of the enaminones 1 using the diazo compounds 112 for multi-substituted furanones 113.64 This method proceeded through C(sp2)–H bond activation of olefinic bond/insertion of carbene/hydrolysis/annulation mechanism of enaminones 1. This protocol was functionally simple, used easily available starting materials, and had good scope of substrates and high tolerance towards functional groups. Also, an experiment at a gram-scale was carried out for furanone derivatives to establish its possible applicability. For example, the morpholinyl group of the synthesized product 113a could be acidified with H2SO4 to give the 5-hydroxy furanone 114. 5-Hydroxyfuranones 114 could undergo transformations to compounds 115 and 116, respectively, in good yields (shown in Scheme 55).
 | ||
| Scheme 55 Cu-catalyzed α,β-selective C–H functionalization and annulation reaction of enaminones 1 with diazo compounds 112. | ||
The proposed possible reaction pathway for product 113a is shown in Scheme 56: at the start, the Cu(I) cation underwent oxidation in Ag(I) and formed Cu(II) cation and Ag(0).
 | ||
| Scheme 56 Proposed reaction pathway for the product 113a. | ||
Then, enaminone 1a reacted with Cu(II) cation and carbonate anion forming copper–olefin intermediate I, which reacted with 112a to give copper–olefin–carbene adduct II. Next, copper–olefin–carbene adduct II was converted to copper-intermediate IIIvia migration insertion. Subsequently, removal of copper from intermediate III by ester hydrolysis led to the intermediate IV liberating Cu(II) for the next cycle. Lastly, 2(5H)-furanone 113a was obtained by the cyclization of intermediate IV.
In 2023, Shi and co-workers developed an extraordinary [2 + 3] annulation reaction of N-sulfonyl-triazoles 117 with the enaminones 1 for synthesis of polysubstituted furans 118via Rh(II)-Brønsted acid cooperative catalytic α-selective C–H functionalization.65 This protocol implied that Rh(II)–azavinyl carbene performed a twin function, permitted by the breaking of the C(sp2)–N bond. Experimental and mechanistic studies suggested that the TSNH-group underwent an intermolecular rearrangement that led to formation of products 118 and 119 (Scheme 57).
 | ||
| Scheme 57 Rh(II)–Brønsted acid cooperative catalytic α-selective C–H functionalization/annulation of N-sulfonyl-triazoles 117 with enaminones 1. | ||
The proposed plausible reaction mechanism for the formation of products 118a and 119a is shown in Scheme 58: initially, N-sulfonyl-triazoles 117 reacted with Rh2(esp)2 and CSA to form Rh(II)–azavinyl carbene intermediate I. Then, Rh(II)–azavinyl carbene intermediate I reacted with enaminone 1avia nucleophilic addition to generate intermediate II. Subsequently, the keto-group intramolecularly attacked the carbene II to form the low-energy oxonium ylide III. Then, intermediate III reacted with in situ-generated TsNH2 to give intermediate IV and V, respectively, which was subsequently converted to intermediate VI. Next, the intermediate VI was converted to intermediate VII with the release of product 119a. Finally, intermediate VII was dissociated to regenerate the Rh catalyst and release desired product 118a.
 | ||
| Scheme 58 Proposed reaction pathway for the formation of product 118a and 119a. | ||
In 2023, Yu and co-workers reported a synthesis of functionalized 2-spirocyclo-pyrrol-3-ones 121 using enaminones 93 with iodonium ylides 120.66 The reaction proceeded by α-selective C–H functionalization with Rh(III) as a catalyst followed by [3 + 2] annulation and then pinacol rearrangement to get extremely substituted 2-spirocyclopyrrol-3-ones. Furthermore, the gram-scale experiment could readily be performed using 5.0 mmol scale and ring opening/hydrolysis of the synthesized product 121a to afford 1H-pyrrol-3-ol carboxylate 122 (Scheme 59).
 | ||
| Scheme 59 Rh(III) catalyst α-selective C–H functionalization/annulation protocol of enaminones 93 with the iodonium ylides 120. | ||
The proposed reaction pathway for product 121a is shown in Scheme 60: [Cp*RhCl2]2 reacted with enaminone 93a through α-selective C–H activation/metalation reaction, forming four-member-containing cyclometalated species I, that reacted with the iodonium ylide 120a giving metal carbene II. Subsequently, II was converted to five-member-containing intermediate III by migratory insertion, then protonolysis occurred to produce IV with the release of [Cp*RhCl2]2. Intermediate IV was converted to Vvia oxidative dehydrogenation and water attack took place at the CC bond forming intermediate species VI. Next, intramolecular cyclization of intermediate VI produced intermediate VII. Lastly, 121a was attained by pinacol rearrangement reaction.
 | ||
| Scheme 60 Proposed mechanism for product 121a. | ||
Recently, Zhu's group demonstrated a Co(III)-catalyzed protocol for imidazoles 124 using enaminones 1 and oxadiazolones 123 as coupling partners via appendage speciation-oriented synthesis.67 In this method, the α, β–C–H bond was activated instead of the normal reaction where Ar–C–H activation is observed in the aromatic ring. This protocol provided a versatile synthetic handle for extended reactivity, structural development scope, and productive organic synthesis (Scheme 61).
 | ||
| Scheme 61 Co(III) catalyst α,β-C–H functionalization/annulation process of enaminones 1 with oxadiazolones 123. | ||
The proposed reaction route for 124a is shown in Scheme 62:68 firstly, cationic species [Cp*Co]2+I was prepared in the reaction cycle from [CoCp*(CH3CN)3](SbF6)2, which coordinated with the alkenyl moiety of enaminone giving intermediate II. Subsequently, II reacted with 123a giving intermediate III. Nucleophilic cyclization led to the liberation of CO2, followed by deaminative aromatization of intermediate IV giving the desired product 124a.
 | ||
| Scheme 62 Proposed mechanism for formation of product 124a. | ||
4. Conclusions
In conclusion, this review summarizes recent advancements in site-selective transformations via C–H functionalization and annulation reactions, focusing on enaminones as key building blocks. β-Enaminones serve as ideal synthons for activating C–H bonds owing to their highly polarized carbonyl and electrophilic alkene functionalities. The CO and amine groups of enaminones act as directing-groups, facilitating the functionalization of C–H bonds at the aryl, α-, and β-C–H positions. The review is structured into two primary sections that highlight site selectivity in C–H functionalization and annulation using transition metal catalysts: (1) transition-metal-catalyzed transformations of β-enaminones via aryl-selective C–H functionalization/annulation and (2) transition-metal-catalyzed transformations of β-enaminones via α/β-selective C–H bond functionalization/annulation. Each section provides a detailed description of enaminone-based organic reactions utilizing transition metal catalysis for C–H functionalization and annulation. This review systematically and thoroughly summarizes illustrative examples from recent years, detailing reaction conditions, substrate scopes, transformations, limitations, and mechanistic insights wherever appropriate. Furthermore, the mechanisms of these reactions are presented to provide researchers with a deeper understanding of these methodologies and to foster the exploration of further transformation patterns. We hope this review will be instrumental in accelerating research on transition metal-catalysed transformations of enaminones through site-selective C–H functionalization and annulation.
Data availability
As this is a review article, we are not reporting any new data in this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (2021R1A2B5B02002436). The authors thank the Core Research Support Center for Natural Products and Medical Materials (CRCNM) for technical support regarding the 600MHz FT-NMR.References
- (a) Y.-N. Lu, C. Ma, J.-P. Lan, C. Zhu, Y.-J. Mao, G.-J. Mei, S. Zhang and F. Shi, Org. Chem. Front., 2018, 5, 2657–2667 RSC; (b) D. Bhattacherjee, S. Ram, A. S. Chauhan, Y. Sheetal and P. Das, Chem. – Eur. J., 2019, 25, 5934–5939 CrossRef CAS; (c) X. He, R. Li, P. Y. Choy, J. Duan, Z. Yin, K. Xu, Q. Tang, R.-L. Zhong, Y. Shang and F. Y. Kwong, Chem. Sci., 2022, 13, 13617–13622 RSC; (d) Y. Zhou, L.-S. Wang, S.-G. Lei, Y.-X. Gao, J.-T. Ma, Z.-C. Yu, Y.-D. Wu and A.-X. Wu, Org. Chem. Front., 2022, 9, 4416–4420 RSC; (e) H. Sheng, Z. Chen, X. Li, J. Su and Q. Song, Org. Chem. Front., 2022, 9, 3000–3005 RSC; (f) L. Fu, J.-P. Wan, L. Zhou and Y. Liu, Chem. Commun., 2022, 58, 1808–1811 RSC; (g) J. Ying, T. Zhou, Y. Liu, L. Zhou and J.-P. Wan, J. Org. Chem., 2024, 89, 9078–9085 CrossRef CAS PubMed.
- (a) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433–2480 CrossRef CAS; (b) G. Li, K. Watson, R. W. Buckheit and Y. Zhang, Org. Lett., 2007, 9, 2043–2046 CrossRef CAS; (c) S. Ma and Z. Yu, Angew. Chem., Int. Ed., 2002, 41, 1775–1778 CrossRef CAS; (d) J. D. White and D. C. Ihle, Org. Lett., 2006, 8, 1081–1084 CrossRef CAS PubMed.
- (a) S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629–2632 CrossRef CAS PubMed; (b) A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2010, 12, 372–374 CrossRef CAS; (c) S. Hirai, Y. Horikawa, H. Asahara and N. Nishiwaki, Chem. Commun., 2017, 53, 2390–2393 RSC; (d) X.-H. Xu, J.-H. Weng, Y. Chen and Z.-B. Dong, Adv. Synth. Catal., 2024, 366, 1649–1653 CrossRef CAS.
- (a) S. Zhong, Y. Lu, Y. Zhang, Y. Liu and J.-P. Wan, Org. Biomol. Chem., 2016, 14, 6270–6273 RSC; (b) J. Chen, P. Guo, J. Zhang, J. Rong, W. Sun, Y. Jiang and T.-P. Loh, Angew. Chem., Int. Ed., 2019, 58, 12674–12679 CrossRef CAS; (c) Z. Zhong, L. Liao, Y. Liu, M. Zhang and J.-P. Wan, Chem. Commun., 2023, 59, 6885–6888 RSC; (d) L. Fu, Y. Liu and J.-P. Wan, Org. Lett., 2021, 23, 4363–4367 CrossRef CAS PubMed; (e) Y. Li, W. Jiang, J. Lin, J. Ma, B.-H. Xu, Y.-G. Zhou and Z. Yu, Org. Lett., 2022, 24, 7123–7127 CrossRef CAS PubMed; (f) W. Song, Y. Liu, N. Yan and J.-P. Wan, Org. Lett., 2023, 25, 2139–2144 CrossRef CAS.
- (a) H. Guo, Y. Liu, C. Wen and J.-P. Wan, Green Chem., 2022, 24, 5058–5063 RSC; (b) Q. Huang, J. Liu and J.-P. Wan, Org. Lett., 2024, 26, 5263–5268 CrossRef PubMed.
- (a) M. Li, Z. He, W. Zhao, Y. Yu, F. Huang and J. B. Baell, J. Org. Chem., 2023, 88, 8257–8267 CrossRef CAS PubMed; (b) Y. Han, L. Y. Zhou, C. Y. Wang, S. T. Feng, R. Ma and J.-P. Wan, Chin. Chem. Lett., 2024, 35, 108977 CrossRef CAS.
- (a) H. Xu, H.-W. Liu, H.-S. Lin and G.-W. Wang, Chem. Commun., 2017, 53, 12477–12480 RSC; (b) I. V. Efimov, M. D. Matveeva, R. Luque, V. A. Bakulev and L. G. Voskressensky, Eur. J. Org. Chem., 2020, 1108–1113 CrossRef CAS; (c) X.-H. Xu, J.-H. Weng, Y. Chen and Z.-B. Dong, Adv. Synth. Catal., 2024, 366, 1649–1653 CrossRef CAS; (d) H. Wu, K. Chen, Y. Liu and J.-P. Wan, J. Org. Chem., 2024, 89, 216–223 CrossRef CAS PubMed; (e) X. Y. Chen, X. Zhang and J.-P. Wan, Org. Biomol. Chem., 2022, 20, 2356–2369 RSC.
- (a) A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16450–16467 RSC; (b) H. M. Gaber, M. C. Bagley, Z. A. Muhammad and S. M. Gomha, RSC Adv., 2017, 7, 14562–14610 RSC; (c) I. J. Amaye, R. D. Haywood, E. M. Mandzo, J. J. Wirick and P. L. Jackson-Ayotunde, Tetrahedron, 2021, 83, 131984 CrossRef CAS; (d) J. Huang and F. Yu, Synthesis, 2021, 587–610 CAS; (e) Z. Wang, B. Zhao, Y. Liu and J.-P. Wan, Adv. Synth. Catal., 2022, 364, 1508–1152 CrossRef CAS; (f) Y. Wang, C. Zhang, S. Li, L. Liu and X. Feng, ChemistrySelect, 2022, 7, e202103345 CrossRef CAS; (g) D. Li and L. Chen, Eur. J. Org. Chem., 2024, e202400895 CrossRef.
- (a) U. Dhawa, N. Kaplaneris and L. Ackermann, Org. Chem. Front., 2021, 8, 4886–4913 RSC; (b) C. Sivaraj, A. Ramkumar, N. Sankaran and T. Gandhi, Org. Biomol. Chem., 2021, 19, 8165–8183 RSC; (c) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed; (d) S. K. Sinha, P. Ghosh, S. Jain, S. Maiti, S. A. Al-Thabati, A. A. Alshehri, M. Mokhtar and D. Maiti, Chem. Soc. Rev., 2023, 52, 7461–7503 RSC.
- (a) V. K. Tiwari and M. Kapur, Org. Biomol. Chem., 2019, 17, 1007–1026 RSC; (b) R. Das, N. P. Khot, A. S. Deshpande and M. Kapur, Chem. – Eur. J., 2020, 26, 927–938 CrossRef CAS PubMed; (c) Q. Zhanga and B.-F. Shi, Chem. Sci., 2021, 12, 841–852 RSC; (d) R. Gramage-Doria, Chem. – Eur. J., 2020, 26, 9688–9709 CrossRef CAS PubMed; (e) B. Li, M. Elsaid and H. Ge, Chem, 2022, 8, 1254–1360 CrossRef CAS; (f) T. A. Shah, T. Sarkar, S. Kar, P. K. Maharana, K. Talukdar and T. Punniyamurthy, Chem. – Asian J., 2024, 19, e202300815 CrossRef CAS PubMed.
- S. Zhou, J. Wang, L. Wang, C. Song, K. Chen and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 9384–9388 CrossRef CAS PubMed.
- (a) S. Keskin and M. Balci, Org. Lett., 2015, 17, 964–967 CrossRef CAS; (b) C. J. Lim, J. Y. Choi, B. H. Lee, K.-S. Oh and K.-Y. Yi, Chem. Pharm. Bull., 2013, 61, 1239–1247 CrossRef CAS; (c) M. Potowski, C. Golz, C. Strohmann, A. P. Antonchick and H. Waldmann, Bioorg. Med. Chem., 2015, 23, 2895–2903 CrossRef CAS PubMed.
- J. Wang, M. Wang, K. Chen, S. Zha, C. Song and J. Zhu, Org. Lett., 2016, 18, 1178–1181 CrossRef CAS PubMed.
- (a) F. Wang, L. Jin, L. Kong and X. Li, Org. Lett., 2017, 19, 1812–1815 CrossRef CAS PubMed; (b) P. Shi, L. Wang, K. Chen, J. Wang and J. Zhu, Org. Lett., 2017, 19, 2418–2421 CrossRef CAS PubMed.
- G. Liang, J. Rong, W. Sun, G. Chen, Y. Jiang and T.-P. Loh, Org. Lett., 2018, 20, 7326–7331 CrossRef CAS PubMed.
- (a) R. A. Novikov, V. P. Timofeev and Y. V. Tomilo, J. Org. Chem., 2012, 77, 5993–6006 CrossRef CAS PubMed; (b) R. S. Pathare, S. Sharma, K. Gopal, D. M. Sawant and R. T. Pardasani, Tetrahedron Lett., 2017, 58, 1387–1389 CrossRef CAS; (c) T. Itoh, K. Kude, S. Hayase and M. Kawatsura, Tetrahedron Lett., 2007, 48, 7774–7777 CrossRef CAS; (d) X.-D. Jia, P.-F. Li, Y. Shao, Y. Yuan, H.-H. Ji, W.-T. Hou, X.-F. Liu and X.-W. Zhang, Green Chem., 2017, 19, 5568–5574 RSC.
- Z. Wang and H. Xu, Tetrahedron Lett., 2019, 60, 664–667 CrossRef CAS.
- (a) C. Zhou, F. Fang, Y. Cheng, Y. Li, H. Liu and Y. Zhou, Adv. Synth. Catal., 2018, 360, 2546–2551 CrossRef CAS; (b) Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534–4542 CrossRef CAS.
- Z. Jiang, J. Zhou, H. Zhu, H. Liu and Y. Zhou, Org. Lett., 2021, 23, 4406–4410 CrossRef CAS PubMed.
- P. N. Reddy, P. Padmaja, B. R. Reddy and S. J. Singh, Med. Chem. Res., 2017, 26, 2243–2259 CrossRef.
- A. K. Pandey, D. Kang, S. H. Han, H. Lee, N. K. Mishra, H. S. Kim, Y. H. Jung, S. Hong and I. S. Kim, Org. Lett., 2018, 20, 4632–4636 CrossRef CAS.
- M. Liu, K. Yan, J. Wen, W. Liu, M. Wang, L. Wang and X. Wang, Adv. Synth. Catal., 2022, 364, 512–517 CrossRef CAS.
- A. Nagireddy, Dattatri, R. Kotipalli, J. B. Nanubolu and M. S. Reddy, J. Org. Chem., 2022, 87, 1240–1248 CrossRef CAS PubMed.
- Y. Li, X.-Y. Liu, Y.-J. Xu and L. Dong, Org. Chem. Front., 2019, 6, 2457–2461 RSC.
- D. Chen, L. Zhou, Y. Liu and J.-P. Wan, Chem. Commun., 2023, 59, 4036–4039 RSC.
- D. Chen, C. Wan, Y. Liu and J.-P. Wan, J. Org. Chem., 2023, 88, 4833–4838 CrossRef CAS.
- V. Suresh, M. N. Kumar, A. Nagireddy and M. S. Reddy, Adv. Synth. Catal., 2023, 365, 1770–1776 CrossRef CAS.
- C. Yang, X. Zhang and X. Fan, Org. Chem. Front., 2023, 10, 4282–4288 RSC.
- C. Yang, B. Li, X. Zhang and X. Fan, Org. Lett., 2024, 26, 6602–6607 CrossRef CAS PubMed.
- (a) F. Xie, S. Yu, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2016, 55, 15351–15355 CrossRef CAS; (b) T. Yuan, C. Pi, C. You, X. Cui, S. Du, T. Wan and Y. Wu, Chem. Commun., 2019, 55, 163–166 RSC; (c) Y. Liu, Y. Tian, K. Su, P. Wang, X. Guo and B. Chen, Org. Chem. Front., 2019, 6, 3973–3977 RSC.
- W. Wu, X. Wu, S. Fan and J. Zhu, Org. Lett., 2022, 24, 7850–7855 CrossRef CAS PubMed.
- X. Yu, K. Chen, Q. Wang, S. Guo, S. Zha and J. Zhu, Angew. Chem., Int. Ed., 2017, 56, 5222–5226 CrossRef CAS PubMed.
- Z. Yang, C. Liu, J. Lei, Y. Zhou, X. Gao and Y. Li, Chem. Commun., 2022, 58, 13483–13486 RSC.
- Z. Dong, P. Li, X.-W. Li and B.-X. Liu, Chin. J. Chem., 2021, 39, 2489–2494 CrossRef CAS.
- (a) Q.-C. Gao, Y.-F. Li, J. Xuan and X.-Q. Hu, Beilstein J. Org. Chem., 2023, 19, 100–106 CrossRef CAS PubMed; (b) Q. Wang, Y. Li, J. Sun, S. Chen, H. Li, Y. Zhou, J. Li and H. Liu, J. Org. Chem., 2023, 88, 5348–5358 CrossRef CAS PubMed; (c) M. Zhang, L. Chen, D. Liu, Z. Liu, J. Huang, X. Li and F. Yu, New J. Chem., 2023, 47, 12274–12278 RSC.
- W. Wu, S. Fan, X. Wu, L. Fang and J. Zhu, J. Org. Chem., 2023, 88, 1945–1962 CrossRef CAS PubMed.
- D. Kumar, P. S. Jadhavar, M. Nautiyal, H. Sharma, P. K. Meena, L. Adane, S. Pancholia and A. K. Chakraborti, RSC Adv., 2015, 5, 30819–30825 RSC.
- (a) J. Yang, X. Hu, Z. Liu, X. Li, Y. Dong and G. Liu, Chem. Commun., 2019, 55, 13840–13843 RSC; (b) K. Uehata, N. Kimura, K. Hasegawa, S. Arai, M. Nishida, T. Hosoe, K.-I. Kawai and A. Nishida, J. Nat. Prod., 2013, 76, 2034–2039 CrossRef CAS.
- S. Mondal, C. K. Giri and M. Baidya, Chem. Commun., 2023, 59, 13187–13190 RSC.
- P. Roy, D. Shrestha, M. S. Akhtar and Y. R. Lee, Org. Lett., 2024, 26, 142–147 CrossRef CAS PubMed.
- (a) A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS; (b) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 CrossRef CAS PubMed; (c) H. M. Gaber, M. C. Bagley, Z. A. Muhammad and S. M. Gomha, RSC Adv., 2017, 7, 14562–14610 RSC; (d) T. B. Poulsen, Acc. Chem. Res., 2021, 54, 1830–1842 CrossRef CAS PubMed; (e) L. Fu and J.-P. Wan, Tetrahedron Lett., 2023, 130, 154766 CrossRef CAS.
- (a) J. P. Wan, Y. Zhou, Y. Liu and S. Sheng, Green Chem., 2016, 18, 402–405 RSC; (b) G. Fang, J. Liu, J. Fu, Q. Liu and X. Bi, Org. Lett., 2017, 19, 1346–1149 CrossRef CAS; (c) J. Duan, G. C. Xu, B. Rong, H. Yan, S. Zhang, Q. Hangwu, N. Zhu and K. Guo, Green Synth. Catal., 2021, 2, 237–240 CrossRef; (d) V. Bodala, R. L. Podugu, K. Yettula, P. Gollamudi, S. Vidavalur and S. Pulipaka, Chem. – Asian J., 2023, 18, e202201004 CrossRef CAS.
- (a) A. Noole, M. Borissova, M. Lopp and T. Kanger, J. Org. Chem., 2011, 76, 1538–1545 CrossRef CAS PubMed; (b) J.-P. Wan, S. Cao, C. Hu and C. Wen, Asian J. Org. Chem., 2018, 7, 328–331 CrossRef CAS; (c) A. Ghosh, S. Upadhyay, D. J. Dahatonde, R. Kant and S. Batra, New J. Chem., 2024, 48, 6902–6910 RSC.
- (a) H.-Y. Liu, J.-R. Zhang, G.-B. Huang, Y.-H. Zhou, Y.-Y. Chen and Y.-L. Xu, Adv. Synth. Catal., 2021, 363, 1656–1661 CrossRef CAS; (b) L. Lu, X. Zhao, W. Dessie, X. Xia, X. Duan, J. He, R. Wang, Y. Liu and C. Wu, Org. Biomol. Chem., 2022, 20, 1754–1758 RSC.
- (a) C. Yuan, X. Huang, Y. Qiao, W. Guan, C. Liu, Z. Fang, Y. Li and K. Guo, Org. Chem. Front., 2023, 10, 2310–2317 RSC; (b) Z. Chen, G. Shi, W. Tang, J. Sun and W. Wang, Eur. J. Org. Chem., 2021, 951–955 CrossRef CAS; (c) D. Li, L. Chen, Y. Jin, X. Wang, L. Liu, Y. Li, G. Chen, G. Wu, Y. Qin, L. Yang, M. Wang, L. Zhao, Z. Xu and J. Wen, Green Chem., 2023, 25, 4656–4661 RSC.
- (a) J.-P. Wan, Y. Zhou and S. Cao, J. Org. Chem., 2014, 79, 9872–9877 CrossRef CAS PubMed; (b) F. Ahmad, P. K. Ranga, S. Fatma, A. Kumar and R. V. Anand, Adv. Synth. Catal., 2023, 365, 3271–3276 CrossRef CAS; (c) S. Zhou, S.-F. Dong, X. Zhang, S.-Y. Zhang, T.-P. Loh and J.-S. Tian, Org. Chem. Front., 2024, 11, 100–105 RSC; (d) X. Zhang, L. Song, Y. Jin and K. Luo, ACS Catal., 2024, 14, 13509–13519 CrossRef CAS.
- J.-P. Wan, D. Hu, Y. Liu, L. Li and C. Wen, Tetrahedron Lett., 2016, 57, 2880–2883 CrossRef CAS.
- D. C. Mohan, R. R. Donthiri, S. N. Rao and S. Adimurthy, Adv. Synth. Catal., 2013, 335, 2217–2221 CrossRef.
- M. O. Akram, S. Bera and N. T. Patil, Chem. Commun., 2016, 52, 12306–12309 RSC.
- U. Dutta, D. W. Lupton and D. Maity, Org. Lett., 2016, 18, 860–863 CrossRef CAS PubMed.
- T. Luo, J.-P. Wan and Y. Liu, Org. Chem. Front., 2020, 7, 1107–1112 RSC.
- L. Fu, Z. Xu, J.-P. Wan and Y. Liu, Org. Lett., 2020, 22, 9518–9523 CrossRef CAS PubMed.
- D. Cao, J.-P. Wan and Y. Liu, Adv. Synth. Catal., 2024, 366, 3670–3675 CrossRef CAS.
- K. Luo, S. Mao, K. He, X. Yu, J. Pan, J. Lin, Z. Shao and Y. Jin, ACS Catal., 2020, 10, 3733–3740 CrossRef CAS.
- (a) Z. Liu, P. Sivaguru, G. Zanoni, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2018, 57, 8927–8931 CrossRef CAS; (b) L. Kotzner, M. Leutzsch, S. Sievers, S. Patil, H. Waldmann, Y. Y. Zheng, W. Thiel and B. List, Angew. Chem., Int. Ed., 2016, 55, 7693–7967 CrossRef; (c) T. Hashimoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 8834–8837 CrossRef CAS PubMed.
- S. Zhou, D.-Y. Liu, S. Wang, J.-S. Tian and T.-P. Loh, Chem. Commun., 2020, 56, 15020–15023 RSC.
- (a) T. Zhou, Y. Wang, B. Li and B. Wang, Org. Lett., 2016, 18, 5066–5069 CrossRef CAS PubMed; (b) B. Jiang, M. Z. Shu, S. L. Yun, H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2018, 57, 555–559 CrossRef CAS; (c) S. Jambu, R. Sivasakthikumaran and M. Jeganmohan, Org. Lett., 2019, 21, 1320–1324 CrossRef CAS PubMed.
- L. Fu, W. Xu, M. Pu, Y.-D. Wu, Y. Liu and J.-P. Wan, Org. Lett., 2022, 24, 3003–3008 CrossRef CAS PubMed.
- S. R. Neufeldt, G. Jiménez-Osés, J. R. Huckins, O. R. Thiel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 9843–9854 CrossRef CAS PubMed.
- Dattatri, M. K. R. Singam, J. B. Nanuboluc and M. S. Reddy, Org. Biomol. Chem., 2022, 20, 6363–6367 RSC.
- K. Chen, J. Lv, J. Chen, J. Zhang, L. Li, M. Zhao and Y. Jiang, Org. Lett., 2023, 25, 4688–4693 CrossRef CAS PubMed.
- V. Mouriès-Mansuy and L. Fensterbank, Isr. J. Chem., 2018, 58, 586–595 CrossRef.
- (a) J. Chen, P. Guo, J. Zhang, J. Rong, W. Sun, Y. Jiang and T.-P. Loh, Angew. Chem., Int. Ed., 2019, 58, 12674–12679 CrossRef CAS; (b) J. Chen, J. Han, J. Zhang, L. Li, Z. Zhang, Y. Yang and Y. Jiang, ACS Catal., 2022, 12, 14748–14753 CrossRef CAS.
- Y. Wang, Y. Yu, Y. Yu, F. Huang and J. B. Baell, Adv. Synth. Catal., 2023, 365, 2601–2606 CrossRef CAS.
- X. Lei, J. Feng, Q. Guo, Y. Li and J. Shi, Org. Lett., 2023, 25, 7338–7343 CrossRef CAS PubMed.
- M. Zhang, L. Chen, H. Sun, Z. Liu, S.-J. Yan and F. Yu, Org. Lett., 2023, 25, 7214–7219 CrossRef CAS PubMed.
- S. Fan, W. Wu, Y. Su, X. Han, Z. Wang and J. Zhu, Org. Lett., 2024, 26, 7620–7625 CrossRef CAS PubMed.
- T. Gläsel, B. N. Baumann and M. Hapke, Chem. Rec., 2021, 21, 3727–3745 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |