
Gold-catalyzed fluorination of alkynes/allenes: mechanistic explanations and reaction scope
Deblina Singha
Roy
a,
Yogesh Bhaskar Singh
Tanwer
a,
Snigdha Rani
Patra
a,
Shivam
Kumar
a,
Sabyasachi
Bhunia
 *a and
Debjit
Das
*b
*a and
Debjit
Das
*b
aDepartment of Chemistry, Central University of Jharkhand, Ranchi-835222, Jharkhand, India. E-mail: sabyasachi.bhunia@cuj.ac.in
bDepartment of Chemistry, Triveni Devi Bhalotia College, Raniganj-713347, India. E-mail: debjitofchem@gmail.com
First published on 30th October 2024
Abstract
Since the beginning of this century, there has been a great deal of research on homogeneous gold-catalyzed alkyne fluorination due to the precious values of fluorinated scaffolds in many bioactive natural products, drugs, and agrochemicals. This area of research, which originally took advantage of gold's mild Lewis acidity and tendency to form π-complexes with alkynes, has gained new momentum after Sadighi's discovery in 2007 of Au-catalyzed hydrofluorination of internal alkynes. The methods have enabled direct access to valuable fluoroalkanes, fluoroalkenes, α-fluorocarbonyls, and fluorinated carbo- and hetero-cycles in one pot from readily available alkyne precursors. Both nucleophilic and electrophilic fluorination modes with versatile reactivity have been used to achieve several new cascade reactions. This study covers the literature reports published since 2007 and provides a comprehensive summary of the methods, applications, and mechanistic insights into gold-catalyzed alkyne fluorination using electrophilic and nucleophilic fluorinating reagents.
 Deblina Singha Roy | Deblina Sinha Roy studied chemistry at Calcutta University (Raja Peary Mohan College), where she received her B.Sc. degree. She joined as a master's student under Dr Sabyasachi Bhunia in 2022 at the Central University of Jharkhand. Her research interest includes transition metal-catalyzed organic transformations and green chemistry. |
 Yogesh Bhaskar Singh Tanwer | Yogesh Bhaskar Singh Tanwer received his M.Sc. degree from Guru Ghasidas Central University in 2020. Thereafter he joined as a doctoral student under Dr Sabyasachi Bhunia at the Central University of Jharkhand. His research interest includes transition metal-catalyzed organic transformations and the development of sustainable chemistry. |
 Snigdha Rani Patra | Snigdha Rani Patra received her M.Phil. degree in chemistry from VSSUT, India, and PhD in chemistry from the Central University of Jharkhand under the supervision of Dr Sabyasachi Bhunia in 2023. Her current research topic is transition metal based organic transformations. |
 Shivam Kumar | Shivam Kumar studied chemistry at the Central University of Jharkhand, where he received his M.Sc. under Dr S. Bhunia in 2021. His research interest includes heterogeneous catalysis-based synthesis, nanoparticles and their applications, organometallic chemistry and green chemistry. |
 Sabyasachi Bhunia | Sabyasachi Bhunia received his MSc degree from Jadavpur University, India, and PhD in chemistry from National Tsing-Hua University under the supervision of Prof. Rai-Shung Liu. After his PhD, he carried out postdoctoral research under Prof. Rai-Shung Liu at National Tsing-Hua University and with Prof. Peter H. Seeberger at the Max Planck Institute of Colloids and Interfaces, Germany. He then moved to GVK BIO, India, as a scientist. In 2016, he started his academic career as an assistant professor at the Central University of Jharkhand, India. His research interest includes TM-catalyzed carbo- and heterocyclizations involving activation of C–C multiple bonds and organic transformations using various waste materials. |
 Debjit Das | Debjit Das received his PhD in the area of organic and organometallic reactivity of palladium–tin heterobimetallic systems from the Indian Institute of Technology, Kharagpur, under the supervision of Prof. Sujit Roy. He then joined the Central University of Jharkhand, Ranchi, as an assistant professor. In 2017, he moved to the Department of Chemistry, Triveni Devi Bhalotia College, Raniganj, India. His research interests include organometallics, green organic synthesis and catalysis. |
1. Introduction
The development of novel and efficient methods to synthesize organofluorine compounds1 from readily available starting precursors has long been a goal in organic chemistry. The application of fluorine in medicinal chemistry,2 drug design,3 agrochemicals,4 and high-performance materials has grown significantly over the last one and a half decades as scientists have explored its distinct properties and more sophisticated techniques for its utilization. A broad spectrum of medical applications, including anticancer, antiviral, antibacterial, anaesthetics, cholesterol inhibitors, and anti-inflammatory drugs (Fig. 1), is greatly impacted by the unique physicochemical characteristics of the fluorine atom in the molecules. In fact, one or more fluorine atoms can be found in approximately 35% of agrochemicals and 25% of drugs.3a,5 Being the second smallest element in the periodic table and due to its inherent properties,1a,6 fluorine possesses remarkable capabilities that can alter the biological and physical characteristics of molecules. Considering its more robust C–F bond (105.4 kcal mol−1) and high electronegativity (4.0 as per the Pauling scale), it is a valuable isostere of the oxygen atom. Selective addition of fluorine can have a significant effect on several aspects2 of the molecule, including intrinsic potency, lipophilicity, pKa (acidity or basicity of the neighbouring functional groups), conformation, metabolic stability, membrane permeability, and pharmacokinetic features. The numerous uses of fluorine in drug development have stimulated the growth of novel synthetic procedures that provide more straightforward access to a wide array of fluorinated molecules. Alternatively, the importance of fluorine-containing compounds was also expanded by the use of radioactive 18F nuclei in PET (positron emission tomography) labelling investigations, as this technique has applications in a variety of medical fields, including oncology. As a result, organofluorine chemistry has contributed significantly to the advancement of medicinal chemistry and has made substantial advancements in this field. As the natural abundance of fluorine atoms is relatively low, they are rare in naturally occurring organic compounds. Since most of the organic substances containing fluorine are manufactured, there is always a need for novel techniques that provide access to fluorinated derivatives. Systematic fluorination of organic molecules at specific positions can be a challenging endeavour and is always considered frontier research. Currently, research is being done in many labs across the globe to develop novel selective fluorinating agents that can generate new C–F bonds from functional groups, C–H bonds, or C–C multiple bonds. | ||
| Fig. 1 Selected examples of fluorine-containing therapeutic drugs. | ||
Alkynes are a dynamic class of easily accessible substances that are frequently employed as starting precursors in the synthesis of more complex molecules.7 In view of the widespread popularity and significance of alkynes, considerable effort has gone into developing methods that allow for the generation of molecular complexity or their transformation into precious complex fluorinated molecules.8 One of the most popular applications of alkyne substrates is gold-catalysed alkyne functionalization in the presence of an internal or external nucleophile, which has revolutionized organic chemistry and brought in a new era.7c,9 The field of homogeneous gold catalysis has dramatically advanced since the year 2000. Gold always shows reactivity towards alkynes only in its cationic form. A lower LUMO and the poor electron back donation of gold(I) make it the most potent Lewis acid for electrophilic activation of alkynes, which leads to the formation of a π-complex9aA1 for a facile nucleophilic attack. As a result, trans-alkenyl gold complex A2 is generated, which can then undergo protodeauration to react with an electrophile, typically a proton, to produce the desired end product (Scheme 1).
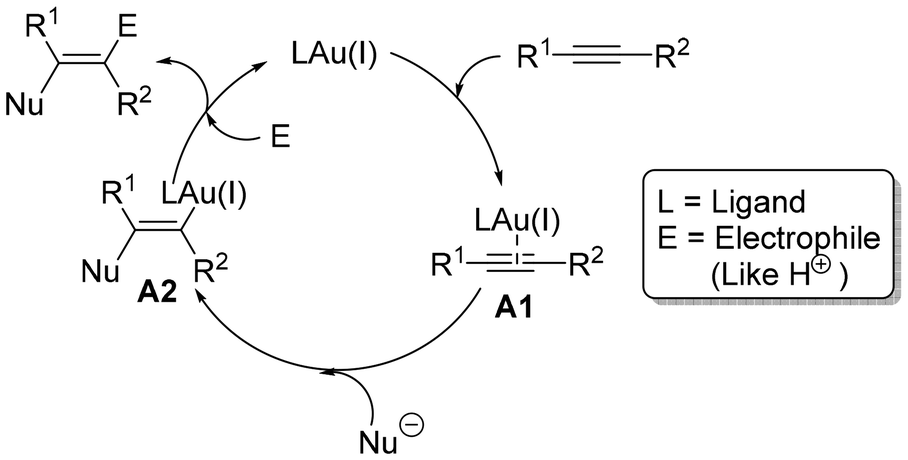 | ||
| Scheme 1 Traditional catalytic cycle for gold-catalyzed alkyne activation. | ||
Indeed, the combination of fluorine, alkyne, and gold(I) salt proved to be quite beneficial, since many valuable reactivities have been developed. When fluorinating reagents or fluorinated building blocks are combined with a distinct alkyne-activated gold complex, a new prospect for developing novel organofluorine scaffolds arises. Research in this area is still ongoing and has produced various new synthetic approaches every day. Thus, by employing Au(I) catalysis, numerous fluorination methods have been developed, wherein versatile fluorinating reagents and ligands have been chosen for selective fluorination. In general, two primary approaches have been studied in gold catalysis to access fluorinated organic compounds. Using either nucleophilic (Fig. 2) or electrophilic (Fig. 3) fluorinating reagents is probably the most straightforward method. The most widely used fluorinating reagents for gold-catalyzed nucleophilic fluorination reactions are aqueous hydrofluoric acid and tert-amine hydrofluoride salts. Conversely, Selectfluor is most effective in gold-catalyzed electrophilic fluorinations.
 | ||
| Fig. 2 Some representative nucleophilic fluorinating reagents. | ||
 | ||
| Fig. 3 Some representative electrophilic fluorinating reagents. | ||
The fluoride ions are the effective nucleophiles in cases of all nucleophilic fluorinating reagents, which react efficiently with Au(I) activated π-alkynes to form a vinyl gold intermediate, A3 (Scheme 2). A successive protodeauration furnishes functionalised vinyl fluorides. Alternatively, electrophilic fluorination offers two distinct paths after the formation of a similar vinyl gold intermediate A4 through alkyne activation (Scheme 3). In path A, protodeauration of intermediate A4 produces an olefin that is activated by Selectfluor (A5), and subsequent nucleophilic addition affords fluoroalkanes.
 | ||
| Scheme 2 Gold-catalyzed fluorination by nucleophilic fluorinating reagents. | ||
 | ||
| Scheme 3 Gold-catalyzed fluorination by electrophilic fluorinating reagents. | ||
Another approach (path B) involves oxidation first, followed by reductive elimination, leading to functionalised vinyl fluorides.
Instead of various Au-catalyzed electrophilic and nucleophilic fluorinations of C–C multiple bonds, there have been several developments where stoichiometric amounts of gold complexes were used in C–F bond formation reactions. These stoichiometric studies of gold C–F bond formation focused mainly on several methods like insertion of carbenes into Au–F bonds,10 oxidation of Au(I) to produce Au(III)–F,11 and reductive elimination.11b,12 However, in this review, we concentrated only on the fluorination of C–C multiple bonds for various transformations using catalytic amounts of Au salts in the presence of external electrophilic or nucleophilic fluorinating agents.
Very few reviews have long since been published in the literature,13 but they are not explicitly focused on the present topic. To ensure a comprehensive analysis of the title, this review has been arranged according to the different modes of reaction, mechanistic features, and nature of the different fluorinating reagents employed in the reactions. This summary also covers the art of choosing various substrates for novel developments and their reactivities.
2. Gold-catalyzed fluorination with nucleophilic fluorinating reagents (F−)
2.1. Hydrofluorination of alkynes
In 2007, Prof. Sadighi first developed14 a reversible carbon–fluorine bond formation by reacting an (NHC)gold(I) fluoride complex, [(SIPr)AuF] [SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene] A6, with excess hex-3-yne to form a β-(fluorovinyl)gold(I) species A7 (eqn (1), Scheme 4). The reaction was accomplished within 10 min at 20 °C in DCM and characterized by 19F NMR spectroscopy for the existence of β-(fluorovinyl)gold(I) A7. The (NHC)gold(I) fluoride complex A6 was quantitatively regenerated following gradual evaporation (over a period of 30 min) of the solvent and excess alkynes. However, rapid (≤5 min) evaporation afforded the mixture of (NHC)gold(I) fluoride complex A6 and β-(fluorovinyl)gold(I) A7 which regained equilibrium in CD2Cl2 within 2 h. In this context, a more stable β-(fluorovinyl)gold(I) complex of trans-{(SIPr)Au[(Ph)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(F)CH3]} was prepared from 1-phenyl-1-propyne and later characterized by crystallography. The methodology has also been applied for (NHC)gold(I) catalyzed trans-hydrofluorination of various internal alkynes with the mild HF source Et3N·3HF at room temperature (Scheme 4). The method provides a broad substrate scope, such as diaryl-, dialkyl-, aryl/alkyl-, or thienyl/alkyl-substituted compounds, with good yields.
C(F)CH3]} was prepared from 1-phenyl-1-propyne and later characterized by crystallography. The methodology has also been applied for (NHC)gold(I) catalyzed trans-hydrofluorination of various internal alkynes with the mild HF source Et3N·3HF at room temperature (Scheme 4). The method provides a broad substrate scope, such as diaryl-, dialkyl-, aryl/alkyl-, or thienyl/alkyl-substituted compounds, with good yields.
 | ||
| Scheme 4 Au-catalyzed hydrofluorination of alkynes. | ||
Prof. Miller developed15 a comparable Au(I)-catalyzed hydrofluorination of alkynes, employing a carbonyl group as a director. Here, significant regio- and stereoselectivity was observed in the formation of fluoroalkenes (Scheme 5). It has been realized that troc-carbamates are better directing groups than esters and give better regioselectivity. The substrate variation has been checked with an (NHC)gold(I) tetrafluoroborate complex [(SIPr)AuBF4] [SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene].
 | ||
| Scheme 5 Synthesis of fluoroalkenes from alkynes. | ||
A related protocol for gold-catalyzed direct addition of HF to alkynes was reported16 by the groups of Xu and Hammond, where β-fluorovinylsulfones were developed from readily accessible alkynylsulfones (Scheme 6). The vinyl gold intermediate A8 was presumably generated via the addition of HF to gold-activated alkynylsulfones and its successive protodeauration furnished β-fluorovinylsulfones. Here, the sulfonyl group acts as an activator as well as a director in this regioselective gold-catalyzed nucleophilic fluorination. The reaction conditions were mild and the reaction was completed at an ambient temperature that tolerated a number of functional groups.
 | ||
| Scheme 6 Synthesis of fluoro-vinyl sulfones. | ||
In 2018, Prof. Toste first disclosed17 a diastereoselective synthesis of β-alkyl/aryl, (Z)-β-fluoro Michael acceptors from easily accessible electron-deficient alkynes (Scheme 7). The reaction was catalyzed by a RuPhos-ligated gold(I) complex, and trimethylamine trihydrogen fluoride (Et3N·3HF) was used for hydrofluorinations. In order to generalize this technique, various alkyl and aryl groups were investigated, and (Z)-vinyl fluorides were generated in moderate to good yields with high diastereoselectivity. Thus, a wide variety of β-fluoro-α,β-unsaturated ketones, esters, nitriles, amides, and aldehydes could be prepared using this method.
 | ||
| Scheme 7 Synthesis of β-fluoro Michael acceptors from electron-deficient alkynes. | ||
Prof. Nolan has developed18 a novel N-heterocyclic carbene gold bifluoride complex from the corresponding N-heterocyclic carbene gold hydroxide, which worked efficiently for hydrofluorination of symmetrical and unsymmetrical alkynes (Scheme 8). The hydrofluorination of simple alkynyls and alkynyl sulfides was examined, and the results included good to excellent yields of fluorinated stilbene analogues and fluorovinyl thioethers with high stereo- and regioselectivity. Electronically diverse substituents were tolerated under optimized conditions, and in the case of symmetrical alkynes, the (Z) isomer was selectively obtained. The difficulty of regioselectivity always arises in the case of asymmetrical alkynes. Better selectivity was obtained by carrying out the reactions for 5 days at room temperature.
 | ||
| Scheme 8 Hydrofluorination of internal alkynes and alkynyl sulfides. | ||
Iodoalkynes are generally useful organic synthons for coupling reactions to generate various molecular scaffolds.19 In 2016, Professor Nolan and his coworkers developed20 an Au(I)-catalyzed efficient protocol for hydrofluorination of iodoalkynes to synthesize 2-fluoro-1-iodoalkenes (Scheme 9). The reaction is highly diastereoselective and delivered the Z-isomer as the major product at ambient temperature with a variety of aromatic substitutions.
 | ||
| Scheme 9 Synthesis of (Z)-2-fluoro-1-iodoalkenes. | ||
Prof. Crimmin has developed21 a novel Au(I)-catalyzed HF transfer reaction that allows the simultaneous hydrofluorination of alkynes and the functionalization of fluoroarenes (Scheme 10). This reaction involved both defluorination and hydrofluorination in the same catalytic system. Consequently, handling the HF-based compound directly was not required since the perfluoroarene interacted with a nucleophile (phenol and aniline based) to generate HF, which made the approach simpler, safer and very cost-effective. DFT simulations, competition experiments, and kinetics studies have been performed in-depth to rationalize the entire catalytic network and understand the HF transfer process. The entire plausible HF transfer process is depicted in Scheme 10 and there are two ways through which the reaction may happen, depending on the type of nucleophile. It is likely that aniline-based nucleophiles will have a turnover-limiting protodeauration step, while phenol-based nucleophiles may exhibit a turnover-limiting cSNAr step.
 | ||
| Scheme 10 Hydrofluorination of internal alkynes via HF transfer. | ||
Being one of the best H-bond acceptors, 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) can form a stable complex with hydrogen fluoride (HF) by hydrogen bonding. Using this new concept, Prof. Hammond has developed22 a DMPU/HF complex that is highly acidic and stable in the presence of a gold(I)-catalyst for nucleophilic fluorination (Scheme 11). Moreover, since DMPU is a weak nucleophile, it cannot compete with HF in nucleophilic processes. Additionally, most of the metal catalysts are not effectively coordinated to DMPU, so considerable interference with these catalysts is unlikely. Hence, the HF/DMPU complex ought to be a perfect fluorination reagent, particularly in processes catalyzed by transition metals. By utilizing these unique properties of the DMPU/HF reagent, mono- and dihydrofluorination of internal and terminal alkynes were accomplished in a highly regioselective manner. As illustrated in Scheme 11, KHSO4 was added as a co-catalyst for dihydrofluorination, and several substrates were explored to generalize both approaches.
 | ||
| Scheme 11 Synthesis of fluoroalkenes and gem-difluoromethylenes. | ||
In the context of hydrofluorinations, several groups have used aqueous hydrofluoric acid for Au(I)-catalyzed hydrofluorination of alkynes. This concept is unique to the aforementioned developments, since competitive gold(I)-catalyzed alkyne hydration in aqueous media is also known.23 However, according to the Mayr nucleophilicity scale,24 fluoride in water is more nucleophilic in nature than water itself; therefore, alkyne hydrolysed products were completely suppressed.
In view of the above concept, Prof. Paquin has established25 a gold(I)-catalyzed hydrofluorination reaction of alkynes using the most economical fluoride source of 48% aqueous hydrofluoric acid (Scheme 12). This hydrofluorination reaction is unique and the corresponding monofluoroalkenes are achieved efficiently from easily accessible alkyne substrates. Both internal and terminal alkynes were utilized to generalize this reaction in aqueous medium and the alkynes associated with a fluorinated group at the propargylic position were better for this regioselective hydrofluorination.
 | ||
| Scheme 12 Hydrofluorination of terminal and internal alkynes. | ||
Profs Xu and Li also described an Au(I)-catalyzed efficient protocol26 for regioselective hydrofluorination of propargyl amines using 40% aq. HF (Scheme 13). Here, the amine group acts as an activator and regioselective director that causes a sharp decrease in entropy loss27 in the transition state. It is most likely caused by the protonated amine's hydrogen bonding with the fluorine anion, which causes the nucleophilic attack to be quasi-intramolecular (A9) and to happen more quickly than the intermolecular attack of water. Both alkyl and aryl substituted propargyl amines were utilized to generalize the reaction in an aqueous medium and in each case fluorine appeared at the location distal to the amino group. This method was also applicable for gram scale synthesis and offered an effective way to synthesize the fluorine-containing allylamine-based antifungal drug naftifine analogue (Scheme 13, eqn (2)).
 | ||
| Scheme 13 Hydrofluorination of propargyl amines. | ||
Nolan et al. achieved an efficient and chemoselective method28 for the hydrofluorination of terminal alkynes with aqueous HF using gold-N-heterocyclic carbene (NHC) complexes (Scheme 14). Mechanistic investigations provided insight into an in situ generated catalyst that demonstrated the maximum chemoselectivity and reactivity. It was created by reacting Brønsted basic gold pre-catalysts with aqueous HF. Attempts were made to identify many gold species involved in the catalytic cycle (path 1 and path 2) through catalytic interactions with specifically designed gold pre-catalysts. The computational studies provided a rationale for the high efficiency and chemoselectivity of gold catalysts, where chemoselectivity appeared to be largely determined by the identity of the counter ions. Based on both experimental and computational analyses, it has been proposed that under these catalytic circumstances, a transformation of A10 into a combination of A11 and A12 is anticipated, with complex A12 making up the majority of this conversion. Complex A12 quickly leads to A13 in the presence of the terminal alkyne (path 1). Complex A13 was then converted to complex A16 and the reactive gold bifluoride species A14, which would then allow the formation of the gold-fluorovinyl intermediate A15via the suggested transition state (TS1). The desired vinyl fluoride was then formed by protodeauration of intermediate A15 with HF or another form of HF present in the reaction medium, which led to the regeneration of the active gold-bifluoride species A14. A number of aliphatic terminal and internal alkynes have been employed successfully, ensuring the general applicability of this approach.
 | ||
| Scheme 14 Hydrofluorination of terminal alkynes. | ||
2.2. Oxo-fluorination of alkynes
Gold(I)-catalyzed alkyne oxidative functionalization29 has widely been used to achieve novel transformations and synthesize complex molecular frameworks. This alkyne oxidation process enabled direct access to gold alkynal complexes or α-oxo gold carbenes with versatile reactivity, expanding the scope and applications of gold alkyne chemistry. One major breakthrough in this regard was the gold-catalyzed oxidative fluorination of alkynes, which accomplished both alkyne oxygenation and further fluorination in one pot.Prof. Xu and Hammond developed16 an efficient protocol for the stereoselective synthesis of α-fluorosulfones from readily accessible alkynyl sulfone (Scheme 15). The reaction was catalyzed by cationic gold, for which N-oxide was used as an external oxidant at ambient temperature.30 In the presence of an Au(I)-catalyst, N-oxide was oxidatively added to the alkynyl sulfone, generating the reactive gold carbene intermediate A17. Here, HF addition to the gold-activated alkyne is entirely prevented in the presence of N-oxide. Thus, it has been presumed that the nucleophilicity of HF is reduced significantly due to the strong H-bonding interaction. On the other hand, N-oxide is one of the stronger nucleophiles which reacted with activated alkyne to generate the gold carbene intermediate. Finally, HF insertion into the gold carbene intermediate A17 produced α-fluorosulfones. Overall, the reaction concluded under mild conditions and permitted a number of functional groups under an ambient atmosphere.
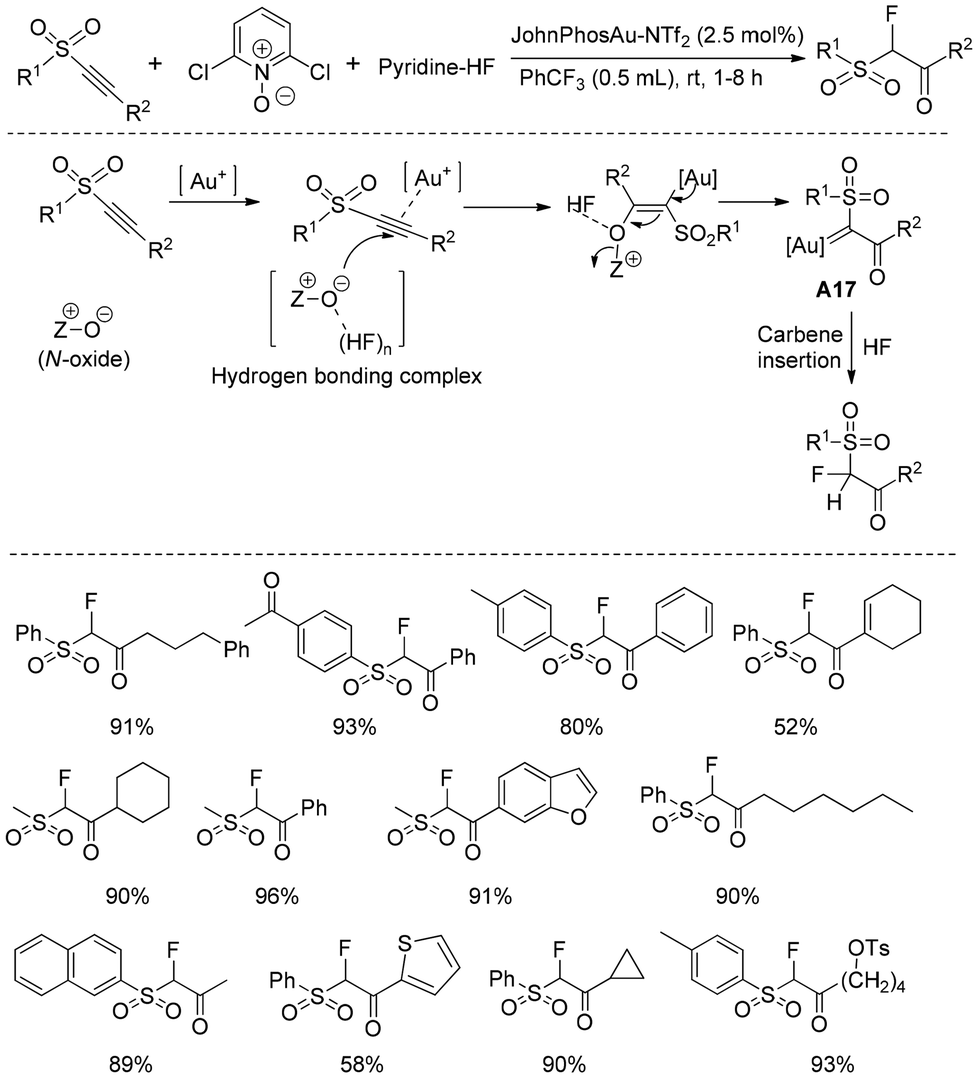 | ||
| Scheme 15 Synthesis of α-fluorosulfones from alkynyl sulfones. | ||
The same group further extended31 the above alkyne oxidative fluorination to readily available alkynyl ketone and ester systems (Scheme 16). Here, 2,6-dibromopyridine N-oxide was used as an external oxidant. The gold(I)-catalyzed intermolecular oxygen-atom transfer reactions between alkynyl ketones/esters and N-oxides probably generated the α-oxo gold carbene intermediate A18. Finally, intermolecular HF insertion into gold carbene A18 produced fluorinated 1,3-dicarbonyl compounds. The mild reaction conditions tolerated many functional groups with excellent yield and regioselectivity.
 | ||
| Scheme 16 Synthesis of fluorinated 1,3-dicarbonyl compounds from alkynyl esters and ketones. | ||
In 2016, Prof. Xu and coworkers developed32 a novel gold(I)-catalyzed synthesis of α-fluoroketones from readily available terminal alkynes (Scheme 17). This is quite an efficient method for the conversion of terminal alkynes to α-fluoroketones where pyridine N-oxide was used as an external oxidant. The reaction was proposed to proceed through the α-oxo gold carbene intermediate A19, which forms regioselectively at the terminal position of the alkyne. The intermediate A19 is highly efficient at inserting into the hydrogen fluoride bond to provide synthetically challenging α-fluoroketones. However, under these optimized conditions, internal alkynes are inactive in the oxofluorination process. The reaction was completed at ambient temperature as well, under mild conditions that tolerated many functional groups.
 | ||
| Scheme 17 Synthesis of α-fluoro ketones from terminal alkynes. | ||
2.3. Fluoroarylation of allenoates
Prof. Feng developed33 a gold-catalyzed unprecedented protocol for the fluoroarylation of allenic esters in the presence of arenediazonium salts prompted by visible light (Scheme 18). In this novel method, carbofluorination occurred in the absence of electrophilic fluorinating agents, and new fluoro-containing quaternary centres were generated efficiently. The reaction proceeded in a highly regio- and stereoselective manner and aryl groups were incorporated proximal to the ester functionality. Extensive control experiments were carried out to provide greater insight into the activation mode of the allenic ester. It was observed that light irradiation was beneficial for the reaction's efficiency, and the transformation was inhibited when the ester group was replaced with cyano, sulfone, or phenyl groups, or by using aliphatic allenes. The absence of the arenediazonium salt also prevented the formation of the corresponding hydrofluorination product (Scheme 19, eqn (3)). This suggests that both ester functionality and arenediazonium salt play crucial roles in the Au(I) activation scenario. A potentially active Ar–AuIII species was generated to determine the precise activation mechanism (Scheme 19, eqn (4) and (5)). Only in the presence of extra AgBF4, the involvement of the Ar–AuIII species was extremely supportive in reacting with the allenic ester and also served as an effective catalyst in this transformation. This also suggests the crucial cationic nature of the high-valent gold(III) complex during the transformation. In order to learn more about the reaction profile, an enantio-enriched allenic ester was examined, which produced the desired product with good selectivity (Scheme 19, eqn (6)). This result indicated that the distal double bond of the allene moiety was functionalized by a concurrent activation on the side opposite the ester group by the Ar–AuIII species, while the HF was accompanied by an interaction between hydrogen bonding with the ester functionality and nucleophilic fluorination observed from another side. To rationalize the isomerization process, fluorinated (Z)-olefin was added to the standard catalytic cycle (Scheme 19, eqn (7)). The fact that (Z)-olefin was recovered after completion without any changes to its geometry suggested that isomerization occurred during the catalytic cycle. Based on the extensive controlled experiments, it was proposed that light-promoted oxidation of the Au(I) complex with the arenediazonium salt generated the high-valent active Ar–AuIII species34A20 for possible allene activation. The subsequent nucleophilic fluorination, followed by isomerization and reductive elimination sequences, afforded the desired fluoroarylation products. This transformation stands out for its high stereo- and regioselectivity, the use of Et3N·3HF as an economical nucleophilic fluorination reagent, excellent tolerance of an extensive range of functionalities, and ease of assembling structurally stabile motifs with fluoro-containing sp3 carbon centers. | ||
| Scheme 18 Fluoroarylation of allenic esters. | ||
 | ||
| Scheme 19 Controlled experiments for the fluoroarylation of allenic esters. | ||
3. Gold-catalyzed fluorination with electrophilic fluorinating reagents (F+)
3.1. Oxo-fluorination of alkynes
In 2010, Prof. Nevado synthesized35 α-fluoro acetals and α-fluoro ketones from alkynes with a high degree of chemo- and regioselectivity. In alcohol medium, catalytic amounts of gold(I) and Selectfluor selectively converted the alkynes into α-fluorinated acetals or ketones, depending on the reaction parameters. Based on some control experiments, it was revealed that the corresponding ketones were not effective intermediates for the transformation of alkynes into α-fluorinated acetals. A different mechanistic aspect was presumed, which has been depicted in Scheme 20. The reaction was proposed to proceed through an alkoxylation/hydration–fluorination protocol. Initially, alkynes are activated by gold(I); as a result, a facile nucleophilic attack takes place by alcohol, which generates the vinyl gold intermediate A21 and subsequent protodeauration furnishes enol ether A22.36 Additional experimental results confirmed that either in the presence (path a) or absence (path b) of gold(I), Selectfluor is effective for fluorination of enol ether A22. However, in the presence of gold(I), better yields were observed for the conversion of enol ether A22 to α-fluoro acetals. These results indicated that the mechanism may proceed through enol ether intermediate A22 and the reaction does not entirely proceed via Selectfluor-mediated fluorination of the in situ generated enol ether A22. As proposed, enol ether A22 is further activated by gold(I) and the simultaneous addition of another molecule of alcohol generates intermediate A23, which is probably oxidized in the presence of Selectfluor-generated Au(III) species A24. The C(sp3)–F bond is thus generated via reductive elimination at the metal center. The reaction is applicable for both internal and terminal alkynes and the formation of the corresponding fluorinated acetals or ketones depends on the reaction conditions.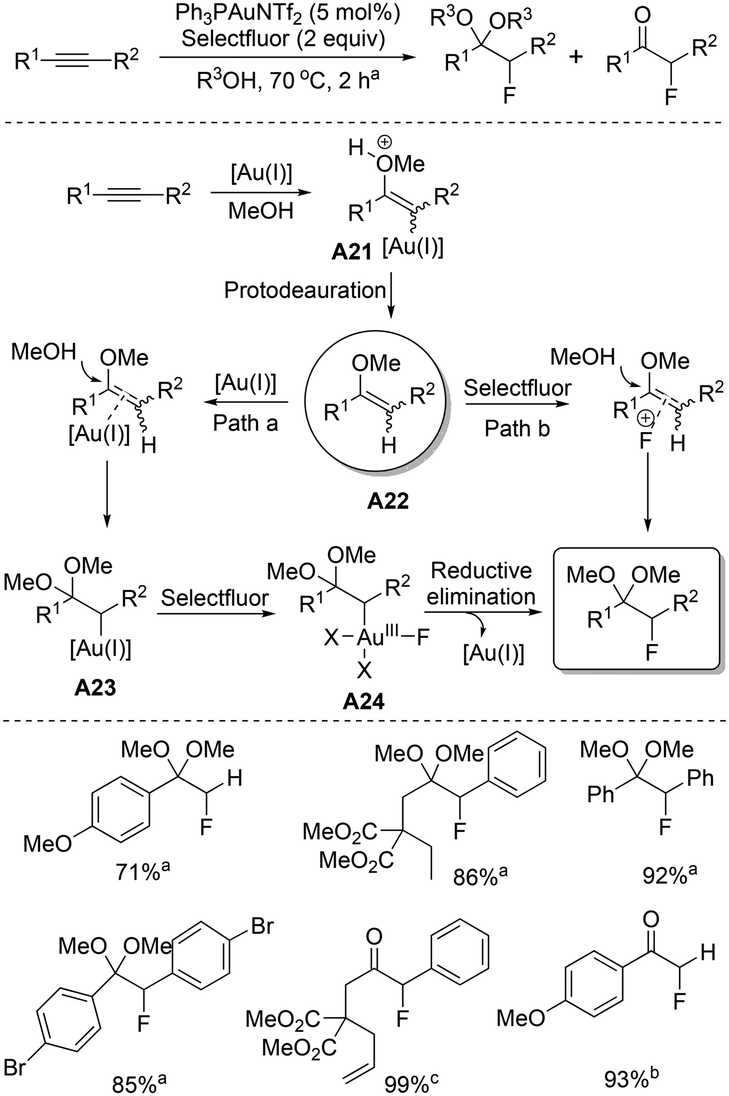 | ||
| Scheme 20 Synthesis of α-fluoro acetals and α-fluoro ketones from alkynes. | ||
Recently, Prof. Xie developed37 a gold-catalyzed oxo-arylfluorination of internal aliphatic and aromatic alkynes in which a site-directing functional group in the alkyne moiety governs the reaction divergence (Scheme 21). This four-component multifunctionalization reaction successfully breaks the alkyne functionality and forms four new chemical bonds simultaneously for fluorination, arylation and oxygenation of alkynes. The presence of a phosphonate unit favours the regioselective oxo-arylfluorination through an Au(I)/Au(III) redox coupling technique with arylboronic acid and employing Selectfluor as an oxidant as well as a fluorinating agent. Water also plays a significant role in this multi-component reaction. Extensive labelling experiments were carried out in an attempt to determine the origin of the oxygenation in the alkyne moiety (Scheme 21, eqn (8)). When the reaction was performed in the presence of labelled H218O, the 18O atom was not connected with the newly generated carbonyl group. Additionally, when the desired α-oxo-fluorinated product was specifically treated using H218O, no 18O atom was incorporated (Scheme 21, eqn (9)). Therefore, it has been presumed that a five-member cyclic phosphonate intermediate A25 is generated through the activation of alkynes followed by an intramolecular nucleophilic attack by the more nucleophilic oxygen atom of the PO bond. HRMS data also support the existence of the newly generated five-membered ring intermediate A25. Selectfluor assisted oxidation of phosphonate Au(I) A25a to vinyl-Au(III) species, followed by arylboronic acid-promoted bimolecular reductive elimination,38 produced intermediate A26 and regenerated the active Au(I) catalyst. The desired oxo-arylfluorination product was then produced through a non-metallic procedure by nucleophilic ring opening of the cyclic phosphonate ester in excess water and simultaneous electrophilic fluorination with Selectfluor. The method facilitates gram-scale synthesis, exhibits high regioselectivity and endures a wide range of functional groups.
 | ||
| Scheme 21 Oxo-arylfluorination of internal aliphatic and aromatic alkynes. | ||
A mild and practical route to prepare functionalized α-fluoroketones was presented39 by Prof. Michelet in the presence of a gold catalyst/Selectfluor association in aqueous ethanol at room temperature (Scheme 22). The α-fluoroketones were obtained in good to excellent yields without any acids or bases using simple aldehyde-yne or ketone-yne derivatives as starting precursors in green medium. Interestingly, this oxofluorination was a domino two-step process of formal alkyne hydration and simultaneous fluorination. The reaction was highly selective for the aldehyde-yne derivative, while the opposite reigoselectivity was also observed for the ketone-yne adducts. Numerous control studies and literature reports have led to the proposal that the reaction is initiated by electrophilic Au(I)-activation of alkynes, which aids in the 6-endo-dig cyclization (path a) by the carbonyl moiety to form isobenzopyrylium intermediate A27. This intermediate functions very well for nucleophilic addition of water and generates an unstable intermediate A28, followed by ring opening to produce enol A29. The desired α-fluoroketones were obtained using a simultaneous demetalation and fluorination process with Selectfluor. An additional approach (path b) that has been postulated would create Au(III) activated alkyne intermediate A30 using an oxidative addition approach. Once A30 is hydrated, an intermediate A31 is produced. This intermediate would then progress to a σ-gold(III) complex A32, which would eventually yield the required α-fluoroketones through reductive elimination. The methodology has broadened the reaction scope by showing its applicability on a larger scale and preparing challenging 4-fluoroisoquinoline derivatives in excellent yields.
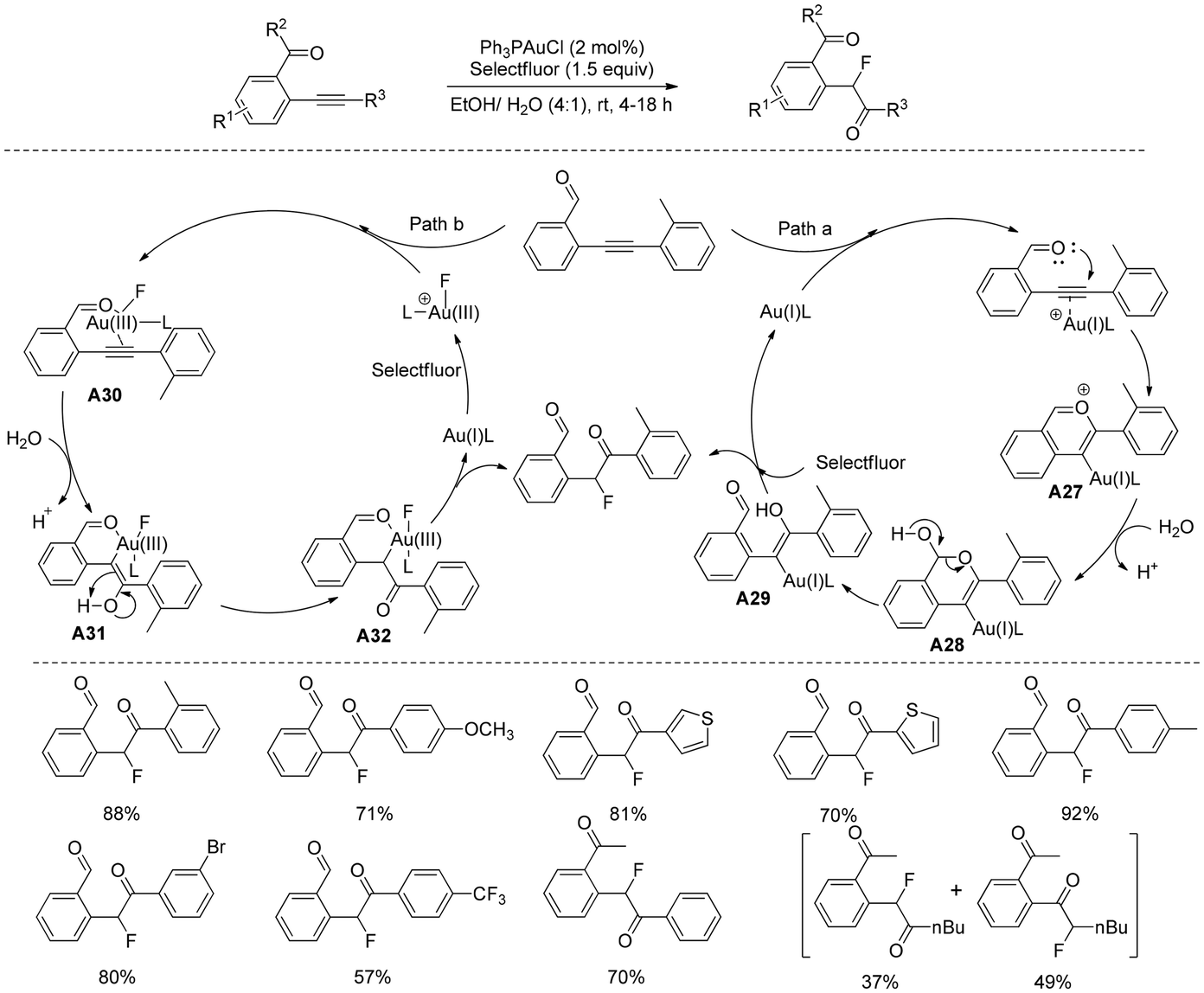 | ||
| Scheme 22 Synthesis of functionalized α-fluoroketones. | ||
Recently, the group of Michelet and Arcadi reported40 the gold-catalyzed oxyfluorination/oxydifluorination reactions for the selective synthesis of α-fluoro, β-phthalimido ketones and α,α-difluoro β-phthalimido ketones from the corresponding alkyl- and aryl-substituted N-propargyl phthalimides (Scheme 23). To clarify the mechanism of this regioselective cascade of oxyfluorination/oxydifluorination reactions, several control experiments were performed. The desired non-deuterated oxyfluorination product was only isolated in the presence of D2O (Scheme 23, eqn (10a)), which means that the proton exchange with EtOH becomes slow under these reaction conditions. However, the desired deuterated fluoro derivative was isolated through a combination of MeOD and D2O (Scheme 23, eqn (10b)). In addition to the desired product, a lower yield of 2-(2,2-difluoro-3,3-dimethoxy-3-phenylpropyl)isoindoline-1,3-dione was also obtained at 50 °C in MeOH without the presence of water (Scheme 23, eqn (11)). Based on these results, it has been hypothesised that the alkyne derivatives are subjected to an alkoxyauration reaction, producing the intermediate A33, which transformed into the fluoro vinyl ether A34 and regenerated the catalyst. Hydrolysis of fluoro vinyl ether A34 afforded the desired α-fluoroketone. In another path with an excess of Selectfluor, ethoxyfluorination of intermediate A35 yielded the α,α-difluoroketones through the ketal intermediate.
 | ||
| Scheme 23 Synthesis of α-fluoro, β-phthalimido ketones and α,α-difluoro β-phthalimido ketones. | ||
Prof. Xu and Hammond presented41 a potential novel approach for fluorine-enabled cationic gold catalysis, wherein internal alkynes are hydrated in the presence of aromatic boronic acids and water in a single pot to create α-substituted α-fluoroketones (Scheme 24). This newly proposed fluorine-enabled cationic gold species was produced through oxidative fluorination of a low valence gold salt (e.g. [ClAuIL]) using an ammonium [N–F]+ fluorination agent. It is anticipated that such species in a higher oxidation state has more robust Lewis acidity during the fluorination process compared to the precursor Au catalyst. The reaction was initiated through alkyne activation by the newly proposed fluorine-enabled cationic gold(III) species, followed by the attack of water and a transmetalation process with boronic acid generated intermediate A36. The weak Au–F bond and the strong B–F bond are believed to be the primary factors causing this transmetalation. The two plausible routes can then be followed to form the desired product. Fluorination of intermediate A36 by Selectfluor and sequential reductive elimination afforded the functionalized ketone A37 (path a). Another approach involved fluorination first, followed by reductive elimination, leading to the functionalized α-fluoroketones A37 (path b). It has also been suggested that fluorine facilitates the processes for cationic gold catalysis and can even be employed as a catalytic activator in this application. The regioselectivity is determined by the steric and electronic effects of the substituents and may be impacted by the participation of internal alkynes with a nearby nucleophilic site in surrounding groups.
 | ||
| Scheme 24 Synthesis of α-substituted-α-fluoroketones from internal alkynes. | ||
3.2. Fluorination by 1,3-acyloxy migration
In 2010, Prof. Nevado developed42 a highly diastereoselective gold(I) catalyzed synthesis of α-fluoroenones from readily accessible propargyl acetate (Scheme 25). The reaction was catalysed by the IPr-gold(I) complex in acetonitrile–water medium, where gold(I)-catalysed facile rearrangement takes place efficiently. Initially, propargyl acetates are activated by cationic gold(I), and as a result, facile [3,3] sigmatropic shift takes place to create the allenyl intermediate A38 followed by acetate hydrolysis affording vinyl gold(I) A39, which is probably oxidized in the presence of Selectfluor. The C(sp2)–F bond is thus generated via reductive elimination at the metal centre to afford the desired product. As per the experimental results, this domino gold(I) catalyzed rearrangement and fluorination of propargylic acetate proceeded with more E selectivity. In the same year, Prof. Gouverneur also developed43 similar α-fluoroenones from readily accessible propargyl acetates (Scheme 25, eqn (12)), where the SIPr-Au(I) complex was used in acetonitrile medium. As the Selectfluor approaches the allene from the least hindered face, the reaction proceeds with more preferential E selectivity.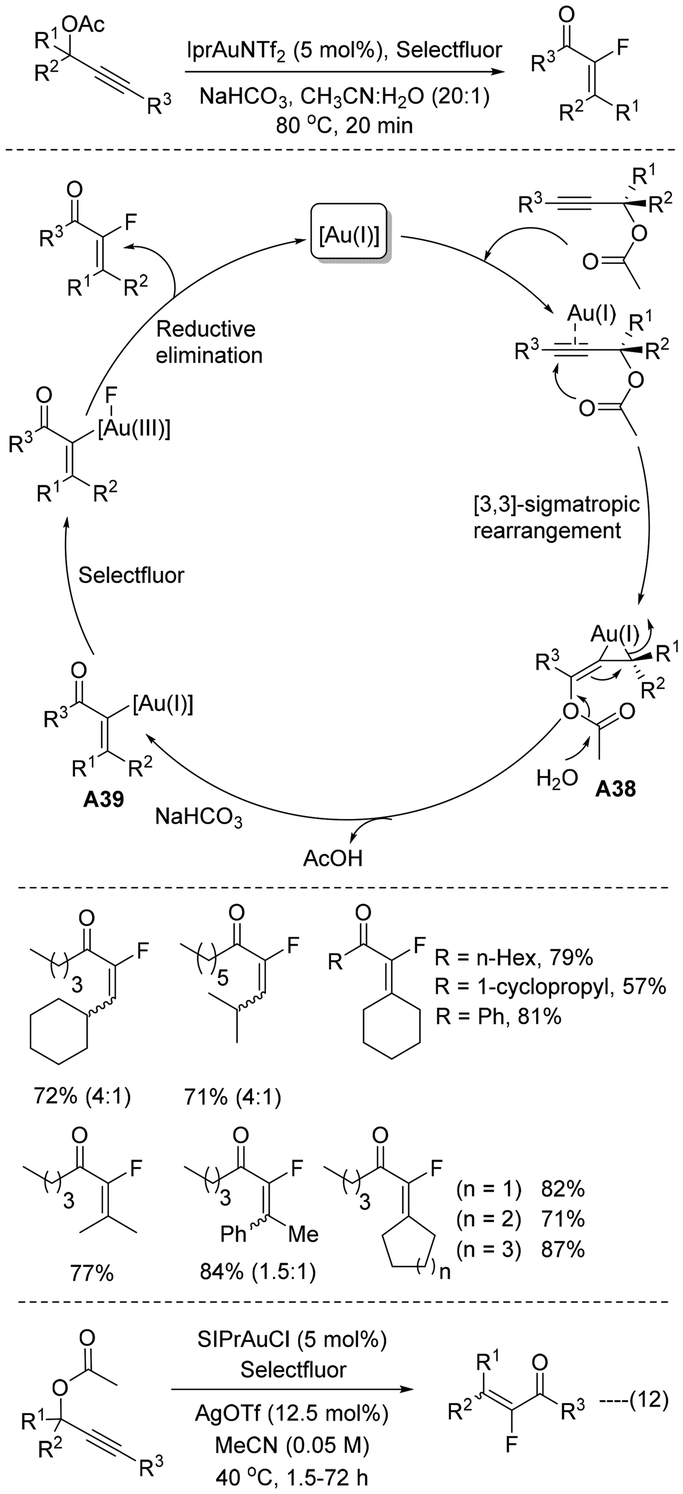 | ||
| Scheme 25 Synthesis of α-fluoroenones from propargyl acetate. | ||
Inspired by the developments of Nevado and Gouverneur,42,43 Profs Hammond and Xu reported44 a gold(I)- and Selectfluor-mediated stereoselective synthesis of monofluorinated α,β-unsaturated ketones from electron-rich allenyl carbinol esters under mild conditions (Scheme 26). It was proposed that, in the presence of a gold catalyst, the allenyl carbinol ester isomerizes to the electron-rich 1,3-butadiene-2-ol ester A40 with moderate to fair yield. Without any gold catalysts, the electron-rich 1,3-butadiene-2-ol ester was fluorinated with Selectfluor to form the allylic cation species A41, which was isomerized to the more stable A42. Finally, the fluoroalkyl α,β-unsaturated ketone was generated with exclusive E stereoselectivity through the hydrolysis of intermediate A42 using water, which was present in trace amounts in the reaction solvent. This transformation stands out for its high stereo- and regioselectivity, with an excellent tolerance of an extensive range of functionalities.
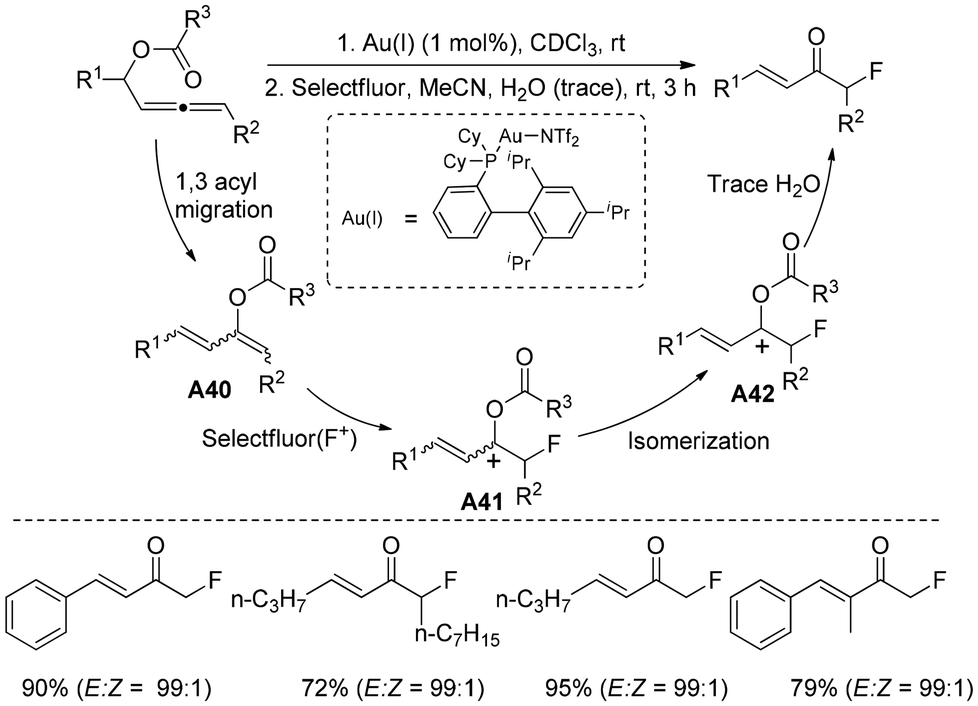 | ||
| Scheme 26 Synthesis of monofluorinated α,β-unsaturated ketones. | ||
3.3. Heterocyclization fluorination
Indoles represent a significant class of heterocycles that are present in numerous biologically active natural products and have been used enormously in drugs and medicinal chemistry.45 Prof. Michelet revealed46 a gold(I)-catalyzed aminoalkylation/fluorination cascade and a one-pot two-step gold(III)-catalyzed aminoalkylation/electrophilic fluorination for the generation of 3,3-difluoro-2-substituted-3H-indoles under mild reaction conditions (Scheme 27). The reaction was conducted smoothly in green ethanol that did not require any additional acids and bases. In this investigation, unprotected 2-alkynylanilines were utilized, and the protocol displayed better flexibility over the other reported procedures. As the gold(I) complex might oxidize into the gold(III) complex in the presence of Selectfluor, it was hypothesized that both gold(I) and gold(III) complexes catalyzed the process. Scheme 27 shows the two key mechanistic pathways involved in the tandem gold-catalyzed aminofluorination of 2-alkynylanilines. First, the reaction may have involved the formation of intermediate A44 by Au(I)-assisted activation of π-alkyne A43 (path I), followed by oxidation to the Au(III)-complex with Selectfluor and sequential reductive elimination of intermediate A45 produced 2-substituted-3-fluoroindoles. Another possibility was the redox Au(I)/Au(III) catalytic cycle, which probably involved the oxidation of the Au(I) catalyst to Au(III) A46 for effective cyclization (path II). Additionally, after protodeauration of intermediate A44, it might have generated 2-substituted indoles A47. In the presence of an excess of Selectfluor, the 2-substituted indole or 2-substituted-3-fluoroindole derivatives might undergo further fluorination to produce 3,3-difluoro-2-substituted-3H-indoles. | ||
| Scheme 27 Synthesis of 3,3-difluoro-2-substituted-3H-indoles. | ||
The group of Liu and Xu discovered47 a gold(I)-catalyzed mild and efficient approach for the synthesis of fluorinated pyrazoles via tandem aminofluorination of alkynes where Selectfluor has been used as a fluorinating agent (Scheme 28). The mild reaction conditions permit a wide range of substituents in different positions with high yields. To establish the mechanism, the formation of pyrazoles A50 and fluorinated pyrazoles A49 over time has been studied. It has been observed that within 15 minutes, starting material A48 was completely consumed, yielding the mixture of pyrazoles and fluorinated pyrazoles with 96% total yield (A49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A50 = 54:42, R1, R3 = Ph, R2 = Me). However, over a prolongated period, pyrazoles get fluorinated, resulting in a total yield of 81% (A49:A50 = 6.2:1, R1, R3 = Ph, R2 = Me). In a different experiment, pyrazoles were allowed to react with Selectfluor in the presence and absence of the gold catalyst. A significantly greater yield of fluorinated pyrazoles was observed when the gold catalyst was present (Scheme 28, eqn (13)). Based on these experimental results, it has been presumed that both Au(I) and Au(III) are equally active for this conversion and Scheme 27 provides a detailed representation of every possible path.
:A50 = 54:42, R1, R3 = Ph, R2 = Me). However, over a prolongated period, pyrazoles get fluorinated, resulting in a total yield of 81% (A49:A50 = 6.2:1, R1, R3 = Ph, R2 = Me). In a different experiment, pyrazoles were allowed to react with Selectfluor in the presence and absence of the gold catalyst. A significantly greater yield of fluorinated pyrazoles was observed when the gold catalyst was present (Scheme 28, eqn (13)). Based on these experimental results, it has been presumed that both Au(I) and Au(III) are equally active for this conversion and Scheme 27 provides a detailed representation of every possible path.
 | ||
| Scheme 28 Synthesis of fluorinated pyrazoles. | ||
In the presence of Selectfluor, Prof. Wu and his group developed48 a gold(I)-catalyzed synthesis of fluorinated imidazoles from readily accessible propargyl amidines (Scheme 29). The method of this aminofluorination of alkyne is quite uncommon and generates a new Csp3–F bond through a cascade cyclization/fluorination sequence. To understand the actual reaction path, a gold(I)-alkyl species A52 was prepared separately (Scheme 29, eqn (14)). However, after treatment with Selectfluor in acetonitrile, species A52 failed to generate any Csp3–F bonds (Scheme 29, eqn (15)). This observation reveals that the monofluorinated imidazoles are probably produced from the vinyl-gold intermediate A51, which has been generated directly from parent propargyl amidines through an intramolecular nucleophilic amination of alkynes in a 5-exo-dig manner. Finally, the fluorination of the vinyl-gold intermediate A51 and simultaneous aromatization afforded the desired fluorinated imidazole derivatives. The method permitted a wide variety of functional groups; however strongly electron withdrawing substituents on propargyl amidines led to lower yields. Additionally, steric hindrance had a significant impact on the yield of the products.
 | ||
| Scheme 29 Synthesis of fluorinated imidazole. | ||
Isoxazole is a significant heteroaromatic framework that exhibits various biological behaviours, including anticancer, antibiotic, analgesic, and antidepressant effects.49 Prof. Ryu has successfully established50 a mild gold-catalyzed tandem cyclization–fluorination process of (Z)-2-alkynone O-methyl oxime to produce biologically important fluoroisoxazole (Scheme 30). This process can be applied to a variety of substrates and offers an effective one-pot cascade path to fluoroisoxazoles with high selectivity and high yields. Several controlled experiments and in situ NMR studies were conducted to establish the reaction mechanism. Under standard reaction conditions, the starting (E)-O-methyl oximes were unable to produce the desired fluoroisoxazoles (Scheme 30, eqn (16)), indicating that (E)- and (Z) isomers were not interconvertible in the reaction medium. At room temperature 3,5-disubstituted isoxazoles failed to undergo direct fluorination under optimized reaction conditions (Scheme 30, eqn (17)), and even with Selectfluor only (Scheme 30, eqn (18)). Thus, the proposed mechanism states that the initial coordination of the gold complex assisting the nucleophilic cyclization would produce intermediate A53. The cross-coupled fluoroisoxazole was then produced by demethylation and Selectfluor-assisted oxidation to cationic gold(III) and reductive elimination sequences.
 | ||
| Scheme 30 Synthesis of biologically important fluoroisoxazole. | ||
The development of new strategies for the synthesis of pyrrolidines and piperidines is very significant because of their enormous applications in the field of medicinal chemistry and the pharmaceutical industry.51 In 2011, the group of Fensterbank and Malacria established52 an efficient approach for synthesizing gold catalyzed fluorinated pyrrolidine skeletons from readily available 1,5-aminoalkynes (Scheme 31). The presence of Selectfluor favours the oxidation of Au(I) to Au(III) species A56 as well as the electrophilic fluorination of the cyclic enamine intermediates to access fluorinated pyrrolidines. The 19F NMR data also support the existence of the newly generated Au(III) species A56. It has been proposed that a five-membered vinyl Au(III) intermediate A57 was generated through the activation of alkyne followed by the nucleophilic attack of the NH moiety in a 5-exo-dig manner. Based on the literature41,53 and experimental evidence, two possible routes (fluorodeauration vs. protodeauration) have been postulated from the cyclic vinyl Au(III) intermediate A57 to the formation of pyrrolidine A54, A55 and A59. Control experiments suggested that the protodeauration path appears to be the major as in the presence of Selectfluor the mixture of isolated tautomers A60 and A61 can produce the desired A54 and A59 with PPh3AuCl and also in the absence of [Au]. Alternatively, A58 may be generated via reductive elimination from the vinyl Au(III) intermediate A57. This would then lead to tautomerization and electrophilic fluorination by Selectfluor, yielding pyrrolidine A55, which could potentially be synthesized from pyrrolidine A54 as supported by the experimental results. Under the same optimized conditions, the method was further extended to prepare fluorinated piperidine skeletons (Scheme 31, eqn (19)).
 | ||
| Scheme 31 Synthesis of gold-catalyzed fluorinated pyrrolidine skeletons. | ||
Profs Shi and Tang developed54 a straightforward and efficient protocol for the synthesis of α-fluorobenzofuranones from 2-alkynylphenols through an Au(I)/Au(III) redox catalytic cycle that enables simultaneous cyclization, fluorination, and hydration in a single operation (Scheme 32). This work addresses novel applications for the heterocyclization–fluorination cascade and offers a new method for generating C–O, CO, and C–F bonds. Based on an asymmetric version of this methodology, several controlled experiments with oxygen labelling, and 19F NMR investigations, a probable mechanism was suggested, identifying the HF generation in this redox catalytic cycle. In the presence of Na2CO3, sodium phenolate was first generated and this was followed by the formation of the vinyl-gold intermediate A62via an intramolecular Au(I)-promoted cyclization process. Afterward, Selectfluor-assisted oxidation formed F–AuIII species A63 followed by fluorination through a Friedel–Crafts reaction, generating the oxocarbenium intermediate A64. Then, through a sequential process of nucleophilic water additions, reductive eliminations, and simultaneous HF release, the desired α-fluorobenzofuranones were obtained (path a; fluorination–hydration process). In another path b, intermediate A63 was directly converted into intermediate A65 following a reductive elimination process. Subsequently, fluorination, nucleophilic water addition and simultaneous HF release produced the same α-fluorobenzofuranones.
 | ||
| Scheme 32 Synthesis of α-fluorobenzofuranones. | ||
In 2014, a novel gold-catalyzed tandem cycloisomerisation of alkynylic alcohol was reported55 by Hammond and his co-workers to synthesize difluoro hydroxyl tetrahydrofurans (Scheme 33). Here, the more electrophilic F-TEDA-PF6 yielded better results than Selectfluor. Based on the experimental results, it was proposed that the reaction proceeded through a complex mechanism rather than a simple two-step cyclization. Salient features of this efficient methodology are easily available starting substrates, wider reaction scopes, functional group tolerance, and high yields under mild reaction conditions. The method provided a good substrate scope including aromatic and aliphatic substitutions with moderate to good yields. This reaction has also been extended to synthesize fluoro-lactones through the monofluorolactonization of alkynylic acids (Scheme 34).55
 | ||
| Scheme 33 Synthesis of difluoro hydroxyl tetrahydrofurans. | ||
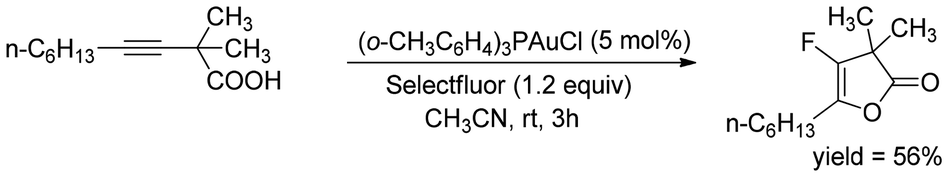 | ||
| Scheme 34 Monofluorolactonization of alkynylic acids. | ||
Prof. Dembinski developed56 a convergent synthesis of highly substituted 3-fluorofurans using alkenynyl silyl ethers as synthetic precursors. Here, the reaction proceeded through a dual catalysed cascade electrophilic cycloisomerisation (Scheme 35). Based on control experiments and 19F NMR investigations, a probable mechanism has been suggested, identifying the electrophilic halogen generation in this redox catalytic cycle. Under standard reaction conditions, the starting silyloxy enynes converted to the fluoroalkynone A66. The initial coordination of the gold complex assisted in nucleophilic cyclization/aromatisation, producing the vinyl gold intermediate A67 which was then trapped by the electrophilic halogen. Here, probably zinc may also potentially enhance the electrophilic cycloisomerisation cascade.
 | ||
| Scheme 35 Synthesis of substituted 3-fluorofurans. | ||
Prof. Gouverneur developed57 a gold(I)-catalyzed alkoxyfluorination for the synthesis of 5-fluoro-3,3′-difluorodihydropyranones from β-hydroxy-α,α-difluoroynones under mild reaction conditions (Scheme 36). Control experiments demonstrated that in the absence of gold, Selectfluor was unsuitable for this cyclization/fluorination. Furthermore, it was observed that the protodeaurated product A69 was unsuitable for electrophilic fluorination, whether gold was present or not. Therefore, it has been assumed that the transient vinyl gold species A68 was the effective intermediate for electrophilic fluorination. Following a 6-endo-dig cyclization that generated the cyclic vinyl gold species A68, simultaneous fluorination produced 5-fluoro-3,3′-difluorodihydropyranones. A lower yield of the protodeauration product was also observed from difluorinated ynones during this cyclization–fluorination cascade. However, unfluorinated ynones afforded difluorinated tetrahydropyranones along with the ring-opened monofluorinated ketones in this catalytic process.
 | ||
| Scheme 36 Synthesis of 5-fluoro-3,3′-difluorodihydropyranones. | ||
Homopropargylic derivatives are generally useful organic synthons to generate various molecular complexities. In 2014, Prof. Fiksdahl and his coworkers reported58 a one-pot gold(I) catalyzed transformation from homopropargyl ketal A70 to the highly substituted 3-fluoro-3,6-dihydro-2H-pyran derivative A71 (Scheme 37). The mechanism is based on the gold(I) catalyzed Petasis–Ferrier rearrangement of homopropargyl ketal A70 by the inclusion of the ketal moiety, followed by subsequent fluorination with Selectfluor in one pot. The reaction was completed at ambient temperature under mild conditions within 15 min.
 | ||
| Scheme 37 Synthesis of substituted 3-fluoro-3,6-dihydro-2H-pyran. | ||
3.4. Carbocyclization fluorination
Prof. Rao and Zhang developed59 an efficient procedure for the synthesis of 5-fluorocyclopentenone by employing the gold(I)-catalyzed cycloisomerization/electrophilic fluorination sequence of 1,3(4)-enyne esters in the presence of NFSI (Scheme 38). This one-pot novel strategy generated a fluorine-substituted carbon stereocenter with good to exceptional yields under mild reaction conditions, exhibiting a wide range of functional group compatibility. Based on the outcomes of controlled experiments and the literature,60 it has been suggested that the reaction was initiated through Au(I)-catalyzed 1,3-acyloxy migration/Nazarov cyclization leading to the formation of cyclopentadiene A72. Thereafter, the electron-rich cyclopentadiene intermediate A72 can be electrophilically fluorinated with NFSI in the absence of any gold catalysts, yielding the allylic cation species A73 or its resonance form A74. Finally, 5-fluorocyclopentenone A75 was generated through the hydrolysis of intermediate A74 using water that was present in trace amounts in the reaction solvent. After this achievement, the technique was extended to fluorinate 1,4-enyne esters61 with NFSI to yield 5-fluorocyclopentenones under the same optimal conditions (Scheme 39). | ||
| Scheme 38 Synthesis of 5-fluorocyclopentenone from 1,3(4)-enyne esters. | ||
 | ||
| Scheme 39 Synthesis of 5-fluorocyclopentenones from 1,4-enyne esters. | ||
Prof. Liu and Xu reported62 a tandem three-component reaction involving allene esters, Selectfluor, and water catalyzed by gold(I) salts, resulting in the synthesis of fluorinated indenes (Scheme 40). This reaction involved an Au(I)/Au(III) redox catalytic cycle and allowed for the concurrent generation of C–C, CO, and C–F bonds in one pot. Based on the H218O-labelling experiment, the oxygenation occurred from the water itself (Scheme 40, eqn (20)). A deuterium labeling experiment also supported the primary kinetic isotope effect (kH/kD = 2.3), which also indicated that cycloauration was most likely a rate-determining step (Scheme 40, eqn (21)). According to these results and previous literature reports41,47,63 it was presumed that the reaction proceeded through two different paths. In path I, initial gold(I) promoted allene activation and simultaneous regioselective water addition generated the vinyl gold intermediate A77. The intermediate A77 probably oxidized in the presence of Selectfluor, forming higher oxidation Au(III) species A78. As an alternative, A78 might also be produced by the hydroxyauration of allene esters A76 with water and a cationic gold(III) species, which might form from Au(I) by the oxidation of Selectfluor (path II). Cycloauration of vinyl gold(III) A78 and simultaneous reductive elimination generated the indene intermediate A79. Finally, a sequential 1,3-H shift64 in the indene intermediate A79, hydroxyl oxidation to carbonyl65 followed by α-fluorination66 afforded the desired fluoroindene.
 | ||
| Scheme 40 Synthesis of fluorinated indenes. | ||
4. Conclusion
The advancements in gold-catalyzed fluorination of C–C multiple bonds have built momentum in the field of homogeneous gold catalysis. The efficient single-step formation of vinyl gold species from alkynes has opened vast areas of research related to gold fluorine chemistry. The development of several novel approaches allowed a one-step conversion of simple alkyne substrates to various fluoroalkanes, fluoroalkenes, α-fluorocarbonyls, and fluorinated carbo- and hetero-cycles, which are found in many bioactive natural products, drugs, and agrochemicals. The gold-catalyzed alkyne activation, simultaneous nucleophilic addition, various rearrangements and an array of cascade transformations illustrate the “economies of synthesis”. Also, significant success has been achieved in developing electrophilic and nucleophilic fluorinating agents and their utilizations in varieties of alkyne substrates which remains a frontier area of research in organic synthesis. The most economical fluoride source of aqueous hydrofluoric acid has also been widely used in the fluorination of alkynes. The efficiency of electrophilic and nucleophilic modes of fluorination to build the diverse molecular complexity has also been covered. However, there are still issues and opportunities for improvement in a number of these approaches. In most cases, the fluorinating agent has a crucial role in the transformations, and there is scope for widening the types of fluorinating reagents, which are currently limited to Selectfluor. The mechanisms of various alkyne fluorinations are still uncertain. Experimental validation might be beneficial for mechanisms that are developed through theoretical calculations. It requires a lot of effort to overcome challenges associated with asymmetric fluorinations of alkyne substrates. Novel asymmetric fluorinating agents may open up new possibilities for asymmetric fluorinations of alkynes. Nonetheless, the tremendous advancements made so far and the prospects in the gold-catalyzed fluorination of C–C multiple bonds have laid a strong platform for more exciting developments.Data availability
Data sharing is not applicable to this article as no data were created or analyzed in this study.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the financial support from the Science and Engineering Research Board (grant CRG/2023/001214 and YSS/2015/001425) and the UGC [F.4-5(207-FRP)/2015(BSR)].References
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) T. Furuya, C. A. Kuttruff and T. Ritter, Curr. Opin. Drug Discovery Dev., 2008, 11, 803–819 CAS; (c) J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 8610–8614 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (b) H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, Wiley-VCH, Weinheim, Germany, 2004 CrossRef; (b) R. D. Chambers, Fluorine in Organic Chemistry, Blackwell Publishing, Oxford, UK, 2004 CrossRef; (c) K. Uneyama, Organofluorine Chemistry, Blackwell Publishing, Oxford, UK, 2006 CrossRef; (d) J.-P. Bégué and D. Bonnet-Delpon, Chimie bioorganique et medicinal du fluor, CNRS Editions, Paris, 2005 Search PubMed.
- (a) H. Schobert, Chem. Rev., 2014, 114, 1743–1760 CrossRef CAS; (b) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2980 CrossRef CAS; (c) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed; (d) I.-T. Trotus, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761–1782 CrossRef CAS PubMed; (e) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (f) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2310 CrossRef CAS PubMed.
- (a) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830–16845 CrossRef CAS PubMed; (b) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC; (c) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS.
- (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS; (c) A. S. K. Hashmi, Top. Organomet. Chem., 2013, 44, 143–164 CrossRef; (d) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS PubMed; (e) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS.
- G. S. Rachor, M. Ahrens and T. Braun, Angew. Chem., 2022, 134, e202212858 CrossRef.
- (a) N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859–12861 CrossRef CAS; (b) D. Vesseur, S. Li, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2024, 146, 11352–11363 CAS.
- (a) N. P. Mankad and F. D. Toste, Chem. Sci., 2012, 3, 72–76 RSC; (b) J. W. McDaniel, J. M. Stauber, E. A. Doud, A. M. Spokoyny and J. M. Murphy, Org. Lett., 2022, 24, 5132–5136 CrossRef CAS PubMed; (c) A. Portugués, M. A. Martínez-Nortes, D. Bautista, P. González-Herrero and J. Gil-Rubio, Inorg. Chem., 2023, 62, 1708–1718 CrossRef PubMed.
- (a) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC; (b) J. Miro and C. Del Pozo, Chem. Rev., 2016, 116, 11924–11966 CrossRef CAS; (c) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Isr. J. Chem., 2010, 50, 675–690 CrossRef CAS; (d) L. Yang, T. Dong, H. M. Revankar and C. P. Zhang, Green Chem., 2017, 19, 3951–3992 RSC.
- J. A. Akana, K. X. Bhattacharyya, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 7736–7737 CrossRef CAS.
- B. C. Gorske, C. T. Mbofana and S. J. Miller, Org. Lett., 2009, 11, 4318–4321 CrossRef CAS.
- X. Zeng, S. Liu, G. B. Hammond and B. Xu, Chem. – Eur. J., 2017, 23, 11977–11981 CrossRef CAS PubMed.
- T. J. O'Connor and F. D. Toste, ACS Catal., 2018, 8, 5947–5951 CrossRef.
- F. Nahra, S. R. Patrick, D. Bello, M. Brill, A. Obled, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, ChemCatChem, 2015, 7, 240–244 CrossRef CAS PubMed.
- (a) P. Fernández-Canelas, E. Rubio and J. M. González, Org. Lett., 2019, 21, 6566–6569 CrossRef; (b) A. Gómez-Herrera, I. I. Hashim, M. Porré, F. Nahra and C. S. Cazin, Eur. J. Org. Chem., 2020, 6790–6794 CrossRef; (c) N. Müller, B. S. Schreib, S. U. Leutenegger and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 61, e202204535 CrossRef PubMed; (d) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed; (e) J. V. Alegre-Requena, A. Valero-Tena, I. G. Sonsona, S. Uriel and R. P. Herrera, Org. Biomol. Chem., 2020, 18, 1594–1601 RSC.
- A. Gómez-Herrera, F. Nahra, M. Brill;, S. Nolan and C. S. Cazin, ChemCatChem, 2016, 8, 3381–3388 CrossRef.
- D. Mulryan, J. Rodwell, N. A. Phillips and M. R. Crimmin, ACS Catal., 2022, 12, 3411–3419 CrossRef CAS PubMed.
- O. E. Okoromoba, J. Han, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2014, 136, 14381–14384 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS.
- (a) H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584–595 CrossRef CAS; (b) C. Nolte, J. Ammer and H. Mayr, J. Org. Chem., 2012, 77, 3325–3335 CrossRef CAS; (c) S. Minegishi, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2004, 126, 5174–5181 CrossRef CAS PubMed.
- R. Gauthier, M. Mamone and J. F. Paquin, Org. Lett., 2019, 21, 9024–9027 CrossRef CAS.
- H. Yang, J. Wang, C. Jin, X. Li and X. Xu, J. Org. Chem., 2023, 88, 12074–12078 CrossRef CAS PubMed.
- X. Cheng and L. Zhang, CCS Chem., 2021, 3, 1989–2002 CrossRef CAS.
- R. Gauthier, N. V. Tzouras, Z. Zhang, S. Bédard, M. Saab, L. Falivene, K. V. Hecke, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2022, 28, e202103886 CrossRef CAS PubMed.
- (a) S. Bhunia, P. Ghosh and S. R. Patra, Adv. Synth. Catal., 2020, 362, 3664–3708 CrossRef CAS; (b) Z. Zheng, X. Ma, X. Cheng, K. Zhao, K. Gutman, T. Li and L. Zhang, Chem. Rev., 2021, 121, 8979–9038 CrossRef CAS PubMed; (c) L. W. Ye, X. Q. Zhu, R. L. Sahani, Y. Xu, P. C. Qian and R. S. Liu, Chem. Rev., 2020, 121, 9039–9112 CrossRef.
- (a) L. Ye, L. Cui, G. Zhang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed; (b) L. A. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed.
- X. Zeng, Z. Lu, S. Liu, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2017, 359, 4062–4066 CrossRef CAS.
- X. Zeng, S. Liu, Z. Shi, G. Liu and B. Xu, Angew. Chem., Int. Ed., 2016, 10032–10036 CrossRef CAS.
- H. J. Tang, X. Zhang, Y. F. Zhang and C. Feng, Angew. Chem., Int. Ed., 2020, 59, 5242–5247 CrossRef CAS PubMed.
- (a) B. Sahoo, M. N. Hopkinson and F. Glorius, J. Am. Chem. Soc., 2013, 135, 5505–5508 CrossRef CAS PubMed; (b) X. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS.
- T. de Haro and C. Nevado, Adv. Synth. Catal., 2010, 352, 2677–2846 CrossRef.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS PubMed.
- S. Fang, J. Han, C. Zhu, W. Li and J. Xie, Nat. Commun., 2023, 14, 3551 CrossRef CAS.
- W. E. Brenzovich Jr, D. Benitez, A. D. Lackner, H. P. Shunatona, E. Tkatchouk, W. A. Goddard and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 5519–5522 CrossRef PubMed.
- X. Chen, S. Martini and V. A. Michelet, Adv. Synth. Catal., 2019, 361, 3612–3618 CrossRef CAS.
- V. Marsicano, A. Arcadi and V. Michelet, Chem. – Eur. J., 2022, 2022, e202101524 CAS.
- W. Wang, J. Jasinski, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2010, 49, 7247–7252 CrossRef CAS.
- T. de Haro and C. Nevado, Chem. Commun., 2011, 47, 248–249 RSC.
- M. N. Hopkinson, G. T. Giuffredi, A. D. Gee and V. Gouverneur, Synlett, 2010, 2737–2742 CAS.
- Z. Jin, R. S. Hidinger, B. Xu and G. B. Hammond, J. Org. Chem., 2012, 77, 7725–7729 CrossRef CAS PubMed.
- (a) D. Das, Tetrahedron Lett., 2020, 61, 152298 CrossRef CAS; (b) D. Das, Monatsh. Chem., 2021, 152, 987–991 CrossRef CAS.
- A. Arcadi, E. Pietropaolo, A. Alvino and V. Michelet, Org. Lett., 2013, 15, 2766–2769 CrossRef CAS.
- J. Qian, Y. Liu, J. Zhu, B. Jiang and Z. Xu, Org. Lett., 2011, 13, 4220–4223 CrossRef CAS PubMed.
- S. Li, Z. Li, Y. Yuan, Y. Li, L. Zhang and Y. Wu, Chem. – Eur. J., 2013, 19, 1496–1501 CrossRef CAS PubMed.
- S. Pal, D. Das and S. Bhunia, Org. Biomol. Chem., 2024, 22, 1527–1579 RSC.
- Y. Jeong, B. I. Kim, J. K. Lee and J. S. Ryu, J. Org. Chem., 2014, 79, 6444–6455 CrossRef CAS PubMed.
- (a) G. L. Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Top. Curr. Chem., 2021, 379, 34 CrossRef; (b) K. K. Rajbongshi, B. Dam and B. K. Patel, Pyridines, dihydropyridines and piperidines: an outline on synthesis and biological activities, in N-Heterocycles, ed. K. L. Ameta, R. Kant, A. Penoni, A. Maspero and L. Scapinello, Springer, Singapore, 2022 Search PubMed.
- A. Simonneau, P. Garcia, J. P. Goddard, V. M. Mansuy, M. Malacria and L. Fensterbank, Beilstein J. Org. Chem., 2011, 7, 1379–1386 CrossRef CAS PubMed.
- N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859–12861 CrossRef CAS.
- Q. Wang, Y. Jiang, R. Sun, X. Y. Tang and M. Shi, Chem. – Eur. J., 2016, 22, 14739–14745 CrossRef CAS PubMed.
- D. Malhotra, L. Liu, W. Wang, M. Durham, G. B. Hammond and B. Xu, J. Fluor. Chem., 2014, 167, 179–183 CrossRef CAS.
- Y. Li, K. A. Wheeler and R. Dembinski, Org. Biomol. Chem., 2012, 10, 2395–2408 RSC.
- M. Schuler, F. Silva, C. Bobbio, A. Tessier and V. Gouverneur, Angew. Chem., Int. Ed., 2008, 47, 7927–7930 CrossRef CAS PubMed.
- J. E. Aaseng, N. Iqbal, C. A. Sperger and A. Fiksdahl, J. Fluor. Chem., 2014, 161, 142–148 CrossRef CAS.
- X. Chen, Y. Zhou, M. Hong, Y. Ling, D. Yin, S. Wang, X. Zhang and W. Rao, Adv. Synth. Catal., 2018, 360, 3700–3708 CrossRef CAS.
- (a) D. Scarpi, M. Petrović, B. Fiser, E. Gómez-Bengoa and E. G. Occhiato, Org. Lett., 2016, 18, 3922–3925 CrossRef CAS; (b) W. Rao, D. Susanti, B. J. Ayers and P. W. H. Chan, J. Am. Chem. Soc., 2015, 137, 6350–6355 CrossRef CAS PubMed; (c) G. Lemiàre, V. Gandon, K. Cariou, A. Hours, T. Fukuyama, A.-L. Dhimane, L. Fensterbank and M. Malacria, J. Am. Chem. Soc., 2009, 131, 2993 CrossRef; (d) G. Lemiàre, V. Gandon, K. Cariou, T. Fukuyama, A.-L. Dhimane, L. Fensterbank and M. Malacria, Org. Lett., 2007, 9, 2207–2209 CrossRef; (e) L. Zhang and S. Wang, J. Am. Chem. Soc., 2006, 128, 1442–1443 CrossRef CAS.
- X. Shi, D. J. Gorin and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 5802–5803 CrossRef CAS PubMed.
- Y. Liu, J. Zhu, J. Qian and Z. Xu, J. Org. Chem., 2012, 77, 5411–5417 CrossRef CAS PubMed.
- (a) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248–8262 CrossRef CAS; (b) H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS PubMed; (c) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493–497 CrossRef CAS; (d) M. N. Hopkinson, A. Tessier, A. Salisbury, G. T. Giuffredi, L. E. Combettes, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2010, 16, 4739–4743 CrossRef CAS; (e) G. Zhang, Y. Luo, Y. Wang and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 4450–4454 CrossRef CAS PubMed; (f) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232–5241 CrossRef CAS.
- A. Hussenius and O. Matsson, Acta Chem. Scand., 1990, 44, 845–850 CrossRef CAS.
- C. D. Pina, E. Falletta and M. Rossi, Oxidation of alcohols and carbohydrates, in Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and D. F. Toste, Wiley-VCH, Weinheim, 2012, pp. 309–330 Search PubMed.
- G. Stavber and S. Stavber, Adv. Synth. Catal., 2010, 352, 2838–2846 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |