
Facile access to trifluoromethyl propargyl alcohol by metal-free transfer hydrogenation and cyanation of alkynyl ketones†
Devadkar
Ajitrao Kisan
 ,
Ishita
Paul
,
Soumyadip
Dey
,
Abhijit
Sau
* and
Tarun K.
Panda
*
,
Ishita
Paul
,
Soumyadip
Dey
,
Abhijit
Sau
* and
Tarun K.
Panda
*
Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy 502284, Telangana, India. E-mail: tpanda@chy.iith.ac.in; asau@chy.iith.ac.in; Fax: +91 40 2301 6032; Tel: +91 40 2301 6254
First published on 1st November 2024
Abstract
We report an efficient synthetic route to the metal-free hydroboration and cyanosilylation of a wide range of alkynyl trifluoromethyl ketones using pinacolborane (4,4,5,5-tetramethyl-1,3,2-dioxaborolane) and trimethylsilyl cyanide under mild reaction conditions at ambient temperature. These highly effective hydroboration and cyanosilylation reactions lead to the corresponding alkynyl trifluoromethyl propargyl alcohols after hydrolysis. In addition, trifluoromethyl (CF3) group-based pharmaceutically active enflicoxib analogs were synthesized from propargyl alcohol.
Introduction
The addition of trifluoromethyl (CF3) groups to organic molecules can modify their chemical and metabolic stability, lipophilicity, and selectivity in binding significantly, given their strong electron-withdrawing nature and large hydrophobic domain.1 Alkynyl trifluoromethyl ketone compounds are used as synthons in modern organic synthesis. Alkynes have been employed widely in preparing valuable compounds in the fields of chemistry, biology, medicine, and materials science.2–10 Approximately 25% of drug molecules contain at least one fluorine in their structures. As such, the efficient synthesis of new organofluoride derivatives is an important objective in organic chemistry.11–15 Optically active propargylic alcohols are important building blocks in the synthesis of complex bioactive compounds.16 In addition, trifluoromethylated nitrogen-containing heterocycles have attracted considerable attention in pharmaceutical and agrochemical industries, as shown in Scheme 1. | ||
| Scheme 1 CF3-containing pharmaceutically active drugs. | ||
Catalytic hydrogenation of alkynyl ketones is one of the most efficient ways to produce propargylic alcohols. However, only a limited number of catalytic systems can effectively hydrogenate alkynyl ketones, as several are unstable under basic ketone hydrogenation conditions.17 The traditional method involves the use of stoichiometric reducing agents such as H2 under high pressure, and the reaction of hazardous metal hydride reductants or boron precursors (BH3) (in equimolar ratio) with carbonyl compounds to form alcohols after hydrolyzing borates.18 In addition, catalysts with transition metals, such as titanium,19,20 manganese,21 iron,22 molybdenum,23 rhodium,24 ruthenium,25 copper,26 and zinc,27 as well as main group metals such as aluminum,28 gallium,29 and germanium,30 have been proved to be instrumental in the hydroboration of carbonyl compounds. Shibasaki and Kanai et al. devised an excellent technique for synthesizing CF3-functionalized propargylic alcohols and they employed Cu-OtBu-xanthous or phenanthroline with alkyne substrates at 60–100 °C.31
Gilman and Straley described the reactions between an organocopper complex (RCu) and organic functional groups (e.g., acid chlorides, allylic halides, aldehydes, etc.).32 In 1986, Gibson et al. reported the cyanosilylation of alkynyl ketones using copper(I) cyanide catalysts as cyanide donors.33,34 In organic synthesis, the cyanosilylation of carbonyl compounds is a simple carbon–carbon bond-forming reaction. The use of oxidants as additives has already been reported in the literature, based on numerous and effective pathways using transition metals.
In 2018, Houk et al. developed a ruthenium-catalyzed asymmetric alkynylation reaction methodology using trifluoromethyl ketones and alkyne as synthons (Scheme 2a).35 An iridium-catalyzed reaction was also reported to synthesize chiral methyl propargyl ketones by asymmetric transfer hydrogenation (Scheme 2b).36 Recently, the Funabiki group developed an elegant approach for the synthesis of CF3-containing propargyl alcohol through two sequential reactions in one pot using alkyne and cyclopentyl magnesium bromide with trifluoroacetic acid esters (Scheme 2c).37
 | ||
| Scheme 2 Previous studies and this work. | ||
In this paper, we present a metal-free and solvent-free protocol for the hydroboration and cyanosilylation of trifluoro-methylated ketones. Employing the hydride from HBpin furnished a boronic ester that led to propargyl alcohol after hydrolysis, whereas using TMSCN resulted in cyano propargyl alcohol via a cyanohydrin intermediate. To the best of our knowledge, this is the first report in which the hydroboration and cyanosilylation of alkynyl trifluoromethyl ketones are demonstrated under efficient metal-free conditions.
Results and discussion
We aimed to synthesize alkynyl trifluoromethyl ketones using simple and commercially available materials and trifluoromethyl ketones as model substrates. Initially, the reaction was performed in the presence of alkynyl trifluoromethyl ketones with the addition of HBpin (2 equiv.) at room temperature for one hour, which afforded a boronic ester in 70% yield (Table 1, entry 1). The yield improved to 95% when the reaction time was increased to two hours (Table 1, entry 2).|
|
||||
|---|---|---|---|---|
| Entry | HBpin (equiv.) | Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: ketones (0.2 mmol, 1.0 equiv.) and HBpin (0.2 mmol, 1.0 equiv.). b Yields have been calculated based on the products isolated by extraction in n-pentane. | ||||
| 1 | 2 | Neat | 1 | 70 |
| 2 | 2 | Neat | 2 | 95 |
| 3 | 1.5 | Neat | 2 | 95 |
| 4 | 1 | Neat | 2 | 95 |
| 5 | 1 | Toluene | 2 | 90 |
| 6 | 1 | THF | 2 | 87 |
| 7 | 1 | CH3CN | 2 | 80 |
| 8 | 1 | MeOH | 2 | 73 |
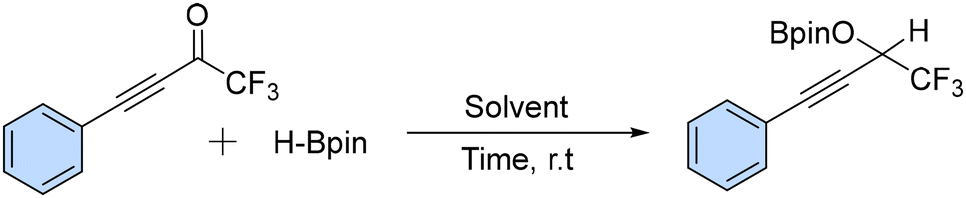
Subsequently, we reduced the HBpin loading (to 1.5 equiv. and 1.0 equiv.), which led to boronic esters in yields of 95% in both cases (Table 1, entries 3 and 4). The reaction was performed using common organic solvents such as toluene, THF, CH3CN, and MeOH (Table 1, entries 5–8) but the best result was obtained under neat conditions.
After identifying the optimal reaction conditions, we explored the substrate scope using electron-donating alkynyl trifluoromethyl ketones with HBpin, which afforded CF3-substituted propargylic boronate esters in excellent yields (Scheme 3, compounds 2b, 2c, 2d and 2e).
 | ||
| Scheme 3 Substrate scope of hydroboration. Reaction conditions: ketones (0.2 mmol, 1.0 equiv.) and HBpin (0.2 mmol, 1.0 equiv.). Yields have been calculated based on the products isolated by extraction in n-pentane. | ||
Afterward, we examined the reaction with electron-withdrawing alkynyl trifluoromethyl ketones, resulting in excellent yields of corresponding propargylic boronate esters (Scheme 3, compounds 2f and 2g).
Remarkably, we observed that thiophene-containing and cyclohexene-containing alkynyl trifluoromethyl ketones afforded an excellent isolated yield of boronate esters (Scheme 3, compounds 2h and 2i). After successfully synthesizing the propargylic boronate esters, we further investigated the cyanosilylation of alkynyl trifluoromethyl ketones under similar conditions. First, the reaction of alkenyl trifluoromethyl ketones with TMSCN (2 equiv.) at room temperature for one hour was performed, which afforded the cyanosilylation product with a yield of 34% (Table 2, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | TMSCN (equiv.) | Solvent | Time (h) | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: ketones (0.2 mmol, 1.0 equiv.) and TMSCN (0.3 mmol, 1.5 equiv.). b Yields have been calculated based on the products isolated by extraction in n-pentane. | |||||
| 1 | 2 | Neat | 1 | rt | 34 |
| 2 | 2 | Neat | 1 | 60 | 90 |
| 3 | 1.5 | Neat | 2 | 60 | 92 |
| 4 | 1 | Neat | 2 | 60 | 85 |
| 5 | 1.5 | Toluene | 2 | 60 | 82 |
| 6 | 1.5 | THF | 2 | 60 | 79 |
| 7 | 1.5 | MeOH | 2 | 60 | 67 |
| 8 | 1.5 | CH3CN | 2 | 60 | 73 |
| 9 | 1.5 | Dioxane | 2 | 60 | 69 |

Next, we conducted the reaction at an elevated temperature of 60 °C for one hour, which afforded the cyanohydrin product exclusively with a yield of 90% (Table 2, entry 2).
Later, when the TMSCN loading was lowered (to 1.5 equiv.), the desired product was obtained with a similar yield (Table 2, entry 3). We also carried out the reaction using various organic solvents such as toluene, THF, MeOH, CH3CN, and 1,4-dioxane, and observed decreased yields (Table 2, entries 5–9).
After optimizing the cyanosilylation reaction conditions, we focused on the substrate scope. Electron-donating groups (OMe and Me) containing ketones resulted in the corresponding cyanohydrin products in excellent yields (Scheme 4, compounds 3b and 3c). 4-Butyl- and t-butylbenzene-containing alkynyl trifluoromethyl ketones led to the formation of propargyl cyanohydrin products in yields of 69% and 91%, respectively (compounds 3d and 3e).
 | ||
| Scheme 4 Substrate scope of cyanosilylation. Reaction conditions: ketones (0.2 mmol, 1.0 equiv.) and TMSCN (0.3 mmol, 1.5 equiv.). Yields have been calculated based on the products isolated by extraction in n-pentane. | ||
Furthermore, we investigated electron-withdrawing groups (F, Br, and Cl) and obtained the desired products in yields of up to 90% (Scheme 3, compounds 3f, 3g, and 3h). Under the optimal reaction conditions, heterocyclic thiophene-containing alkynyl trifluoromethyl ketone was reacted with TMSCN to obtain a 78% yield of the propargyl cyanohydrin product (Scheme 3, compound 3i). Next, we performed the cyanosilylation reaction of cyclohexene propargyl ketone, which proceeded smoothly to form the propargyl cyanohydrin product, affording a yield of 85% (Scheme 4, compound 3j).
In addition, hydroboration compounds were hydrolyzed to their corresponding propargyl alcohols using silica and methanol at 60 °C for two hours and isolated in good yields (Scheme 5, compounds 4a–4e).38–40 Furthermore, cyanosilylation compounds were also hydrolyzed to their corresponding cyano alcohols using silica and methanol at 60 °C for two hours and isolated in good yields (Scheme 5, compounds 5a–5e).
 | ||
Scheme 5 One-pot hydrolysis of fluorinated ketone to propargyl alcohol. Reaction conditions: (1) ketone (0.2 mmol, 1.0 equiv.), HBpin (0.2 mmol, 1.0 equiv.), and TMSCN (0.3 mmol, 1.5 equiv.) under neat conditions. (2) Silica and MeOH at 60 °C in 2 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Gram-scale reaction: ketone (5.0 mmol), HBpin (5.0 mmol) and TMSCN (7.5 mmol) under neat conditions. Isolated yields. Gram-scale reaction: ketone (5.0 mmol), HBpin (5.0 mmol) and TMSCN (7.5 mmol) under neat conditions. Isolated yields. | ||
After exploring the substrate scope, we extended our study to demonstrate the utility of propargyl alcohol derivatives by synthesizing bioactive molecules (enflicoxib analogs), which are important in the pharmaceutical industry. To obtain these compounds, we used a one-step reaction involving propargyl alcohol and phenylhydrazine, followed by cyclization.37 We were able to successfully synthesize four different enflicoxib analogs – 6a, 6b, 6c, and 6d – using DBU as a catalyst in toluene, affording yields of 96%, 45%, 32%, and 92% respectively (Scheme 6).
 | ||
| Scheme 6 Synthesis of enflicoxib analogs. Reaction conditions: propargyl alcohol (1.0 mmol), phenylhydrazine (1.2 mmol), and DBU (1.0 mmol). Isolated yields. | ||
In addition, we investigated the one-pot synthesis of trifluoromethyl propargyl alcohol using tandem reactions of n-BuLi with commercially available ethyl trifluoroacetate, terminal alkynes, and HBpin (Scheme 7). This synthetic method involves one-pot reactions, deprotonation of terminal alkynes with n-BuLi, and ethyl trifluoroacetate nucleophilic addition of in situ-generated alkynyl trifluorinated ketone. Further addition of HBpin led to the formation of a boronic ester which afterward hydrolyzed using silica and methanol at 60 °C for two hours, giving the corresponding trifluoromethyl propargyl alcohol (4a) in 60% yield.
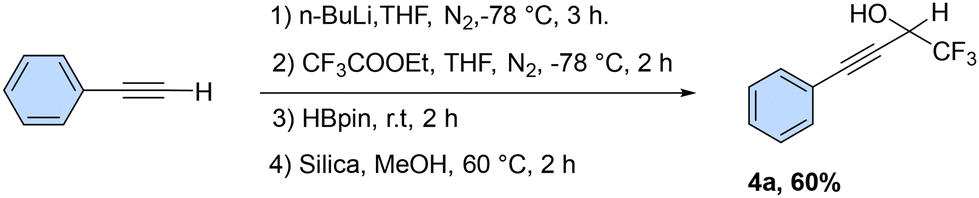 | ||
| Scheme 7 Alkyne to propargyl alcohol via a one-pot reaction. Reaction conditions: phenylacetylene (10 mmol), n-BuLi (12 mmol), CF3COOEt (12 mmol), HBpin (10 mmol), THF (5 ml), silica and MeOH at 60 °C in 2 h. Isolated yields. | ||
Conclusions
To summarize, we have developed a simple and feasible method for the hydroboration and cyanosilylation of alkynyl trifluoromethyl ketones to access the corresponding boronate esters and cyanohydrin. The catalyst-free and solvent-free conditions are ecofriendly and the sustainable reaction process affords trifluoromethyl propargyl alcohols. We successfully synthesized different analogs of the drug molecule, enflicoxib, using the core propargyl alcohols in a one-pot reaction. Trifluoromethyl propargyl alcohol can also be obtained using one-pot synthesis. Thus, our research group is committed to further exploring the potential pharmaceutical applications of fluorinated propargyl alcohol compounds and their derivatives.Author contributions
DAK: data collection, data analysis, methodology, investigation, and writing the original draft. IP: data collection, data analysis, methodology, and investigation. SD: data analysis, methodology, and investigation. AS: project supervision, review, and editing of the manuscript. TKP: project supervision, review, editing of the manuscript, and acquisition of funding.Data availability
The data that support the findings of this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from SERB, India under project no. CRG/2022/001941 and Indian Institute of Technology Hyderabad (IITH). D. A. K. and I. P. thank UGC India. S. D. thanks IIT Hyderabad India for their respective PhD fellowships.References
- P. Kirsch, Modern fluoroorganic chemistry: synthesis, reactivity, applications, Wiley-VCH, Weinheim, 2004 Search PubMed.
- E. A. Zakharova, O. I. Shmatova and V. G. Nenajdenko, Russ. Chem. Rev., 2018, 87, 619–635 CrossRef CAS.
- D. V. Vidhani, J. W. Cran, M. E. Krafft, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2013, 78, 2059–2073 CrossRef CAS.
- I. V. Alabugin and E. Gonzalez-Rodriguez, Acc. Chem. Res., 2018, 51, 1206–1219 CrossRef CAS.
- V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Ananikov, Molecules, 2018, 23, 2442 CrossRef.
- G. Larson, Synthesis, 2018, 2433–2462 CAS.
- D. S. Ryabukhin and A. V. Vasilyev, Russ. Chem. Rev., 2016, 85, 637–665 CrossRef CAS.
- A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471–4503 RSC.
- B. A. Trofimov and E. Y. Schmidt, Russ. Chem. Rev., 2014, 83, 600–619 CrossRef.
- A. V. Vasilyev, Russ. Chem. Rev., 2013, 82, 187–204 CrossRef.
- R. Sanz, A. Martínez, J. M. Álvarez-Gutiérrez and F. Rodríguez, Eur. J. Org. Chem., 2006, 1383–1386 CrossRef CAS.
- R. Sanz, D. Miguel, A. Martínez, M. Gohain, P. García-García, M. A. Fernández-Rodríguez, E. Álvarez and F. Rodríguez, Eur. J. Org. Chem., 2010, 7027–7039 CrossRef CAS.
- S. Antony Savarimuthu, D. G. Leo Prakash and S. Augustine Thomas, Tetrahedron Lett., 2014, 55, 3213–3217 CrossRef CAS.
- S. Gujarathi, H. P. Hendrickson and G. Zheng, Tetrahedron Lett., 2013, 54, 3550–3553 CrossRef CAS.
- P. Srihari, J. Reddy, S. Mandal, K. Satyanarayana and J. Yadav, Synthesis, 2008, 1853–1860 CrossRef CAS.
- H. Qian, D. Huang, Y. Bi and G. Yan, Adv. Synth. Catal., 2019, 361, 3240–3280 CrossRef CAS.
- N. Arai, H. Satoh, N. Utsumi, K. Murata, K. Tsutsumi and T. Ohkuma, Org. Lett., 2013, 15, 3030–3033 CrossRef CAS PubMed.
- X. Wang, Y. Li, Y. D. Wu, M. N. Paddon-Row, N. G. Rondan and K. N. Houk, J. Org. Chem., 1990, 55, 2601–2609 CrossRef CAS.
- A. A. Oluyadi, S. Ma and C. N. Muhoro, Organometallics, 2013, 32, 70–78 CrossRef CAS.
- Z. Huang, D. Liu, J. Camacho-Bunquin, G. Zhang, D. Yang, J. M. López-Encarnación, Y. Xu, M. S. Ferrandon, J. Niklas, O. G. Poluektov, J. Jellinek, A. Lei, E. E. Bunel and M. Delferro, Organometallics, 2017, 36, 3921–3930 CrossRef CAS.
- G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., Int. Ed., 2016, 55, 14369–14372 CrossRef CAS PubMed.
- S. R. Tamang and M. Findlater, J. Org. Chem., 2017, 82, 12857–12862 CrossRef CAS PubMed.
- A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455–457 RSC.
- D. A. Evans and G. C. Fu, J. Org. Chem., 1990, 55, 5678–5680 CrossRef CAS.
- A. Kaithal, B. Chatterjee and C. Gunanathan, Org. Lett., 2015, 17, 4790–4793 CrossRef CAS PubMed.
- S. Bagherzadeh and N. P. Mankad, Chem. Commun., 2016, 52, 3844–3846 RSC.
- S.-G. Roh, Y.-C. Park, D.-K. Park, T.-J. Kim and J. H. Jeong, Polyhedron, 2001, 20, 1961–1965 CrossRef CAS.
- V. K. Jakhar, M. Kr. Barman and S. Nembenna, Org. Lett., 2016, 18, 4710–4713 CrossRef CAS PubMed.
- S. Rajput, R. K. Sahoo, N. Sarkar and S. Nembenna, ChemPlusChem, 2024, e202300737 CrossRef CAS.
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed.
- R. Motoki, M. Kanai and M. Shibasaki, Org. Lett., 2007, 9, 2997–3000 CrossRef CAS PubMed.
- H. Gilman and J. M. Straley, Recl. Trav. Chim. Pays-Bas, 1936, 55, 821–834 CrossRef CAS.
- S. H. Bertz and C. P. Gibson, J. Am. Chem. Soc., 1986, 108, 8286–8288 CrossRef CAS.
- N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed.
- S. Chen, Y. Zheng, T. Cui, E. Meggers and K. N. Houk, J. Am. Chem. Soc., 2018, 140, 5146–5152 CrossRef CAS PubMed.
- Y.-M. Zhang, M.-L. Yuan, W.-P. Liu, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2018, 20, 4486–4489 CrossRef CAS PubMed.
- R. Kani, T. Inuzuka, Y. Kubota and K. Funabiki, Asian J. Org. Chem., 2022, 11, e202100700 CrossRef CAS.
- N. Sarkar, M. Mahato and S. Nembenna, Eur. J. Inorg. Chem., 2020, 2295–2301 CrossRef CAS.
- R. Kumar, P. Rawal, I. Banerjee, H. Pada Nayek, P. Gupta and T. K. Panda, Chem. – Asian J., 2022, 17, e202200013 CrossRef CAS PubMed.
- G. S. Kumar, R. Kumar, A. Sau, V. Chandrasekhar and T. K. Panda, J. Org. Chem., 2023, 88, 12613–12622 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C{1H} NMR spectra of products. CCDC 2349834–2349836. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00844h |
| This journal is © The Royal Society of Chemistry 2025 |