
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical restructuring of H2O2 activated copper selenide for CO2 reduction†
Wenjian
Hu
 ab,
Deema
Balalta
c,
Zhiyuan
Chen
b,
Imran
Abbas
a,
Jia
Song
b,
Balázs
Barhács
de,
Márton
Guba
de,
Tibor
Höltzl
def,
Francesco
D'Acapito
g,
Thomas
Altantzis
h,
Jan
Vaes
b,
Sara
Bals
c,
Didier
Grandjean
*a,
Deepak
Pant
*bi and
Ewald
Janssens
*a
ab,
Deema
Balalta
c,
Zhiyuan
Chen
b,
Imran
Abbas
a,
Jia
Song
b,
Balázs
Barhács
de,
Márton
Guba
de,
Tibor
Höltzl
def,
Francesco
D'Acapito
g,
Thomas
Altantzis
h,
Jan
Vaes
b,
Sara
Bals
c,
Didier
Grandjean
*a,
Deepak
Pant
*bi and
Ewald
Janssens
*a
aQuantum Solid-State Physics, Department of Physics and Astronomy, KU Leuven, Celestijnenlaan 200 D, 3001 Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be; didier.grandjean@kuleuven.be
bElectrochemistry Excellence Centre, Materials & Chemistry Unit, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400 Mol, Belgium. E-mail: deepak.pant@vito.be
cElectron Microscopy for Materials Science (EMAT), University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
dDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szent Gellért tér 4, 1111 Budapest, Hungary
eFurukawa Electric Institute of Technology, Késmárk utca 28/A, 1158 Budapest, Hungary
fHUN-REN-BME Computation Driven Research Group, Budapest University of Technology and Economics, Szent Gellért tér 4, H-1111 Budapest, Hungary
gCNR-IOM-OGG c/o ESRF LISA CRG – The European Synchrotron, 71 Av. des Martyrs, 38000 Grenoble, France
hApplied Electrochemistry & Catalysis (ELCAT), University of Antwerp, Universiteitsplein 1, 2610 Wilrijk, Belgium
iCenter for Advanced Process Technology for Urban Resource Recovery (CAPTURE), Frieda Saeysstraat 1, 9052 Zwijnaarde, Belgium
First published on 3rd July 2025
Abstract
Copper chalcogenides such as Cu2−xSe, acknowledged as efficient CO2 reduction catalysts, do not represent the active phases but rather are precursors or pre-catalysts as they undergo significant transformations under reaction conditions. In this work we have tailored the initial structure of Cu2−xSe to steer structural evolution under catalytic conditions and facilitate the generation of the active phases. As-prepared Cu2−xSe nanowires were reconstructed through H2O2 and electrochemical treatments, yielding distinct pre-catalysts. Their electrochemical reduction was found to be an effective strategy to enhance the formation of active metallic Cu nanoparticles. Chemical pretreatment with H2O2 further accelerates this process by inducing a structural loosening and partial oxidation of the Cu2−xSe phase. Supported by in situ Raman spectroscopy, quasi-in situ X-ray diffraction, X-ray absorption fine structure spectroscopy and high-angle annular dark-field scanning transmission electron microscopy analysis, it is suggested that structural transformation is a common feature of many copper-based catalysts during CO2 electroreduction. The as-prepared Cu2−xSe nanowires, with diameters of about 300 nm, exhibit a 23% methanol selectivity and a low CO2 selectivity of only 4% at −1.4 V versus the reversible hydrogen electrode. In contrast, 50–90 nm Cu2O cubes obtained after H2O2 oxidation and electro-activation treatments, also acting as pre-catalysts, have a CO selectivity up to 82%. Density functional theory computations demonstrate lower binding energy of reaction intermediates, including *CO, on metallic Cu (110) than on Cu2Se (220), which may account for the enhanced CO production of the electro-activated catalyst. Our work sheds light on the dependence of the catalytic performance of copper selenide on its initial restructuration and provides guidance for the development of efficient and selective CO2 conversion catalysts.
Introduction
The process of CO2 electroreduction using Cu catalysts1,2 has garnered tremendous attention, because Cu demonstrates remarkable activity for the production of high-valued products such as alcohols. However, obtaining a specific product with high selectivity remains a challenge.2 Several strategies are being followed to improve the efficiency and selectivity of the electrochemical CO2 reduction reaction (CO2RR) on copper catalysts, such as morphology control,3 surface modification,4 oxide-derived copper,5 single Cu atoms,6 and alloying.7 Alloying Cu with other metals can enhance the selectivity of CO2RR toward different products by modulating the catalyst's electronic structure and thereby the adsorption of reaction intermediates.8Similarly, combining copper with non-metallic elements to form copper chalcogenides, such as Cu2−xSe, has gained attention.9–11 The presence of selenium atoms modulates the electronic structure, in particular the Cu d-band center. This alters the adsorption behavior of reaction intermediates and thereby the catalyst's specificity and effectiveness. Recent studies on copper selenide catalysts demonstrated significant enhancements in CO2 electroreduction towards methanol and ethanol.11 Optimizing the composition and structure of copper selenide by potassium or vanadium doping,12,13 or by creating selenium vacancies,14,15 also has proven effective to increase its selectivity and efficiency for ethanol.
While excellent results were reported using copper chalcogenides for CO2RR, the identification of the active states remain incomplete.16–22 This is largely due to the structural reconstruction under CO2RR conditions that occurs even in Cu-based catalysts with well-defined nanostructures.23,24 In particular, most active sites on the surface of metal compounds experience irreversible reconstruction induced by electro-derived reduction or oxidation processes, which critically affects product selectivity.25,26
Recent operando and in situ method developments provide powerful non-destructive tools to probe the active sites and structural changes in Cu-based electrocatalysts under reaction conditions. Grazing incident X-ray diffraction reveals that CuO nanoplate catalysts are transformed into a mixture of metallic copper and Cu2O after a single minute of electrochemical reduction at −0.6 VRHE.27 An in situ Raman study has shown that typical Cu2O peaks (∼620 cm−1) disappear below −0.2 VRHEversus the reversible hydrogen electrode (RHE),5 indicating a reduction of surface Cu+ to metallic Cu. Thermodynamically, CuOx phases should be removed under CO2RR conditions along with the active Cuδ+ species.28 However, the underlying reason for the existence of dynamical Cuδ+ species remains to be uncovered.29 More recently an operando analytical and four-dimensional electrochemical liquid-cell scanning transmission electron microscopy (STEM) investigation has shown that a Cu nanoparticle ensemble evolves into metallic Cu nanograins during electrolysis, before complete oxidation to single-crystal Cu2O nanocubes following post-electrolysis air exposure.2 Similarly, Yoon et al. demonstrated that CuI, formed via iodide pre-treatment, transforms into filament-like metallic Cu nanostructures under cathodic potentials in an iodide-free KHCO3 electrolyte. When the applied bias was removed and the system returned to open circuit potential, partial re-oxidation occurred, resulting in the re-formation of Cu+-based CuI and Cu2O particles.30 These studies enhance understanding about the intricate characteristics of active Cu sites for CO2RR. It also triggers the question whether post-electrolysis complete oxidation to single-crystal Cu2O nanocubes is a common phenomenon for Cu-based materials.
The electrolyte has a role in the restructuring behavior of copper-based catalysts during CO2 electroreduction. In conventional aqueous electrolytes, Cu-based catalysts such as Cu2O typically undergo rapid and complete reduction to metallic Cu0 under CO2 reduction potentials.31 In contrast, intermediate oxidation states like Cu+ can be stable in an ionic liquid, thus suppressing the reduction kinetics. Such stabilization effect has been reported in prior studies, where Cu2−xSe nanowires retained their structure in an ionic liquid electrolyte, even after CO2 reduction,32,33 although several studies have shown that some as-prepared catalysts are not the actual reactive species, but rather their precursors or pre-catalysts.34 Modification of their initial structure may influence the formation and availability of the actual reactive species.
In this work, we investigate the restructuring process of Cu2−xSe with the aim to enhance understanding about the active sites and ultimately control the product selectivity. To this end, the restructuring of as prepared Cu2−xSe nanowires was induced through hydrogen peroxide (H2O2) chemical activation and electro-activation. The H2O2 pre-treatment facilitates oxidation and loosening of the Cu2−xSe framework, enhancing its susceptibility to electrochemical activation.11 We utilize an ionic liquid electrolyte to stabilize Cu+ intermediates and suppress reduction kinetics. By integrating in situ with quasi-in situ spectroscopic characterization, we demonstrate that Cu2−xSe dynamically restructures to highly active metallic Cu under CO2RR conditions. Cu2O monocrystals, ranging in size from 50 to 90 nm, obtained by the activation processes were also found to function as pre-catalysts rather than being the actual active species. While the as-prepared Cu2−xSe nanowires favour methanol production, the restructured Cu2O monocrystals mixed with Cu2−xSe exhibit an 82% faradaic efficiency (FE) for CO at −1.4 VRHE.
Results and discussion
Synthesis and characterization of Cu2−xSe pre-catalysts
Cu2−xSe catalysts were synthesized by a water evaporation-induced self-assembly method, applying the procedure described in ref. 35. Those as-prepared Cu2−xSe nanowires (AP-CuSe) were subsequently restructured using two distinct methodologies. As shown in Scheme 1, the first method entails direct electro-activation, where Cu2−xSe was directly exposed to reductive (−1.6 VRHE) electrochemical conditions (EA-CuSe). The second method is a sequential approach, starting with a chemical oxidative activation using hydrogen peroxide for 2 minutes (CA-CuSe), followed by electro-activation (EA-CA-CuSe). This approach results in four distinct and reproducible materials and allows to assess the impact of an initial superficial oxidative chemical treatment on the electrochemical reconstruction of Cu2−xSe nanowires.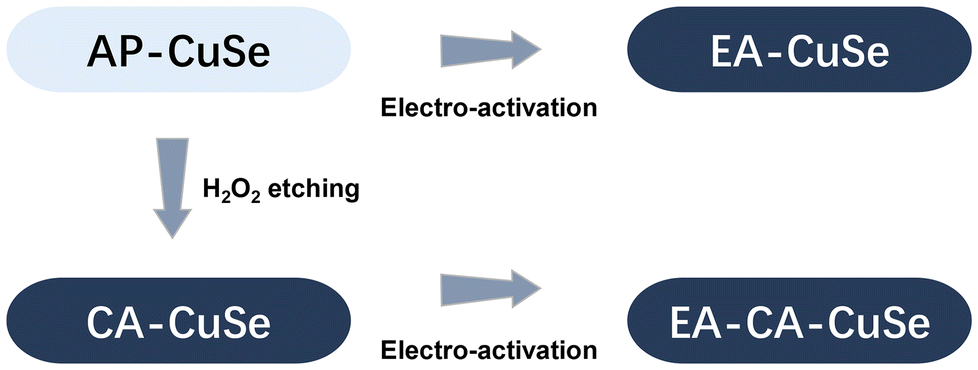 | ||
| Scheme 1 Schematic overview of the relation between the four CuSe-based catalyst materials studied in this work. | ||
The four different materials were extensively characterized by electron microscopy. The Field Emission Scanning Electron Microscopy (FE-SEM) image of AP-CuSe presented in the ESI (Fig. S2a†), shows bundles of one-dimensional Cu2−xSe nanowires with lengths of several micrometers and diameters of 100–500 nm. These 1D nanostructures have a relatively smooth surface. On the other hand, the CA-CuSe wires treated with H2O2 for varying durations (10 s, 2 min, and 10 min) have a rough, flocculent surface. Fig. S2b–d† show that their rough surface structure develops further upon longer H2O2 treatment. High Angle Annular Dark Field (HAADF) STEM, Energy Dispersive X-ray Spectroscopy (EDS) and Electron Energy Loss Spectroscopy (EELS) characterization provides deeper insights into the morphology, structure, and composition of the individual nanowires. Fig. 1a shows that the AP-CuSe nanowire bundles have a porous nature and are comprised of thin loosely-packed nanowires with few nanosheets located at their edges (Fig. S3a and S3b in the ESI†). EELS of the thin nanosheets confirms their mixed chemical composition of Cu and Se (Fig. S8a†). The interatomic distances measured from high resolution HAADF STEM images and the corresponding Fast Fourier Transform pattern (FFT) (Fig. 1e) are in agreement with the cubic structure (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) of Cu2−xSe.36
m) of Cu2−xSe.36
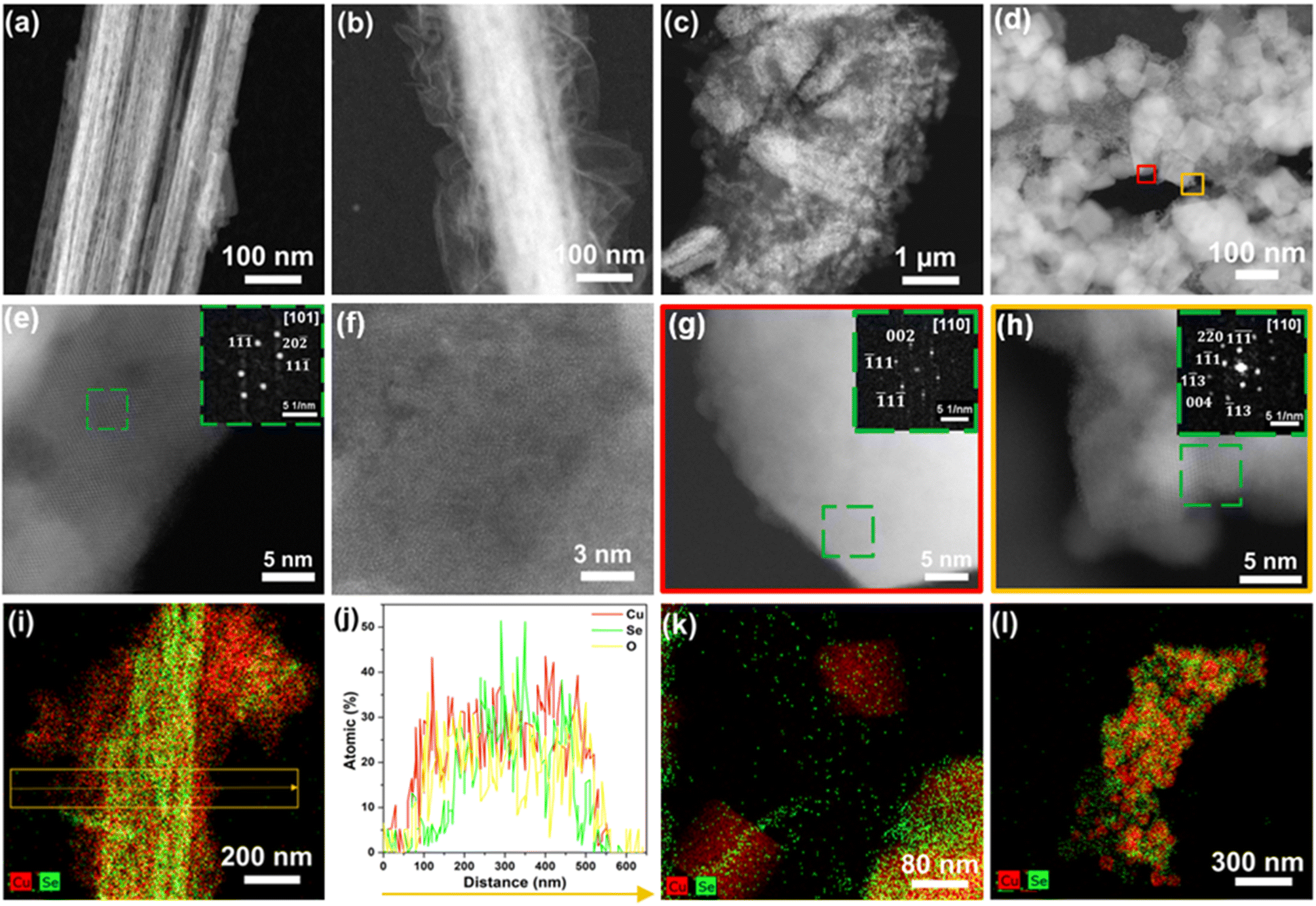 | ||
| Fig. 1 HAADF-STEM images of (a and e) AP-CuSe, (b and f) CA-CuSe, (c) EA-CuSe, (d, g and h) EA-CA-CuSe. FFT pattern insets in (e, g and h) show the zone axis and the crystalline planes of the crystal structures in the green dashed squares. EDS elemental maps of (i) CA-CuSe, (k) EA-CuSe, and (l) EA-CA-CuSe. (j) Line scan along the cross-section of the CA-CuSe nanowire in (i), which shows that Cu is preferentially present at the surface. | ||
The rough flocculent surface of CA-CuSe (Fig. 1b) consists of thin low-crystalline wrinkled sheets as highlighted by the high magnification HAADF STEM images (Fig. 1f). The thin outer surface is rich in copper and oxygen but depleted in selenium as observed in the EDS elemental mapping (Fig. 1i) and the atomic ratios in the line profile analysis along the nanowire cross section (Fig. 1j). These observations suggest that the hydrogen peroxide treatment has led to the formation of a thin superficial copper oxide layer.
Some of the basic nanowire morphology is retained after direct electrochemical reconstruction (EA-CuSe), as seen in HAADF STEM and SEM images (Fig. 1c, S3e, S3f, and S4†). However, the interior of the nanowire exhibits a significant degree of fragmentation into cube-like particles. High resolution HAADF STEM reveals its structural transformation into Cu2O nanocubes (space group: Pnm)37 and fragmented pieces of Cu2−xSe, as confirmed by the interatomic distances (Fig. S5†) and the EDS elemental distribution maps (Fig. 1k and S6†).
Upon combination of chemical and electro-activation, EA-CA-CuSe (Fig. 1d, S3g, and S3h†), Cu2−xSe exposed to the ambient displays a structure similar to that of EA-CuSe but with further fragmentation of the nanowires into single crystalline Cu2O nanocube aggregates, surrounded by small Cu2−xSe particles as shown in Fig. 1g and h and confirmed by the corresponding FFT and the EELS analysis (Fig. S8b†). We note that the contrast variations in HAADF-STEM images arise mainly from differences in atomic number and local thickness, rather than from unequal beam penetration. The observed morphological heterogeneity, including wire-like, sheet-like, and blocky features, consistently appearing across different areas of each sample reflects the diversity of Cu2−xSe structures and their process-dependent restructuring. We conclude that the original Cu2−xSe nanowire structure has post electro-activation (both EA-CuSe and EA-CA-CuSe) transformed into 50–90 nm single-crystal Cu2O nanocubes (cubic, Pnm) surrounded by Cu2−xSe nanoparticles. Analysis of the EDS maps in Fig. 1l and S7† give an atomic Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Se ratio close to 9:1 (Table S1 in the ESI†), supporting the transformation of CuSe into separate Cu- and Se-based phases, which are further characterized below.
:Se ratio close to 9:1 (Table S1 in the ESI†), supporting the transformation of CuSe into separate Cu- and Se-based phases, which are further characterized below.
The crystalline structure and composition of the bulk Cu2−xSe-based samples were characterized by X-ray diffraction (XRD), summarized in Fig. 2a. XRD patterns of AP-CuSe and CA-CuSe show diffraction peaks at 31.4°, 36.4°, 52.4°, 62.4°, 77.3° and 85.5°, which are ascribed to the (111), (200), (220), (311), (400) and (331) crystal planes of Cu1.71Se (ICDD: 04-024-2132), respectively and no impurity peaks are detected. The relatively large width of the diffraction peaks confirms that the CuSe nanowire bundles are made up of thin nanofibers with diameters of less than 10 nm, in line with previous work.38 Although CA-CuSe features the same diffraction pattern as AP-CuSe, all XRD peaks are notably shifted towards higher angles. Using Bragg's law, those shifts correspond to a lattice contraction of about 3%, which can be attributed to the migration of copper to the surface of the material, accompanied by the creation of Cu vacancies in the CuSe phase and the formation of a copper oxide phase. Similar shifts of the diffraction peaks of copper selenide, corresponding to lattice contraction, have been reported previously.39 The formation of the copper oxide phase is confirmed by the emergence of a peak at 45°, associated with the (111) plane of CuO (ICDD: 04-006-4186). After electro-activation, all Cu2−xSe and CuO peaks disappeared, while both EA-CuSe and EA-CA-CuSe have narrow diffraction peaks at 49.8°, 73.1° and 88.6°, ascribed to the (200), (220), and (222) crystal planes of Cu2O (ICDD: 00-005-0667). The absence of the Cu2O (111) plane at 42.7° could indicate that crystal growth is restricted in this direction, with a preference for other growth orientations. Such suppression of the (111) plane and preferential growth along the (200) direction has been observed before in thin Cu2O films.40 The Cu2O grain size in the EA-CA-CuSe sample, calculated using Scherrer's equation, is 90 nm, which agrees with the size of the Cu2O cubic structures observed with STEM.
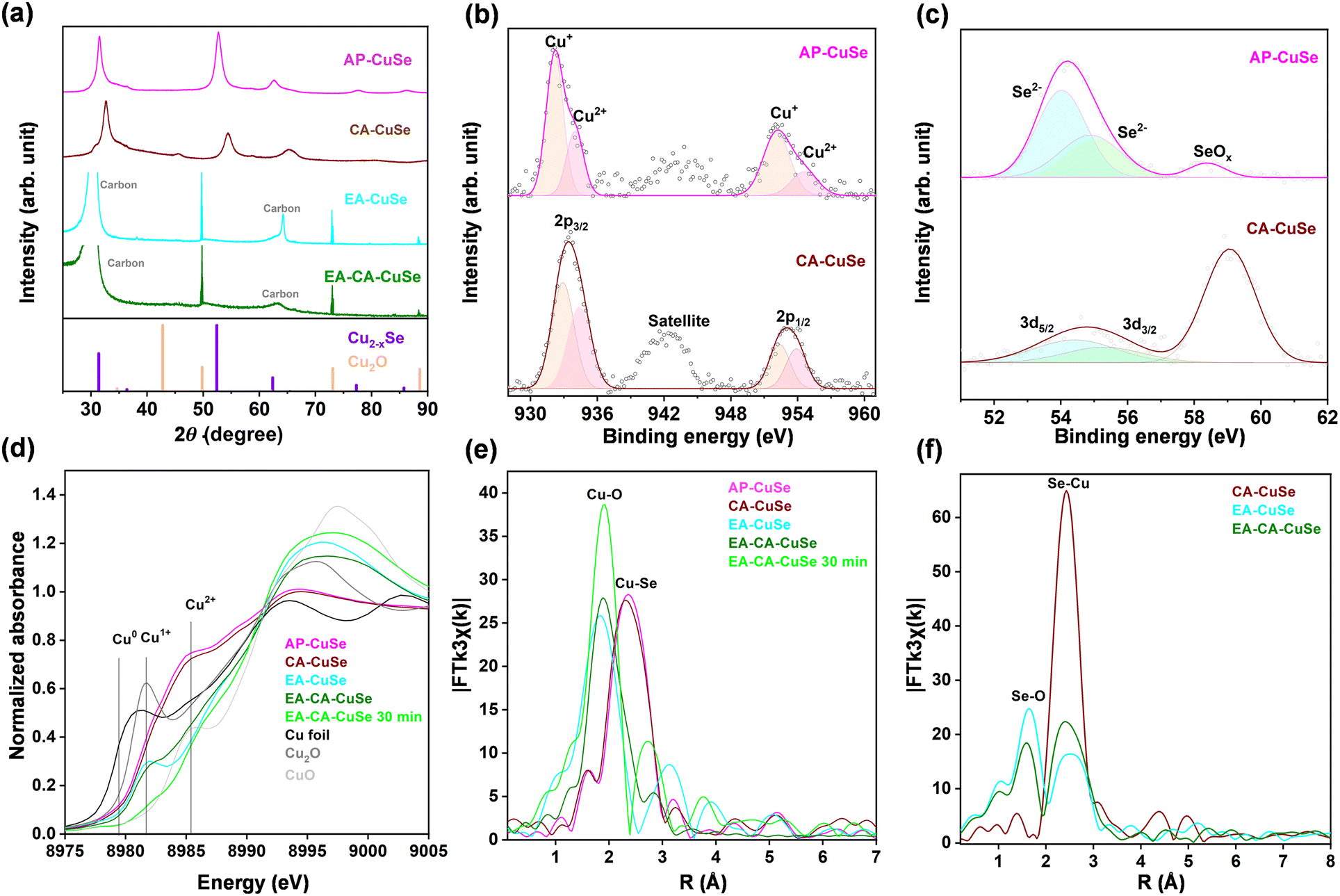 | ||
| Fig. 2 (a) XRD spectra of the different Cu2−xSe-based samples. XPS of AP-CuSe and CA-CuSe samples at the (b) Cu 2p and (c) Se 3d levels. (d) Cu K-edge XANES. Fourier Transforms of (e) Cu K-edge and (f) Se K-edge k3-weighted EXAFS spectra. | ||
The effect of H2O2 chemical activation on the surface was investigated by X-ray photoelectron spectroscopy (XPS), through comparison of Cu 2p (Fig. 2b), Se 3d (Fig. 2c), and O 1s (Fig. S9†) valence states of AP-CuSe and CA-CuSe. In the high-resolution Cu 2p spectra of the AP-CuSe, Cu 2p1/2 and 2p3/2 peaks at 952.2 and 932.1 eV are attributed to Cu+ and the shoulders at 954.3 and 934.1 eV to Cu2+. In CA-CuSe, all Cu 2p peaks shift to higher binding energies (maxima at 952.4 and 933.0 eV), indicating a general increase in the Cu oxidation state. After H2O2 treatment, the Cu2+ to Cu+ ratio increases from 0.38 to 0.90, in line with the presence of a CuO phase as observed in the CA-CuSe XRD spectra. The O 1s spectrum of the AP-CuSe nano wires has a Cu–O peak at 530.1 eV and that of CA-CuSe can be deconvoluted in two peaks at approximately 529.4 and 531.3 eV, which can be attributed to Cu–O and Cu–OH, respectively. The copper hydroxide likely originates from the interaction between the catalyst and the aqueous solution. The deconvoluted AP-CuSe Se 3d peaks at 54.0 and 55.0 eV correspond to Se2− 3d5/2 and 3d3/2. Another peak located at 58.3 eV is assigned to SeOx, which is likely caused by oxidation of the samples in air. In CA-CuSe, the shift of the Se 3d5/2 and 3d3/2 peaks (54.3 and 55.4 eV) to higher energies, implies an increase in the Se oxidation state. The SeOx peak is now at 59.0 eV and much more intense, which is due to the oxidizing H2O2 treatment.
The Raman spectrum of AP-CuSe (Fig. S10†) shows a peak at 259 cm−1, which can be assigned to a Se–Se vibrational mode in copper selenide.41,42 The absence of other peaks confirms the formation of a single Cu2−xSe phase. The CA-CuSe sample has an additional peak at 241 cm−1 that may be associated with intrinsic host lattice defects.43 These defects could arise from superficial oxygen doping in Cu2−xSe or the formation of SeO bonds.44 This further suggests that Se on the surface of the nanowires is oxidized following H2O2 treatment.
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra, measured on ambient-exposed samples at Cu and Se K-edges, allow to further investigate the electronic and structural properties of the CuSe catalysts before and after their activation. XANES spectra at Cu and Se K-edges are presented in Fig. 2d and Fig. S11,† respectively. XANES at Cu K-edge shows that AP-CuSe and CA-CuSe feature similar profiles. The significant structural and chemical transformation after electro-activation (EA-CuSe and EA-CA-CuSe), results in a shift of the edge position towards higher energy as well as the increase of the white line intensity. The characteristic profile of Cu2−xSe seen for AP-CuSe and CA-CuSe, transforms into profiles resembling a mixture of Cu2O and CuO after electro-activation. The pre-edge peak corresponding to the 1s → 4p transition in Cu2O that appears at approximately 8981.7 eV in EA-CuSe indicates that this sample contains a significant fraction of Cu2O, while EA-CA-CuSe mainly consists of Cu2+ oxide whose fraction further increases when the latter treatment was prolonged to 30 min. The absence of the pre-edge located at around 8985.4 eV corresponding to the 1s → 4p transition in CuO, suggests that amorphous CuO combined with a hydroxide-like phase has formed in the EA-CA-CuSe sample. This is in line with the Se K-edge XANES that also exhibits two very distinct profiles for CA-CuSe on the one hand and EA-CuSe and EA-CA-CuSe on the other hand. The blue shift in energy and the increase of the white-line feature both indicate a significant oxidation of the Se phase following the electro-activation. XANES at both edges confirms the electro-activated transformation of a significant fraction of the original copper selenide phase into copper and selenium oxides.
Fourier transforms (FTs) before phase correction of k3 weighted EXAFS at the Cu (Fig. 2e and S12†) and Se K-edges (Fig. 2f and S13) further confirm the drastic transformation of Cu2−xSe, by segregation of copper and selenium atoms after electro-activation that forms large fractions of separate oxide phases after the samples are exposed to the ambient. The main peak at around 2.3 Å in FT at Cu K-edges of AP-CuSe and CA-CuSe was fitted by 2.6 and 2.8 Se at 2.395 Å, corresponding to the Cu2−xSe phase, while the shoulder at 1.7 Å was fitted by contributions of 0.8 and 1.0 O at 1.92 and 1.93 Å, respectively, corresponding to a mixture of Cu2O, CuO and possibly Cu(OH)2 (Table S2†). Using the copper oxygen coordination in Cu2O (2 O at 1.85 Å) and CuO (4 O at 1.95 Å) as references, it is found that AP-CuSe and CA-CuSe consist of ca. 26% and 32% of oxide/hydroxide, respectively, while the rest of the samples remain in the Cu2−x Se phase. Se K-edge analysis (Table S3†) confirms that CA-CuSe primarily consists of Cu2−xSe, as indicated by two main FT peaks, which are fitted with 0.24 O at 1.66 Å and 5.1 Se at 2.41 Å, respectively. This composition includes a minor fraction, approximately 8% (0.24/3), of SeO2, in which Se is coordinated with three O atoms.
Electro-activation of the AP-CuSe and CA-CuSe samples further transforms the CuSe phase into pure Cu and Se oxides as confirmed by the fits of the Cu K-edge EXAFS, highlighting a sharp increase of the O shells coordination to 2.1 and 2.5 atoms in EA-CuSe and EA-CA-CuSe samples and up to 3.7 O when the electro-activation is prolonged. The concomitant reduction of the Se shell coordination down to 2.0 in EA-CuSe and EA-CA-CuSe and 0.6 after 30 min of EA-CA-CuSe electro-activation is consistent with the segregation of Cu2−xSe into separate Cu and Se oxides. EA-CuSe and EA-CA-CuSe have similar fractions of copper oxide/hydroxide; 62% and 68%, respectively, which increases up to 92% after prolonged electro-activation of EA-CA-CuSe. Remarkably, a significant fraction of 31% of Cu2O is still present in EA-CuSe, despite its exposure to the ambient. In the latter sample, the original Cu2−xSe cubic phase has been entirely transformed as even the tiny remaining part of copper selenide corresponds to the copper-poor CuSe hexagonal phase, characterized by shorter Cu–Se distances of 2.29 Å.38 These results are in line with the analysis of Se K-edge EXAFS that highlights the presence of ca. 50% of SeO2 (1.5–1.6 O at 1.66 Å), complemented by a mixture of Cu2−xSe (2.1–2.2 Se at 2.39–2.40 Å) and CuSe (1.0–0.9 Se at 2.19 Å) phases in EA-CuSe and EA-CA-CuSe.
The EXAFS results demonstrate, in good agreement with those obtained with the other characterization techniques, that the chemical and electrochemical treatments of Cu2−xSe nanowires produce highly distinctive materials through various surface reconstruction processes. Chemical activation of as-prepared samples induces the formation of a thin mostly amorphous copper oxide/hydroxide layer at the surface of the Cu2−x Se nanowires, while the electro-activation transforms the material for a large part into SeO2 (50% of Se) and copper oxide/hydroxide (62% of Cu) including up to 31% of Cu2O in the form of single-crystal Cu2O cubes (cubic, Pnm)32 with 50 to 90 nm sizes. These cubes are surrounded by smaller particulates consisting of the remainder of the original Cu2−xSe as well as a copper-poor CuSe phase. Combined CA and EA treatment further completes the copper/selenium segregation (mixture of 92% of CuOx and 8% Cu-poor CuSe) when the EA treatment is prolonged to 30 min.
Electrochemical CO2RR performance
The electrochemical CO2RR performance was investigated in an ionic liquid, namely 1-butyl-3-methylimidazolium tetrafluoroborate/acetonitrile/water electrolyte ([Bmim]PF4-CH3CN-H2O), which enhances CO2 solubility compared to aqueous solution. The different CuSe catalysts were dispersed in a Nafion-acetone solution and drop-cast onto Toray carbon paper (TGP-H-60), which served as the gas diffusion layer and working electrode (see SEM images in Fig. S14†). All electrochemical measurements were conducted in a two-compartment H-type cell separated by a Nafion 117 membrane.As reference, polycrystalline Cu foil was tested under identical conditions at −1.4 VRHE (Fig. S15†). The Cu foil reference exhibited a current density of around −2 mA cm−2 and produced 89.3% H2, 8.1% HCOOH, and only 1.5% CO, indicating a modest CO2 reduction activity in the ionic liquid electrolyte. In contrast, AP-CuSe treated for 2 minutes with H2O2 exhibited a relatively high current density in the Linear Sweep Voltammetry (LSV) measurements after achieving material stabilization (see Fig. S16 in the ESI†), making it a suitable candidate for further study. Current densities of the as-prepared and activated CuSe materials ranged from 1 to 36 mA cm−2 depending on the applied potentials. Chronoamperometry measurements of the four samples at different potentials shows that the activity of CA-CuSe decreases over time (Fig. S17†), which may be due to a rapid transformation of this catalyst under CO2RR conditions. It is worth noting that at potentials of −1.2 VRHE and lower, the current densities of EA-CA-CuSe and EA-CuSe samples increase over time, which may be attributed to a higher conductivity caused by the formation of metallic Cu at the surface.
The current density increases with the applied potential and is peaking for EA-CA-CuSe (36.7 mA cm−2 at −1.5 VRHE), which is 9.7 and 4.3 times that of AP-CuSe (3.8 mA cm−2) and CA-CuSe (8.4 mA cm−2), respectively. EA-CA-CuSe exhibits also the highest partial current density for CO (Fig. 3a). Fig. 3b shows the FE of CH3OH over the four catalysts, while Fig. 3c gives the FE of all products. AP-CuSe exhibits the highest selectivity towards methanol production, with values exceeding 22% in the −1.3 to −1.5 VRHE potential range, while EA-CA-CuSe and CA-CuSe reach a maximum for methanol at −1.2 VRHE, with values of 9.1% and 11.1%, respectively. The higher methanol selectivity of AP-CuSe indicates that untreated Cu2−xSe nanowires favour methanol production. On the other hand, EA-CA-CuSe yields the highest CO selectivity in a wide potential window with a maximum FECO of 82.1% ± 2.0% at −1.4 VRHE.
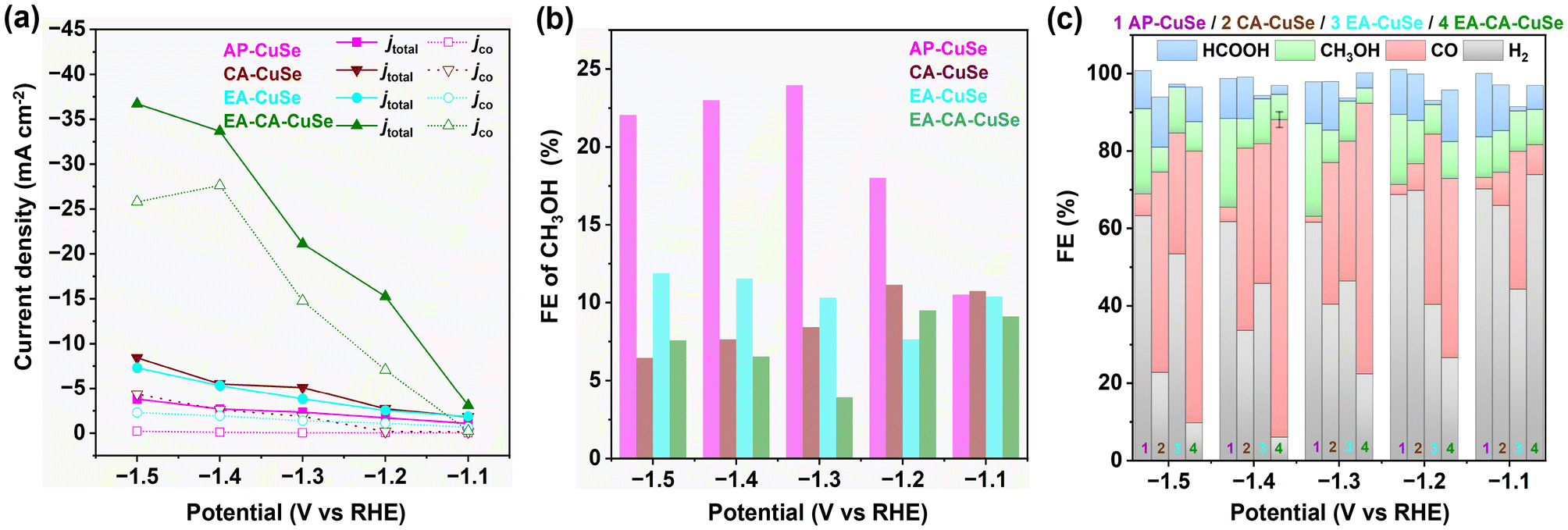 | ||
| Fig. 3 Variation with the applied potential of (a) total current density and CO partial current density, (b) FE of CH3OH and (c) FE of all products for AP-CuSe, CA-CuSe, EA-CuSe, and EA-CA-CuSe. The error bar on FECO for EA-CA-CuSe at −1.4 VRHE is the standard deviation of triplicate measurements. For other samples and bias voltages only single measurements were performed. | ||
Structure–reactivity relationship
To gain insight into the Cu2−xSe reconstruction mechanism and the reaction pathways under CO2RR conditions, quasi in situ XRD patterns of the EA-CA pre-catalyst were measured immediately after electro-activation (Fig. 4a). The broad shoulder visible around 50.6° corresponds to the (111) plane of metallic copper, while the peak at 49.5° can be assigned to the (200) plane of Cu2O (Cu1+), confirming the reduction and segregation of Cu2+ from the original Cu2−xSe phase into Se-free reduced Cu phases. This provides strong evidence for the existence of Cu nanoparticles in the reduced phase. According to Scherrer's equation, the Cu2O nanocrystallites formed after electrochemical activation exhibited an average size of approximately 14.3 ± 0.3 nm, representing a 20% increase compared to the metallic copper nanoparticles (11.9 ± 0.5 nm). This measured size increase corresponds well with theoretical value of 18%, indicating that Cu2O likely forms through the oxidation of these metallic Cu nanoparticles.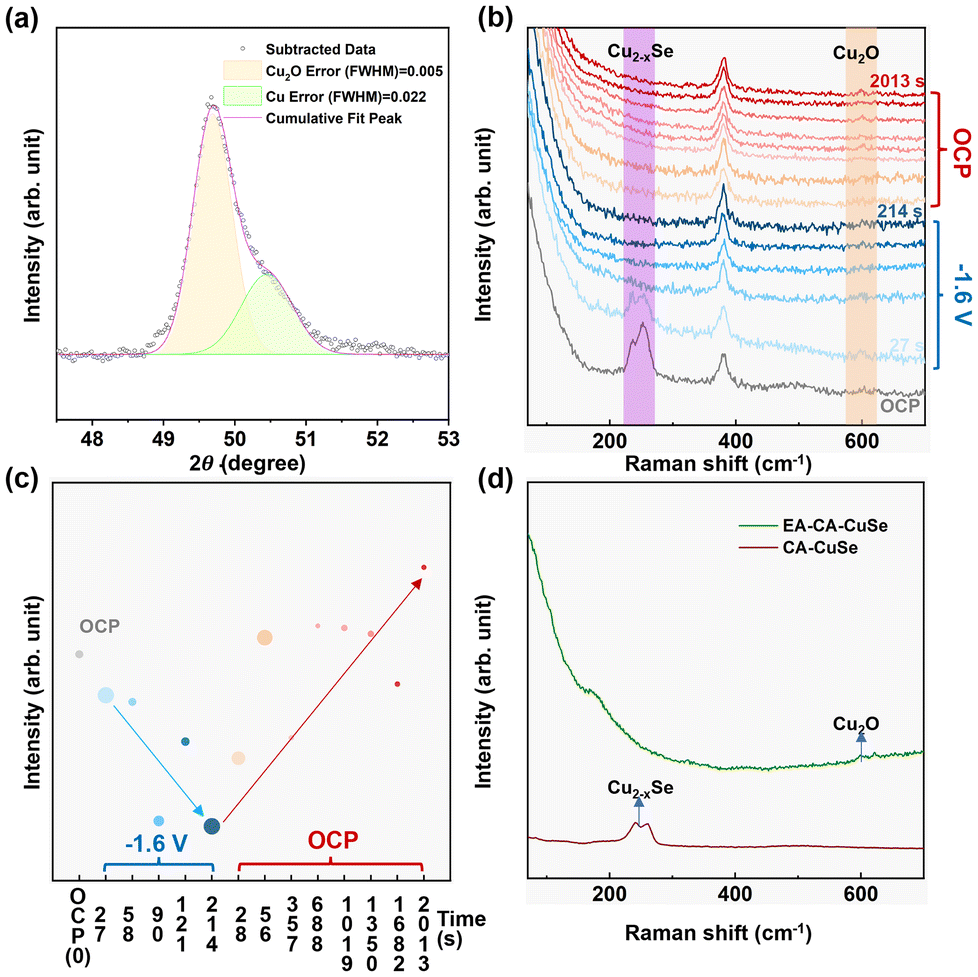 | ||
| Fig. 4 EA-CA-CuSe (a) Quasi in situ XRD spectra. (b) In situ Raman spectra and (c) intensity of the Raman peak at 600 cm−1 in (b) as a function of the time during which −1.6 VRHE is applied (blue symbols) or at OCP (red symbols). The size of the symbol represents the error bar. (d) Ex situ Raman spectra of CA-CuSe and EA-CA-CuSe. | ||
The presence of Cu metal nanocrystallites under electroactivation conditions is further supported by operando and ex situ Raman spectra recorded for EA-CA-CuSe under −1.6 VRHE (Fig. 4b and d) that shows a drastic reduction of the Cu–Se bond signal with time, indicating degradation of the Cu2−xSe phase and the build-up of pure metallic copper. The metallic phase is re-oxidized to Cu1+ as soon as the potential is returned to open circuit potential (OCP) as indicated by the increase of the Cu2O signal in line with the results of the quasi in situ XRD analysis. Under the very negative potential, Cu is thus reduced and segregates from the original Cu2−xSe phase into metallic copper nanoparticles, while Se-rich copper selenide and pure Se oxide phases are formed. More specifically, the intensity of the Raman peak of CA-CuSe decreases rapidly once −1.6 VRHE is applied and disappears after 1 minute (Fig. 4c).
XRD and STEM analyses conducted after exposure to ambient conditions reveal that the Cu2O phase in the electro-activated material appears as large (50–90 nm) single-crystal cubes in EA and EA-CA-CuSe. This suggests that their formation results from the migration and agglomeration of numerous smaller nanoparticles, specifically 11.9 nm metallic Cu and/or 14.3 nm Cu2O. Such agglomeration has been reported earlier in similar nanoparticle systems.45,46 Additionally, surface oxidation of remaining metallic Cu could further contribute to gradual particle growth, even at OCP, driven by the residual oxidative species in solution.47
A recent operando STEM work hypothesized that catalytically active metallic copper nanograins form through the agglomeration of smaller Cu metal nanoparticles upon negative potentials.2 The grain boundaries between the Cu nanoparticles serve as active sites for the formation of C1 and C2 products from CO2RR. The porous structure and high reactivity of these nanograins would explain their fast transformation into Cu2O cubes upon exposure to the ambient. Since the transformation of nanosized Cu into larger ordered Cu2O nanocubes following exposure to ambient conditions is structurally irreversible, we hypothesize that transient Cu metal nanograins formed under CO2RR activation—due to the segregation of Cu from the Cu2−xSe phase—serve as the active sites responsible for the enhanced methanol selectivity in the AP-CuSe catalyst. Moreover, in this study 11.9 nm Cu metal nanoparticles are expected to produce mostly C1 products,2 which is in line with the current results.
According to the characterization results from quasi-XRD and operando Raman spectroscopy, the high CO selectivity of EA-CA-CuSe may originate from the reduction of the large Cu2O nanocubes formed by electro-activation.
Under CO2RR conditions, these nanocubes form metallic surfaces that favor CO production. This observation aligns with documented research,48 which describes the Cu2O nanocubes undergo a multi-step reconstruction, developing rough surfaces, transitioning into hollow shells, and aggregating into mixed Cu/Cu oxide nanoparticles. It has been shown that Cu2O nanocubes with sizes around 80 nm—comparable to the sizes observed in this study—achieve higher CO selectivity than larger particles, such as 170 nm and 390 nm cubes, supporting the importance of the particle size in optimizing CO formation.49
Upon treatment and exposure to the ambient, all samples contain a significant amount of copper oxide, highlighting Cu2−xSe degradation and the various degrees of segregation of Cu and Se phases into pure CuOx and SeO2 with a remaining small Cu2−xSe fraction.
AP-CuSe that comprises 13% Cu2O and 13% CuO and 74% of Cu2−xSe, exhibits methanol and CO selectivities of 23% and 4%, respectively at −1.4 VRHE. Upon oxidation with H2O2, CA-CuSe is obtained, which contains 14% Cu2O, 18% CuO and 68% of Cu2−xSe demonstrates a significant shift in product selectivity towards CO (FECO of 47%). The electrochemically activated EA-CuSe, with a much higher oxide content (68% CuxO), yields a CO selectivity of 36% at −1.4 VRHE. Remarkably, the fourth material, EA-CA-CuSe, which underwent both peroxide oxidation and electro-activation, increasing the CuxO content to 92%, achieves the highest CO selectivity of 82% at the same potential. Across all derivatives, the selectivity for methanol decreased after any activation treatment of the original Cu2−xSe, accompanied by a shift in catalytic behaviour favouring CO production.
Density functional theory calculations
To enhance our insight in the CO2RR selectivity of the different Cu2−xSe-derived catalyst materials, density functional theory calculations were performed on two model systems: Cu2Se (220) and Cu (110), whose starting structures were based on ref. 11 and 50, respectively. The selection of the Cu (110) surface is driven by its previously reported high activity in CO2 reduction51 and its efficient C–O splitting capabilities,52 which are relevant for CO formation. Both the Cu (110) and the Cu2Se (220) structures were reoptimized by keeping the positions of the bottom atomic layer fixed, a commonly used method for slab models. Reaction free energies were calculated at −1 V and without voltage applied. More details of the computational methodology are provided in the ESI.† The Cu2Se (220) slab is a proxy for the AP-CuSe sample because of its intense (220) diffraction peak (Fig. 2a) and the Cu (110) slab is a proxy for the reduced electro-activated catalysts (EA-CuSe and EA-CA-CuSe) under reaction conditions. Structural relaxation of the models with a fixed bottom layer indicated that Cu (110) underwent relatively small geometrical changes, while the surface atoms of Cu2Se (220) significantly reorganized. This, combined with the known preference of the material for non-stoichiometric compositions, is characteristic of its structural flexibility that may lead to significant instability under reaction.The computed reaction paths are consistent with those presented in literature.11,50 The CO2 reduction pathways towards CO and methanol under an applied bias of −1 VRHE are presented in Fig. 5. In the applied computational hydrogen electrode model,53 the effect of the cell voltage is taken into account after the quantum chemical computations. It downshifts the free energy, proportionally to the number of electrons needed to reach each intermediate. Methanol, which is the thermodynamically favoured product for both catalysts without applied cell voltage (Fig. S18 and S19†), is even more favoured at −1 VRHE. CO desorption is endergonic, but CO hydrogenation is exergonic due to the applied voltage, making methanol formation thermodynamically more favoured. However, the selectivity towards different products on each catalyst is influenced strongly by the reaction kinetics. It has been shown that the CO binding energy is an important descriptor for the product selectivity.54
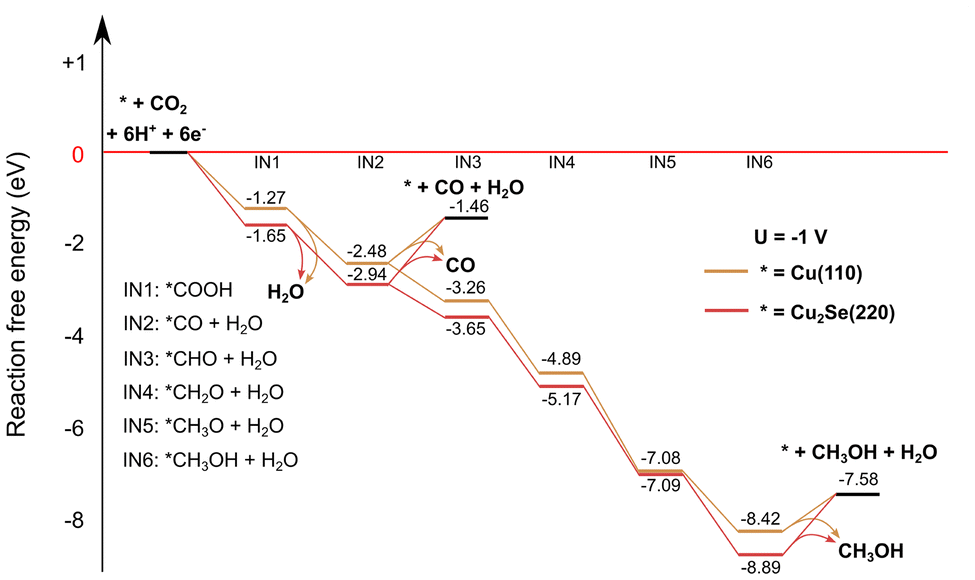 | ||
| Fig. 5 Computed reaction CO2RR pathways on Cu2Se (220) and Cu (110) slab models to CO and methanol with an applied cell voltage of −1 V. | ||
A notable difference between the computed reaction paths of the two model catalysts is the free energy change for CO desorption. On the Cu (110) surface, the desorption free energy is 1.0 eV, compared 1.5 eV for the Cu2Se (220) surface (in computational hydrogen electrode the desorption free energy is independent of the applied bias). This can be attributed to the presence of less stable intermediates (except IN5) on the pure copper surface. Since CO desorption is irreversible, the easier desorption observed on the Cu surface implies a higher CO formation rate, consistent with the experimental results on both EA-Cu and EA-CA-CuSe.
However, it is important to note that oxide-derived copper possesses a highly complex structure. In fact, the catalysts studied here are chemically complex systems, and the simple models do not explain the non-monotonous potential-dependence of the product ratios shown in Fig. 3. Nevertheless, these simplified models allow for a qualitative assessment of the impact of distinct local chemical environments on CO binding and product selectivity. In this sense, the DFT results support the experimental observation that CO formation is favored on Cu-rich surfaces. Detailed reaction mechanism studies that lead to different products are currently the subject of intensive research. On the other hand, we noted that the detailed structure of our model system has a minimal impact on the energetics of the reaction, suggesting that the primary factor influencing the reaction is the local chemical environment at the reaction site, and the main factor to qualitatively understand the reactivity is the difference of the CO binding free-energies on the Cu (110) and Cu2Se (220).
Conclusions
We showed that Cu2−xSe, which is undergoing major restructuring under CO2RR conditions, behaves rather as a pre-catalyst and developed a facile reconstruction strategy to transform it into active phases with tailored selectivity. Without H2O2 pre-treatment, direct in situ electroreduction of Cu2−xSe nanowires produces a small amount of metallic Cu nanoparticles, which reoxidize into Cu2O nanocubes upon returning to open circuit, yielding a mixture of Cu2−xSe nanowires and Cu2O cubes. In contrast, H2O2 pretreatment causes significant surface oxidation and nanowire fragmentation, destabilizing the as prepared material and resulting in the creation of metallic Cu nanoparticles during electroreduction. Those are largely converted to Cu2O nanocubes after reaction. The resulting catalyst material consists mainly of Cu2O nanocubes and residual Cu2−xSe nanoparticles, while the nanowire morphology is nearly lost. In situ and ex situ characterizations confirm that dynamic restructuring of Cu2−xSe involves Cu segregation from the selenide matrix.A clear structure–selectivity relationship of copper-based catalysts was found. The as-prepared Cu2−xSe (AP-CuSe), which dynamically transforms into Cu nanograins during CO2RR, exhibits 23% methanol selectivity at −1.4 VRHE. In contrast, the H2O2-etched and electrochemically activated catalyst (EA-CA-CuSe), initially composed of Cu2O nanocubes, achieves 82% CO selectivity at a partial current density of 27.7 mA cm−2. These findings not only corroborate previous observations on structural evolution in pure Cu catalysts but also extend them to Cu–Se systems, offering deeper insights into active phase formation.
Our results demonstrate that controlled pre-treatment and activation of Cu2−xSe pre-catalysts can steer the formation of desirable active sites, thereby guiding the rational design of efficient and selective copper-based electrocatalysts for CO2 electroreduction and potentially other electrochemical transformations.
Author contributions
W. Hu prepared the samples and performed the electrochemical experiments, D. Balalta and T. Altantzis carried out the electron microscopy, J. Song and W. Hu did the XRD, Z. Chen and W. Hu performed the Raman characterization, I. Abbas, F. D'Acapito and D. Grandjean carried out the XAFS measurements and analysis. W. Hu and D. Balalta analyzed the STEM data. B. Barhács, M. Guba and T. Höltzl conducted the calculations. T. Höltzl, S. Bals, D. Grandjean, D. Pant and E. Janssens secured funding. D. Grandjean, D. Pant and E. Janssens directed the research project. W. Hu and D. Grandjean prepared the first version of the manuscript. All authors discussed the results and participated in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data of this article are shown in the Figures and the ESI.† The data of the figures presenting experimental results are available on the KU Leuven's institutional research data repository at https://rdr.kuleuven.be/dataverse/rdr with https://doi.org/10.48804/CYCRZD. The cartesian coordinates of all calculated reaction intermediates can be found on Zenodo: https://doi.org/10.5281/zenodo.12745044.Acknowledgements
This project has received funding from the European Union's Horizon 2020 MSCA-ITN programme under grant agreement no. 955650 (CATCHY) and by the Moonshot program of Catalisti and VLAIO (Flanders Innovation & Entrepreneurship) under grant number HBC.2021.0586 (CLUE). T. H. is grateful for the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (grant number BO/00642/21/7). B.B. is grateful to the University Research Scholarship Programme – Cooperative Doctoral Programme of the Hungarian National Research, Development and Innovation Office (grant number: EKÖP_KDP-24-1-BME-8) M.G. is grateful to the Cooperative Doctoral Programme for Doctoral Scholarships of the Hungarian National Research, Development and Innovation Office (KDP-23, grant number: C2273140). The authors are grateful to Zviadi Zarkua for his help with the XPS measurements and thank the ESRF for providing beamtime (CH-6247) and the staff of LISA-BM08 for their assistance.References
- B. Seger, M. Robert and F. Jiao, Best practices for electrochemical reduction of carbon dioxide, Nat. Sustainability, 2023, 6, 236–238 CrossRef.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P. C. Chen, H. Wang, C. J. Pollock, X. Huang, Y. T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Operando studies reveal active Cu nanograins for CO2 electroreduction, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- Y. Wang, H. Shen, K. J. T. Livi, D. Raciti, H. Zong, J. Gregg, M. Onadeko, Y. Wan, A. Watson and C. Wang, Copper Nanocubes for CO2 Reduction in Gas Diffusion Electrodes, Nano Lett., 2019, 19, 8461–8468 CrossRef CAS.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard III and A. T Bell, Effects of Surface Roughness on the Electrochemical Reduction of CO2 over Cu, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Hydroxyl radicals dominate reoxidation of oxide-derived Cu in electrochemical CO2 reduction, Nat. Commun., 2022, 13, 3694 CrossRef CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, Scalable Production of Efficient Single-Atom Copper Decorated Carbon Membranes for CO2 Electroreduction to Methanol, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Synergistic geometric and electronic effects for electrochemical reduction of carbon dioxide using gold–copper bimetallic nanoparticles, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- X. Zhang, X. Sun, S.-X. Guo, A. M. Bond and J. Zhang, Formation of lattice-dislocated bismuth nanowires on copper foam for enhanced electrocatalytic CO2 reduction at low overpotential, Energy Environ. Sci., 2019, 12, 1334–1340 RSC.
- M. Wang, H. Chen, M. Wang, J. Wang, Y. Tuo, W. Li, S. Zhou, L. Kong, G. Liu, L. Jiang and G. Wang, Tuning C1/C2 Selectivity of CO2 Electrochemical Reduction over in–Situ Evolved CuO/SnO2 Heterostructure, Angew. Chem., Int. Ed., 2023, 62, e202306456 CrossRef CAS PubMed.
- W. Hu, D. Grandjean, J. Vaes, D. Pant and E. Janssens, Recent advances in copper chalcogenides for CO2 electroreduction, Phys. Chem. Chem. Phys., 2023, 25, 30785 RSC.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Selective electroreduction of carbon dioxide to methanol on copper selenide nanocatalysts, Nat. Commun., 2019, 10, 677 CrossRef CAS.
- L. Ding, N. Zhu, Y. Hu, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, Over 70% Faradaic Efficiency for CO2 Electroreduction to Ethanol Enabled by K Dopant-Tuned Cu Sites-Intermediates Interaction, Angew. Chem., Int. Ed., 2022, 61, e202209268 CrossRef CAS PubMed.
- W. Sun, P. Wang, Y. Jiang, Z. Jiang, R. Long, Z. Chen, P. Song, T. Sheng, Z. Wu and Y. Xiong, V–Doped Cu2Se Hierarchical Nanotubes Enabling Flow–Cell CO2 Electroreduction to Ethanol with High Efficiency and Selectivity, Adv. Mater., 2022, 34, e2207691 CrossRef.
- H. Wang, X. Bi, Y. Yan, Y. Zhao, Z. Yang, H. Ning and M. Wu, Efficient Electrocatalytic Reduction of CO2 to Ethanol Enhanced by Spacing Effect of Cu-Cu in Cu2-xSe Nanosheets, Adv. Funct. Mater., 2023, 33, 2214946 CrossRef CAS.
- S. Li, J. Yu, S. Zhang, W. Qiu, X. Tang, Z. Lin, R. Cai, Y. Fang, S. Yang and X. Cai, Operando Reconstruction of Porous Carbon Supported Copper Selenide Promotes the C2 Production from CO2RR, Adv. Funct. Mater., 2023, 34, 2311989 CrossRef.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, The role of in situ generated morphological motifs and Cu(I) species in C2+ product selectivity during CO2 pulsed electroreduction, Nat. Energy, 2020, 5, 317–325 CrossRef.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Engineering Cu surfaces for the electrocatalytic conversion of CO2: Controlling selectivity toward oxygenates and hydrocarbons, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. M. Pettersson and A. Nilsson, Subsurface oxygen in oxide-derived copper electrocatalysts for carbon dioxide reduction, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper, Nature, 2014, 508, 504–507 CrossRef CAS.
- K. W. Kimura, R. Casebolt, J. Cimada DaSilva, E. Kauffman, J. Kim, T. A. Dunbar, C. J. Pollock, J. Suntivich and T. Hanrath, Selective Electrochemical CO2 Reduction during Pulsed Potential Stems from Dynamic Interface, ACS Catal., 2020, 10, 8632–8639 CrossRef CAS.
- C.-J. Chang, S.-C. Lin, H.-C. Chen, J. Wang, K. J. Zheng, Y. Zhu and H. M. Chen, Dynamic Reoxidation/Reduction-Driven Atomic Interdiffusion for Highly Selective CO2 Reduction toward Methane, J. Am. Chem. Soc., 2020, 142, 12119–12132 CrossRef CAS.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Copper adparticle enabled selective electrosynthesis of n-propanol, Nat. Commun., 2018, 9, 4614 CrossRef PubMed.
- S. Yang, H. An, S. Arnouts, H. Wang, X. Yu, J. de Ruiter, S. Bals, T. Altantzis, B. M. Weckhuysen and W. van der Stam, Halide-guided active site exposure in bismuth electrocatalysts for selective CO2 conversion into formic acid, Nat. Catal., 2023, 6, 796–806 CrossRef CAS.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- Y. Yuan, Q. Wang, Y. Qiao, X. Chen, Z. Yang, W. Lai, T. Chen, G. Zhang, H. Duan, M. Liu and H. Huang, In Situ Structural Reconstruction to Generate the Active Sites for CO2 Electroreduction on Bismuth Ultrathin Nanosheets, Adv. Energy Mater., 2022, 12, 2200970 CrossRef CAS.
- Y. Jiang, X. Wang, D. Duan, C. He, J. Ma, W. Zhang, H. Liu, R. Long, Z. Li, T. Kong, X. J. Loh, L. Song, E. Ye and Y. Xiong, Structural Reconstruction of Cu2O Superparticles toward Electrocatalytic CO2 Reduction with High C2+ Products Selectivity, Adv. Sci., 2022, 9, 2105292 CrossRef CAS.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Electrochemical CO2 reduction to ethylene by ultrathin CuO nanoplate arrays, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- B. Beverskog and I. Puigdomenech, Revised Pourbaix diagrams for copper at 25 to 300 °C, J. Electrochem. Soc., 1997, 144, 3476 CrossRef CAS.
- S. Mu, H. Lu, Q. Wu, L. Li, R. Zhao, C. Long and C. Cui, Hydroxyl radicals dominate reoxidation of oxide-derived Cu in electrochemical CO2 reduction, Nat. Commun., 2022, 13, 3694 CrossRef CAS PubMed.
- A. Yoon, J. Poon, P. Grosse, S. W. Chee and B. Roldan Cuenya, Iodide-mediated Cu catalyst restructuring during CO2 electroreduction, J. Mater. Chem. A, 2022, 10, 14041–14050 RSC.
- R. Xiong, H. Xu, H. Zhu, Z. Zhang and G. Li, Recent progress in Cu-based electrocatalysts for CO2 reduction, Chem. Eng. J., 2025, 505, 159210 CrossRef CAS.
- X. Li and X. Li, Promotion of Cu-Based Catalyst by Supported Ionic Liquid for Efficient Acetylene Hydrochlorination, Mol. Catal., 2025, 584, 115262 Search PubMed.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Atomic-scale surface restructuring of copper electrodes under CO2 electroreduction conditions, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- Y. Zhu, H. Chen, C. Hsu, T. Lin, C. Chang, S. Chang, L. Tsai and H. Chen, Operando Unraveling of the Structural and Chemical Stability of P-Substituted CoSe2 Electrocatalysts toward Hydrogen and Oxygen Evolution Reactions in Alkaline Electrolyte, ACS Energy Lett., 2017, 4, 987–994 CrossRef.
- J. Xu, W. Zhang, Z. Yang, S. Ding, C. Zeng, L. Chen, Q. Wang and S. Yang, Large - Scale Synthesis of Long Crystalline Cu2-xSe Nanowire Bundles by Water–Evaporation–Induced Self–Assembly and Their Application in Gas Sensing, Adv. Funct. Mater., 2009, 19, 1759–1766 CrossRef CAS.
- S. A. Danilkin, A. N. Skomorokhov, A. Hoser, H. Fuess, V. Rajevac and N. N. Bickulova, Crystal structure and lattice dynamics of superionic copper selenide Cu2− δSe, J. Alloys Compd., 2003, 361, 57–61 CrossRef CAS.
- M. C. Neuburger, Präzisionsmessung der Gitterkonstante von Cuprooxyd Cu2O, Z. Phys., 1931, 67, 845–850 CrossRef CAS.
- H. Peng, J. Ren, Y. Wang, Y. Xiong, Q. Wang, Q. Li, X. Zhao, L. Zhan, L. Zheng, Y. Tang and Y. Lei, One-stone, two birds: Alloying effect and surface defects induced by Pt on Cu2− xSe nanowires to boost C-C bond cleavage for electrocatalytic ethanol oxidation, Nano Energy, 2021, 88, 106307 CrossRef CAS.
- Y.-X. Zhang, T.-Y. Yang, Z.-H. Ge and J. Feng, High-performance copper selenide nanocomposites for power generation, J. Eur. Ceram. Soc., 2023, 43, 5255–5262 CrossRef CAS.
- T. H. Yin, B. J. Liu, Y. W. Lin, Y. S. Li, C. W. Lai, Y. P. Lan, C. Choi, H. C. Chang and Y. M. Choi, Electrodeposition of Copper Oxides as Cost-Effective Heterojunction Photoelectrode Materials for Solar Water Splitting, Coatings, 2022, 12, 1839 CrossRef CAS.
- S. D. Sharma, K. Bayikadi, S. Raman and S. Neeleshwar, Structural, morphological and thermoelectric properties of self-decorated copper selenide nanosheets synthesized at room temperature, Curr. Appl. Phys., 2022, 40, 74–82 CrossRef.
- W. Lee, N. Myung, K. Rajeshwar and C.-W. Lee, Electrodeposition of Cu2Se Semiconductor Thin Film on Se-Modified Polycrystalline Au Electrode, J. Electrochem. Sci. Technol., 2013, 4, 140–145 CrossRef CAS.
- A. Abdalla, S. Bereznev, N. Spalatu, O. Volobujeva, N. Sleptsuk and M. Danilson, Pulsed laser deposition of Zn (O, Se) layers in nitrogen background pressure, Sci. Rep., 2019, 9, 17443 CrossRef PubMed.
- G. D. Brabson, L. Andrews and C. J. Marsden, Reactions of Selenium and Oxygen. Matrix Infrared Spectra and Density Functional Calculations of Novel SexOy Molecules, J. Phys. Chem., 1996, 100, 16487–16494 CrossRef CAS.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. Luca De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction, Nat. Commun., 2018, 9, 3117 CrossRef.
- M. Bernal, A. Bagger, F. Scholten, I. Sinev, A. Bergmann, M. Ahmadi, J. Rossmeisl and B. Roldan Cuenya, CO2 electroreduction on copper-cobalt nanoparticles: Size and composition effect, Nano Energy, 2018, 53, 27–36 CrossRef CAS.
- A. Yoon, J. Poon, P. Grosse, S. W. Chee and B. Roldan Cuenya, Iodide-mediated Cu catalyst restructuring during CO2 electroreduction, J. Mater. Chem. A, 2022, 10, 14041–14050 RSC.
- Q. Ren, N. Zhang, Z. Dong, L. Zhang, X. Chen and L. Luo, Structural evolution of Cu2O nanocube electrocatalysts for the CO2 reduction reaction, Nano Energy, 2023, 106, 108080 CrossRef CAS.
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. Roldan Cuenya, Dynamic transformation of cubic copper catalysts during CO2 electroreduction and its impact on catalytic selectivity, Nat. Commun., 2021, 12, 6736 CrossRef CAS.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Structure effects on the energetics of the electrochemical reduction of CO2 by copper surfaces, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- O. J. Wahab, M. Kang, E. Daviddi, M. Walker and P. R. Unwin, Screening Surface Structure–Electrochemical Activity Relationships of Copper Electrodes under CO2 Electroreduction Conditions, ACS Catal., 2022, 12, 6578–6588 CrossRef CAS.
- T. Yang, T. Gu, Y. Han, W. Wang, Y. Yu, Y. Zang, H. Zhang, B. Mao, Y. Li, B. Yang and Z. Liu, Surface Orientation and Pressure Dependence of CO2 Activation on Cu Surfaces, J. Phys. Chem. C, 2020, 124, 27511–27518 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. Hussain, H. Jónsson and E. Skúlason, Calculations of Product Selectivity in Electrochemical CO2 Reduction, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr02511g |
| This journal is © The Royal Society of Chemistry 2025 |