
Recent developments in difunctionalization of unsaturated hydrocarbons with organosilicon reagents
Jiawei
Mao
ab,
Ming
Chen
*a,
Yao
Zhong
b and
Ren-Jie
Song
 *bc
*bc
aCollege of Bioengineering, Dalian Polytechnic University, Dalian 116034, China. E-mail: chenming@dlpu.edu.cn
bKey Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resource Reuse, Nanchang Hangkong University, Nanchang 330063, China. E-mail: srj0731@hnu.edu.cn
cState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, China
First published on 29th October 2024
Abstract
Organosilicon compounds have attracted considerable attention because of their special biological activities. Direct difunctionalization of unsaturated hydrocarbons with organosilicon reagents for the efficient construction of synthetically valuable silicon-functionalized compounds are featured with a high step and atom economy, which could form carbon–silicon/carbon–carbon bonds or carbon–silicon/carbon-hetero bonds in one step. This review summarizes the recent advances on this topic based on different unsaturated hydrocarbons along with typical examples and mechanisms.
1. Introduction
During the past few decades, the introduction of silicon groups to unsaturated hydrocarbons for the efficient construction of synthetically valuable silicon-functionalized compounds has attracted great attention from chemists mainly because silicon-containing compounds exhibit special physical and chemical properties.1 The ability of silicon-containing molecules to undergo a range of organic transformations is well documented, including the Hosomi–Sakurai allylation, Hiyama–Denmark cross-coupling, and Brook rearrangement.2 Many organosilicon compounds display diverse biological activities, making them an emerging and leading research field in the pharmaceutical industry.3 For example, a camptothecin derivative, karenitecin (BNP 1350), substituted by silane at the C7 position, which is designed by the BioNumerik company, has good lipophilicity and greatly improved anti-cancer activity and plasma stability (Scheme 1).4 In the context of industry, silicon-containing molecules also occupy a significant position. For example, oligosilanes, such as silicon carbide, serve as precursors for the synthesis of numerous high-value materials, and cationic silicone Lewis acids offer a unique set of advantages as catalysts for organic conversions.5 Traditionally, these valuable compounds are synthesized by hydrosilylation of unsaturated bonds, free-radical-promoted C–Si formation, transition-metal-catalyzed C–H silylation, C–Si bond activation via a direct transition metal insertion and so on.6 | ||
| Scheme 1 Selected drugs comprising silicon motif. | ||
In recent years, the difunctionalization of unsaturated hydrocarbons with organosilicon reagents has developed into a powerful tactic for the generation of highly valued silicon-containing compounds. In this case, new carbon–silicon bonds/carbon–carbon bonds or carbon–silicon/carbon-heteroatom bonds were formed in a single operation, which dramatically shortens the synthetic steps. Yoshihiko Ito's work in 1991 represents an early example of the silylation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.
C bonds.
Recently, different reaction types of difunctionalization of unsaturated hydrocarbons with organosilicon reagents have been reported, including 1,2-bis-silylation, 1,2-arylsilylation, 1,2-silacarboxylation, 1,2-hydroxysilylation, 1,2-aminosilylation, 1,2-acyl silylation, cyclization and cycloaddition. This review outlines recent efforts to investigate difunctionalization reactions between organosilicon reagents and unsaturated compounds. Meanwhile, the hydroxysilylation of alkenes is reviewed in this section, and we focus on the other functional groups, except for H.
2. Simple alkenes as substrates
In the past few years, alkene hydrosilylation has emerged as a powerful and atom-economic tool to produce alkylsilanes.7 Transition metal-catalyzed hydrosilylation of unsaturated hydrocarbons was the most representative method for the synthesis of organosilicon reagents, which adds H and SiR3 groups to unsaturated carbon–carbon bonds. Recently, the construction of more complex and valuable silicon-functionalized derivatives via cascade functionalization reactions has attracted significant scientific interest. In 1991, Ito's group reported a palladium-catalyzed intramolecular bis-silylation of CC bonds.8 After that, the addition of a silicon group and another functional group to unsaturated hydrocarbons has been simultaneously demonstrated to be an efficient method for the construction of functionalized organosilicon compounds. Moreover, most of the approaches involved a radical process.
A copper-catalyzed radical 1,2-aryl migration in α,α-diaryl allylic alcohols with silanes was initially reported by Cheng and co-workers in 2015 (Scheme 2).9 In this transformation, 10 mol% of Cu2O was utilized as the catalyst, 4 equivalents of di-tert-butyl peroxide (DTBP) were used as the oxidant, and 0.25 equivalents of Et3N was used as the additive, providing a convenient method for the preparation of diverse β-silyl carbonyl compounds.
 | ||
| Scheme 2 Copper-catalyzed radical 1,2-aryl migration in α,α-diaryl allylic alcohols with silanes. | ||
In 2017, our group presented an iron-catalyzed 1,2-difunctionalization of styrenes and conjugated alkenes with silanes and nucleophiles by applying DTBP as an oxidant combined with a suitable iron precursor (Scheme 3).10 This catalytic system worked well for the 1,2-aminosilylation, 1,2-arylsilylation, and 1,2-alkylsilylation of alkenes to produce silicon-containing alkanes. In this transformation, amines, amides, indoles, pyrroles, and 1,3-dicarbonyls could be used as nucleophiles. In this regard, it is worth mentioning that the conjugated alkenes were suitable substrates. However, the aliphatic alkenes could not be converted to the corresponding silicon-containing alkanes based on this catalytic system.
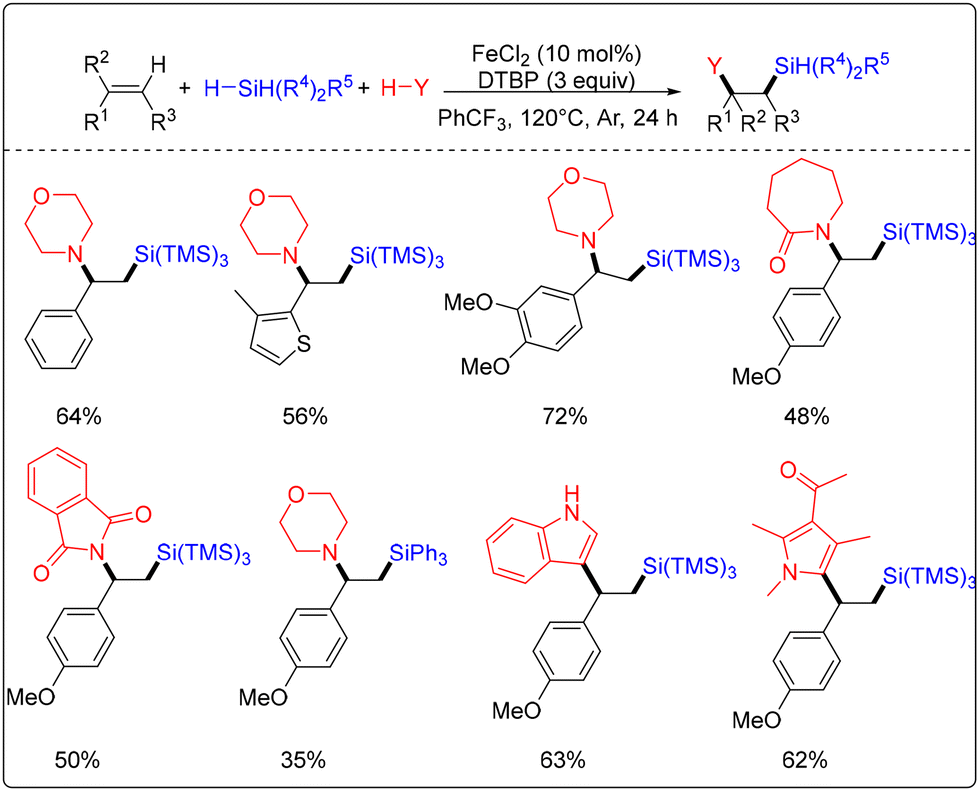 | ||
| Scheme 3 Iron-catalyzed 1,2-difunctionalization of styrenes and conjugated alkenes with silanes. | ||
The catalytically active species and proposed intermediates are shown in Scheme 4. A tert-butoxyl radical was formed from DTBP with the aid of FeII species under heating and gave the FeIII(tBuO) species. The silicon-centered radical was constructed with an active tert-butoxyl radical. The silicon-centered radical reacted with the CC bond of alkenes to obtain the alkyl radical intermediate. Oxidation occurred by the FeIII(tBuO) species to deliver the alkyl cation intermediate. Finally, the reaction between nucleophiles provided the corresponding desired products.
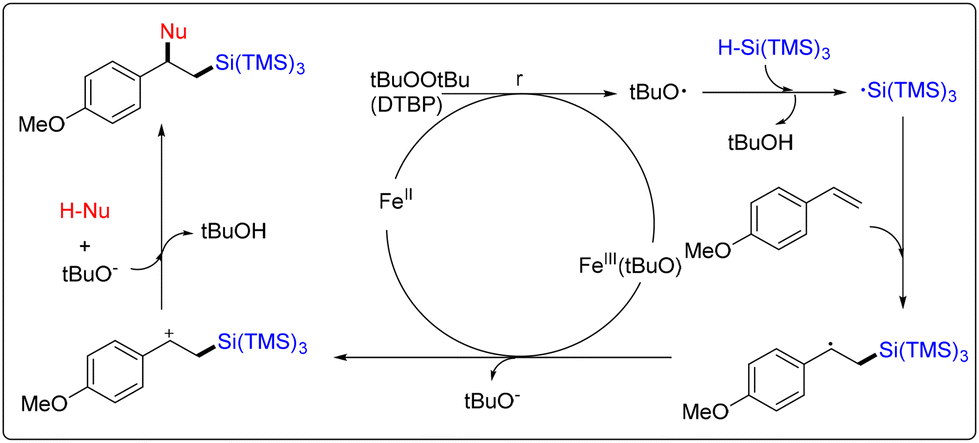 | ||
| Scheme 4 Possible mechanism of iron-catalyzed 1,2-difunctionalization of styrenes and conjugated alkenes with silanes. | ||
Wu and co-workers managed to synthesize various β-silacarboxylic acids under the combination of photoredox and hydrogen atom transfer (HAT) catalysis (Scheme 5).11 The reaction is operationally simple and does not require the presence of any external oxidant or transition metals. In the case of 18 W blue LED irradiation at ambient temperature with CO2 at atmospheric pressure, the method is tolerant of a wide range of functional groups. It was noted that the strategy was extended to the carbocarboxylation of alkenes using unprefunctionalized C(sp3)–H bonds to obtain γ-amino acids.
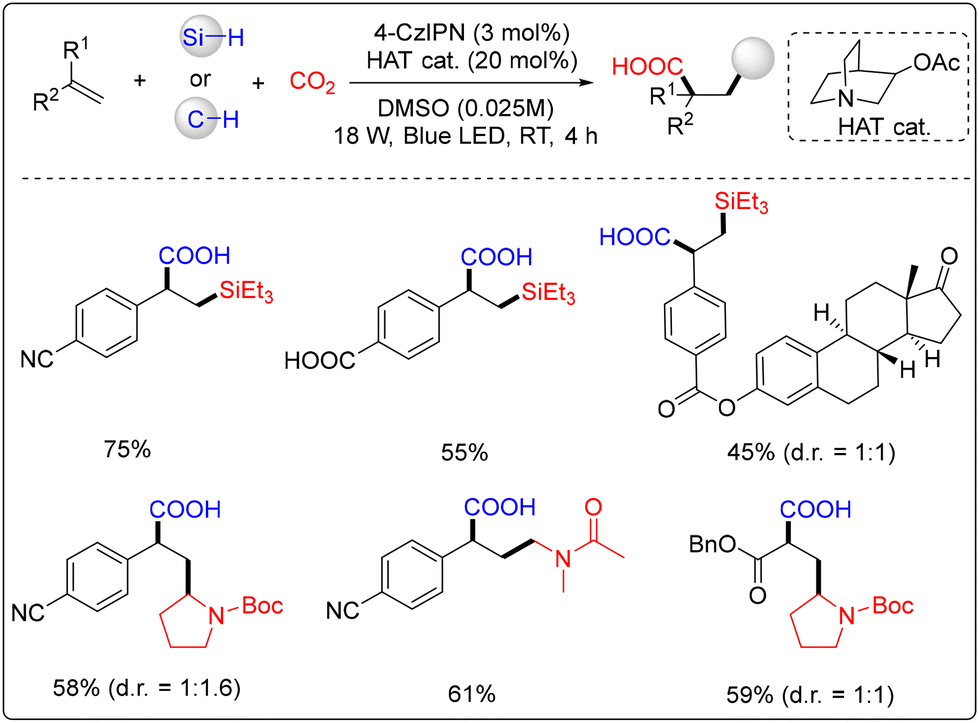 | ||
| Scheme 5 Visible-light-mediated metal-free difunctionalization of alkenes with CO2 and silanes. | ||
Subsequently, Zhu and co-workers used unactivated alkenes as substrates to directly synthesize heteroaryl-substituted alkyl silanes via an intramolecular heteroaryl migration process (Scheme 6).12 The optimal reaction conditions were CuO (10 mol%) as the catalyst, MeCO3tBu (4 equiv.) as the oxidant, and DMAP (25 mol%) as the base under N2 atmosphere in benzene at 130 °C for 15 h. The reaction was tolerant of a wide range of heteroaryl groups, such as the benzothiazolyl, thiazolyl, imidazolyl, and pyridyl groups.
 | ||
| Scheme 6 Copper-catalyzed heteroarylsilylation of unactivated olefins through distal heteroaryl migration. | ||
In 2019, palladium(0)-catalyzed directed 1,2-carbosilyation between alkenes and PhMe2Si-Bpin (Suginome's reagent) was reported by the Engle group (Scheme 7).13 After optimizing the reaction conditions, the authors found that the optimal conditions were acquired when the reaction was carried out at 100 °C using Pd2dba3 as the catalyst, Cy-JohnPhos as the ligand, and i-Pr2Net and 4 Å MS as additives. In this case, alkene substrates and electrophiles were examined for this 1,2-carbosilyation.
 | ||
| Scheme 7 Palladium(0)-catalyzed directed 1,2-carbosilyation between alkenes and PhMe2Si-Bpin. | ||
In the same year, Liu and co-workers demonstrated a radical-initiated asymmetric 1,2-aminosilylation of an alkene with a hydrosilane by the use of Cu(I)/CPA cooperative catalysis (Scheme 8).14 They synthesized a series of silicon-containing azaheterocycles, including pyrrolidine, indoline and isoindoline, bearing an α-tertiary stereocenter with high enantioselectivity. In this work, the authors believe that the silyl group in this transformation stabilizes the generated carbocation intermediate by the β-silicon effect.
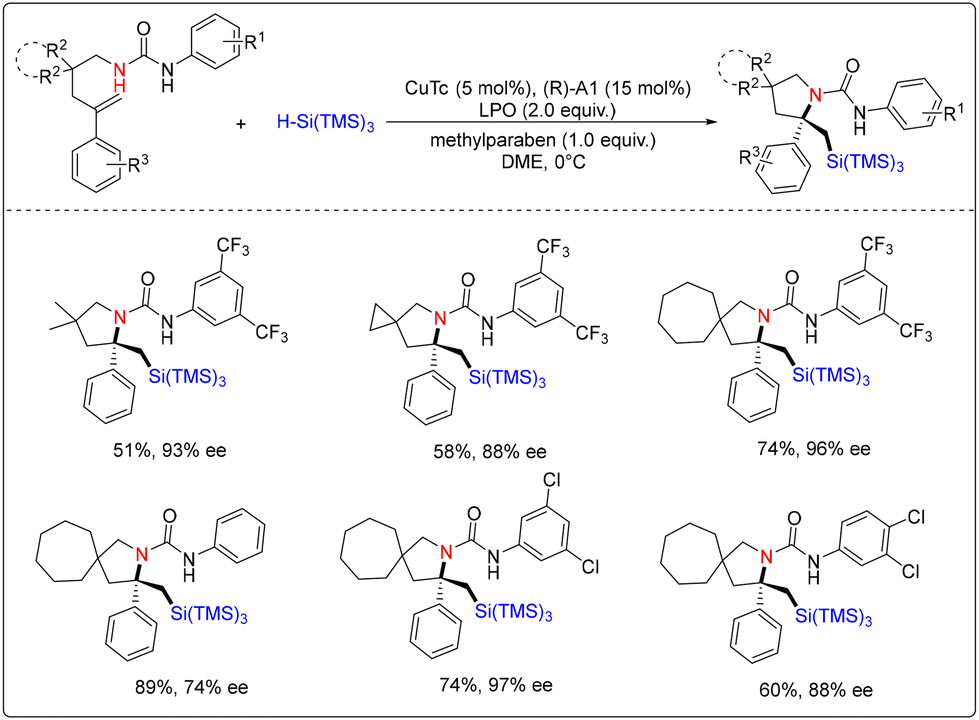 | ||
| Scheme 8 Cu/chiral phosphoric acid-catalyzed radical-initiated asymmetric aminosilylation of alkene with hydrosilane. | ||
Hu and co-workers reported an arylsilylation reaction between electron-deficient terminal alkenes with aryl bromides and TMS3SiH (Scheme 9).15 In this case, they used cooperative photoredox [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and nickel catalysis Ni(COD)2 as the catalytic system. This conversation occurs through a photogenerated silyl radical intermediate, arylation, via Ni-catalyzed cross coupling processes. Notably, this reaction featured mild conditions, broad scope and high functional group tolerance.
 | ||
| Scheme 9 Photoredox and nickel catalysis promoted arylsilylation of electron-deficient alkenes. | ||
In 2019, Wang, Liang, Zhao and co-workers described a visible-light-driven silylative cyclization of aza-1,6-dienes, offering a direct route to various polysubstituted piperidines (Scheme 10).16 In this process, treating 1,6-dienes in the presence of eosin Y, thiol and base additives enabled the radical silylative cyclization reaction to afford the silicon-containing piperidines with cis-stereoselectivity. Remarkably, excellent diastereoselectivity was observed when 1,6-dienes bear a trisubstituted electron-neutral olefin. A density functional theory (DFT) study revealed that a transition state with an ester or cyano group in the axial position of the newly forming six-membered ring might be involved, and the diastereoselectivity was reduced by increasing 1,3-diaxial repulsion as well as the lack of hydrogen-bonding interaction.
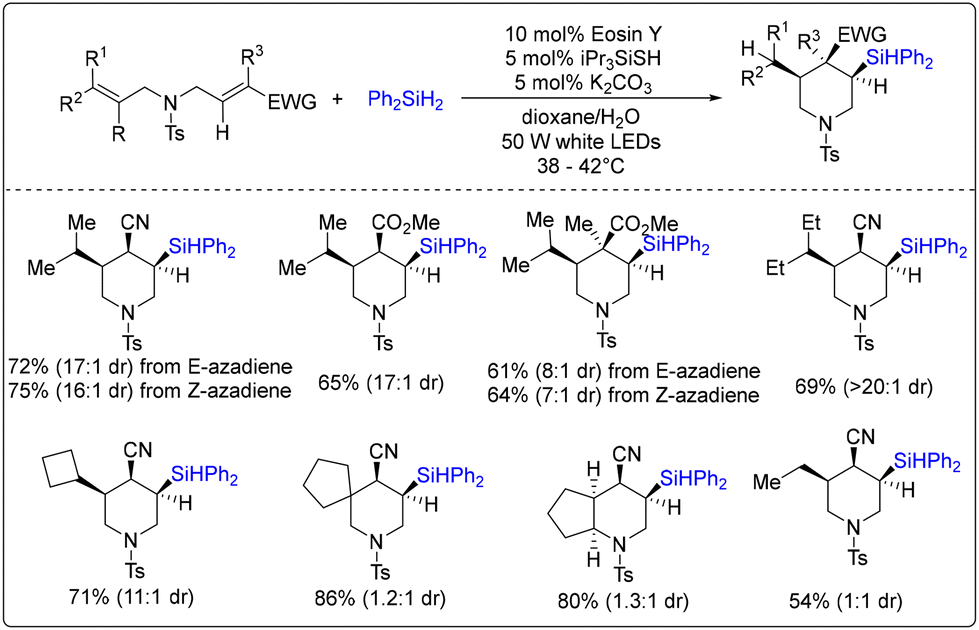 | ||
| Scheme 10 Visible-light-driven radical silylative cyclization of aza-1,6-dienes. | ||
In 2020, Fujihara et al. demonstrated a protocol for β,γ-unsaturated ketones with a (dimethylphenylsilyl)methyl moiety at the α-position via Cu-catalyzed three-component reactions between 1,3-dienes, nitriles and silylboranes (Scheme 11).17 In this protocol, 1,2-difunctionalization occurred between 1,3-dienes, and various nitriles and silylboranes in the presence of copper catalysts to yield the β,γ-unsaturated ketones in 23%–99% yields.
 | ||
| Scheme 11 Cu-catalyzed three-component coupling reactions using nitriles, 1,3-dienes and silylboranes. | ||
Yin's research group employed a bimetallic Co-catalytic strategy based on nickel and copper to complete the arylsilylation of terminal olefins (Scheme 12).18 The reaction was characterized by a simple reaction bar and excellent regioselectivity. By investigating the reaction mechanism, the researchers proposed that the ligand introduced during the reaction served as the pivotal factor in the inhibition of the generation of C-Heck reaction by-products. Additionally, they posited that the copper catalyst plays a role in promoting the metal conversion process of the silyl reagent. It was demonstrated that the spatial resistance of the ligand's oxazoline component significantly impacts the chain-walking process. However, the precise mechanism underlying this phenomenon remains to be elucidated.
 | ||
| Scheme 12 Ni/Cu-catalyzed arylation–silylation of terminal olefins and mechanistic diagrams. | ||
In 2020, Uchiyama's group reported the inaugural instance of the decarboxylative addition of a silyl carboxylic acid with an olefin via a photocatalytic strategy. The strategy exhibits broad substrate adaptability and high yields, with the capacity to maintain high yields even when scaled up to the gram scale. Notably, in the absence of water, the carboxyl group acts as a source of hydrogen, while in the presence of water, water becomes a new hydrogen source (Scheme 13).19 Consequently, this strategy can be employed to perform the silylation-deuteration reaction of olefins with D2O as the deuterium source.
 | ||
| Scheme 13 Photocatalytic decarboxylative silylation of olefins with silyl carboxylic acids. | ||
Studer's group employed a photoredox strategy to cleave the Si–Si bond of trimethylsilyl–polysilyl groups, thereby forming silyl radicals. These were then added to the electron-deficient olefins, resulting in the formation of a range of trisilanes (Scheme 14).20 The versatility of this strategy, coupled with its relative simplicity, proves invaluable for synthesizing oligosilanes.
 | ||
| Scheme 14 Photocatalytic silane hydrosilylation reaction with electron-deficient olefins. | ||
Lin's research group devised a strategy for the electrocatalytic bisilylation reaction of olefins (Scheme 15).21 In the mechanism put forth by the researchers, chlorosilane, acting as the reaction substrate, is cleaved into silane radicals at the elevated reduction potential of the negative electrode. This generates radical intermediates following the addition of olefins. The intermediate is subsequently oxidised by the cathode material, resulting in the formation of an anionic intermediate. Ultimately, nucleophilic substitution occurs in chlorosilane, leading to the formation of the target product.
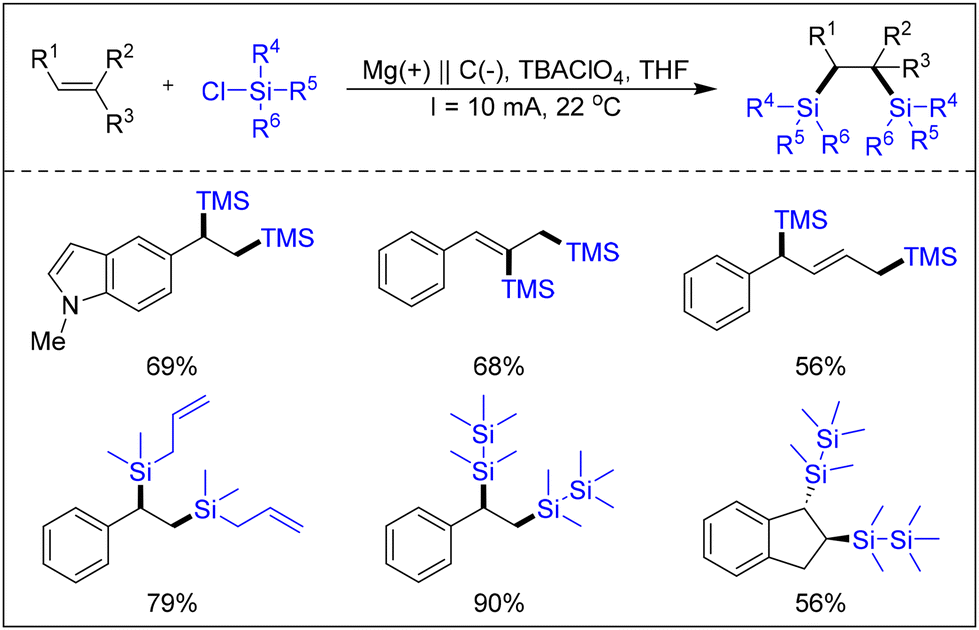 | ||
| Scheme 15 Electrocatalytic bisilylation of silanes with olefins. | ||
In 2021, He's group achieved the silylation oxidation of electron-deficient olefins by electrochemically activating the Si–H bond to obtain silyl radicals (Scheme 16).22 This strategy is characterized by its mildness, the absence of the need for transition metal catalysts and chemical oxidants, and the attainment of favorable yields, along with excellent chemoselectivity and regioselectivity.
 | ||
| Scheme 16 Electrocatalytic silylation oxidation of activated olefins with hydrosilanes and N-hydroxyphthalimides. | ||
Li's group successfully achieved the 1,2-silyl functionalisation of three-component olefins by photoreduction utilising (TMS)3SiH as a source of silyl radicals (Scheme 17).23 The catalytic system exhibits excellent compatibility with a wide range of nucleophilic reagents, including alcohols, H2O, thiols, and indoles, delivering high yields.
 | ||
| Scheme 17 Photocatalytic silanization of olefins with silanes. | ||
Hajra's group also synthesized 3-(2-(1,1,1,3,3,3-hexamethyl-2(trimethylsilyl)trisilan-2-yl)-1-phenylethyl)-2-phenylimidazo [1,2-a] pyridine derivatives for the first time by applying a photocatalytic/Fe-catalytic synergistic method using (TMS)3SiH as the silicon source (Scheme 18).24 The process is mild, efficient, high-yield, and a one-pot synthesis of the corresponding products, which is economically advantageous.
 | ||
| Scheme 18 Photocatalytic/Fe-catalysed synergistic three-component silylation of olefins. | ||
Brown's research group developed a novel silylation acylation reaction of olefins (Scheme 19).25 This reaction employs olefins, acid chlorides, and ClZnSiR3 as reaction substrates and NiBr2 as the catalyst. The method employs [Ni]-SiR3 as an intermediate to induce the reaction, which is a highly unusual occurrence. Additionally, the researchers conducted further functionalization studies on the β-silicone products obtained from the reaction and discussed the comprehensive application pathways of these products.
 | ||
| Scheme 19 Nickel-catalyzed three-component silylation acylation of olefins with silicon reagents and acid chlorides. | ||
A strategy for the silylation of olefins with silicoboronates and organofluorides, conducted without the use of a catalyst, was reported by Shibata's research group (Scheme 20).26 The reaction conditions of this strategy are notably mild, allowing for the synthesis of a diverse range of β-functionalized silyl compounds through the addition of 4 equivalents of bases and a 2.5-hour reaction at room temperature. Furthermore, the researchers have synthesized silyl compounds with quaternary carbon centers using this strategy. Additionally, partial reactions of 1,3-dienes have been achieved, demonstrating this strategy's excellent functional group tolerance and stereoselectivity. The researchers concluded that the selective activation of the C–F bond is crucial in enabling the reaction to proceed successfully. Furthermore, this strategy can potentially expand the application of organosilicon compounds in various fields.
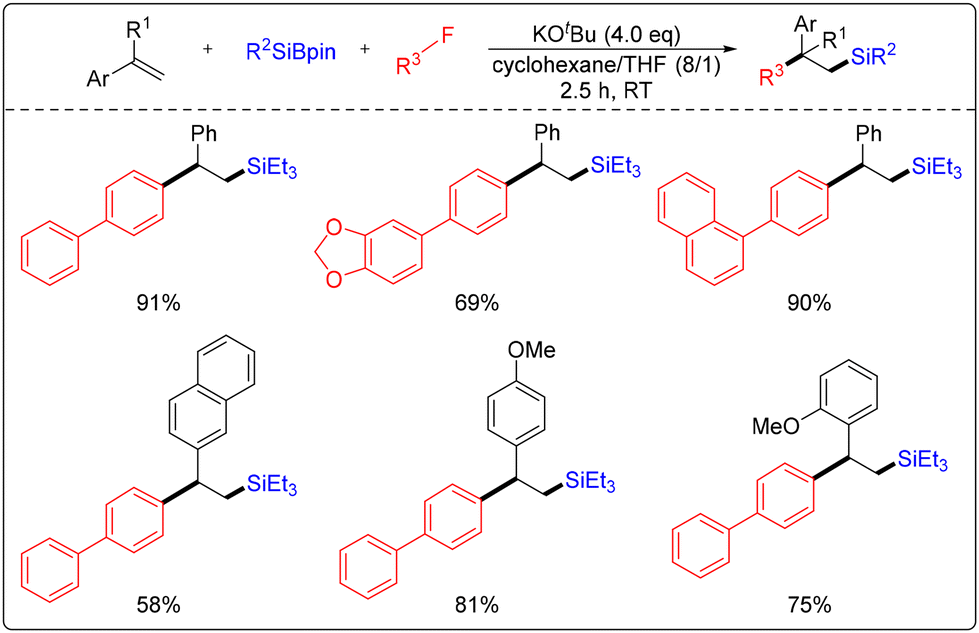 | ||
| Scheme 20 Three-component silylation of olefins with silicoboron hydrochloric acid and organofluorides. | ||
In 2022, Wang's group used a photocatalytic/Fe-catalytic synergistic catalytic strategy to achieve the hydrogenosilylation reaction of electron-deficient olefins (Scheme 21).27 The reaction has a wide range of substrates, and many structurally complex natural cyclic compounds can be synthesized using this strategy. Through mechanism validation experiments, the researchers also demonstrated that the chloride ions generated by the ligand-to-metal charge transfer (LMCT) process play a role in promoting the formation of silyl radicals.
 | ||
| Scheme 21 Photocatalytic/Fe-catalysed hydrosilane alkylation of electron-deficient olefins. | ||
Li's group reported a nickel-catalyzed three-component silylation addition reaction of olefins with zinc–silicon reagents and halogenates (Scheme 22).28 The lithium silicate reagent was transformed into zinc silicate through transmetallation, acting as the reaction precursor. Subsequently, the nickel catalyst was introduced with the remaining two reactants, and dimethyl sulfoxide was employed as the reaction solvent, resulting in the target product after 16 hours at room temperature. This strategy is characterized by a high degree of mildness and ease of operation, effectively addressing the shortcomings of the zinc silicate reagent: low reactivity, and difficult handling.
 | ||
| Scheme 22 Nickel-catalyzed three-component addition reactions of olefins with silica-zinc reagents and halides. | ||
Oestreich's research group reported a selective addition reaction of alkynylsilanes with unactivated terminal olefins (Scheme 23).29 The reaction was completed in 16 hours at room temperature with the terminal olefin acting as the acceptor reagent, alkynylsilanes as the carbon-nucleophilic and silicon-electrophilic reagents, Me3Si(HCB11H5Br6) as the initiator, and chlorobenzene as the reaction solvent. This represents a previously unreported strategy for the two-component construction of C(sp3)–C(sp) bonds and C(sp3)–Si bonds, exhibiting excellent regioselectivity and atom economy.
 | ||
| Scheme 23 Addition reactions of terminal olefins with alkynylsilanes. | ||
Jing's group achieved the synthesis of ketodifluoropropylsilanes without the involvement of transition metals using hydrosilanes as the silicon source through a photoredox/HAT strategy (Scheme 24).30 This method has excellent functional group tolerance and allows for the late modification of bioactive molecules.
 | ||
| Scheme 24 Photocatalytic defluorosilylation of α-trifluoromethylstyrene with hydrogenated silanes. | ||
Lin's group also devised a photo/HAT dual catalysis methodology for the silylation arylation of olefins (Scheme 25).31 The corresponding organosilicon compounds were synthesized under visible light irradiation via a radical–radical coupling process utilizing olefins, hydrosilanes, and aryl cyanides as substrates. Notably, the reaction can be conducted without adding an organic photocatalyst (PC) due to the formation of an electron donor–acceptor (EDA) complex through the mutual coordination of the components.
 | ||
| Scheme 25 Photocatalytic reaction of arylsilylation of alkenes with hydrosilanes. | ||
Furthermore, Ohmiya's research group documented a photocatalyzed three-component silylation addition reaction (Scheme 26).32 The researchers employed visible light to generate silicon radicals, and introducing a mild base to the system also enhanced this process. Adding silicon radicals to olefins results in the formation of radical intermediates, which are subsequently coupled with acyl radicals to form the target product. This strategy effectively addresses the challenge of completing the acyl silylation reaction of olefins through NHC radical binding. It successfully facilitates the silylation acylation reaction of olefins using a photocatalytic method.
 | ||
| Scheme 26 Photocatalytic three-component silylation of olefins. | ||
Li and his team successfully employed the silylpyridylation of olefins using silylboronate as the silicon source and p-cyanopyridine as the acceptor reagent (Scheme 27).33 This strategy has a broad substrate scope and good functional group tolerance, allowing the synthesis of a diverse range of drug and bioactive molecules. The computational and experimental evidence provided by the authors demonstrates that p-cyanopyridine, when employed in this strategy, can induce not only the breakage of Si–B bonds, resulting in the formation of silyl radicals, but also the homolytic cleavage of B–B bonds in B2pin2, leading to the generation of pyridinyl-boronyl radicals. This will provide further guidance for future related research.
 | ||
| Scheme 27 Photocatalytic reaction of aryl olefins with 1,2-silylpyridylation of silylborates. | ||
Zhang's group successfully completed the three-component silylation of olefins using nickel catalysis (Scheme 28).34 The researchers completed the alkylation-silylation reaction with high regioselectivity by employing readily available alkyl bromo-substituents and chlorosilanes as electrophilic reagents, along with acrylonitrile as the acceptor reagent. The method is distinguished by its low cost, good functional group tolerance, and the ability to synthesize bioactive molecules, which renders the strategy a promising prospect for practical industrial applications.
 | ||
| Scheme 28 Nickel-catalyzed silylation of olefins with alkyl bromo substituents and chlorosilanes. | ||
In 2023, Shu's group also reported a nickel-catalyzed three-component silylation reaction of olefins (Scheme 29).35 The utilization of 1,2-diene, aryl bromo substituents and chlorosilanes as reaction substrates enabled the attainment of 1,2-linear silylation products through nickel catalysis. The reaction mechanism is markedly distinct from the conventional one, wherein the chlorosilane initially reacts with the 1,3-diene and subsequently couples with the aryl bromo substituent rather than forming the traditional metal–allyl intermediate to complete the reaction. This work demonstrates the feasibility of non-radical three-component cross-electrophilic reactions.
 | ||
| Scheme 29 Nickel-catalyzed arylation–silylation of three components and a mechanistic diagram. | ||
In 2024, Hajra's research group reported a ruthenium/iron Co-catalysed three-component arylation–silylation reaction strategy for olefins (Scheme 30).36 This strategy achieves the activation of C(sp2)–H bonds, and the researchers provided experimental and computational evidence to elucidate the underlying reaction mechanism. By employing diffusion-corrected density generalized spectral theory, the researchers investigated the selectivity and potential mechanistic pathways of synergistic 3d/4d transition metal catalysis, which was absent in previous studies.
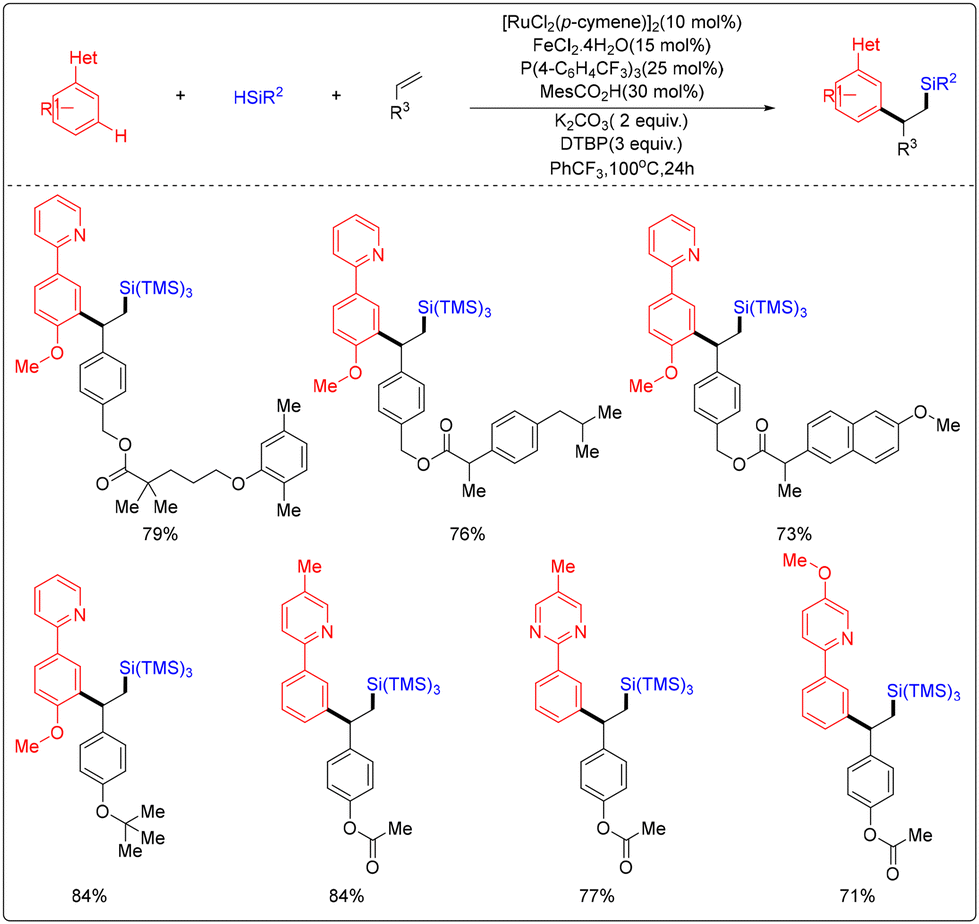 | ||
| Scheme 30 Synergistic catalytic arylation–silylation of three components by two metals, representing a significant advancement in the field of chemical synthesis. | ||
3. Acrylamides as substrate
Oxindoles and quinolinones are more common structural scaffolds widely found in biologically active molecules and natural products.37 Radical cascade addition/cyclization reactions of acrylamides are straightforward and effective methods for synthesizing such compounds.38In 2015, Liu and co-workers developed the first example of copper-catalyzed free radical cascade silylarylation of N-arylacrylamide with silanes (Scheme 31).39 First, a cumyloxyl radical was formed from DCP through a single-electron reduction by Cu(I) delivery. Subsequently, the silyl radical was obtained through H atom abstraction by the cumyloxyl radical and/or methyl radical generated from the β-cleavage of the cumyloxyl radical. The addition of the silyl radical to the alkene, followed by cyclization to the aromatic core, produced another radical intermediate. Deprotonation of the radical intermediate and release of an electron finally afforded the silylated oxindole (Scheme 32). In this case, a series of silylated oxindoles can be obtained in moderate to high yields, and this reaction tolerates functional groups, including alkyl, phenyl, methoxyl, and halogen groups.
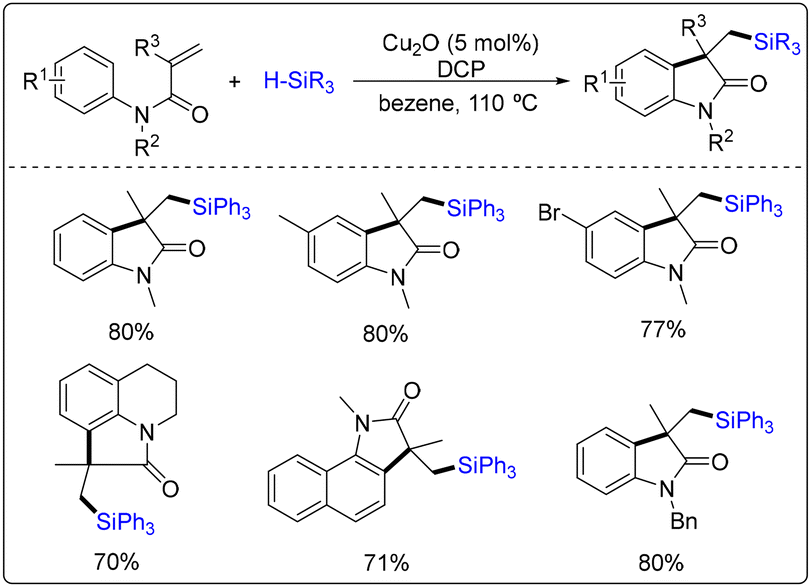 | ||
| Scheme 31 Copper-catalyzed free radical cascade silylarylation of N-arylacrylamide with silanes. | ||
 | ||
| Scheme 32 Possible mechanism of radical cascade silylarylation of N-arylacrylamide. | ||
Our group used tertiary silanes as a silicon radical precursor for the radical heteroannulation of N-(2-cyanoaryl)-acrylamides (Scheme 33).40 The use of DTBP as an oxidant was shown to be necessary. The yield of this cascade could be increased using a catalytic amount of MnCl2 as a Lewis acid. Various N-(2-cyanoaryl)acrylamides were competent for forming 1,3-azasiline-fused quinolinones through silyl C(sp3)–H functionalization using an oxidative radical strategy. The scope of this transformation with respect to the silane is restricted to tertiary silanes with an aryl group, in which tertiary silanes with three linear alkyl groups were not compatible with the optimal conditions.
 | ||
| Scheme 33 Mn-promoted intermolecular oxidative radical heteroannulation of N-(2-cyanoaryl)-acrylamides and tertiary silanes. | ||
Using similar N-(2-cyanoaryl)-acrylamides as substrates, Sun and co-workers later also achieved radical cascade addition/cyclization reactions for the synthesis of silyl-functionalized pyrido[4,3,2-gh]phenanthridin derivatives under metal-free conditions (Scheme 34).41 Their efficient alkene difunctionalization protocol features a broad substrate scope and provides the desired products with 49%–81% yields under metal-free conditions.
 | ||
| Scheme 34 Silyl radical-initiated radical cascade addition/cyclization of N-arylacrylamides. | ||
Very recently, Jia and co-workers achieved copper-catalyzed arylsilylation of N-(2-iodoaryl)acrylamides with PhMe2Si-Bpin using CuOAc as the sole catalyst (Scheme 35).42 It is worth noting that the reaction occurred at room temperature through intermolecular olefin silylcupration, followed by the intramolecular coupling of an alkyl-Cu intermediate with aryl iodide. A range of silylated 3,3′-disubstituted oxindoles is obtained in 52%–90% yields.
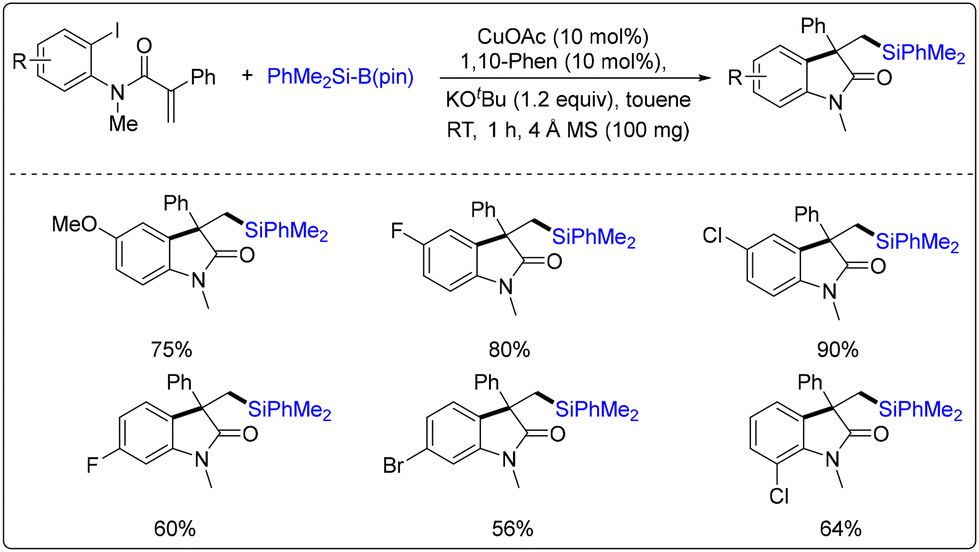 | ||
| Scheme 35 Copper-catalyzed arylsilylation of N-(2-iodoaryl)acrylamides with PhMe2Si-Bpin. | ||
The advancement of technology has enabled chemists to pursue research in the fields of photochemistry and electrochemistry. In this context, chemists have attempted to utilize a photocatalytic/electrocatalytic synergistic catalytic strategy to facilitate the cyclization reaction of acrylamide derivatives. In 2021, Zeng's team synthesized benzimidazole-thickened isoquinolines bearing silyl groups using a photocatalytic and electrocatalytic co-catalysis approach (Scheme 36).43 This was achieved through electron oxidation, photoinduced LMCT, and a radical-mediated HAT strategy. The reaction employs CeCl3·7H2O as a photocatalyst to facilitate the generation of methoxy radicals. Subsequently, the Si–H bond is selectively deactivated via the HAT pathway, forming Si radicals. This approach was adopted to ensure high selectivity, functional group tolerance, and favorable yields for the reaction.
 | ||
| Scheme 36 Photocatalytic and electrocatalytic co-catalyzed cyclisation reaction of N-substituted benzimidazole with hydrogenated silanes. | ||
In 2023, Wang's research group successfully completed the cyclisation reaction of CF3-substituted N-arylacrylamides through the application of photocatalytic and electrocatalytic co-catalysis utilising silane as a source of silicon radicals and 9,10-phenanthrenequinone (PQ) as both a photocatalyst and a HAT reagent (Scheme 37).44 The results of the mechanistic study demonstrated that the completion of the reaction was contingent upon the crucial involvement of the photocatalyst. The first role of the photocatalyst is to promote the production of silyl radicals, and the second is to facilitate the combination of the catalyst with intermediates of the silylation products within the catalytic cycle, which assumes the functions of both a HAT reagent and a chemical oxidant.
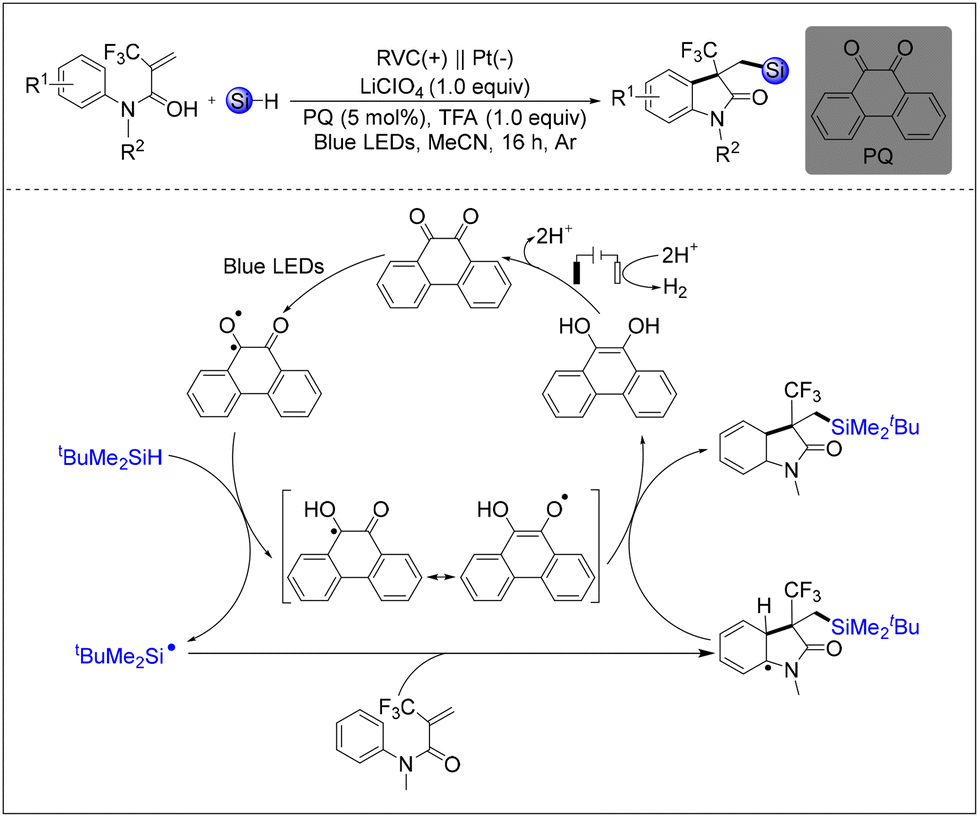 | ||
| Scheme 37 Photocatalytic and electrocatalytic co-catalyzed cyclisation reaction of CF3-substituted N-arylacrylamides to silylated 3-CF3-2-oxindoles. | ||
Ackermann's group reported a synergistic photocatalytic and electrocatalytic catalysis of the silyl functionalisation reaction (Scheme 38).45 In this strategy, iron complexes are induced by visible light to oxidise hydrogen silanes to silicon radicals via LMCT and HAT processes, which are then subjected to addition reactions to generate the target products. The scheme demonstrates excellent chemoselectivity and regioselectivity and does not require expensive metal catalysts, while no additional hydrogen atom transfer reagents are required.
 | ||
| Scheme 38 Photoelectrochemical iron-catalyzed silyl arylation of N-aryl acrylamides. | ||
4. Allenes as substrate
Allenes exist in natural products and pharmaceuticals and are often used in organic synthesis because they are highly reactive.46 The difunctionalization of allenes with organosilicon reagents also provided a direct method for synthesizing the silicon-functionalized compounds. PhMe2Si-B(pin) has recently been reported as a commonly used silicon precursor for the difunctionalization of allenes.In 2014, Tsuji and co-workers achieved a copper-catalyzed regiodivergent silacarboxylation of allenes (Scheme 39).47 Using CO2 as the carboxylated reagent and PhMe2Si-B(pin) as a silicon source, the reaction occurred selectively on allenes, and they assumed that the structure of the products was probably controlled by the ligand. Their results proved that carboxylated vinylsilanes were obtained when rac-MeDuPhos was used as the ligand. However, carboxylated allylsilanes were afforded when the PCy3 ligand was used in this silacarboxylation.
 | ||
| Scheme 39 Copper-catalyzed regiodivergent silacarboxylation of allenes. | ||
Based on their mechanistic study, Tsuji group proposed a plausible mechanism, as shown in Scheme 40. In the first step, the Cu precursor activates PhMe2Si-B(pin) to produce a silylcopper species using a ligand. The silylcopper species were added to the terminal double bond of the allene to afford the allylcopper intermediate when Me-DuPhos was used as the ligand. After that, CO2 was inserted at the γ-position of the allylcopper intermediate to provide copper carboxylate species, which underwent σ-bond metathesis with PhMe2Si-B(pin) to afford the difunctionalization products. The other vinylcopper intermediate was formed when ligand PCy3 was used. The results showed that the regioselectivity in the transformation might be attributed to the difference in the relative steric bulk of the Cu-ligand and SiMe2Ph moieties.
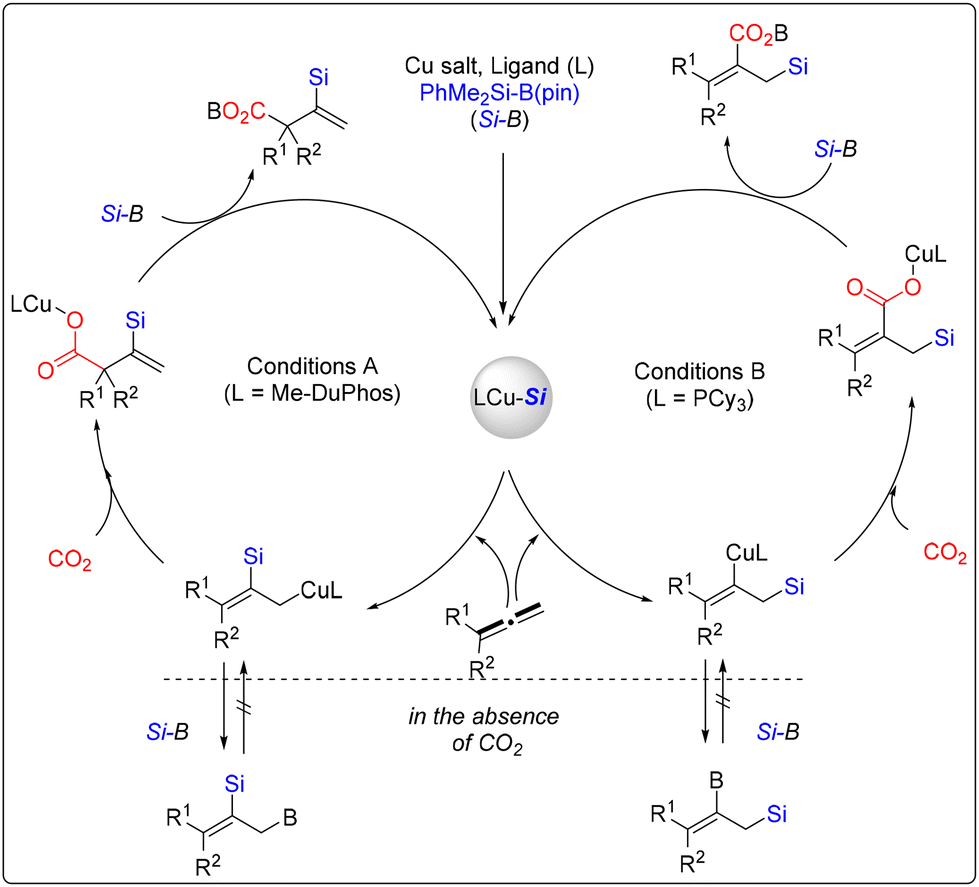 | ||
| Scheme 40 Plausible mechanism for the copper-catalyzed regiodivergent silacarboxylation of allenes. | ||
A similar copper-catalyzed silaformylation of allenes with PhMe2SiB(pin) and hexyl formate was achieved by the same group in 2016 using CuOAc/DTBM-dppbz catalysis system (Scheme 41).48 Various functional groups, including terminal olefin, chlorophenyl, iodophenyl, acetal, and ester, were tolerated and gave the corresponding silaformylation product β-silyl β,γ-unsaturated aldehyde derivatives in 34%–81% yield. Notably, the gram-scale silaformylation could be achieved and provided the corresponding product with up to 95% yield.
 | ||
| Scheme 41 Copper-catalyzed silaformylation of allenes with PhMe2SiB(pin) and hexyl formate. | ||
In 2015, Yoshida and co-workers developed Cu-catalyzed silylstannylation of various terminal allenes (Scheme 42).49 In this transformation, PhMe2Si-B(pin) was used as a silicon precursor. Using the Bu3SnOMe reagent as the acceptor, various 1-silyl-2-stannyl-2-alkenes bearing allylsilane and alkenylstannane units could be obtained from the corresponding terminal allenes in the presence of the ClIMesCuCl catalyst.
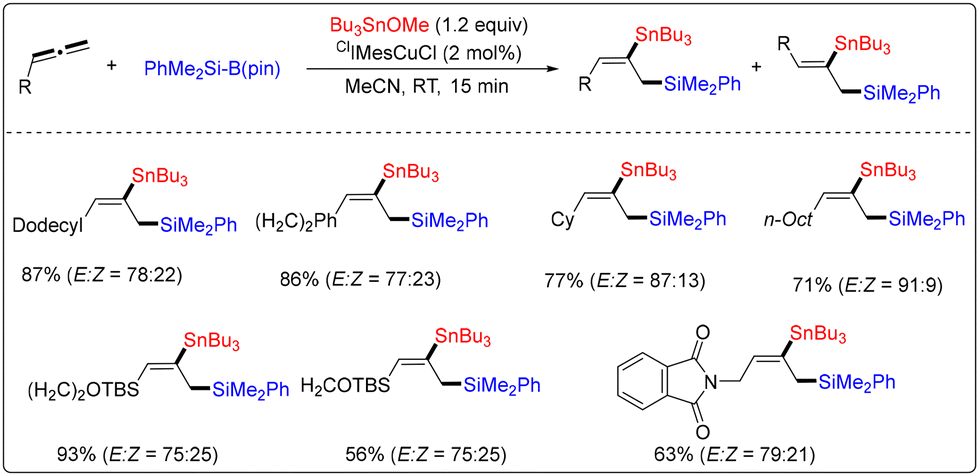 | ||
| Scheme 42 Cu-catalyzed silylstannylation of various terminal allenes. | ||
The Tian group reported a copper-catalyzed asymmetric silylative cyclization of cyclohexadienone-tethered allenes with PhMe2Si-Bpin (Scheme 43).50 PhMe2Si-Bpin as the silylation reagent provides the desired bicyclo[4.3.0]nonane frameworks 3, including cishydrobenzofuran, cis-hydroindole, and cis-hydroindene in high yields (80%–98%) with excellent enantioselectivities (94–98% ee). Mechanistically, this transformation may be through the regioselective β-silylation of the allene and subsequent enantioselective 1,4-addition to the cyclohexadienone pathway. It was noted that the bicyclic products could be further converted to bridged and tricyclic ring structures.
 | ||
| Scheme 43 Copper-catalyzed asymmetric silylative cyclization of cyclohexadienone-tethered allenes with PhMe2Si-Bpin. | ||
PhMe2Si-Bpin, which was previously used only as precursors providing a silicon group for the silylation reaction of allenes, was successfully applied as a difunctional reagent to the silaboration of allenes (Scheme 44).54 This allene silaboration protocol features a high atom economy and proceeds at room temperature using 1 mol% Au/TiO2 to initiate the process.51 In contrast to previous reports, the Bpin moiety was attached to the terminal carbon atom in this transformation. In addition, this conversation differs from the Pd(0)-catalyzed silaboration of terminal allenes, which occurs on the internal double bond. Regrettably, phenyl bearing 1,1-disubstituted allenes was unreactive although many substrates are suitable for this optimal reaction condition.
 | ||
| Scheme 44 Cu-catalyzed silylstannylation of various terminal allenes. | ||
In 2019, the same research group reported the first instance of a bisilylation reaction strategy for 1,1-disubstituted olefins utilizing TiO2 materials loaded with Au nanoparticles as catalysts (Scheme 45).52 The strategy was distinguished by its straightforward reaction conditions and exceptional regioselectivity. The products obtained can be subjected to the same catalytic conditions as water to yield more complex heterocyclic compounds containing silicon. This significantly broadens the scope of the subsequent development of this strategy.
 | ||
| Scheme 45 Au-catalysed bis-silylation of 1,1-disubstituted olefins. | ||
In 2022, Tobisu's research group developed a strategy for palladium-catalyzed silylation acylation reactions of linked olefins (Scheme 46).53 This strategy, which exhibits high regioselectivity, facilitates the loading of silyl groups onto the intermediate carbon atoms of the linked olefins, thereby yielding alkenylsilane-derived compounds with some degree of functionalization. This study provides a foundation for subsequent research on silyl transformations.
 | ||
| Scheme 46 Palladium-catalysed silylation acylation of linked olefins. | ||
5. Alkynes as a substrate
Alkyne difunctionalizations have become new efficient synthetic routes for forming highly functionalized olefins. In 2015, Li and co-workers reported a DTBP-mediated radical cascade reaction between readily available arylhydrosilanes and internal alkynes by C–H and Si–H cleavage (Scheme 47).54 This work provided a novel approach for constructing silaindenes using the DTBP peroxide system under metal-free conditions. Diarylacetylenes containing electron-donating and electron-withdrawing groups were found to be suitable for this transformation and gave the corresponding multiaryl-substituted silaindenes in 56%–85% yields. Moreover, dithiophen-2-ylacetylene and dialkylacetylene could also be tolerated under these reaction conditions but gave low yields. | ||
| Scheme 47 DTBP-mediated radical cascade reaction between readily available arylhydrosilanes and internal alkynes. | ||
An oxidative silyl radical cascade pathway was proposed by the authors for this transformation, as shown in Scheme 48. Initially, a tert-butoxy radical was formed via the homolysis of DTBP. The tert-butoxy radical interacted with arylhydrosilanes to generate the silyl radical. The silyl radical was added to the alkyne triple bond to generate a highly reactive alkenyl radical and then gave another cyclohexadienyl radical through intramolecular addition to the phenyl moiety. Finally, the hydrogen abstraction by the tertbutoxyl radical affords the silaindenes.
 | ||
| Scheme 48 Plausible mechanism of radical cascade reaction between arylhydrosilanes and internal alkynes. | ||
Later, Yoshida and co-workers reported a copper-catalyzed three-component silylstannylation of alkynes with silylborane and tin alkoxide (Scheme 49).49 Under these reaction conditions, various terminal alkynes were applied to prepare the 2-silyl-1-stannyl-1-alkenes, which are important structural units in many further organic conversions.
 | ||
| Scheme 49 Copper-catalyzed three-component silylstannylation of alkynes with silylborane and tin alkoxide. | ||
Dearomatization reactions have emerged as a powerful synthetic technique in recent years, providing core structures for many natural products and pharmaceuticals. In 2016, Gao and co-workers achieved copper-catalyzed oxidative ipso-annulation of activated alkynes with silanes (Scheme 50).55 The reaction proceeded using CuI as a catalyst (5 mol%) and TBHP as an oxidant (7 equiv.) in t-BuOH at 130 °C. This process represents a novel pathway for the synthesis of useful 3-silyl azaspiro[4,5]-trienones through a tandem difunctionalization of alkyne, dearomatization, and oxidation processes.
 | ||
| Scheme 50 Oxidative ipso-annulation of activated alkynes with silanes. | ||
Almost simultaneously, our group also reported the Fe-catalyzed oxidative spirocyclization of N-arylpropiolamides with silanes (Scheme 50).56 This reaction was conducted with a low loading of an iron catalyst (5 mol%), and TBHP was utilized as the oxidant in a mixture of t-BuOH and H2O as solvents at 130 °C.
The above radical cyclization strategy could also be used in the synthesis of silyl-functionalized indenone derivatives. Xie, Zhu and co-workers demonstrated a copper-catalyzed radical silylarylation of ynones with silanes (Scheme 51).57 Various silyl-functionalized indenones were efficiently constructed by the two-component reactions of ynones and silanes using CuCl as the catalyst and dicumyl peroxide (DCP) as the oxidant. The reaction also allowed the synthesis of the desired silylarylation product on a gram scale of up to 2.97 g. The authors also confirmed that the constructed silyl-functionalized indenones could be further post-modification to construct complex target molecules.
 | ||
| Scheme 51 Copper-catalyzed radical silylarylation of ynones with silanes. | ||
In 2018, Stratakis’ group achieved a bisilylation reaction of alkynes using a loaded gold catalyst (Scheme 52).58 The reaction was highly selective and yielded excellent results. Furthermore, the workers employed a one-pot method to react the product with water and another alkyne molecule, thereby achieving functionalized applications of the product.
 | ||
| Scheme 52 Gold-catalysed double reactions of alkynes. | ||
In 2019, Hong, Lu, and co-workers reported a cobalt-catalyzed asymmetric by double hydrosilylation of aliphatic terminal alkynes (Scheme 53).59 Various gem-bis(silyl)alkanes were achieved with excellent chemo-, regio-, and enantioselectivity (up to 99% ee). The possible reaction pathway was proposed by the authors through control experiments, kinetic studies, isotopic labeling experiments, and density functional theory calculations.
 | ||
| Scheme 53 Asymmetric double hydrosilylation of alkynes. | ||
Later, Fu, Xu and co-workers developed a copper-catalyzed regio- and stereo-selective silaboration of alkynes in aprotic solvents (Scheme 54).60 This method provides an effective route to multisubstituted functionalized alkenes through the cis-difunctionalization pathway between alkynes and silaboronate reagents. Various aliphatic alkynes bearing functional groups, including long carbon chain, ester, ketone, silyl ether, ether, tosyl, phthalimidyl, halide, and cyano, were all well tolerated for the 1,2-silaboration and 2,1-silaboration reactions. In this case, the regiodivergent silaborations were controlled by the copper catalysts and phosphine ligands.
 | ||
| Scheme 54 Copper-catalyzed regio- and stereo-selective silaboration of alkynes. | ||
In 2021, Suginome's group developed a silylation reaction of endo-aromatic alkynes using copper catalysis (Scheme 55).61 The silylation reaction of endo-aromatic alkynes with a silicoboron ester as the substrate and CuCl as the catalyst was successfully developed with a high degree of regioselectivity. Furthermore, the researchers observed that when the reaction was conducted in hydrocarbon solvents, the products formed were no longer the result of stereochemical reactions but rather the products of cis-addition reactions. This discovery provides valuable insights for subsequent studies.
 | ||
| Scheme 55 Cu-catalysed silylation of non-terminal aryl alkynes. | ||
Li's group achieved the silylation hydrogenation of terminal alkynes for the first time via a palladium/copper co-catalysis strategy using PhMe2SiBpin as the silicon source and propargyl acetate as the olefinic precursor (Scheme 56).62 This strategy, which exhibits excellent chemoselectivity, regioselectivity, and stereoselectivity, as well as good yields, yielded high-value target products through the synergistic effect of the two metal catalytic systems. Shintani's research group achieved a regioselective addition reaction of silicon borate on an inner alkyne using copper catalysis (Scheme 57).63 The scheme mentioned above exhibits reduced reactivity and more straightforward reaction conditions while maintaining excellent selectivity.
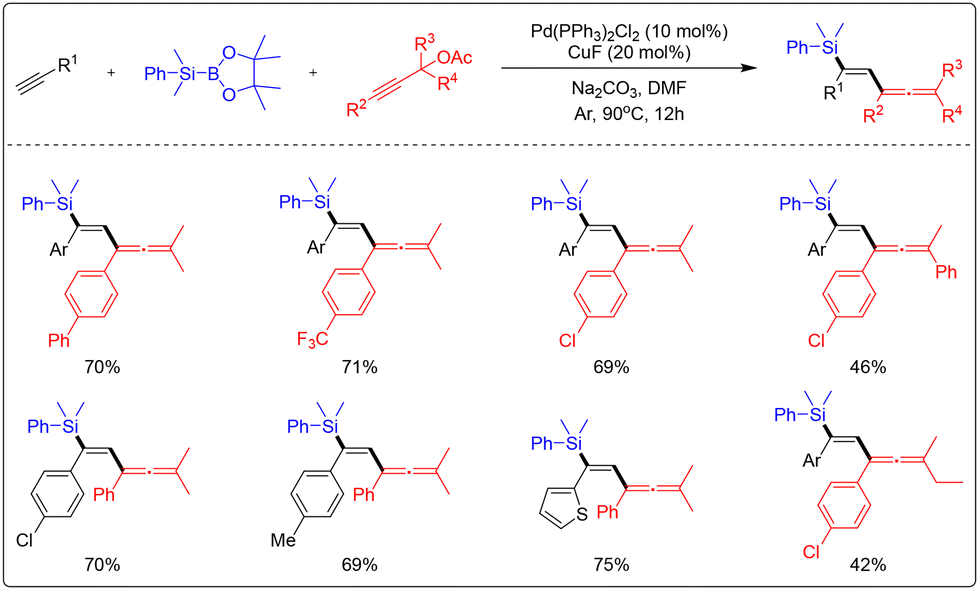 | ||
| Scheme 56 Palladium-/copper-catalyzed anti-selective intermolecular allenylsilylation of terminal alkynes. | ||
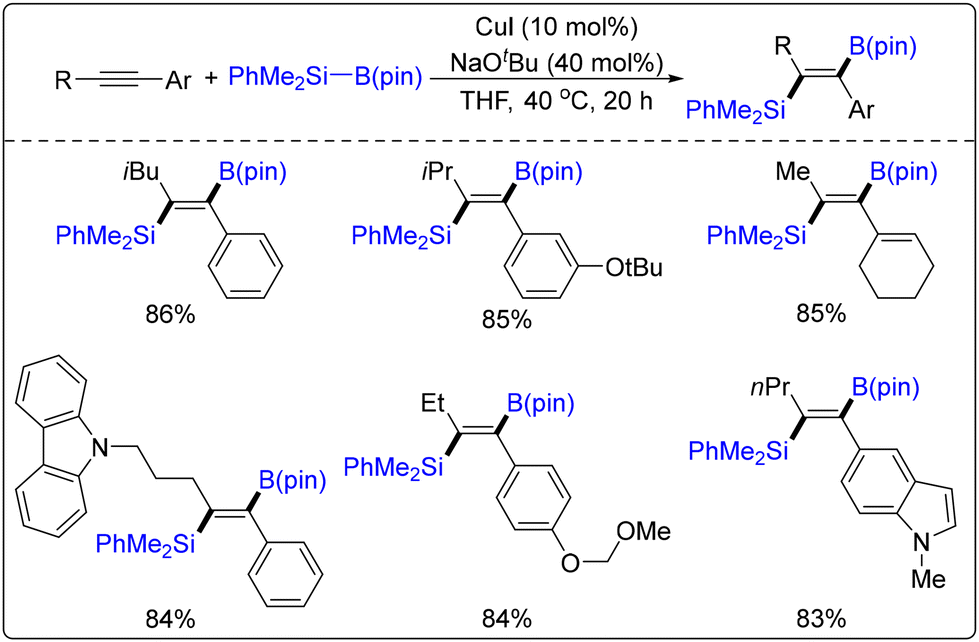 | ||
| Scheme 57 Cu-catalysed addition reactions of non-terminal alkynes with boronic acid silanes. | ||
In 2022, Poisson's team reported a new strategy for carrying out electrocatalytic reactions that involve the hydrogenosilylation of alkynes (Scheme 58).64 The researchers employed PhMe2SiBpin as the source of silicon radicals, stainless steel as the material's positive and negative electrodes, nBu4NBF4 as the electrolyte, and a mixture of acetonitrile and ethanol as the reaction solvent, successfully achieving the reaction at room temperature and in an air environment. The reaction exhibited remarkable regioselectivity and yielded promising outcomes.
 | ||
| Scheme 58 Electrocatalytic hydrosilylation of alkynes. | ||
Li's group accomplished the silylation carbonylation of 2-alkynylaniline by Rh(I) catalytic strategy using silane and CO as silicon and carbon sources (Scheme 59).65 The strategy uses acrylic acid as a transient chelating group to control the stereoselectivity of the reaction, which is a conceptual innovation. Moreover, the reaction has the advantages of mild reaction conditions, a wide range of substrates, and good functional group tolerance.
 | ||
| Scheme 59 Rh(I)-catalyzed silylative aminocarbonylation of 2-alkynylanilines. | ||
In 2023, Li's group synthesized tetra substituted silylpropenes using a cobalt-catalyzed electrocatalytic co-catalysis methodology with chloro(vinyl)silanes and 2-alkynyl-1-propyl acetates as substrates (Scheme 60).66 Through mechanistic studies, the researchers demonstrated that CoCl2 facilitates the alkenyl ionic intermediates in the system via the electron transfer pathway, thereby accelerating the completion of the overall reaction. Meanwhile, the reaction substrate is well suited for synthesizing 1,3-diolefins, alkynes, and trisubstituted methylsilyl alkenes.
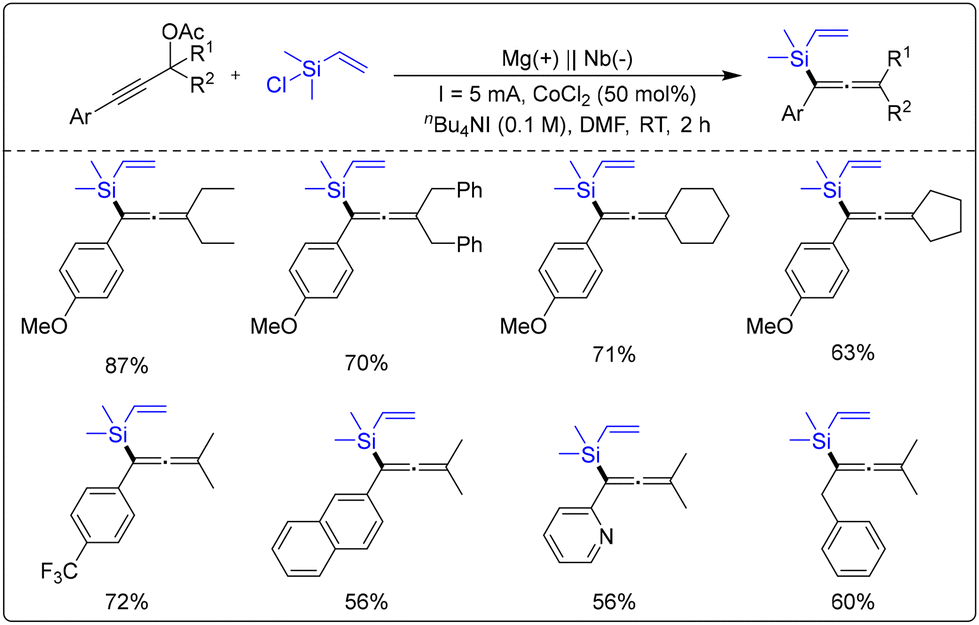 | ||
| Scheme 60 Cobalt-promoted electroreductive cross-coupling of prop-2-yn-1-yl acetates with chloro(vinyl)silanes. | ||
Yamanaka and Tobisu presented a strategy for the palladium-catalyzed selective addition reaction of non-terminal alkynes with acylsilanes (Scheme 61).67 It is noteworthy that when the temperature in the original reaction conditions was increased from 60 °C to 140 °C, the reaction underwent a transformation from the initial silylation reaction to a cyclization addition reaction, resulting in the formation of an indolone product.
 | ||
| Scheme 61 Palladium-catalysed addition reactions of non-terminated olefins with acylsilanes. | ||
6. Enynes as a substrate
Heterocycles, especially nitrogen-containing heterocycles, are widely found in natural products, medicinally relevant compounds, biologically active structures, and organic materials. Recently, the radical cascade cyclization of 1,n-enynes and radical precursors has emerged as a powerful strategy for preparing heterocycle compounds.68Using the Cu(MeCN)4PF6 as the catalyst and TBPB as the oxidant, our group carried out the radical-mediated intermolecular annulation cascade reactions of N-(2-(ethynyl)-aryl)-acrylamides with tertiary silanes to silino[3,4-c]-quinolin-5(3H)-ones. (Scheme 62).69 In this transformation, three new chemical bonds, including one C–Si bond and two C–C bonds, were constructed by simultaneously functionalizing both the Si–H and silyl C(sp3)–H bonds. For the secondary silane substrates, diethylsilane and diphenylsilane reacted smoothly with N-(2-(ethynyl)-aryl)-acrylamide to deliver the 4H-silolo[3,4-c]quinolin-4-ones in 73% and 52% yields via dual Si–H bond functionalization, respectively. HSi(TMS)3 also underwent the Si–H/Si–Si bond cleavage and annulation with various N-(2-(ethynyl)aryl)acrylamides to afford 4H-silolo-[3,4-c]quinolin-4-ones in 50%–86% yields. Notably, this type of conversion did not require a copper catalyst.
 | ||
| Scheme 62 Oxidative radical divergent Si-incorporation of N-(2-(ethynyl)aryl)acrylamides and tertiary silanes. | ||
Based on their mechanistic study, the authors proposed a possible mechanism (Scheme 63). A tert-butoxyl radical was formed in the presence of the active CuI species, giving the CuII(PhCO2) species. Subsequently, Si–H bond cleavage occurred to provide a silicon-centered radical. The in situ-generated silicon-centered radical was added to the alkene to give an alkyl radical intermediate, which underwent annulation to form the vinyl radical intermediate. 1,6-HAT occurred with the silyl C(sp3)–H bond to afford the silyl alkyl radical intermediate, which converted the desired product under single-electron oxidation and deprotonation processes. In addition, when tertiary silanes were changed to secondary silanes, 1,5-HAT with the Si–H bond was involved in this transformation and gave 4H-silolo[3,4-c]quinolin-4-ones. Notably, when HSi(TMS)3 was used as the silane, a Me3Si radical was formed in this reaction.
 | ||
| Scheme 63 Plausible mechanism of oxidative radical divergent Si-incorporation of N-(2-(ethynyl)aryl)acrylamides and silanes. | ||
7. Conclusion and outlook
Difunctionalization reactions of unsaturated hydrocarbons with organosilicon reagents have unique advantages in constructing silicon-containing compounds, especially in synthesizing functionalized linear silane and silicon-containing heterocycle compounds. In this review, we summarized the crucial developments in this field based on the different reagents, including (1) simple alkenes, (2) acrylamides, (3) allenes, (4) alkynes, and (5) enynes. Various C–Si/C–C, C–Si/C–N, C–Si/C–B, C–Si/C–Si and C–Si/C–Sn bonds were formed by transition-metal catalyzed, visible-light-mediated and metal-free difunctionalization reactions between unsaturated hydrocarbons with organosilicon reagents.Although the difunctionalization of unsaturated hydrocarbons with organosilicon reagents are increasingly versatile tools for the synthesis of functional silicon compounds, there are still some challenges in this area, especially it is still a challenging task to achieve high region- and enantio-selectivity due to the difficulty in the stereocontrol of the reactive and unstable radical intermediates. Meanwhile, low reaction efficiency and excessive by-products were the problems in this type of transformation. In the future, the exploration of more efficient catalytic radical cycles, stereoselective control mode, and the development of more types of silicone reagents is highly desirable.
Author contributions
J. M.: investigation, visualization and writing-original draft; M. C.: project administration and supervision; Y. Z.: funding acquisition, visualization and writing-review & editing; R.-J. S.: conceptualization, Funding acquisition and writing-review & editingData availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52270039 and 22408149) and Key Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resource Reuse (No. 2023SSY02061). We are grateful for the financial support of the projects and research platform support provided by the laboratory.References
- (a) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed; (b) J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter, S. R. HarruffRyan and V. Hoveln, ACS Catal., 2019, 9, 8961–8979 CrossRef CAS; (c) X. Shang and Z. Q. Liu, Org. Biomol. Chem., 2016, 14, 7829–7831 RSC; (d) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS; (e) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS; (f) C. H. Li, H. Y. Qiu and C. H. Xu, Chin. Chem. Lett., 2007, 18, 1436–1438 CrossRef CAS; (g) Q. Zhou, R. Meng, J. Xing, W. Yao and X. Dou, ACS Catal., 2023, 13, 8516–8524 CrossRef CAS; (h) G. Pandey, S. K. Tiwari, P. Singh and P. K. Mondal, Org. Lett., 2021, 23, 7730–7734 CrossRef CAS PubMed.
- (a) A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef; (b) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (c) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC; (d) S. Fujii and Y. Hashimoto, Future Med. Chem., 2017, 9, 485–505 CrossRef CAS PubMed; (e) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- (a) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (b) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS; (c) J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Organometallics, 2006, 25, 1188–1198 CrossRef CAS; (d) B. Wang, P. Sukkaew, G. Song, A. Rosenkranz, Y. X. Lu, K. Nishimura, J. Wang, J. Lyu, Y. Cao, J. Yi, L. Ojamäe, H. Li and N. Jiang, Chin. Chem. Lett., 2020, 31, 2013–2018 CrossRef CAS; (e) H. Bian, X. Dong, S. Chen, D. W. Dong and N. Zhang, Chin. Chem. Lett., 2018, 29, 171–174 CrossRef CAS; (f) C. R. Eddy Jr and D. K. Gaskill, Science, 2009, 324, 1398–1400 CrossRef CAS PubMed; (g) K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530–531 CrossRef CAS PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- (a) S. Yajima, J. Hayashi and M. Omori, Chem. Lett., 1975, 4, 931–934 CrossRef; (b) S. Yajima, Philos. Trans. R. Soc., A, 1980, 294, 419–426 CrossRef CAS; (c) R. S. Klausen, J. R. Widawsky, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, J. Am. Chem. Soc., 2012, 134, 4541–4544 CrossRef CAS PubMed; (d) E. A. Marro and R. S. Klausen, Chem. Mater., 2019, 31, 2202–2211 CrossRef CAS; (e) J. C. L. Walker, H. F. T. Klare and M. Oestreich, Nat. Rev. Chem., 2020, 4, 54–62 CrossRef CAS.
- (a) C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188–194 CrossRef CAS; (b) C. Chatgilialoglu, Chem. – Eur. J., 2008, 14, 2310–2320 CrossRef CAS PubMed; (c) S. E. Denmark and J. H. C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978–2986 CrossRef CAS PubMed; (d) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (e) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed; (f) D. A. Powell and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 7788–7789 CrossRef CAS PubMed; (g) S. E. Denmark, J. Org. Chem., 2004, 74, 2915–2927 CrossRef; (h) T. Hiyama and Y. Hatanaka, Pure. Appl. Chem., 1994, 66, 1471–1478 CrossRef CAS.
- (a) X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS; (b) T. Wang, L. Chen, Y. Y. Liu, Z. B. Zhang, P. Han and L. H. Jing, Org. Lett., 2024, 26, 4526–4531 CrossRef CAS PubMed; (c) W. Chen, N. Zhang, Z. Chai, J. N. Wei, G. Luo and W. X. Zhang, ACS Catal., 2024, 14, 5612–5620 CrossRef CAS; (d) D. Kim, C. Chen, B. Q. Mercado, D. J. Weix and P. L. Holland, Organometallics, 2020, 39, 2415–2424 CrossRef CAS; (e) Z. Zhang and X. Hu, ACS Catal., 2020, 10, 777–782 CrossRef CAS.
- M. Murakami, P. G. Andersson, M. Suginome and Y. Ito, J. Am. Chem. Soc., 1991, 113, 3988–3990 CrossRef.
- H. Peng, J.-T. Yu, Y. Jiang and J. Cheng, Org. Biomol. Chem., 2015, 13, 10299–10302 RSC.
- Y. Yang, R. J. Song, X. H. Ouyang, C. Y. Wang, J. H. Li and S. L. Luo, Angew. Chem., Int. Ed., 2017, 56, 7916–7919 CrossRef CAS PubMed.
- J. Hou, A. Ee, H. Cao, H. W. Ong, J. H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- H. Zhang, X. Wu, Q. Zhao and C. Zhu, Chem. – Asian J., 2018, 13, 2453–2457 CrossRef CAS PubMed.
- Z. Liu, J. Chen, H. X. Lu, X. H. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068–17073 CrossRef CAS PubMed.
- Y. Zeng, X. D. Liu, X. Q. Guo, Q. S. Gu, Z. L. Li, X. Y. Chang and X. Y. Liu, Sci. China: Chem., 2019, 62, 1529–1536 CrossRef CAS.
- Z. Zhang and X. Hu, ACS Catal., 2020, 10, 777–782 CrossRef CAS.
- W. C. Cui, W. Zhao, M. Gao, W. Liu, S. Z. Wang, Y. Liang and Z. J. Yao, Chem. – Eur. J., 2019, 25, 16506–16510 CrossRef CAS PubMed.
- Y. Matsuda, Y. Tsuji and T. Fujihara, Chem. Commun., 2020, 56, 4648–4651 RSC.
- B. Zhao, Y. Li, H. Li, M. Belal, L. Zhu and G. Yin, Sci. Bull., 2021, 66, 570–577 CrossRef CAS PubMed.
- N. X. Xu, B. X. Li, C. Wang and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 10639–10644 CrossRef CAS PubMed.
- X. Yu, M. Lübbesmeyer and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 675–679 CrossRef CAS PubMed.
- L. X. Lu, J. C. Siu, Y. H. Lai and S. Lin, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS PubMed.
- J. Ke, W. Liu, X. Zhu, X. F. Tan and C. He, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed.
- M. Zheng, J. Hou, L. L. Hua, W. Y. Tang, L. W. Zhan and B. D. Li, Org. Lett., 2021, 23, 5128–5132 CrossRef CAS PubMed.
- S. Neogi, A. K. Ghosh, S. Mandal, D. Ghosh, S. Ghosh and A. Hajra, Org. Lett., 2021, 23, 6510–6514 CrossRef CAS PubMed.
- D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS PubMed.
- J. Zhou, B. Jiang, Y. Fujihira, Z. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749 CrossRef CAS PubMed.
- L. Ding, K. Niu, Y. Liu and Q. M. Wang, ChemSusChem, 2022, 15, e202200367 CrossRef CAS PubMed.
- J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., 2022, 134, e202202379 CrossRef.
- T. He, Z. W. Qu, H. F. T. Klare, S. Grimme and M. Oestreich, Angew. Chem., Int. Ed., 2022, 61, e202203347 CrossRef CAS PubMed.
- C. Luo, Y. Zhou, H. Chen, T. Wang, Z. B. Zhang, P. Han and L. H. Jing, Org. Lett., 2022, 24, 4286–4291 CrossRef CAS PubMed.
- W. Y. Zheng, Y. J. Xu, H. Luo, Y. H. Feng, J. Q. Zhang and L. Q. Lin, Org. Lett., 2022, 24, 7145–7150 CrossRef CAS PubMed.
- N. Takemura, Y. Sumida and H. Ohmiya, ACS Catal., 2022, 12, 7804–7810 CrossRef CAS.
- L. Gao, X. Liu, G. Li, S. Chen, J. Cao, G. Wang and S. Li, Org. Lett., 2022, 24, 5698–5703 CrossRef CAS PubMed.
- J. Sun, Y. Zhou, R. Gu, X. Li, A. Liu and X. Zhang, Nat. Commun., 2022, 12, 7093 CrossRef PubMed.
- Q. Q. Pan, L. Qi, X. Pang and X. Z. Shu, Angew. Chem., 2023, 135, e202215703 CrossRef.
- S. Neogi, S. Bhunya, A. K. Ghosh, B. Sarkar, L. Roy and A. Hajra, ACS Catal., 2024, 14, 4510–4522 CrossRef CAS.
- (a) A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed; (b) M. Mondal and U. Bora, RSC Adv., 2013, 3, 18716–18754 RSC; (c) H. L. Schmitt, D. Martymianov, O. Green, T. Delcaillau, Y. S. P. Kim and B. Morandi, J. Am. Chem. Soc., 2024, 146, 4301–4308 CrossRef CAS PubMed; (d) P. Xu, H. Xu, S. Wang, T. Z. Hao, S. Y. Yan, D. Guo and X. Zhu, Org. Chem. Front., 2023, 10, 2013–2017 RSC; (e) Y. Hosoya, K. Mizoguchi, H. Yasukochi and M. Nakada, Synlett, 2022, 495–501 CAS; (f) A. D. Marchese, T. Adrianov and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 16750–16762 CrossRef CAS PubMed.
- (a) J. Lin, R. J. Song, M. Hu and J. H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed; (b) Y. Li, G. A. Pan, M. J. Luo and J. H. Li, Chem. Commun., 2020, 56, 6907–6924 RSC; (c) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CrossRef CAS; (d) J. B. Peng, Adv. Synth. Catal., 2020, 362, 3059–3080 CrossRef CAS; (e) T. B. Hua, C. Xiao, Q. Q. Yang and J. R. Chen, Chin. Chem. Lett., 2020, 31, 311–323 CrossRef CAS; (f) K. J. Liu, J. H. Deng, T. Y. Zeng, X. J. Chen, Y. Huang, Z. Cao, Y. W. Lin and W. M. He, Chin. Chem. Lett., 2020, 31, 1868–1872 CrossRef CAS; (g) L. H. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W. M. He, Chin. Chem. Lett., 2019, 30, 1237–1240 CrossRef CAS.
- L. Zhang, D. Liu and Z. Q. Liu, Org. Lett., 2015, 17, 2534–2537 CrossRef CAS PubMed.
- L. J. Wu, Y. Yang, R. J. Song, J. X. Yu, J. H. Li and D. L. He, Chem. Commun., 2018, 54, 1367–1370 RSC.
- C. Zhang, J. Pi, L. Wang, P. Liu and P. P. Sun, Org. Biomol. Chem., 2018, 16, 9223–9229 RSC.
- R. X. Liang, R. Y. Chen, C. Zhong, J. W. Zhu, Z. Y. Cao and Y. X. Jia, Org. Lett., 2020, 22, 3215–3218 Search PubMed.
- Y. Jiang, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1796–1805 CrossRef CAS.
- Q. Wan, C. Y. Huang, Z. W. Hou, H. Jiang and L. Wang, Org. Chem. Front., 2023, 10, 3585–3590 RSC.
- W. Wei, S. L. Homölle, T. Münchow, Y. Li, I. Maksso and L. Ackermann, Cell Rep. Phys. Sci., 2023, 4, 101550 Search PubMed.
- (a) G. Li, X. Huo, X. Jiang and W. B. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC; (b) L. Liu, R. M. Ward and J. M. Schomaker, Chem. Rev., 2019, 119, 12422–12490 CrossRef CAS PubMed; (c) Q. Ran, K. F. Wu and Y. H. Xu, Org. Lett., 2024, 26, 3767–3771 CrossRef CAS PubMed; (d) G. Liu, X. Yang, P. Gu, M. Wang, X. Zhang and X. Q. Dong, J. Am. Chem. Soc., 2024, 146, 7419–7430 CrossRef CAS PubMed; (e) J. Zhou, Z. Zhao, S. Mori, K. Yamamoto and N. Shibata, Chem. Sci., 2024, 15, 5113–5122 RSC; (f) W. Geng, H. Wang, J. Xi, R. Jiang, J. Guo, K. Q. Jiao, M. Y. Tang, Q. Liu, L. Zhang and H. Liu, Org. Biomol. Chem., 2023, 21, 279–283 RSC.
- Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef CAS PubMed.
- T. Fujihara, A. Sawada, T. Yamaguchi, Y. Tani, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2017, 56, 1539–1543 CrossRef CAS PubMed.
- H. Yoshida, Y. Hayashi, Y. Ito and K. Takaki, Chem. Commun., 2015, 51, 9440–9442 RSC.
- Z. T. He, X. Q. Tang, L. B. Xie, M. Cheng, P. Tian and G. Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815–14818 CrossRef CAS PubMed.
- M. Kidonakis and M. Stratakis, ACS Catal., 2018, 8, 1227–1230 CrossRef CAS.
- M. Kidonakis, V. Kotzabasaki, E. Vasilikogiannaki and M. Stratakis, Chem. – Eur. J., 2019, 25, 9170–9173 CrossRef CAS PubMed.
- T. Inagaki, S. Sakurai, M. Yamanaka and M. Tobisu, Angew. Chem., Int. Ed., 2022, 61, e202202387 CrossRef CAS PubMed.
- L. Xu, S. Zhang and P. Li, Org. Chem. Front., 2015, 2, 459–463 Search PubMed.
- P. Gao, W. Zhang and Z. Zhang, Org. Lett., 2016, 18, 5820–5823 CrossRef CAS PubMed.
- L. J. Wu, F. L. Tan, M. Li, R. J. Song and J. H. Li, Org. Chem. Front., 2017, 4, 350–353 RSC.
- Z. Yan, J. Xie and C. Zhu, Adv. Synth. Catal., 2017, 359, 4153–4157 CrossRef CAS.
- I. Saridakis, M. Kidonakis and M. Stratakis, ChemCatChem, 2018, 10, 980–983 Search PubMed.
- J. Guo, H. Wang, S. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881–895 CAS.
- M. Zhao, C. C. Shan, Z. L. Wang, C. Yang, Y. Fu and Y. H. Xu, Org. Lett., 2019, 21, 6016–6020 CrossRef CAS PubMed.
- T. Ohmura, Y. Takaoka and M. Suginome, Chem. Commun., 2021, 57, 4670–4673 RSC.
- L. F. Yang, Q. A. Wang and J. H. Li, Org. Lett., 2021, 23, 6553–6557 CrossRef CAS PubMed.
- H. Moniwa and R. Shintani, Chem. – Eur. J., 2021, 27, 7512–7515 CrossRef CAS PubMed.
- T. Biremond, P. Jubault and T. Poisson, ACS Org. Inorg. Au, 2022, 2, 148–152 CrossRef CAS PubMed.
- Y. F. Han, G. F. Lv, Y. Li, L. J. Wu, X. H. Ouyang and J. H. Li, Chem. Sci., 2022, 13, 9425 RSC.
- C. H. Xu, Z. Q. Xiong, J. H. Qin, X. H. Xu and J. H. Li, Org. Lett., 2023, 25, 7263–7267 CrossRef CAS PubMed.
- T. Inagaki, T. Ando, S. Sakurai, M. Yamanaka and M. Tobisu, Chem. Sci., 2023, 14, 2703–2712 RSC.
- (a) C. H. Xu, Y. Li, J. H. Li, J. N. Xiang and W. Deng, Sci. China: Chem., 2019, 62, 1463–1475 CrossRef CAS; (b) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813–834 CrossRef CAS PubMed; (c) V. Michelet, P. Y. Toullec and J. P. Genêt, Angew. Chem., Int. Ed, 2008, 47, 4268–4315 CrossRef CAS PubMed; (d) R. P. Kelly, D. Toniolo, F. F. Tirani, L. Maron and M. Mazzanti, Chem. Commun., 2018, 54, 10268–10271 RSC; (e) M. H. Huang, W. J. Hao and B. Jiang, Chem. – Asian J., 2018, 13, 2958–2977 CrossRef CAS PubMed.
- Y. Yang, R.-J. Song, Y. Li, X. H. Ouyang, J. H. Li and D. L. He, Chem. Commun., 2018, 54, 1441–1444 RSC.
| This journal is © The Royal Society of Chemistry 2025 |